Back to Journals » Journal of Inflammation Research » Volume 9
Toll-like receptor cascade and gene polymorphism in host–pathogen interaction in Lyme disease
Authors Rahman S, Shering M, Ogden N, Lindsay R, Badawi A
Received 22 January 2016
Accepted for publication 4 February 2016
Published 31 May 2016 Volume 2016:9 Pages 91—102
DOI https://doi.org/10.2147/JIR.S104790
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Shusmita Rahman,1 Maria Shering,2 Nicholas H Ogden,3 Robbin Lindsay,4 Alaa Badawi1
1National Microbiology Laboratory, Public Health Agency of Canada, Toronto, 2Faculty of Arts and Science, University of Toronto, Toronto, ON, 3National Microbiology Laboratory, Public Health Agency of Canada, Saint-Hyacinthe, QC, 4National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, MB, Canada
Abstract: Lyme disease (LD) risk occurs in North America and Europe where the tick vectors of the causal agent Borrelia burgdorferi sensu lato are found. It is associated with local and systemic manifestations, and has persistent posttreatment health complications in some individuals. The innate immune system likely plays a critical role in both host defense against B. burgdorferi and disease severity. Recognition of B. burgdorferi, activation of the innate immune system, production of proinflammatory cytokines, and modulation of the host adaptive responses are all initiated by Toll-like receptors (TLRs). A number of Borrelia outer-surface proteins (eg, OspA and OspB) are recognized by TLRs. Specifically, TLR1 and TLR2 were identified as the receptors most relevant to LD. Several functional single-nucleotide polymorphisms have been identified in TLR genes, and are associated with varying cytokines types and synthesis levels, altered pathogen recognition, and disruption of the downstream signaling cascade. These single-nucleotide polymorphism-related functional alterations are postulated to be linked to disease development and posttreatment persistent illness. Elucidating the role of TLRs in LD may facilitate a better understanding of disease pathogenesis and can provide an insight into novel therapeutic targets during active disease or postinfection and posttreatment stages.
Keywords: Lyme disease, Toll-like receptors, Borrelia lipoproteins, genetic polymorphisms, host–pathogen interaction
Introduction
Lyme disease (LD) is a tick-borne disease caused by spirochetes of the bacterial species complex Borrelia burgdorferi sensu lato.1 Risk from LD, also known as Lyme borreliosis, occurs primarily in forested areas across North America, Asia, and parts of Europe where the tick vectors (black-legged ticks Ixodes scapularis and I. pacificus) and natural wild-animal hosts (eg, mice, chipmunks, gray squirrels, opossums, and raccoons) of the ticks and bacterium are found. A number of species of B. burgdorferi sensu lato cause LD in humans, including B. burgdorferi sensu stricto, B. afzelii, B. bissettii, B. bavariensis, and B. garinii. Among these, B. burgdorferi sensu stricto (hitherto termed B. burgdorferi) is the primary confirmed cause of LD in North America, while the others are responsible for LD in Europe and Asia.2
In the US, there were over 36,000 confirmed and probable cases in 2013,3,4 with an estimated number of approximately 300,000 true cases every year.4 Risk of infection is found where populations of black-legged ticks occur. These ticks are found in distinct regions of eastern, midwestern, and western North America, although the geographic footprint of risk is expanding: northward geographic expansion of the geographic range of infected ticks has recently driven LD expansion in Canada, and this has been attributed to climate change.5–8 Although the incidence of LD is not as high in Canada compared to the USA, there was a twofold rise in cases reported in Canada from 315 in 2012 to 682 in 2013.9,10
Early LD can be manifested as the characteristic erythema migrans rash, with or without a range of common symptoms, such as fatigue, chills, fever, headache, muscle and joint pain, swollen lymph nodes, and numbness or tingling.11 The skin infection can be localized close to the site of the tick bite or become disseminated systemically within days or weeks.12 However, as the diagnostic erythema migrans rash occurs in ~70%–80% of infected subjects, various other early and late symptoms may arise without the rash.12,13 If untreated or not treated adequately, the early form of the disease can progress to the disseminated phase, which is characterized by neurologic complications of the central or peripheral nervous system, cardiac manifestations, and Lyme arthritis.14,15 A small percentage of patients can also be affected by posttreatment LD syndrome despite antibiotic treatment, in which such symptoms as fatigue, neurological problems, disabling musculoskeletal aches, paralysis, and persistent Lyme arthritis are observed and can last from months to years.13
Exposure to foreign microbial, chemical, or physical agents causes the activation of the host innate immune system. This is the first line of defense, and results in a wide range of inflammatory reactions to allow for the repair of incurred damage, isolation, or elimination of the infectious agent and reestablishment of homeostasis.16,17 In order to initiate inflammatory reactions following infection, host innate immunity relies on pattern-recognition receptors (PRRs), such as the Toll-like receptors (TLRs). TLRs are type I transmembrane proteins that have an extracellular domain containing leucine-rich repeats and a cytoplasmic tail with a conserved Toll/IL-1 receptor (TIR) domain, and they are expressed by such cells as macrophages and dendritic cells.18 PRRs recognize structurally conserved pathogen-derived molecules, eg, triacetylated lipoproteins, peptidoglycans, and lipopolysaccharides,19,20 triggering a downstream proinflammatory signaling cascade leading to the nuclear translocation of the transcription factor NFκB. This process initiates cytokine production, the expression of an array of adhesion molecules and generation of reactive oxygen species.21–24 Of the ten human TLRs, TLR2 and TLR4 were the first members of the PRRs to be recognized as involved in antibacterial defense.25
Interaction of TLRs with the immunogenic B. burgdorferi outer-surface proteins (Osps) plays an important role in the initial stages of LD pathogenesis.2,26–28 This interaction was proposed to mediate both short-lasting and severe Lyme arthritis and Lyme carditis in animal models.29–31 More recent evidence, however, suggests that a number of single-nucleotide polymorphisms (SNPs) in the TLR genes may modulate the host response to infection with B. burgdorferi. For example, SNPs in TLR8 can lead to immunodeficiency syndromes and cause an increased risk of severe clinical manifestations associated with B. burgdorferi infection.32,33
This review explores our current knowledge of the interaction between B. burgdorferi surface lipoproteins and the TLR-related signaling cascade in humans. We used such terms as “Lyme disease”, “toll-like receptors”, “Borrelia lipoproteins”, and “genetic polymorphisms” to identify key studies relevant to this article from the Ovid and PubMed databases. Special emphasis is directed to understanding the critical function of TLR genetic polymorphisms in LD manifestations, and their possible role as mediators of pathogen–host interaction in disease symptomatology and severity.
