Back to Journals » ImmunoTargets and Therapy » Volume 5
Toll-like receptors in the pathogenesis of autoimmune diseases: recent and emerging translational developments
Authors Duffy L, O'Reilly S
Received 27 April 2016
Accepted for publication 21 June 2016
Published 22 August 2016 Volume 2016:5 Pages 69—80
DOI https://doi.org/10.2147/ITT.S89795
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Laura Duffy, Steven C O’Reilly
Immunology and Cell Biology Group, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, UK
Abstract: Autoinflammatory diseases are defined as the loss of self-tolerance in which an inflammatory response to self-antigens occurs, which are a significant global burden. Toll-like receptors are key pattern recognition receptors, which integrate signals leading to the activation of transcription factors and ultimately proinflammatory cytokines. Recently, it has become apparent that these are at the nexus of autoinflammatory diseases making them viable and attractive drug targets. The aim of this review was to evaluate the role of innate immunity in autoinflammatory conditions alongside the role of negative regulation while suggesting possible therapeutic targets.
Keywords: autoimmunity, toll-like receptors, danger signals, arthritis
Introduction
The innate immune system serves as the first line of defense in tackling host invasion, recognizing highly conserved pathogenic-associated molecular patterns (PAMPs) found on invading organisms. Subsequent signaling allows the host to mount an immune response without memory. The innate immune system uses a diverse set of pattern recognition receptors (PRRs) to sense the intra- and extracellular environment for pathogens.1 Toll-like receptors (TLRs) are PRRs that play a key role in the recognition of PAMPs, in the initiation of the hosts’ immune response, and interestingly, in neurological development2 and are conserved in species ranging from worms to mammals, denoting their importance.3 The basic structure of mammalian TLRs consists of a leucine-rich repeat extracellular domain, which creates a horseshoe-like solenoid capable of interacting with ligands, and the cytoplasmic toll/interleukin (IL)-1 receptor domain, associated with the TLR signaling cascade, culminating in nuclear factor kappa B (NF-kB) translocation.4,5 Along with NF-kB, TLRs also activate activator protein 1 (AP-1) and interferon regulatory factors (IRFs), such as IRF3 and IRF7; both of these cascades lead to the production of cytokines and pro- and antiinflammatory mediators, which are critical in mounting the host response (Figure 1). There are ten known TLRs in humans, residing on the cell surface to interact with pathogenic ligands or intracellularly in endosomes to interact with pathogenic nucleic acids (Figure 1). TLRs also respond to damage-associated molecular patterns (DAMPs) that are endogenous ligands released at the site of tissue damage (Table 1).
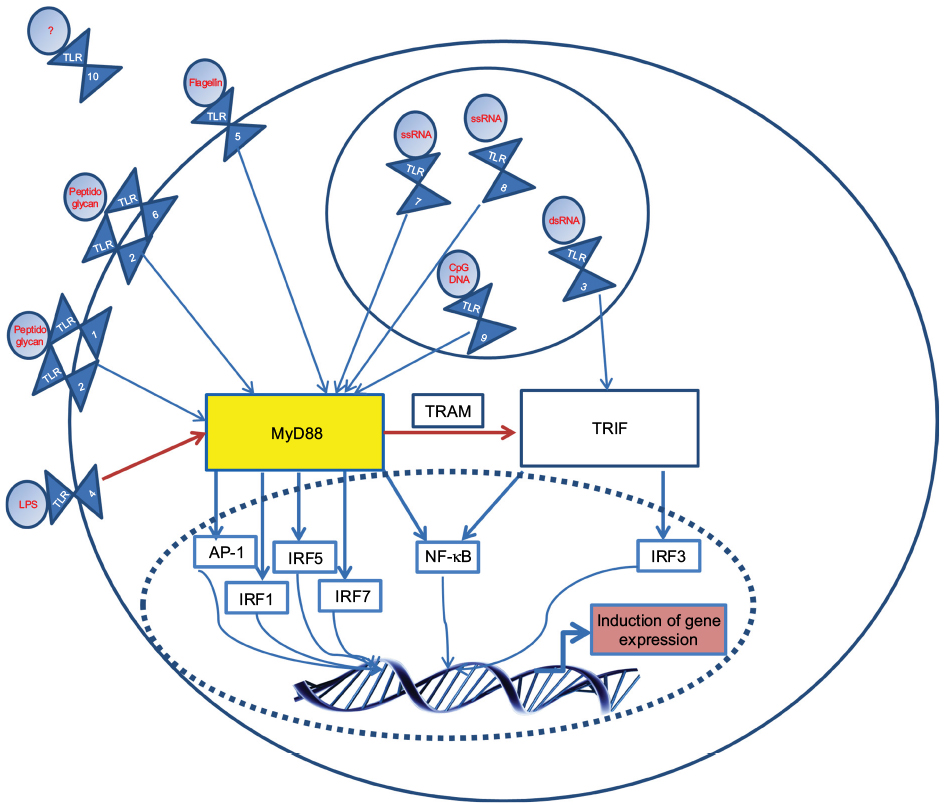 | Figure 1 Basic signaling of TLRs: upon ligation with their associated ligands (common ones are shown in red), TLRs form homo- or heterodimers. Notes: Following ligation, TIR domains engage with adaptor proteins. TLRs 1, 2, 5, 6, 7, 8, 9 signal via the TIR domain-containing adaptor protein MyD88, while TLR3 uses the TRIF. TLR4 can move from the plasma membrane in order to switch signaling from MyD88 to TRIF, with the use of TRAM. A complex cascade, involving molecules such as IRAK-1, results in the induction of key transcription factors such as NF-κB. ‘’?’’, unknown. Abbreviations: dsRNA, double-stranded RNA; IFNβ, interferon-β; IL, interleukin; IRAK-1, IL-1R-associated kinases; IRF, interferon regulatory factor; LPS, lipopolysaccharide; NF-kB, nuclear factor kappa B; TIR, toll IL-1 resistance; ssRNA, single-stranded RNA; TLR, toll-like receptor; TRAM, TRIF-related adapt molecule; TRIF, TIR-domain-containing adaptor protein-inducing IFNβ. |
 | Table 1 Endogenous ligands of human TLRs Note: “-’’, no known endogenous ligand. Abbreviations: ICs, immune complexes; HMGB-1, high mobility group box 1; siRNA, small interfering RNA; ssRNA, single-stranded RNA; TLR, toll-like receptor. |
Autoimmune diseases encompass a wide range of diseases, which can affect a single organ to the whole organ systems. Autoimmune diseases are generally defined as the dysregulation of the immune system causing an immune response to “self”. The etiology of autoimmunity is still a widely debated topic with some claiming environmental factors play a large role in its development,6 while others claiming genetics play the lead role. However, it is generally accepted that it is an interplay between genetic and environmental factors that lead to the development of autoimmunity. Although not fully understood, TLRs have been implicated in the pathogenesis of various autoimmune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and systemic sclerosis (SSc).7–9 The aims of this review were to highlight the roles of TLRs in autoimmune diseases, particularly in rheumatic diseases, and to suggest where these can be targeted for therapeutic intervention and treatment. Finally, further areas of study of the TLRs in relation to autoinflammatory conditions have been suggested.