Borrelia lipoproteins in diseases pathogenesis
B. burgdorferi expresses a wide variety of surface lipoproteins that allow for adaptation to hosts and tick vectors throughout the bacterium’s life cycle, including infectivity and dissemination within hosts. B. burgdorferi is a Gram-negative bacterium that lacks the highly immunogenic surface glycolipid lipopolysaccharide (LPS) present in most other Gram-negative bacteria. Therefore, immune-system modulation can be mediated primarily via the bacterial surface lipoproteins.34 These lipoproteins can be categorized into three main subclasses: 1) Osps interacting with the innate immune system via PRRs,34 2) lipoproteins that mediate attachment to the extracellular matrix (ECM),35 and 3) lipoproteins that influence complement and acquired immune-response inactivation to prolong bacterial survival.36
Outer-surface proteins in B. burgdorferi
When bacteria enter the tick, the new environment activates the Rrp2–RpoN–RpoS pathway, a novel regulatory network that governs the differential expression of numerous genes of B. burgdorferi, including lipoproteins and the two major virulence genes OSPA and OSPC, allowing for bacterial survival within the arthropod vector.37 Following infection of the tick after feeding on an infected host, B. burgdorferi persists in the tick gut, at which time OspA and OspB are expressed, allowing attachment to the midgut endothelium.2 Expression of surface lipoproteins BptA7438 and BB69039 is also critical for bacterial colonization and prolonged residence in the tick midgut. Feeding activates bacterial translocation from the midgut to the tick’s salivary glands, which is facilitated by interaction between such lipoproteins as BBE3140 and BBA52.41 At this stage, OspA is downregulated and replaced with OspC.42 This allows B. burgdorferi to migrate to the salivary glands and bind the Salp15 protein, which has immunosuppressive properties and likely aids bacterial survival during the early stage of infection of vertebrate hosts.43 OspC has been shown to play a significant role in invading the salivary glands and thus the subsequent transmission of B. burgdorferi from the tick to the vertebrate host.44 OspC has plasminogen-binding activity, which likely facilitates the migration of bacteria within the host by destroying intercellular junctions.45 OspC is not the only B. burgdorferi lipoprotein that has plasminogen-binding activity: OspA, ErpA/C/P46 and CRASP147 have also exhibited varying binding affinities to human plasminogen. Hijacking of the plasminogen proteolytic system leads to the secretion of urokinase, which is thought to be synthesized as a result of TLR-related signaling activation.48 Plasminogen activation mediates a downstream cascade of activation of several host proteases that digest the ECM.49
While OspA expression diminishes during the early stages of infection, anti-OspA antibodies have been observed in patients with rheumatoid arthritis, suggesting that OspA may also play a role in the manifestation of persistent disease outcomes.50 Scheckelhoff et al51 reported that when recognized by B. burgdorferi, host adrenergic stress hormones lead to OspA expression for what was hypothesized to be a reentry stage into a tick vector during the bacterial life cycle. The role of OspA in persistent disease was in part determined by its homology to LFA1, an adhesion molecule mediating cell–cell interactions during inflammation and found only in B. burgdorferi sensu stricto. This homology results in autoimmune reactions responsible for some posttreatment LD syndromes.52 IgG antibody titers to OspA in persistent Lyme arthritis patients correlate with the severity of this condition.53
B. burgdorferi attachment to extracellular matrix
In order to establish initial infection, B. burgdorferi binds to various components of the host ECM. One of the pathways involves bacterial binding to glycosaminoglycans (GAGs) on the host cell surface. The GAG-binding protein Bgp attaches to host cells and inactivates toxic metabolites, allowing bacterial growth.35 The DbpA and DbpB proteins bind decorin, which is a type I collagen-associated GAG.54 Since the Bgp and Dbp proteins complement each other during establishment of infection within the host, constant expression of either is not necessary.35
The P66 adhesin protein has been shown to bind the cell-surface integrins αIIβ3 and αvβ3, causing upregulation of surface-adhesion molecules.55 Fibronectin, another component of the ECM, has been identified as an additional ligand for the B. burgdorferi lipoproteins BBK3256 and RevA/RevB. Furthermore, RevA,57 along with ErpX58 and Bmp,59 bind laminin within the host ECM.60 BmpA can be directly involved in Lyme arthritis, due to its spatial and temporal upregulation in the joints, which coincides with the development of inflammation.60 Not all adhesin proteins are expressed to the same extent, and lack of one type is usually compensated for by upregulated expression of another.61
Complement inactivation in Lyme disease
The complement system is a major component of the innate immune system, which acts as a first defense against pathogenic invasion. Complement comprises a complex of more than 30 proteins that through different pathways lead to the conversion of C3 to C3a and C3b by C3-convertase proteins. C3b aids in opsonization of pathogens, thereby attracting immune cells that directly kill pathogens. The complement system is tightly regulated by employing several C3-convertase regulators that either directly dissociate C3 convertase or degrade the reaction products.62 B. burgdorferi uses such complement C3-convertase regulators to its own advantage, where various surface lipoproteins bind factor H, which is responsible for mediating cleavage of C3b.63,64 By binding B. burgdorferi factor H, the bacteria facilitate further cleavage of C3b and downregulate the overall activation of the immune system.36 CRASP1 or CspA has been identified as a critical component in immune-system evasion by B. burgdorferi, where complement sensitivity was increased in human sera with CspA-knockout bacteria.36 Although binding of host complement regulators is the main mechanism of bypassing complement-mediated killing, CspA has also been found to bind late complement products like C7, C8, and C9 in the absence of regulators and interfere with assembly of the membrane attack complex.65 Furthermore, CspA has also been shown to take part in host cell adhesion by binding to various ECM proteins. This seems to occur, however, at a different binding site, and thereby CspA can simultaneously bind factor H, downregulate host defenses, and enhance tissue invasion.47