TLRs in RA
RA is an autoimmune disease, which primarily affects the synovial joints, causing chronic inflammation leading to the destruction of articular tissues. RA affects ~1% of the population. The exact molecular pathogenesis of the disease remains unclear; however, it is widely accepted that the activation of synovial fibroblasts, by self-reactive immune cells, causes inflammation, which is a critical step in disease pathogenesis. Synovial inflammation and consequent destruction are mediated via an increased expression of inflammatory cytokines and matrix metalloproteinases (MMPs). Elevated levels of TLRs and their associated endogenous ligands have been shown to contribute to the pathogenesis of the disease.10,11
The events leading to the activation of TLRs in RA are still a discussed topic; the ligands associated with septic inflammation are less clear than their exogenous counterparts. It is believed that PAMPs, derived from commensal flora or infectious bacteria, are involved in the initiation of RA.12 Initial insult causes an autocrine loop involving increased MMPs, leading to increased damage (Figure 2). Intra-articular injection of streptococcal cell wall fragments that contain TLR2 ligands induces an inflammation and arthritis that is MyD88 dependent. It has been revealed that DNA and peptidoglycans released from intestinal flora are present in the synovium of patients with RA,13 and oral microbiota may be important also. The presence of bacterial DNA, peptidoglycans, and lipopolysaccharides (LPSs) in the synovium will ultimately result in the activation of TLRs. Necrotic cells in the joints of patients suffering from RA have also been shown to activate TLR3 through the release of endogenous RNA.14 The release of single-stranded RNA (ssRNA) from the patient’s cell during cellular stress or injury can activate TLR8 in the synovial fluid.15 Recently, IL-29, released early in viral infection, has been shown to affect the release of TLR-mediated cytokines and TLR expression, possibly contributing to disease progression.16 It has been found that several endogenous ligands, such as high mobility group box 1 (HMGB-1), are released from damaged cells or induced in the synovial tissue of patients with RA.17–19 HMGB is a nuclear architectural protein and an endogenous TLR4 ligand; HMGB-1 and S100-A8 levels are increased in patients with RA20. In combination, these studies show the complexity and various systems that may be involved in initiating and maintaining RA.
 | Figure 2 Role of TLRs in RA joint damage: initial cellular damage causes the release of DAMPs that are upregulated during inflammation, possibly caused by PAMPs. Notes: Consequent TLR activation on cells residing in the synovium results in increased production of key inflammatory cytokines such as TNF-α and IL-1, which in turn can activate FLS, leading to increased MMPs. Increased MMPs lead to the loss of homeostasis between TIMPs and MMPs leading to increased destruction of ECM and cartilage, causing the release of DAMPs and activation of TLRs on FLS. Abbreviations: DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; FLS, fibroblast-like synoviocytes; HMGB-1, high-mobility group box 1; IL, interleukin; INF, interferon; miRNA, microRNA; MMPs, matrix metalloproteinases; PAMPs, pathogenic-associated molecular patterns; RA, rheumatoid arthritis; TIMPs, tissue inhibitor of metalloproteinases; TLR, toll-like receptor; TNF-α, tumor necrosis factor-α. |
TLR4 is important in the pathogenesis of RA, as shown by enhanced expression in the synovial tissue of patients with RA and protection from experimental models of RA in TLR4 knockout mice.21,22 TLR signaling has been implicated in an indirect mechanism of inducing inflammation via the protein kinase inositol-requiring enzyme 1α (IRE1α).23 Active IRE1α unconventionally splices Xbox-binding protein 1 (XBP1).24 Interestingly, sXBP1 encodes an enhanced transcription factor involved in the expression of proinflammatory molecules, such as tumor necrosis factor (TNF)-α and IL-6. Increased sXBP1, mediated by the TLR activation of IRE1α, can increase proinflammatory molecules contributing to inflammation in patients with RA.
The presence of unfolded proteins in the lumen of the endoplasmic reticulum activates the unfolded protein response (UPR) pathway. The UPR pathway is a highly conserved mechanism, which leads to the activation of IRE1α. The translation of IRE1α is comparable in patients with RA and osteoarthritis, meaning that the expression between the two diseases is unchanged. However, the detection of mRNA coding for sXBP1 is increased only in patients with RA. As IRE1α exclusively splices XBP1, the study showed that it is the activation, not the expression, of IRE1α which is altered between the diseases.23 An in vivo mouse model of RA found that the deletion of IRE1α, in myeloid cells, delayed the onset and severity of the disease.23 The role of sXBP1 was further clarified when it was found that the activation of IRE1α was uncoupled from the UPR pathways in patients with RA. Another study found the secretion of TNF-α was not only was induced by sXBP1, but TNF-α-induced sXBP1 expression, was even more prominent when combined with IL-6 and IL-1β.25 From a therapeutic standpoint, targeting TLR-dependent IRE1α or TNF-α could disrupt the autocrine loop described by reducing sXBP1 expression and in turn reducing the expression of proinflammatory cytokines.
The hampering of effective therapies to combat RA is largely due to an incomplete knowledge of the endogenous ligands associated with chronic “sterile” inflammation. TLR4 signaling is associated with the exogenous ligand LPS; however, the activation of TLR4 signaling has also been shown via the endogenous ligand tenascin C. Tenascin C is not usually produced in adult tissue but is transiently expressed upon tissue injury or persistently expressed in chronic inflammation. Interestingly, the domains within tenascin C share homology with those found in DAMPs, which is associated with TLR signaling. Wild-type and TenC-/- mice have been used to dissect tenascin C’s role in TLR signaling. It was found that tenascin C plays a part in the maintenance of inflammation, more specifically its fibrinogen-like globe domain.26 The challenge experienced with wild-type mice with recombinant fibrinogen-like globe was dose-dependent stimulation of inflammation, which required functioning MyD88 and is dependent on TLR4.26 Once inflammation is initiated, tenascin C acts in a destructive cycle by amplifying the inflammatory response and inducing other endogenous proinflammatory molecules.