B. burgdorferi and corresponding TLR signaling
There are ten different human TLRs (Table 1),16,17,32 but only a few of these are capable of recognizing and mounting a response to B. burgdorferi. TLR3, TLR7, and TLR8 do not respond to B. burgdorferi lipoproteins, as they are intracellular sensors that recognize viral single- and double-stranded nucleic acids. Nonetheless, some studies have indicated a link between B. burgdorferi and TLR766 and TLR867 that induce the synthesis of type I IFNs.68 TLR9 recognizes bacterial CpG DNA, and might play a role in B. burgdorferi infection when the bacterial DNA is made accessible via lysis or degradation.26 TLR2 and TLR4 are extracellular receptors that recognize bacterial ligands, and have been demonstrated to play a role in B. burgdorferi infection.68
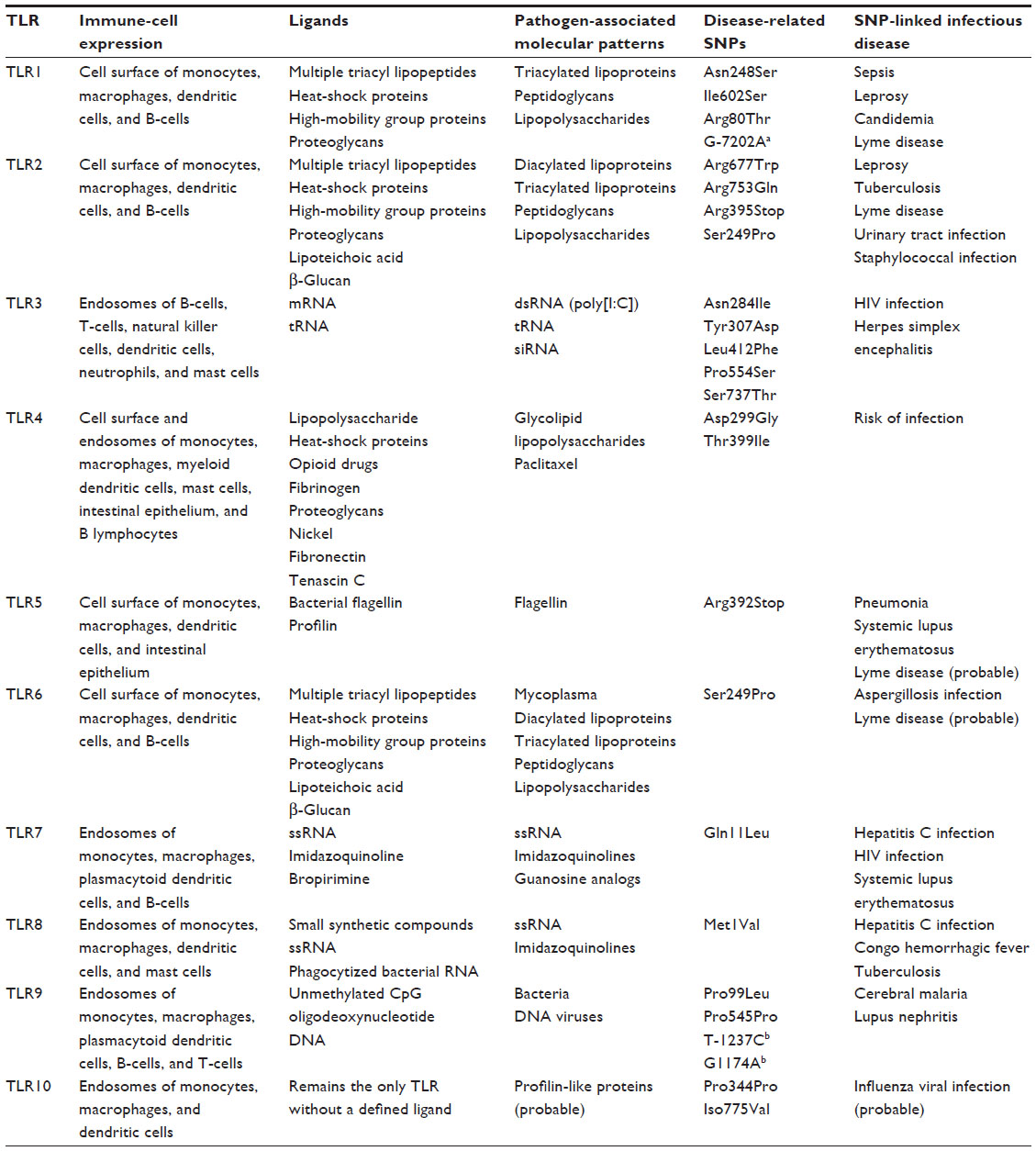 | Table 1 Characteristics of the Toll-like receptors and their gene polymorphism-related infectious diseases |
TLR2 has a broad range of ligands, due to its ability to form heterodimer receptors with other TLRs. When TLR2 associates as a heterodimer with TLR6, it can recognize peptidoglycan and diacylated lipopeptides. The TLR2/1 heterodimer, on the other hand, recognizes bacterial lipoproteins, such as OspA. However, an innate immune response to B. burgdorferi was still observed in TLR1- and TLR2-knockout mice,69 indicating an alternative mechanism(s) for B. burgdorferi recognition. TLR4 normally responds to the highly immunogenic surface glycolipid LPS of Gram-negative bacteria (see Table 1) that is lacking in B. burgdorferi. TLR2 and TLR4 downstream cascades are similar, where TIRAP is involved in the MyD88-dependent pathway via both receptors,16 which may account for the similarity in innate immune response to B. burgdorferi lipoproteins and response to LPS. Both TLR2 and TLR4 employ the CD14 co-receptor to enhance the detection of LPS and recognize other pathogen-associated molecular patterns (PAMPs), such as lipoteichoic acid.34 CD14 is a surface receptor on neutrophils and monocytes/macrophages, binds to and facilitates signaling by LPS, and is also the signaling receptor for B. burgdorferi lipoproteins and can bind to both OspA and OspC.70 A loss of this CD14 or its function was found to result in a severe inflammatory response to B. burgdorferi infection.34 The role of TLR5 in Borrelia infection is somewhat unclear. Although this TLR is known to respond to the flagellin protein (Table 1), B. burgdorferi has endoflagella that are not surface-exposed. In other similar pathogenic spirochetes, eg, Treponema phagedenis, TLR5 activation could occur due to transient gaps in the outer membrane.71 Despite the fact that the presence of these gaps is yet to be fully characterized in B. burgdorferi, they might provide an explanation for the reported involvement of TLR5 in modulating innate immune responses to the pathogen.72
TLR cascade in Lyme disease
The innate immune system can be activated by a wide range of pathogenic molecules, due to its ability to recognize PAMPs by PRRs (Table 1). PAMPs are molecular signatures of pathogens that are conserved for infectivity and/or survival across various classes of microorganisms that are not expressed by host cells.73 Although their downstream cascades and terminal effectors are quite similar, the ten human TLRs respond differently to different PAMPs.68
Each TLR has an extracellular domain, which contains binding sites for ligands, and a cytoplasmic domain involved in signal transmission. However, the cytoplasmic domains of TLRs are too short to transduce signals, and require additional proteins to carry out this function.16 The TIR component of cytoplasmic domains functions by recruiting adapter proteins like MyD88, TIRAP, TRIF, or TRAM, and facilitates dimerization of different TLRs.16,17 The first step in signal transduction is recruitment of the MyD88 adapter, which triggers the assembly of several IRAK scaffold proteins to form an MyD88–IRAK complex (myddosome). The IRAK complex subsequently recruits TRAF6 and TAKs to phosphorylate IKK.16 Upon phosphorylation, IKK dissociates from the NFºB transcription factor, allowing for its translocation into the nucleus and activation of a number of genes that code for a variety of proinflammatory cytokines, chemokines, and other effectors.17,74 Expression of TNF and IL-1β lead to activation of the endothelium and local upregulation of cell surface-adhesion molecules, allowing for tethering of immune cells to the site of infection.17 IL-1β and IL-6 act together in liver cells to produce acute-phase reactants, which in turn activate the complement cascade and enhance opsonization of the pathogen. Signaling through TLRs also leads to direct killing of the pathogen via production and secretion of antimicrobial peptides into the intracellular phagocytic compartments of immune cells.75 The following subsections emphasize on and explain the role of TLRs and related signaling, as well as pathogen–host interaction along this pathway, in LD susceptibility and early and long-term complications.
TLRs relevant to Lyme disease
A number of TLRs have been shown to play a significant role in the susceptibility and risk of LD. The involvement of TLRs in LD pathogenesis occurs through: 1) proinflammatory reactions mediated by the endosomal TLRs (TLR3, TLR7, TLR8, and TLR9) upon phagocytosis of the bacterium, or 2) the interaction of B. burgdorferi lipoproteins with plasma membrane TLRs (TLR1, TLR2, TLR4, TLR5, and TLR6).67,76 The ensuing inflammatory reactions induced by these processes differ both quantitatively and qualitatively.67,76 For example, internalization of live spirochetes leads to a higher synthesis of different proinflammatory cytokines than when the pathogen lipoproteins interact with cell-surface TLRs.77 Furthermore, different cytokines can be induced by different host TLRs.67 Taken together, these observations may at least partially explain interindividual variability in response to B. burgdorferi infection and in LD symptomatology.
Numerous studies have examined the involvement of TLR2 in the response to B. burgdorferi independently or as a heterodimer with TLR1 and TLR6.78,79 TLR2 recognizes B. burgdorferi OspA, as demonstrated in human peripheral blood mononuclear cells (PBMCs)66 and in OspA-vaccinated mice,68 leading to the expression of NFκB-dependent cytokines. Neutrophils from patients with LD exhibited elevated expression of TLR2 messenger RNA and protein together with an excessive synthesis of IL-6 and IL-1β.80 In contrast, TLR2-deficient mice had an impaired innate immune response to B. burgdorferi OspA and a diminished cytokine synthesis from their bone marrow-derived macrophages.81,82 Moreover, B. burgdorferi-infected TLR2–/– mice had 50- to 250-fold higher numbers of spirochetes and severe arthritis81,83,84 and inflammatory carditis85 compared to their wild-type homozygous or heterozygous counterparts. This effect was also noted to a lesser extent in TLR2–/+ mice,86 demonstrating that one nonfunctional allele of TLR2 may be enough to impair cytokine synthesis and influence disease susceptibility upon exposure to B. burgdorferi. Some evidence suggests that TRIF and MYD88 adapters are essential to the inflammatory function of TLR2, and may have a more critical role than TLR2 in LD pathogenesis. TRIF-deficient mice had lower cytokine production in response to B. burgdorferi compared to their wild-type counterparts,87 whereas MyD88 had a more marked effect on phagocytosis of the bacterium than TLR2.67,82
Other members of the TLR family were also implicated in the susceptibility to LD and its complications. A status of enhanced pathogen lipoprotein recognition was elicited upon the heterodimerization of TLR1 with TLR2.88 In addition, TLR5 exhibits a transcriptional upregulation upon B. burgdorferi phagocytosis, although a receptor for bacterial flagellin is lacking in B. burgdorferi.26 Moreover, the expression of endosomal TLR7 and TLR9, mediators of type I IFN synthesis in dendritic cells, was downregulated upon exposure to B. burgdorferi,66 together with the synthesis of IFNα,89 IFNγ1,66 and the transcription of IFN-induced genes.89 Similarly, downregulation of TLR8, which influences the synthesis of IFNβ, inhibits cytokine release in cells that have phagocytosed B. burgdorferi.67,90
Genetic polymorphisms of TLRs in Lyme disease etiology
A number of SNPs have been identified in the TLR genes and their downstream factors91,92 (Table 1). These polymorphisms are known to alter TLR-signaling patterns, and have been linked to changes in clinical manifestations caused by bacterial, fungal, and viral infections.76 For example, TLR1 Ile602Ser was linked to elevated synthesis of proinflammatory cytokines and a more effective TH1-like response in patients with LD.72 On the other hand, TLR2 Arg677Trp and Arg753Gln were associated with lepromatous leprosy93 and tuberculosis.94–96 In LD, however, the TLR2 Arg753Gln polymorphism provides protection against the development of late-stage disease.86 Compared to wild-type PBMCs from healthy individuals, cells with TLR1 Arg80Thr, Asn248Ser, Ile602Ser, and TLR6 Ser249Pro had a significantly lower synthesis of proinflammatory cytokines when heterodimerized with TLR2.97
TLR1 T1805G (Ile602Ser), TLR2 G2258A (Arg753Gln), and TLR5 C1174T (Arg395Stop) were examined in patients with different LD-associated clinical symptoms, including erythema migrans, antibiotic-responsive arthritis, and antibiotic-refractory arthritis.72 These SNPs were associated with an impairment of downstream signaling and reduced cytokine production by decreasing the number of plasma membrane TLRs (TLR1 T1805G and TLR2 G2258A) or with abrogation of the cellular flagellin-signaling pathway (TLR5 C1174T). Subjects with antibiotic-refractory arthritis had approximately twofold-higher frequency of TLR1 Ile602Ser (T1805G) compared to subjects with erythema migrans (odds ratio [OR] 1.9, P=0.05)72 (Table 2). The status of antibiotic-refractory Lyme arthritis occurs when there is persistence of synovitis for at least 3 months after antibiotic treatment, despite expulsion of viable B. burgdorferi from the affected area.98
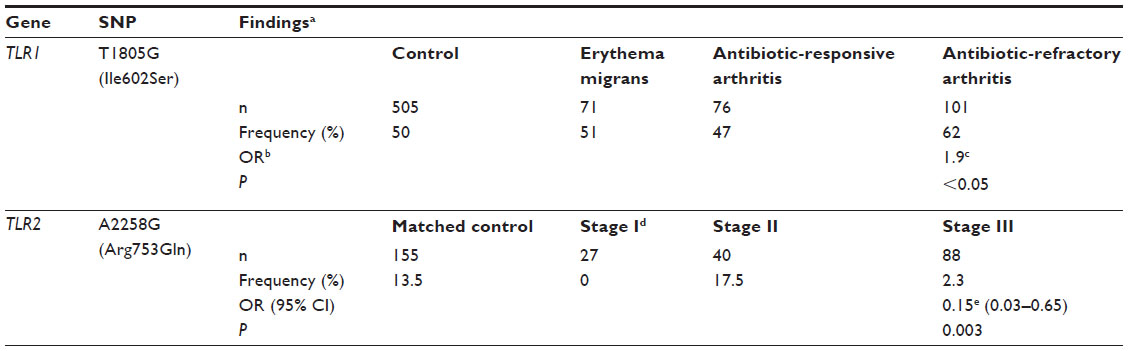 | Table 2 Impact of TLR1 T1805G (Ile602Ser) and TLR2 A2258G (Arg753Gln) alleles on late-stage Lyme disease |
In contrast to the possible increased risk of Lyme arthritis associated with TLR1 Ile602Ser (T1805G), TLR2 Arg293Gln (A2258G) was shown in one study to be protective.86 The arginine residue at the 753 position of TLR2, located within the signal-transduction domain of the receptor, is critical for the stable formation of the TLR1–TLR2 heterodimer.86,91 The frequency of TLR2 Arg753Gln (A2258G) was lower in LD patients compared to matched controls (OR 0.39, 95% confidence interval 0.17–0.89; P=0.03). In this study, patients with stage III LD (late persistent disease with Lyme arthritis) had a further lower frequency of Arg753Gln (A2258G) compared to their matched controls (OR 0.15, 95% confidence interval 0.03–0.65; P=0.003)86 (Table 2). These findings substantiated a protective effect of TLR2 Arg293Gln against Lyme arthritis, a clinical symptom in the late stages of LD.