Unlike tenascin C, the IL-17-IL-23 axis has been shown to be important in both the onset phase and destruction phase of RA. The axis can also be linked to the expression of key TLRs such as TLR2, TLR3, and TLR4 in fibroblast-like synoviocytes, which are obtained from patients with RA. Of therapeutic importance is the inhibition of the STAT3 pathway, which has been shown to reduce the expression of TLR3 in fibroblast-like synoviocytes.27 It has also been suggested that TLR3 expression is increased in the onset stage of RA.28 A research group has documented a role for microRNA (miRNA)-26a to downregulate TLR3 production,29 which could act as a possible therapeutic agent for the treatment of RA and other inflammatory diseases associated with TLR signaling. In these studies, repression of TLR signaling via targeting of their ligands or downstream mediators has been useful over direct targeting of TLR themselves, which are essential for immunity. Nonspecific targeting of TLRs, and indeed their downstream mediators, will leave the patient open to invasion via pathogenic organisms. However, generalized inhibition may be overcome due to a high degree of cross talk between TLR-initiated signaling pathways. For example, patients with defunct MyD88 are usually resistant to common bacterial and fungal infection, meaning that the repression of this key molecule does not leave the patient completely immunocompromised.30 It is also important to note that protective roles of TLRs have been defined, such as TLR9 in SLE, and that increased levels of other TLRs in autoimmunity may be to offer a protective role; thus, complete inhibition would be counterintuitive. In the case of autoimmunity, it is rather the overzealous activity of TLRs causing damage. TLRs also regulate each other, for example, TLR9 regulates TLR7; thus any therapeutic agent should not completely inhibit TLR signaling but rather the increased activity.
A novel role for TLR5 in RA has recently been detailed.31 The study found that TLR5 expression was higher in macrophages and epithelial cells derived from the lining and sublining of synovial tissue were obtained from patients with RA, when compared to the control.31 More importantly, it was found that not only TLR5 regulated TNF-α in RA monocytes but also the expression levels of TLR5 strongly correlated with TNF-α production and the disease severity score.31 Therefore, effective targeting of TLR5 has the potential to decrease the expression of TNF-α and lower the disease severity score.
Endosomal TLRs (3, 7, 8, and 9; Figure 1) have been implicated in RA progression. TLRs 7 and 8 respond to ssRNA. Genetic deletion of murine TLR7, which is comparable to human TLR8 (huTLR8), has been shown to decrease RA disease parameters, such as inflammation.32 Accurate murine models of TLR8 have proven somewhat difficult as murine TLR8 does not recognize the same ligands as huTLR8 due to a sequence difference of five amino acids. However, this has been addressed with the production of huTLR8-expressing mice; this development will allow further research into the exact role of huTLR8 in RA. Generation of the mice was not trivial; mice that were expressing high levels of huTLR8 had a shorter life span than those expressing low levels of huTLR8.33 Development of the mice led to the suggestion that TLR8 may be implicated in the maintenance and not the onset of the disease, acting after ligation with “self” ssRNA, which was released after initial damage. Thus, therapeutic targeting of TLR8 may be useful in patients with chronic RA. In support of the role of TLRs and downstream interferon induction, a polymorphism has been described in IRF5.34
TLRs in SLE
SLE has various pathogenic factors leading to an array of clinical manifestations across multiple organs. Autoantibodies, produced by hyperactive B-cells, are often found against double-stranded DNA and/or DNA/RNA-bound proteins and appear to play a crucial role in the disease, although the molecular mechanisms remain elusive. The autoantibodies are both diagnostic and pathologic in the disease, and the disease is characterized by a high type I interferon. The clearance of apoptotic cells in patients with SLE is deficient; this can result in the formation of the immune complex (IC). The structural and sequential similarity of “self” and microbial nucleic acids can result in the binding of “self” DNA to anti-DNA antibodies; this recognition will form an IC (DNA and anti-DNA antibody). The IC can interact with TLR9 resulting in dendritic cell activation, consequently activating B- and T-cells to release proinflammatory cytokines, ultimately resulting in inflammation.7 Conflicting studies have been reported about the role of TLR9 in SLE, with some claiming TLR9 has a protective or regulatory role, and it is in fact TLR7 that enhances the autoimmune response.35,36 The ambiguity surrounding TLR9 may be in part due to the methylation of mammalian DNA that may not be recognized by TLR9. These contradictory reports should be considered in terms of the murine models used as they differ between studies; also, the exact molecular detail of the protective response needs to be properly detailed. In contrast to TLR9, the role of TLR7 in SLE appears to be proinflammatory and may also be regulated by TLR9 itself.37 The activation of TLR7 in plasmacytoid dendritic cells (pDC) can be caused by decreased clearance or increased number of autoantibodies that misdirect self RNA to the pDC to activate TLR7 and elicit an immune response. It is hypothesized that TLR9 interacts with TLR7 in the endosome inhibiting its response;35 this has the potential to explain the increase in disease in TL9-/- murine models.
The mechanism by which TLR7 and TLR9 translocate to endosomes has recently been suggested to be dependent on heat shock protein 90 (HSP90).38 The study found that the inhibition of HSP90 alleviated the disease activity in murine models.38 Thus, targeting of HSP90 may be of therapeutic benefit. Although there are reports of reduced clearance of apoptotic cells in SLE and that this may be the stimulus for the autoantibodies, precisely which cells are apoptotic is unclear. It has recently been found that RNA complexes containing antimicrobial peptides trigger TLR7- and TLR8-mediated inflammation in dendritic cells.39 It is now known that this is due to neutrophil extracellular traps (NETs); that it is neutrophils releasing DNA (nuclear and mitochondrial) in a net type fashion in a controlled cell death manor. NETs contain antimicrobial peptides and may be a host response, but patients with SLE trigger an immune response via dendritic cells via TLR9 activation. Interestingly SLE patients’ neutrophils released more NETs on stimulation than healthy controls.40 Furthermore, patients with SLE have a reduced ability to degrade NETs.41
The sex bias in SLE has been well documented, with an array of possible causes. Gene analyses of peripheral blood mononuclear cells treated with 17β-estradiol (E2) increased the secretion of key proinflammatory cytokines as well as gene expression of TLR8 mediated through estrogen receptor alpha. In fact, the expressions of all of the endosomal TLRs were increased upon treatment with E2.42 Further work on the exact TLR response to estrogen may produce a viable therapeutic target for SLE, alongside a molecular justification for sex bias.
TLRs in SSc
SSc is an autoimmune idiopathic connective tissue disease, which has three clearly defined hallmarks: small vessel vasculopathy, production of autoantibodies, and fibroblast dysfunction leading to increased collagen accumulating in the skin and major organs.43
The symptoms of SSc are mainly caused by the deposition of excess extracellular matrix (ECM), which is seen when MMPs and their inhibitors, such as tissue inhibitor of metalloproteinases (TIMP-1), lose homeostasis. Circulating monocytes play an important role in the development of SSc, and a link has been shown between TLR8 signaling and excessive TIMP-1 production in these cells.44 Monocytes are also found in close proximity to myofibroblasts.
It has also recently been found that the AP-1 family member, Fos-related antigen 2 (Fra2), facilitates TLR8-mediated overexpression of TIMP-1, in ex vivo monocytes derived from patients with SSc.45 Skin-infiltrating monocytes from patients with SSc showed an increase in Fra2; treatment of these monocytes with a histone demethylation agent caused an increase in both Fra2 and TIMP-1 production when challenged with the TLR8 agonist ssRNA; and increased TIMP-1 can directly induce myofibroblast transdifferentiation.45 The factors affecting transdifferentiation of myofibroblast is complex; once transdifferentiation is complete, the myofibroblasts are able to upregulate the expression of many fibrotic proteins, such as collagen 1. The revealing of the transdifferentiation pathway, involving Fra2 and TLR8, may evoke further study into the epigenetic regulation involved in myofibroblast transdifferentiation. The TLR8-mediated induction of TIMP-1 and the subsequent fibrosis, while important in revealing mechanisms, does not clarify where the ssRNA is derived. Synthetic ssRNA was used to stimulate cells, but incubation of monocytes with the serum of patients diluted in media incited the same response as the synthetic RNA ligand and this could be blocked by the treatment of the serum with RNAase; thus, patients have elevated ssRNA in their circulation. It is speculated but not proved that this is from damaged endothelium.
DAMPs associated with SSc include ligands for TLR4, both of which are elevated in SSc skin and lung tissue.46 The endogenous ligands for TLR4 can be released as the consequence of cellular stress or damage, oxidative stress, and ECM remodeling. Stimulation of TLR4 in healthy fibroblasts was associated with the induction of genes linked with ECM remodeling. Although TLR4 signaling in SSc is complex, it is believed that enhanced Smad signaling, downregulation of TGF-β antagonist, and suppression of anti-fibrotic miRNAs lead to prolonged fibroblast activity,46 resulting in pathological fibrosis. Murine models also show that a point mutation in the TLR4 gene attenuated bleomycin-induced skin fibrosis.47 The exact mechanism for the attenuation in the murine model remains uncertain; however, the model clarified the pivotal role of TLR4 in the development of the disease. It has recently been demonstrated that TLR4 induction of fibrosis in the bleomycin model is mediated via IRF5. In this paradigm, the activation of IRF5 via TLR4 ligation through the standard MyD88 signaling pathway leads to the activation of fibrosis-related genes as IRF KO mice are protected and IRF-/- fibroblasts are anti-fibrotic; of interest is the fact that immune abnormalities are absent in the IRF KO mice including B-cell activation reductions and IL-6 levels; it is this loss of IL-6 induction that is important here.48,49
The exact molecular initiation of SSc remains unknown; however, it is believed that disruption in the vasculature leads to epithelial damage. This damage in turn can release “danger” molecules such as serum amyloid A.50 In dermal fibroblasts, TLR2 plays a role in enhancing the signal produced by serum amyloid A in order to secrete proinflammatory cytokines such as IL-6; this system is dependent on the downstream molecule IRAK4. Therefore, targeting of TLR2 may be of therapeutic benefit. Interestingly, it has also been suggested that infection with Epstein–Barr virus may cause a patho-immunological response in patients with SSc and contribute to fibrosis via aberrant TLR7 and TLR9 signaling.51 It has also been found that NF-κB regulates epidermal homeostasis and can promote fibrosis in in vivo models.52
Cross talk between the complex pathways involved in fibrosis is poorly understood; the roles TLRs play in the sustained expression of profibrotic molecules, as well as the transdifferentiation of myofibroblasts, appear to be pivotal and may open up new therapeutic options for a disease with little available therapy. In addition, is there a hierarchy among DAMPs?
TLRs in Sjogren’s syndrome
Sjogren’s syndrome (SS) is an autoimmune disorder in which the immune system attacks fluid-secreting glands, for example, the lacrimal and salivary glands, causing hypofunction that leads to dry eyes and dry mouth (xerostomia). There is a lymphocytic infiltrate in the glands leading to destruction and also autoantibodies, implying a role of the adaptive immune system, but the innate immune system may also have a role to play. In support of this hypothesis, it has been shown that salivary epithelial gland cells (line the glands) from patients with SS have higher levels of TLR expression compared to controls,53 and secretion of cytokines, from B-cells, increased upon TLR7 and TLR9 stimulation in primary SS.53,54
Interestingly, there is an increased occurrence of glandular destruction that could be releasing endogenous TLR ligands (DAMPs). In support of this hypothesis, it was shown that mouse glandular cells express TLR3 and that the injection of TLR3 ligand directly induces salivary gland hypofunction and that this was reversible on the cessation of TLR3 ligand, suggesting that it is independent of autoantibody production.55 This was followed up with another study demonstrating reduced salivary flow in PolyI:C-treated mice.56 TLR9 is also elevated in SS and promotes B-cell germinal center formation.57
Impaired phagocytosis of apoptotic salivary epithelial cells by monocytes has been reported in patients with SS, thus indicating that if increased apoptosis and reduced clearance are occurring this would lead to elevated concentrations of antigenic nucleic acids.58 Interestingly, the microRNA146a is dysregulated in SS compared to controls; one of the targets of miR146a is IRAK, which is a critical component in TLR signaling.59