Functional effects of TLR polymorphisms
A number of genotype–phenotype matching studies were carried out to elucidate the functional effects of gene polymorphisms in TLRs on host cytokine synthesis in response to B. burgdorferi infection (Table 3). Patients showing erythema migrans who were carriers of the TLR1 T1805G allele and infected with the RST1 strain of B. burgdorferi had higher levels of IL-6 and the IFNγ-inducible chemokines CXCL9 and CXCL10 in their PBMCs compared to wild-type carriers.72 These subjects also exhibited elevated serum cytokine and chemokine levels than patients infected with the RST2 or RST3 strains. Similar findings were observed for the TLR2 Arg753Gln and TLR5 Arg395Stop SNPs (Table 3). The RST1 strain of B. burgdorferi-carrying genes for OspC major group A is known to be associated with stimulation of macrophages to release higher levels of IL-6, IL-8, CCL3, CCL4, TNF, and IL-1β than RST2 and RST3 strains.99,100
 | Table 3 Summary of TLR-gene polymorphisms’ functional significance in Lyme disease and their role in host–pathogen interactions |
Similarly to patients with erythema migrans, subjects with Lyme arthritis exhibited elevated TH1-like adaptive cytokines and chemokines in the synovial fluid of individuals with the TLR1 T1805G compared to wild-type carriers.72 Evidence for the association of the TLR1 T1805G with this stage of LD was substantiated from observations demonstrating that patients with antibiotic-refractory arthritis had approximately 15- and twofold-higher CXCL9 and CXCL10 chemokine levels, respectively, than subjects with erythema migrans and antibiotic-responsive arthritis.72 CXCL9 and CXCL10 chemokines act as chemoattractants for CD4+ and CD8+ T-cells, the main infiltrating cell types that cause chronic synovial inflammation in antibiotic-refractory Lyme arthritis,101 suggesting a role for these factors in symptom persistence despite antibiotic therapy at the late disease stages. Further studies on three different TLR1 polymorphisms in PBMCs from individuals homozygous for Arg80Thr, Asn248Ser, and Ser602Ile demonstrated that stimulation with the Pam3Cys ligand can significantly lower cytokine production compared to that in the wild-type carrier cells.97 Arg80Thr influenced the synthesis of IL-1β, whereas Asn248Ser and Ile602Ser affected that of IL-6, IL8, TNFα, and IL-10. The three TLR1 SNPs affected the production of IFNγ and IL-17.72,97
The effect of the TLR2 Arg753Gln SNP on the inflammatory responses in blood samples from heterozygous and wild-type carrier subjects has been studied.86 Upon stimulation with B. burgdorferi lysate, the SNP carriers exhibited decreased synthesis of TNFα and IFNγ compared to their wild-type counterparts.86 This observation was thought to be related to the influence of the nonfunctional allele on stabilizing the TLR2–TLR1 heterodimer that interacts with triacylated B. burgdorferi lipoproteins. Further studies on the human kidney embryo epithelial cell line HEK293 (heterozygous for TLR2 Arg293Gln) stimulated with the synthetic diacylated lipopeptide Pam2Cys or with B. burgdorferi lysate demonstrated the ability of TLR2 Arg293Gln to induce ~50% lowering in cytokine synthesis and downregulate the innate immune response compared to the wild-type allele.86 The nonsynonymous Ser249Pro in TLR6, another coreceptor of TLR2, causes a malfunction of its extracellular domain.97 Although stimulation with the specific TLR2–TLR6 ligand FSL1 in PBMCs from Ser249Pro carriers did not change the levels of IL-1β, IL-6, IL-8, IL-10, or TNFα, lower levels were observed with B. burgdorferi treatment.97 This observation indicates a limited role of TLR6 in cytokines response to B. burgdorferi infection and LD risk, severity, and complications.
Conclusion
Although several in vitro and animal studies have demonstrated a role of TLRs 1, 2, and 4–9 in LD, the consensus of available information particularly implicates TLRs 1 and 2 in the process of disease risk and severity. The nature of TLR interaction with pathogen lipoproteins and the ensuing pattern of cytokine synthesis differs depending on the cellular location of the TLRs67,77 and the presence or absence of a polymorphism in their regulating genes.72,86 This effect may be linked to the involvement of receptors and their downstream signaling at different stages of LD.
A number of functional TLR SNPs were recently reported to include differential cytokine synthesis and level, altered pathogen recognition, and disruption of the downstream signaling cascade.72,97 However, contradictory results may preclude a definitive conclusion about the effect of a particular TLR SNP(s) on host–pathogen interaction in LD etiology. For example, following cell exposure to B. burgdorferi lysate, TLR2 Arg753Gln did not significantly affect basal chemokine/cytokine levels in one study,72 although it was linked to a marked reduction in the synthesis of these factors in another report.86 The effects of the TLR6 Ser249Pro polymorphism on cytokine synthesis in response to B. burgdorferi were similarly inconsistent.97 While this SNP results in disruption of the extracellular TLR6 domain and influences the TLR2–TLR6 heterodimeric structure, it does not affect cytokine synthesis in cells exposed to FSL1 (a TLR2–TLR6 ligand), but only in those treated with live B. burgdorferi.97 Although the contradictory findings for each of TLR2 Arg753Gln72,86 and TLR6 Ser249Pro97 may be simply attributed to the differences in the types of treated cells and/or administered B. burgdorferi components, it highlights the need to develop further studies to elucidate the genotype–phenotype relationship of various polymorphisms along the TLR cascade and their function in LD risk, severity, and complications.
Several approaches can be considered to identify TLR SNPs and their functional effect in LD. For instance, the candidate-gene approach could be employed, given its ability to evaluate directly the effects of specific gene variants (even those with small effects) in disease causation and progression.102 This approach can be limited by the small number of relevant SNPs that have been already identified in LD in human, animal, and cell-culture models. However, evaluating TLR SNPs examined for their role in other infectious diseases (eg, those mentioned in Table 3) can provide an initial guide for these types of studies. Genome-wide association studies can be similarly considered as a feasible approach to identify novel candidate TLRs and other SNPs throughout the human genome that may play a role in disease risk and severity.103 Developing these types of studies however, can be hampered by the requirement for large sample sizes in order to identify associations between SNPs and disease outcomes. Ideally, meta-analysis would have been used here to study the role of TLR SNPs in LD as an alternative to candidate-gene studies or genome-wide association-study approaches. However, in our literature search we found only three studies addressing the role of TLR polymorphisms in human disease, and there were inconsistent findings and significant limitations in methodologies in these studies.72,86,97 This inadequate number of reports is not sufficient or comprehensive enough for meta-analyses at this time.
A recombinant OspA Lyme vaccine was approved by the US Food and Drug Administration in 1998, and had approximately 80% efficacy in adults. In 2002, the manufacturer voluntarily withdrew this vaccine product for fears of vaccine side effects and declining sales. Currently, no vaccine is available for LD. Antibiotics and extended-treatment plans are the available options now for mitigating early and chronic LD, respectively. Indeed, before attempting to utilize TLRs in the prevention of LD, large-scale studies addressing the role of TLR SNPs in host–pathogen interaction should be generated. In this respect, it is necessary to consider a systematic and more robust research approach in human subjects. One possibility can be adopted from studies that recently defined the role of TLR SNPs in influencing the inflammatory response in obstructive pulmonary disease.104 In that report, nine different TLR2 and 17 TLR4 SNPs were identified in a longitudinal study by employing SNP tagging while analyzing their effect on lung function and inflammatory reactions. In this manner, the roles of the polymorphisms in disease severity and progression were determined. In parallel, it will be imperative to develop prospective cohort studies nationally and/or internationally of patients at different stages of LD. These would enable the design of multiple candidate-gene studies (aided by animal or cell-culture models) or genome-wide association studies to identify particular TLR polymorphisms related to disease risk, severity, and complications. These studies have the potential to provide not only a better understanding of disease pathogenesis but also an insight into novel, therapeutic targets during active disease or postinfection and posttreatment complications, as well as disease-preventive targets aiding in vaccine development.