TLRs in myositis
Myositis is a chronic inflammatory autoimmune disorder, consisting of five conditions that primarily affects skeletal muscles, causing profound muscle weakness and fatigue (Figure 3). There is a presence of an immune cell infiltration; however, this does not always correlate with disease. Incubation of skeletal muscle fibers with the DAMP HMGB-1 results in robust induction of MHC class I expression, which is a histopathological hallmark of myositis. The incubation of isolated muscle fibers with HMGB-1 also resulted in reduced intracellular calcium levels.60
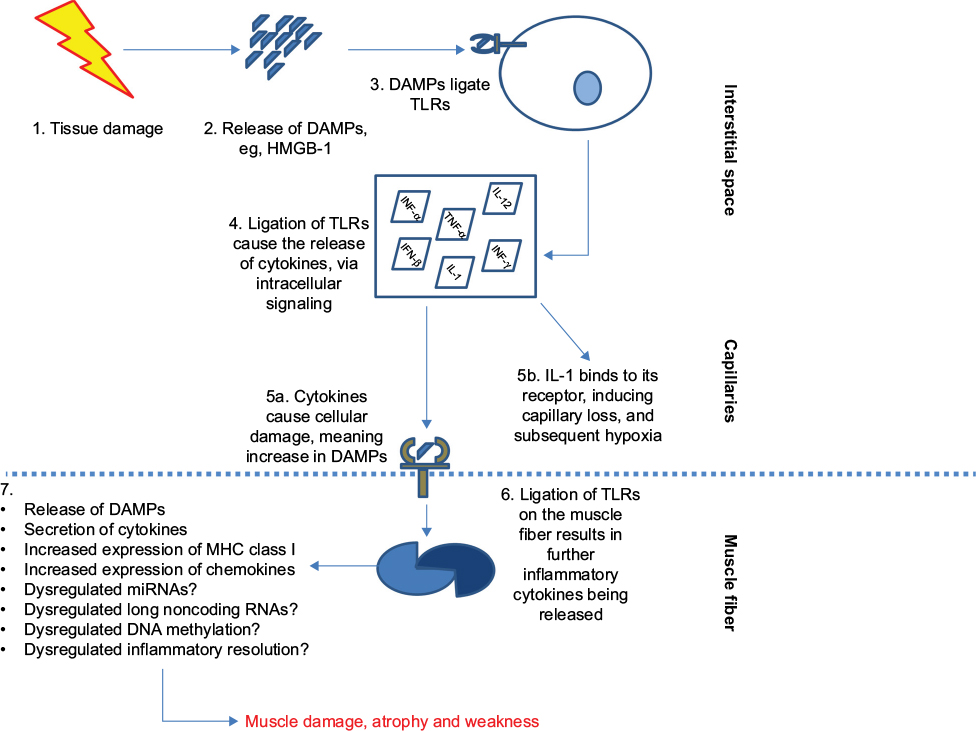 | Figure 3 Basic role of TLRs in myositis: post tissue damage (eg, via infection or exercise), cells release DAMPs. Notes: These DAMPs ligate to TLRs on infiltrating cells such as macrophages and plasmacytoid cells. The ligation ultimately results in the secretion of proinflammatory molecules into the microenvironment. IL-1 binds to its receptor, IL-1R, and exerts its downstream effects, which lead to capillary loss and hypoxia. The damage to the cells caused by the cytokine, in turn, produces more DAMPs that can bind to the TLR and induce further inflammation and increase chemokine release in the muscle fibers. Recent murine models of myositis have shown the importance of TLR8, MyD88, and NF-κB in the development of myositis.84 Possible dysregulation of miRNA and long noncoding RNAs and altered inflammation resolution also contribute. Abbreviations: DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; FLS, fibroblast-like synoviocytes; IL, interleukin; miRNA, microRNA; MMPs, matrix metalloproteinases; NF-kB, nuclear factor kappa B; TLR, toll-like receptor; TNF-α, tumor necrosis factor-α. |
It was further demonstrated that the receptor needed for HMGB-1-induced muscle dysfunction was TLR4, and not RAGE receptor, and that the induction of the muscle dysregulation HMGB-1 was dependent on its redox state.61 In addition, the treatment of patients with corticosteroids reduced the aberrant HMGB-1 expression in myositis. Stimulation of isolated myotubes with the TLR3 ligand, poly I–C, resulted in the production of interferons, which subsequently leads to the upregulation of MHC class I, the hallmark of myositis.62 TLR3, TLR7, and TLR9 have also been demonstrated to be upregulated in myositis tissue.63 Interestingly, TLR7 stimulation in combination with antigen-expressing apoptotic myoblasts in a mouse model, but not TLR7 stimulation alone, resulted in autoantibody production and replicated histopathological findings in human myositis.64 TLR3 and RIG-I have also been recently demonstrated to be upregulated in myositis tissue.65 The link between inflammation and clinical course in myositis is not linear, and much more work is required to dissect the role of innate immunity in the disease.
TLRs in other autoimmune diseases
TLRs are also implicated in an abundance of other autoimmune diseases, such as inflammatory bowel disease (IBD), multiple sclerosis, and diabetes mellitus (Table 2).
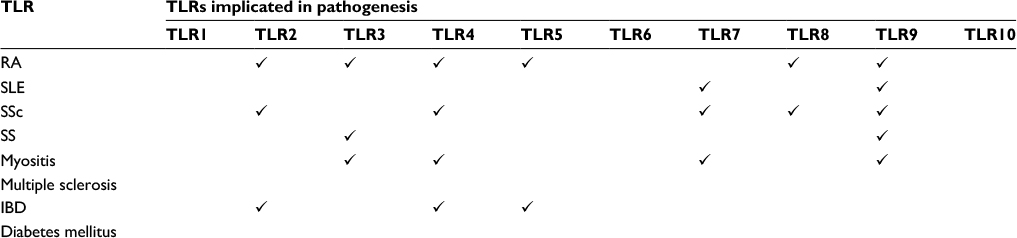 | Table 2 Variety of some TLRs implicated in autoimmune diseases Abbreviations: IBD, inflammatory bowel disease; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SS, Sjogren’s syndrome; SSc, systemic sclerosis; TLR, toll-like receptor. |
In healthy conditions, the expression of TLRs in the intestinal tract remains low in order to reduce the recognition of the commensal intestinal flora.66 However, it has been shown in pediatric IBD samples that mRNA levels of TLR4 and TLR2 are increased in comparison with their non-inflamed counterparts.67 Increased TLR expression will lead to increased TLR-mediated inflammation in the intestines. As patients with IBD suffer from periodic injury and repair, it is of importance to understand the role of innate immunity. An in vivo model of TLR9-deficient mice found that deficient TLR9 leads to an increased susceptibility to intestinal damage and delayed healing, suggesting a regulatory role of TLR9.68
TLRs as therapeutic targets
The complex systems involved in TLR signaling and their pivotal role in autoimmunity remain incompletely understood; despite this, TLRs and their associated pathways remain a key therapeutic target.
Inhibitory oligodeoxynucleotides (ODN) were originally shown to inhibit TLR9 signaling, using B-cells and pDC from patients with SLE.69 It was found that ODN IRS954 inhibited the cells response, measured via autoantibody levels, to synthetic and exogenous TLR7 and TLR9 ligands.70 However, it has recently been shown that a single ODN, ODN 1411, can inhibit all of the endosomal TLRs (3, 7, 8, and 9) in primary human cells.71 The exact mechanism of the ODNs inhibition in TLR is yet to be discovered with some stating extracellular domain binding,71 while others claiming it exerts its effects to disrupt downstream molecules, such as HMGB.72 The study, involving the endosomal TLRs, found it was most likely ODN1411 was acting via direct interaction with the endosomal TLRs that elicited the inhibitory effect.71 Overall, the development of ODNs has the potential to be of benefit in both RA and SLE.
The inhibitor of salt-inducible kinase (SIK), HG-9-9101, has been shown to significantly reduce TLR4- and TLR2-induced TNF-α production in human myeloid cells, while also increasing the secretion of the anti-inflammatory molecule IL-10.73 Interestingly, data presented found that SIK inhibition did not fully suppress the immune system;73 this has clinical relevance as the treatment with the SIK inhibitor would not leave the patient completely immunocompromised.