Author contributions
AB conceived the study idea and design. AB, SR, and MS prepared the first draft of the manuscript. NHO and RL helped in drafting the article and contributed to study design. All authors critically reviewed the manuscript, approved the final draft, and agreed to be accountable for all aspects of the work.
Acknowledgments
The authors thank Dr Paul Arora for discussing the manuscript. This work was supported by a fund from the Public Health Agency of Canada.
Disclosure
The authors report no conflicts of interest in this work.
References
Tilly K, Rosa PA, Stewart PE. Biology of infection with Borrelia burgdorferi. Infect Dis Clin North Am. 2008;22(2):217–234. | |
Berende A, Oosting M, Kullberg BJ, Netea MG, Joosten LA. Activation of innate host defense mechanisms by Borrelia. Eur Cytokine Netw. 2010;21(1):7–18. | |
[No authors listed]. Lyme disease in Canada: Q and A for paediatricians. Paediatr Child Health. 2009;14(2):103–108. | |
Mead PS. Epidemiology of Lyme disease. Infect Dis Clin North Am. 2015;29(2):187–210. | |
Ho K, Melanson M, Desai JA. Bell palsy in Lyme disease-endemic regions of Canada: a cautionary case of occult bilateral peripheral facial nerve palsy due to Lyme disease. CJEM. 2012;14(5):321–324. | |
Ogden NH, Bouchard C, Kurtenbach K, et al. Active and passive surveillance and phylogenetic analysis of Borrelia burgdorferi elucidate the process of Lyme disease risk emergence in Canada. Environ Health Perspect. 2010;118(7):909–914. | |
Koffi JK, Leighton PA, Pelcat Y, et al. Passive surveillance for I. scapularis ticks: enhanced analysis for early detection of emerging Lyme disease risk. J Med Entomol. 2012;49(2):400–409. | |
Ogden NH, Koffi JK, Pelcat Y, Lindsay R. Environmental risk from Lyme disease in central and eastern Canada: a summary of recent surveillance information. Can Commun Dis Rep. 2014;40(5):74–82. | |
Hatchette T, Ian D, Johnston L. Lyme disease: clinical diagnosis and treatment. CCDR. 2014;40(11):194–208. | |
Hatchette TF, Johnston BL, Schleihauf E, et al. Epidemiology of Lyme disease, Nova Scotia, Canada, 2002-2013. Emerg Infect Dis. 2015;21(10):1751–1758. | |
Aucott J, Morrison C, Munoz B, Rowe PC, Schwarzwalder A, West SK. Diagnostic challenges of early Lyme disease: lessons from a community case series. BMC Infect Dis. 2009;9:79. | |
Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004;113(8):1093–1101. | |
Shapiro ED, Auwaerter P. Borrelia burgdorferi (Lyme disease). In: Long SS, Pickering LK, Prober CG, editors. Principles and Practice of Pediatric Infectious Diseases. New York: Churchill Livingstone; 1997. | |
Ogden NH, Lindsay LR, Morshed M, Sockett PN, Artsob H. The emergence of Lyme disease in Canada. CMAJ. 2009;180(12):1221–1224. | |
Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089–1134. | |
Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004; 16(1):3–9. | |
Beutler B. Innate immunity: an overview. Mol Immunol. 2004; 40(12):845–859. | |
Choe J, Kelker MS, Wilson IA. Crystal structure of human Toll-like receptor 3 (TLR3) ectodomain. Science. 2005;309(5734):581–585. | |
Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: focus on the Toll-like receptors. J Intern Med. 2007;261(6):511–528. | |
Kumar S, Ingle H, Prasad DV, Kumar H. Recognition of bacterial infection by innate immune sensors. Crit Rev Microbiol. 2013;39(3):229–246. | |
Wooten RM, Modur VR, McIntyre TM, Weis JJ. Borrelia burgdorferi outer membrane protein A induces nuclear translocation of nuclear factor-κB and inflammatory activation in human endothelial cells. J Immunol. 1996;157(10):4584–4590. | |
Ma Y, Weis JJ. Borrelia burgdorferi outer surface lipoproteins OspA and OspB possess B-cell mitogenic and cytokine-stimulatory properties. Infect Immun. 1993;61(9):3843–3853. | |
Ma Y, Seiler KP, Tai KF, Yang L, Woods M, Weis JJ. Outer surface lipoproteins of Borrelia burgdorferi stimulate nitric oxide production by the cytokine-inducible pathway. Infect Immun. 1994;62(9):3663–3671. | |
Morrison TB, Weis JH, Weis JJ. Borrelia burgdorferi outer surface protein A (OspA) activates and primes human neutrophils. J Immunol. 1997;158(10):4838–4845. | |
Schnare M, Röllinghoff M, Qureshi S. Toll-like receptors: sentinels of host defence against bacterial infection. Int Arch Allergy Immunol. 2006;139(1):75–85. | |
Bernardino AL, Myers TA, Alvarez X, Hasegawa A, Philipp MT. Toll-like receptors: insights into their possible role in the pathogenesis of Lyme neuroborreliosis. Infect Immun. 2008;76(10):4385–4395. | |
Petzke MM, Brooks A, Krupna MA, Mordue D, Schwartz I. Recognition of Borrelia burgdorferi, the Lyme disease spirochete, by TLR7 and TLR9 induces a type I IFN response by human immune cells. J Immunol. 2009;183(8):5279–5292. | |
Radolf JD, Caimano MJ, Stevenson B, Hu LT. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol. 2012;10(2):87–99. | |
Batsford S, Dunn J, Mihatsch M. Outer surface lipoproteins of Borrelia burgdorferi vary in their ability to induce experimental joint injury. Arthritis Rheum. 2004;50(7):2360–2369. | |
DuChateau BK, Munson EL, England DM, et al. Macrophages interact with enriched populations of distinct T lymphocyte subsets for the induction of severe destructive Lyme arthritis. J Leukoc Biol. 1999;65(2):162–170. | |
Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol. 2003;4(12):1230–1237. | |
Lin YT, Verma A, Hodgkinson CP. Toll-like receptors and human disease: lessons from single nucleotide polymorphisms. Curr Genomics. 2012;13(8):633–645. | |
Cervantes JL, Hawley KL, Benjamin SJ, Weinerman B, Luu SM, Salazar JC. Phagosomal TLR signaling upon Borrelia burgdorferi infection. Front Cell Infect Microbiol. 2014;4:55. | |
Benhnia MR, Wroblewski D, Akhtar MN, et al. Signaling through CD14 attenuates the inflammatory response to Borrelia burgdorferi, the agent of Lyme disease. J Immunol. 2005;174(3):1539–1548. | |
Parveen N, Cornell KA, Bono JL, Chamberland C, Rosa P, Leong JM. Bgp, a secreted glycosaminoglycan-binding protein of Borrelia burgdorferi strain N40, displays nucleosidase activity and is not essential for infection of immunodeficient mice. Infect Immun. 2006;74(5):3016–3020. | |