An alternative to novel drug development is the use of a compound already available for other diseases; an example of this is mianserin. Mianserin is a serotonin receptor antagonist, which is usually delivered as an antidepressant. It was found that mianserin was able to not only inhibit endosomal TLRs in primary human cells but also reduce spontaneous TNF and IL-6 production in synovial membrane cultures.15 A further study used the collagen-induced arthritis (CIA) murine model to confirm that mianserin was able to decrease RA disease progression and preserve joint architecture.32 The effect of mianserin, on TLRs, was only elicited at a dose that would be unsafe in humans, limiting its use. An extension of this work found that fluoxetine, a selective serotonin reuptake inhibitor was able to reduce inflammation and bone erosion in CIA murine model and also reduce spontaneous cytokine production in human RA synovial membrane cultures.74 Although the use of selective serotonin reuptake inhibitor appears to hold promise, it is important to note that their effects on TLRs is an off-target effect and further research would be required before the treatment could be delivered to patients suffering from RA.
As mentioned earlier, targeting of associated molecules such as IRE1α may also hold therapeutic potential. A study found that 7-hydroxy-4-methylcoumarin (4µ8C) was a novel inhibitor of IRE1α.75 A further in vivo study found that 4µ8C was able to inhibit IRE1α, along with a significant reduction in TNF-α and IL-6 in the joint tissue of 4µ8C-treated mice.23 This could hold potential in the treatment of RA.
The development of OPN-305, a humanized IgG4 monoclonal antibody, by Opsona Therapeutics also has potential benefits for patients with RA.76 OPN-305 targets TLR2, possibly via binding to ligand sites resulting in an inability to heterodimeric with TLR1 or TLR6. The first in-human Phase I study showed promising results;77 even at the lowest dose (0.5 mg/kg), a 50% reduction in IL-6 was seen in ex vivo samples. Further ongoing studies are required to validate the use of OPN-305.
Although the targeting of TLRs is an exciting prospect, it is important to fully understand TLRs and their signaling pathways before the transfer can be made into clinics to treat autoimmune diseases.
Epigenetic regulation of TLRs
TLR signaling is critical in the immune host defense to pathogens and also to internal DAMPs, and thus tight regulation of TLR signaling must be in place to ensure a return to immune homeostasis and not unresolved inflammation. One mechanism of controlling TLR signaling is that of epigenetic regulation, specifically miRNAs. miRNAs are small, non-coding RNAs that regulate gene expression and are mainly synthesized by RNA polymerase II. miRNAs work by imperfectly binding the 3′ untranslated region of target mRNAs and downregulating their expression through mRNA degradation or mRNA decay. Many miRNAs are induced rapidly after the activation of TLR. One of the most important and first to be described to negatively regulate TLR signaling was miR 155. miR155 is induced upon TLR4 stimulation in macrophages78 and is also regulated by TLR3. miR155 is also dysregulated in arthritis, and miR155 KO mice are resistant to CIA.
Another hugely important miR in TLR signaling is miR146. MiR146a/b is upregulated in a TLR- and NF-KB-dependent mechanism and targets TRAF6 and IRAK1: two key adaptor proteins in TLR signaling and proinflammatory cytokine production.79 Indeed, miR146a is critical in the resolution of inflammation in T-cells.80
In addition, miR21 has emerged as a key regulator of TLR signaling. It was found that upon TLR4 signaling that miR21 is induced; this binds to its cognate target, mRNA programmed cell death 4 (PDCD4) to reduce its expression. PDCD4 is a tumor-suppressor protein that regulates IL-10 levels. Loss of PDCD4 in mice results in protection from LPS-induced lethality.81 PDCD4 is induced by inflammatory signaling and apoptotic debris. Because miR21 appears to be a critical regulator of TLR-negative signaling to restore immune homeostasis after stimulation, any disturbance in this may lead to exacerbated unrestrained inflammation. A recent manuscript has shown that unmanipulated levels of miR21 are reduced in RA patient’s immune cells (macrophages) and importantly, as opposed to control cells, the RA patients’ cells did not upregulate miR21 upon TLR stimulation, suggesting an intrinsic defect. Interestingly, lower miR21 levels correlated with Th17 levels (Th17 cells are pathogenic in RA).82 An interesting question is whether the induced miRs to negatively regulate signaling are different in response to DAMPs and PAMPs, even though they signal through shared TLRs.
Modulation of miRNAs is now possible through both synthetic mimics and antagomirs, and this will alter the levels of the miRNAs and their targets. Enhancing the stability of the miRNAs is important in any therapeutic targeting, but it is likely this will happen and they could be key therapies in the future.
Conclusion
The innate immune system is a critical part of the response to pathogens, tissue damage, and the release of DAMPs. Chief among these systems are TLRs as part of the PRRs. TLRs sense pathogens and consequently activate NF-kB and inflammatory cytokines. However, an anti-inflammatory role has recently been described for TLR10.83 Resolution of inflammation and the return to homeostasis are important and these appear to be mediated, at least in part, by miRs that act to positively or negatively regulate inflammation. In autoinflammatory diseases such as RA, it may be that the feedback loop to restore homeostasis is somehow broken, and miRNA targeting with miR mimics may be one therapeutic option as these are chemically stable. Although TLRs and their associated ligands are an attractive target in the development of new therapies, we do not fully appreciate neither the complexity of signaling yet nor the full consequences of targeting TLRs and their signaling pathways. There are currently clinical trials underway in the use of endosomal TLR inhibitors that may be more valuable in rheumatic diseases. TLR-mediated inductions of miRNAs are now known to negatively regulate their signaling, and a greater understanding of the mechanism and targets of these miRNAs may yield new therapies.
Disclosure
The authors report no conflicts of interest in this work.
References
Fullard N, O’Reilly S. Role of innate immune system in systemic sclerosis. Semin Immunopathol. 2015;37(5):511–517. | ||
Too LK, McGregor IS, Baxter AG, Hunt NH. Altered behaviour and cognitive function following combined deletion of toll-like receptors 2 and 4 in mice. Behav Brain Res. 2016;303:1–8. | ||
Škanta F, Roubalová R, Dvořák J, Procházková P, Bilej M. Molecular cloning and expression of TLR in the Eisenia andrei earthworm. Dev Comp Immunol. 2013;41(4):694–702. | ||
Bell JK, Botos I, Hall PR, et al. The molecular structure of the toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci USA. 2005;102(31):10976–10980. | ||
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–397. | ||
Rook GA. Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol. 2011;42(1):5–15. | ||
Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202(8):1131–1139. | ||
Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, et al. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005;52(8):2313–2322. | ||
Broen JC, Bossini-Castillo L, van Bon L, et al. A rare polymorphism in the gene for toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum. 2012;64(1):264–271. | ||
Ospelt C, Brentano F, Rengel Y, et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008;58(12):3684–3692. | ||
Wähämaa H, Schierbeck H, Hreggvidsdottir HS, et al. High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis Res Ther. 2011;13(4):R136. | ||
Schrijver IA, Melief MJ, Tak PP, Hazenberg MP, Laman JD. Antigen-presenting cells containing bacterial peptidoglycan in synovial tissues of rheumatoid arthritis patients coexpress costimulatory molecules and cytokines. Arthritis Rheum. 2000;43(10):2160–2168. | ||
van der Heijden IM, Wilbrink B, Tchetverikov I, et al. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000;43(3):593–598. | ||
Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via toll-like receptor 3. Arthritis Rheum. 2005;52(9):2656–2665. | ||
Sacre SM, Lo A, Gregory B, et al. Inhibitors of TLR8 reduce TNF production from human rheumatoid synovial membrane cultures. J Immunol. 2008;181(11):8002–8009. | ||
Xu L, Feng X, Tan W, et al. IL-29 enhances toll-like receptor-mediated IL-6 and IL-8 production by the synovial fibroblasts from rheumatoid arthritis patients. Arthritis Res Ther. 2013;15(5):R170. | ||
Roelofs MF, Boelens WC, Joosten LA, et al. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol. 2006;176(11):7021–7027. | ||
Huang QQ, Sobkoviak R, Jockheck-Clark AR, et al. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol. 2009;182(8):4965–4973. | ||
Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277(17):15028–15034. | ||
Hamada T, Torikai M, Kuwazuru A, et al. Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthritis Rheum. 2008;58(9):2675–2685. | ||
Radstake TR, Roelofs MF, Jenniskens YM, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-γ. Arthritis Rheum. 2004;50(12):3856–3865. | ||
Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, et al. Inhibition of toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007;56(9):2957–2967. | ||
Qiu Q, Zheng Z, Chang L, et al. Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013;32(18):2477–2490. | ||
Shen X, Ellis RE, Lee K, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107(7):893–903. | ||
Savic S, Ouboussad L, Dickie LJ, et al. TLR dependent XBP-1 activation induces an autocrine loop in rheumatoid arthritis synoviocytes. J Autoimmun. 2014;50:59–66. | ||
Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15(7):774–780. | ||
Lee SY, Yoon BY, Kim JI, et al. Interleukin-17 increases the expression of toll-like receptor 3 via the STAT3 pathway in rheumatoid arthritis fibroblast-like synoviocytes. Immunology. 2014;141(3):353–361. | ||
Meng L, Zhu W, Jiang C, et al. Toll-like receptor 3 upregulation in macrophages participates in the initiation and maintenance of pristane-induced arthritis in rats. Arthritis Res Ther. 2010;12(3):R103. | ||
Jiang C, Zhu W, Xu J, et al. MicroRNA-26a negatively regulates toll-like receptor 3 expression of rat macrophages and ameliorates pristane induced arthritis in rats. Arthritis Res Ther. 2014;16(1):R9. | ||
Gern JE. Pyogenic bacterial infections in humans with MyD88 deficiency. Pediatrics. 2009;124(suppl 2):S154–S154. | ||
Chamberlain ND, Vila OM, Volin MV, et al. TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF-α levels. J Immunol. 2012;189(1):475–483. | ||
Alzabin S, Kong P, Medghalchi M, Palfreeman A, Williams R, Sacre S. Investigation of the role of endosomal toll-like receptors in murine collagen-induced arthritis reveals a potential role for TLR7 in disease maintenance. Arthritis Res Ther. 2012;14(3):1–11. | ||
Guiducci C, Gong M, Cepika AM, et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J Exp Med. 2013;210(13):2903–2919. | ||
Han SW, Lee WK, Kwon KT, Lee BK, Nam EJ, Kim GW. Association of polymorphisms in interferon regulatory factor 5 gene with rheumatoid arthritis: a metaanalysis. J Rheumatol. 2009;36(4):693–697. | ||
Santiago-Raber M-L, Dunand-Sauthier I, Wu T, et al. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2010;34(4):339–348. | ||
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell Richard A, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–428. | ||
Nickerson KM, Christensen SR, Shupe J, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184(4):1840–1848. | ||
Saito K, Kukita K, Kutomi G, et al. Heat shock protein 90 associates with toll-like receptors 7/9 and mediates self-nucleic acid recognition in SLE. Eur J Immunol. 2015;45(7):2028–2041. | ||
Ganguly D, Chamilos G, Lande R, et al. Self-RNA–antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206(9):1983–1994. | ||
Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3(73):ra19–ra73. | ||
Leffler J, Gullstrand B, Jonsen A, et al. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res Ther. 2013;15(4):R84. | ||
Young NA, Wu L-C, Burd CJ, et al. Estrogen modulation of endosome-associated toll-like receptor 8: an IFNα-independent mechanism of sex-bias in systemic lupus erythematosus. Clin immunol (Orlando, Fla.). 2014;151(1):66–77. | ||
van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72(11):1747–1755. | ||
Ciechomska M, Huigens CA, Hügle T, et al. Toll-like receptor-mediated, enhanced production of profibrotic TIMP-1 in monocytes from patients with systemic sclerosis: role of serum factors. Ann Rheum Dis. 2013;72(8):1382–1389. | ||
Ciechomska M, O’Reilly S, Przyborski S, Oakley F, Bogunia-Kubik K, van Laar JM. Histone demethylation and TLR8-dependent crosstalk in monocytes promotes trans-differentiation of fibroblasts in systemic sclerosis via Fra2. Arthritis Rheum. 2016. | ||
Bhattacharyya S, Kelley K, Melichian DS, et al. Toll-like receptor 4 signaling augments transforming growth factor-β responses: a novel mechanism for maintaining and amplifying fibrosis in scleroderma. Am J Pathol. 2013;182(1):192–205. | ||