Kraiczy P, Skerka C, Kirschfink M, Brade V, Zipfel PF. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and factor H. Eur J Immunol. 2001;31(6):1674–1684. | |
Xu H, He M, He JJ, Yang XF. Role of the surface lipoprotein BBA07 in the enzootic cycle of Borrelia burgdorferi. Infect Immun. 2010;78(7):2910–2918. | |
Revel AT, Blevins JS, Almazán C, et al. BptA (bbe16) is essential for the persistence of the Lyme disease spirochete, Borrelia burgdorferi, in its natural tick vector. Proc Natl Acad Sci U S A. 2005;102(19):6972–6977. | |
Li X, Pal U, Ramamoorthi N, et al. The Lyme disease agent Borrelia burgdorferi requires BB0690, a Dps homologue, to persist within ticks. Mol Microbiol. 2007;63(3):694–710. | |
Zhang L, Zhang Y, Adusumilli S, et al. Molecular interactions that enable movement of the Lyme disease agent from the tick gut into the hemolymph. PLoS Pathog. 2011;7(6):e1002079. | |
Kumar M, Kaur S, Kariu T, et al. Borrelia burgdorferi BBA52 is a potential target for transmission blocking Lyme disease vaccine. Vaccine. 2011;29(48):9012–9019. | |
Tokarz R, Anderton JM, Katona LI, Benach JL. Combined effects of blood and temperature shift on Borrelia burgdorferi gene expression as determined by whole genome DNA array. Infect Immun. 2004;72(9):5419–5432. | |
Marchal CM, Luft BJ, Yang X, Sibilia J, Jaulhac B, Boulanger NM. Defensin is suppressed by tick salivary gland extract during the in vitro interaction of resident skin cells with Borrelia burgdorferi. J Invest Dermatol. 2009;129(10):2515–2517. | |
Pal U, Yang X, Chen M, et al. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J Clin Invest. 2004;113(2):220–230. | |
Lagal V, Portnoï D, Faure G, Postic D, Baranton G. Borrelia burgdorferi sensu stricto invasiveness is correlated with OspC-plasminogen affinity. Microbes Infect. 2006;8(3):645–652. | |
Floden AM, Watt JA, Brissette CA. Borrelia burgdorferi enolase is a surface-exposed plasminogen binding protein. PLoS One. 2011;6(11):e27502. | |
Hallström T, Haupt K, Kraiczy P, et al. Complement regulator-acquiring surface protein 1 of Borrelia burgdorferi binds to human bone morphogenic protein 2, several extracellular matrix proteins, and plasminogen. J Infect Dis. 2010;202(3):490–498. | |
Coleman JL, Benach JL. The urokinase receptor can be induced by Borrelia burgdorferi through receptors of the innate immune system. Infect Immun. 2003;71(10):5556–5564. | |
Coleman JL, Roemer EJ, Benach JL. Plasmin-coated Borrelia burgdorferi degrades soluble and insoluble components of the mammalian extracellular matrix. Infect Immun. 1999;67(8):3929–3936. | |
Hsieh YF, Liu HW, Hsu TC, et al. Serum reactivity against Borrelia burgdorferi OspA in patients with rheumatoid arthritis. Clin Vaccine Immunol. 2007;14(11):1437–1441. | |
Scheckelhoff MR, Telford SR, Wesley M, Hu LT. Borrelia burgdorferi intercepts host hormonal signals to regulate expression of outer surface protein A. Proc Natl Acad Sci U S A. 2007;104(17):7247–7252. | |
Gross DM, Huber BT. Cellular and molecular aspects of Lyme arthritis. Cell Mol Life Sci. 2000;57(11):1562–1569. | |
Ghosh S, Huber BT. Clonal diversification in OspA-specific antibodies from peripheral circulation of a chronic Lyme arthritis patient. J Immunol Methods. 2007;321(1–2):121–134. | |
Salo J, Loimaranta V, Lahdenne P, Viljanen MK, Hytönen J. Decorin binding by DbpA and B of Borrelia garinii, Borrelia afzelii, and Borrelia burgdorferi sensu stricto. J Infect Dis. 2011;204(1):65–73. | |
LaFrance ME, Pierce JV, Antonara S, Coburn J. The Borrelia burgdorferi integrin ligand P66 affects gene expression by human cells in culture. Infect Immun. 2011;79(8):3249–3261. | |
Fikrig E, Feng W, Barthold SW, Telford SR, Flavell RA. Arthropod-and host-specific Borrelia burgdorferi bbk32 expression and the inhibition of spirochete transmission. J Immunol. 2000;164(10):5344–5351. | |
Brissette CA, Bykowski T, Cooley AE, Bowman A, Stevenson B. Borrelia burgdorferi RevA antigen binds host fibronectin. Infect Immun. 2009;77(7):2802–2812. | |
Brissette CA, Verma A, Bowman A, Cooley AE, Stevenson B. The Borrelia burgdorferi outer-surface protein ErpX binds mammalian laminin. Microbiology. 2009;155(3):863–872. | |
Verma A, Brissette CA, Bowman A, Stevenson B. Borrelia burgdorferi BmpA is a laminin-binding protein. Infect Immun. 2009;77(11):4940–4946. | |
Pulzova L, Bhide M. Outer surface proteins of Borrelia: peerless immune evasion tools. Curr Protein Pept Sci. 2014;15(1):75–88. | |
Yang X, Izadi H, Coleman AS, et al. Borrelia burgdorferi lipoprotein BmpA activates pro-inflammatory responses in human synovial cells through a protein moiety. Microbes Infect. 2008;10(12–13):1300–1308. | |
Ekdahl KN, Henningsson AJ, Sandholm K, Forsberg P, Ernerudh J, Ekerfelt C. Immunity in borreliosis with special emphasis on the role of complement. Adv Exp Med Biol. 2007;598:198–213. | |
McDowell JV, Wolfgang J, Tran E, Metts MS, Hamilton D, Marconi RT. Comprehensive analysis of the factor H binding capabilities of Borrelia species associated with Lyme disease: delineation of two distinct classes of factor H binding proteins. Infect Immun. 2003;71(6):3597–3602. | |
Kraiczy P, Hellwage J, Skerka C, et al. Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J Biol Chem. 2004;279(4):2421–2429. | |
Hammerschmidt C, Koenigs A, Siegel C, et al. Versatile roles of CspA orthologs in complement inactivation of serum-resistant Lyme disease spirochetes. Infect Immun. 2014;82(1):380–392. | |
Love AC, Schwartz I, Petzke MM. Borrelia burgdorferi RNA induces type I and III interferons via Toll-like receptor 7 and contributes to production of NF-κB-dependent cytokines. Infect Immun. 2014;82(6):2405–2416. | |
Cervantes JL, Dunham-Ems SM, La Vake CJ, et al. Phagosomal signaling by Borrelia burgdorferi in human monocytes involves Toll-like receptor (TLR) 2 and TLR8 cooperativity and TLR8-mediated induction of IFN-β. Proc Natl Acad Sci U S A. 2011;108(9):3683–3688. | |
Singh SK, Girschick HJ. Toll-like receptors in Borrelia burgdorferi-induced inflammation. Clin Microbiol Infect. 2006;12(8):705–717. | |
Alexopoulou L, Thomas V, Schnare M, et al. Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat Med. 2002;8(8):878–884. | |