Takahashi T, Asano Y, Ichimura Y, et al. Amelioration of tissue fibrosis by toll-like receptor 4 knockout in murine models of systemic sclerosis. Arthritis Rheum. 2015;67(1):254–265. | ||
Saigusa R, Asano Y, Taniguchi T, et al. Multifaceted contribution of the TLR4-activated IRF5 transcription factor in systemic sclerosis. Proc Natl Acad Sci U S A. 2015;112(49):15136–15141. | ||
O’Reilly S, Cant R, Ciechomska M, van Laar JM. Interleukin-6: a new therapeutic target in systemic sclerosis? Clin Transl Immunol. 2013;2(4):e4. | ||
O’Reilly S, Cant R, Ciechomska M, et al. Serum amyloid A induces interleukin-6 in dermal fibroblasts via toll-like receptor 2, interleukin-1 receptor-associated kinase 4 and nuclear factor-κB. Immunology. 2014;143(3):331–340. | ||
Farina A, Cirone M, York M, et al. Epstein–Barr virus infection induces aberrant TLR activation pathway and fibroblast–myofibroblast conversion in scleroderma. Journal of Investigative Dermatology. 2014;134(4):954–964. | ||
Fullard N, Moles A, O’Reilly S, et al. The c-Rel subunit of NF-κB regulates epidermal homeostasis and promotes skin fibrosis in mice. Am J Pathol. 2013;182(6):2109–2120. | ||
Spachidou MP, Bourazopoulou E, Maratheftis CI, et al. Expression of functional toll-like receptors by salivary gland epithelial cells: increased mRNA expression in cells derived from patients with primary Sjögren’s syndrome. Clin Exp Immunol. 2007;147(3):497–503. | ||
Karlsen M, Jonsson R, Brun JG, Appel S, Hansen T. TLR-7 and -9 stimulation of peripheral blood B cells indicate altered TLR signalling in primary Sjögren’s syndrome patients by increased secretion of cytokines. Scand J Immunol. 2015;82(6):523–531. | ||
Deshmukh US, Nandula SR, Thimmalapura PR, Scindia YM, Bagavant H. Activation of innate immune responses through toll-like receptor 3 causes a rapid loss of salivary gland function. J Oral Pathol Med. 2009;38(1):42–47. | ||
Nandula SR, Scindia YM, Dey P, Bagavant H, Deshmukh US. Activation of innate immunity accelerates sialoadenitis in a mouse model for Sjögren’s syndrome-like disease. Oral Dis. 2011;17(8):801–807. | ||
Guerrier T, Le Pottier L, Devauchelle V, Pers JO, Jamin C, Youinou P. Role of toll-like receptors in primary Sjögren’s syndrome with a special emphasis on B-cell maturation within exocrine tissues. J Autoimmun. 2012;39(1–2):69–76. | ||
Hauk V, Fraccaroli L, Grasso E, et al. Monocytes from Sjögren’s syndrome patients display increased vasoactive intestinal peptide receptor 2 expression and impaired apoptotic cell phagocytosis. Clin Exp Immunol. 2014;177(3):662–670. | ||
Pauley KM, Stewart CM, Gauna AE, et al. Altered miR-146a expression in Sjögren’s syndrome and its functional role in innate immunity. Eur J Immunol. 2011;41(7):2029–2039. | ||
Grundtman C, Bruton J, Yamada T, et al. Effects of HMGB1 on in vitro responses of isolated muscle fibers and functional aspects in skeletal muscles of idiopathic inflammatory myopathies. FASEB J. 2010;24(2):570–578. | ||
Zong M, Bruton JD, Grundtman C, et al. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann Rheum Dis. 2013;72(8):1390–1399. | ||
Tournadre A, Lenief V, Eljaafari A, Miossec P. Immature muscle precursors are a source of interferon-β in myositis: role of toll-like receptor 3 activation and contribution to HLA class I up-regulation. Arthritis Rheum. 2012;64(2):533–541. | ||
Cappelletti C, Baggi F, Zolezzi F, et al. Type I interferon and toll-like receptor expression characterizes inflammatory myopathies. Neurology. 2011;76(24):2079–2088. | ||
Sciorati C, Monno A, Ascherman DP, Seletti E, Manfredi AA, Rovere-Querini P. Required role of apoptotic myogenic precursors and toll-like receptor stimulation for the establishment of autoimmune myositis in experimental murine models. Arthritis Rheum. 2015;67(3):809–822. | ||
Li L, Dai T, Lv J, et al. Role of toll-like receptors and retinoic acid inducible gene I in endogenous production of type I interferon in dermatomyositis. J Neuroimmunol. 2015;285:161–168. | ||
Suzuki M, Hisamatsu T, Podolsky DK. Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular toll-like receptor 4-MD-2 complex. Infect Immun. 2003;71(6):3503–3511. | ||
Szebeni B, Veres G, Dezsõfi A, et al. Increased expression of toll-like receptor (TLR) 2 and TLR4 in the colonic mucosa of children with inflammatory bowel disease. Clin Exp Immunol. 2008;151(1):34–41. | ||
Rose WA 2nd, Sakamoto K, Leifer CA. TLR9 is important for protection against intestinal damage and for intestinal repair. Sci Rep. 2012;2:574. | ||
Stunz LL, Lenert P, Peckham D, et al. Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur J Immunol. 2002;32(5):1212–1222. | ||
Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37(12):3582–3586. | ||
Sacre S, Lo A, Gregory B, et al. Oligodeoxynucleotide inhibition of toll-like receptors 3, 7, 8, and 9 suppresses cytokine production in a human rheumatoid arthritis model. Eur J Immunol. 2016;46(3):772–781. | ||
Yanai H, Chiba S, Ban T, et al. Suppression of immune responses by nonimmunogenic oligodeoxynucleotides with high affinity for high-mobility group box proteins (HMGBs). Proc Natl Acad Sci U S A. 2011;108(28):11542–11547. | ||
Lombardi MS, Gilliéron C, Dietrich D, Gabay C. SIK inhibition in human myeloid cells modulates TLR and IL-1R signaling and induces an anti-inflammatory phenotype. J Leukoc Biol. 2015;jlb:A715–A307. | ||
Sacre S, Medghalchi M, Gregory B, Brennan F, Williams R. Fluoxetine and citalopram exhibit potent antiinflammatory activity in human and murine models of rheumatoid arthritis and inhibit toll-like receptors. Arthritis Rheum. 2010;62(3):683–693. | ||
Cross BC, Bond PJ, Sadowski PG, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109(15):E869–E878. | ||
OPN-305: a TLR-specific monoclonal antibody [webpage on the Internet]. Dublin: Opsona Therapeutics; Available from http://www.opsona.com/opn305. Accessed May 3, 2016. | ||
Reilly M, Miller RM, Thomson MH, et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin Pharmacol Ther. 2013;94(5):593–600. | ||
O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104(5):1604–1609. | ||
Taganov KD, Boldin MP, Chang K-J, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–12486. | ||
Yang L, Boldin MP, Yu Y, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med. 2012;209(9):1655–1670. | ||
Sheedy FJ, Palsson-McDermott E, Hennessy EJ, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 2010;11(2):141–147. | ||
Dong L, Wang X, Tan J, et al. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J Cell Mol Med. 2014;18(11):2213–2224. | ||
Oosting M, Cheng SC, Bolscher JM, et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci U S A. 2014;111(42):E4478–E4484. | ||
Zhang H, Kang J, Han W, Hu M, Jia H. [The expression and significance of TLR4, MyD88 and NF-κB mRNA in mouse lymph node of experimental autoimmune myositis]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2012;28(3):272–275. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.