Wooten RM, Morrison TB, Weis JH, Wright SD, Thieringer R, Weis JJ. The role of CD14 in signaling mediated by outer membrane lipoproteins of Borrelia burgdorferi. J Immunol. 1998;160(11):5485–5492. | |
Blanco DR, Radolf JD, Lovett MA, Miller JN. The antigenic interrelationship between the endoflagella of Treponema phagedenis biotype Reiter and Treponema pallidum Nichols strain. I. Treponemicidal activity of cross-reactive endoflagellar antibodies against T. pallidum. J Immunol. 1986;137(9):2973–2979. | |
Strle K, Shin JJ, Glickstein LJ, Steere AC. Association of a Toll-like receptor 1 polymorphism with heightened Th1 inflammatory responses and antibiotic-refractory Lyme arthritis. Arthritis Rheum. 2012;64(5):1497–1507. | |
Teixeira MM, Almeida IC, Gazzinelli RT. Introduction: innate recognition of bacteria and protozoan parasites. Microbes Infect. 2002;4(9):883–886. | |
Badawi A, Klip A, Haddad P, et al. Type 2 diabetes mellitus and inflammation: prospects for biomarkers of risk and nutritional intervention. Diabetes Metab Syndr Obes. 2010;3:173–186. | |
Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449(7164):819–826. | |
Frazão JB, Errante PR, Condino-Neto A. Toll-like receptors’ pathway disturbances are associated with increased susceptibility to infections in humans. Arch Immunol Ther Exp (Warsz). 2013;61(6):427–443. | |
Salazar JC, Duhnam-Ems S, La Vake C, et al. Activation of human monocytes by live Borrelia burgdorferi generates TLR2-dependent and-independent responses which include induction of IFN-β. PLoS Pathog. 2009;5(5):e1000444. | |
Takeuchi O, Sato S, Horiuchi T, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169(1):10–14. | |
Bulut Y, Faure E, Thomas L, Equils O, Arditi M. Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. J Immunol. 2001;167(2):987–994. | |
Jablonska E, Marcinczyk M. TLR2 expression in relation to IL-6 and IL-1 β and their natural regulators production by PMN and PBMC in patients with Lyme disease. Mediators Inflamm. 2006;2006(1):32071. | |
Wooten RM, Ma Y, Yoder RA, et al. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J Immunol. 2002;168(1):348–355. | |
Shin OS, Isberg RR, Akira S, Uematsu S, Behera AK, Hu LT. Distinct roles for MyD88 and Toll-like receptors 2, 5, and 9 in phagocytosis of Borrelia burgdorferi and cytokine induction. Infect Immun. 2008; 76(6):2341–2351. | |
Bolz DD, Sundsbak RS, Ma Y, et al. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J Immunol. 2004;173(3):2003–2010. | |
Liu N, Montgomery RR, Barthold SW, Bockenstedt LK. Myeloid differentiation antigen 88 deficiency impairs pathogen clearance but does not alter inflammation in Borrelia burgdorferi-infected mice. Infect Immun. 2004;72(6):3195–3203. | |
Wang G, Ma Y, Buyuk A, McClain S, Weis JJ, Schwartz I. Impaired host defense to infection and Toll-like receptor 2-independent killing of Borrelia burgdorferi clinical isolates in TLR2-deficient C3H/HeJ mice. FEMS Microbiol Lett. 2004;231(2):219–225. | |
Schröder NW, Diterich I, Zinke A, et al. Heterozygous Arg753Gln polymorphism of human TLR-2 impairs immune activation by Borrelia burgdorferi and protects from late stage Lyme disease. J Immunol. 2005;175(4):2534–2540. | |
Petnicki-Ocwieja T, Chung E, Acosta DI, et al. TRIF mediates Toll-like receptor 2-dependent inflammatory responses to Borrelia burgdorferi. Infect Immun. 2013;81(2):402–410. | |
Farhat K, Riekenberg S, Heine H, et al. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J Leukoc Biol. 2008;83(3):692–701. | |
Satoshi U, Shizuo A. Toll-like receptors and type I interferons. J Biol Chem. 2009;282(21):15319–15324. | |
Cervantes JL, La Vake CJ, Weinerman B, et al. Human TLR8 is activated upon recognition of Borrelia burgdorferi RNA in the phagosome of human monocytes. J Leukoc Biol. 2013;94(6):1231–1241. | |
Gautam JK, Comeau LD, Krueger JK, Smith MF. Structural and functional evidence for the role of the TLR2 DD loop in TLR1/TLR2 heterodimerization and signaling. J Biol Chem. 2006;281(40):30132–30142. | |
Hawn TR, Misch E, Dunstan SJ, et al. A common human TLR1 polymorphism regulates the innate immune response to lipopeptides. Eur J Immunol. 2007;37(8):2280–2289. | |
Kang TJ, Chae GT. Detection of Toll-like receptor 2 (TLR2) mutation in the lepromatous leprosy patients. FEMS Immunol Med Microbiol. 2001;31(1):53–58. | |
Ben-Ali M, Barbouche MR, Bousnina S, Chabbou A, Dellagi K. Toll-like receptor 2 Arg677Trp polymorphism is associated with susceptibility to tuberculosis in Tunisian patients. Clin Diagn Lab Immunol. 2004;11(3):625–626. | |
Dalgic N, Tekin D, Kayaalti Z, et al. Arg753Gln polymorphism of the human Toll-like receptor 2 gene from infection to disease in pediatric tuberculosis. Hum Immunol. 2011;72(5):440–445. | |
Wu H, Yang L. Arg753Gln polymorphisms in Toll-like receptor 2 gene are associated with tuberculosis risk: a meta-analysis. Med Sci Monit. 2015;21:2196–2202. | |
Oosting M, Ter Hofstede H, Sturm P, et al. TLR1/TLR2 heterodimers play an important role in the recognition of Borrelia spirochetes. PLoS One. 2011;6(10):e25998. | |
Sellati TJ, Sahay B, Wormser GP. The toll of a TLR1 polymorphism in Lyme disease: a tale of mice and men. Arthritis Rheum. 2012;64(5):1311–1315. | |
Strle K, Jones KL, Drouin EE, Li X, Steere AC. Borrelia burgdorferi RST1 (OspC type A) genotype is associated with greater inflammation and more severe Lyme disease. Am J Pathol. 2011;178(6):2726–2739. | |
Jones KL, McHugh GA, Glickstein LJ, Steere AC. Analysis of Borrelia burgdorferi genotypes in patients with Lyme arthritis: high frequency of ribosomal RNA intergenic spacer type 1 strains in antibiotic-refractory arthritis. Arthritis Rheum. 2009;60(7):2174–2182. | |
Steere AC, Duray PH, Butcher EC. Spirochetal antigens and lymphoid cell surface markers in Lyme synovitis. Arthritis Rheum. 1988;31(4):487–495. | |
Kwon JM, Goate AM. The candidate gene approach. Alcohol Res Health. 2000;24(3):164–168. | |
Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. | |
Budulac SE, Boezen HM, Hiemstra PS, et al. Toll-like receptor (TLR2 and TLR4) polymorphisms and chronic obstructive pulmonary disease. PLoS One. 2012;7(8):e43124. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.