Back to Journals » International Journal of Nanomedicine » Volume 16
TMTP1-Modified, Tumor Microenvironment Responsive Nanoparticles Co-Deliver Cisplatin and Paclitaxel Prodrugs for Effective Cervical Cancer Therapy
Authors Jiang G, Wang X, Zhou Y, Zou C, Wang L, Wang W, Zhang D, Xu H, Li J ![]() , Li F, Luo D, Ma X, Ma D, Tan S
, Li F, Luo D, Ma X, Ma D, Tan S ![]() , Wei R, Xi L
, Wei R, Xi L
Received 31 December 2020
Accepted for publication 28 April 2021
Published 15 June 2021 Volume 2021:16 Pages 4087—4104
DOI https://doi.org/10.2147/IJN.S298252
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Mian Wang
Guiying Jiang,1,* Xueqian Wang,1,* Ying Zhou,1 Chenming Zou,2,3 Ling Wang,1 Wei Wang,1 Danya Zhang,1 Hanjie Xu,1 Jie Li,1 Fei Li,1 Danfeng Luo,1 Xiangyi Ma,1 Ding Ma,1 Songwei Tan,2 Rui Wei,1 Ling Xi1
1Department of Obstetrics & Gynecology, Tongji Hospital, Tongji Medical College, Huazhong University of Science & Technology, Wuhan, Hubei Province, People’s Republic of China; 2Tongji School of Pharmacy, Tongji Medical College, Huazhong University of Science & Technology, Wuhan, Hubei Province, People’s Republic of China; 3School of Pharmacy, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ling Xi; Rui Wei
Department of Obstetrics & Gynecology, Tongji Hospital, Tongji Medical College, Huazhong University of Science & Technology, Wuhan, Hubei, People’s Republic of China
Tel/Fax +86 27 83662688
Email [email protected]; [email protected]
Background and Purpose: Cisplatin–paclitaxel (TP) combination chemotherapy as the first-line therapy for numerous cancers is hindered by its inadequate accumulation in tumors and severe side effects resulting from non-specific distribution. The aim of this study is to explore whether TMTP1-modified, cisplatin and paclitaxel prodrugs co-loaded nanodrug could improve cervical cancer chemotherapy and relieve its side effects through active and passive tumor targeting accumulation and controlled drug release.
Methods: TDNP, with capacities of active targeting for tumors and controlled drug release, was prepared to co-deliver cisplatin and paclitaxel prodrugs. The characteristics were investigated, including the diameter, surface zeta potential, stability and tumor microenvironment (TME) dependent drug release profiles. Cellular uptake, cytotoxicity, drug accumulation in tumors, antitumor effects and safety analysis were evaluated in vitro and in vivo.
Results: The oxidized cisplatin and the paclitaxel linked to the polymer achieved a high loading effciency of over 80% and TME-dependent sustained drug release. Moreover, TMTP1 modification enhanced cellular uptake of TDNP and further improved the cytotoxicity of TDNP in vitro. In vivo, TDNP showed an extended blood circulation and increased accumulation in SiHa xenograft models with the aid of TMTP1. More importantly, TDNP controlled tumor growth without life-threatening side effects.
Conclusion: Our study provided a novel TP co-delivery platform for targeted chemotherapy of cervical cancer, which was promising to improve the therapeutic effcacy of TP and may also have application in other tumors.
Keywords: TME-responsive, targeted co-delivery, combined chemotherapy, cervical cancer
Introduction
Cervical cancer represents one of the major global health challenges, with an estimated 569,847 new diagnosed cases and 311,365 deaths worldwide in 2018.1 Combination chemotherapy with platinum-based agents and paclitaxel (TP) is the dominant treatment for primary cervical cancer and perhaps the only choice for patients with recurrent cervical cancer who have a challenge on secondary surgery and a poor outcome resulting from the adverse effects of radiotherapy on the bladder, small bowel and rectum.2 However, platinum (PT(II)) applied in clinical medicine has deficiencies of drug resistance and dose-dependent toxicity resulting from non-specific distribution.3 Paclitaxel (PTX) also has a limited efficiency due to the hydrophobicity and poor selectivity for the tumor.4 For further improvement in therapeutic index and biosafety of TP, nanoparticle-based drug delivery systems (NDDSs) have been developed due to its advantages of improved drug pharmacokinetics, reduced drug side effects, alleviated drug resistance and combination therapy.5 Till now, variety of NDDSs have been developed for chemotherapeutics delivery.3,4,6–8 There are some nanodrugs for co-delivery of cisplatin and paclitaxel that have been developed to fight against non-small cell lung cancer, breast cancer, ovarian cancer and cervical cancer.9–16 However, some drawbacks are associated with these nanodrugs, including low loading capacity, insufficient tumor accumulation and uncontrollable release of drugs. Challenges for precise targeted co-delivery of multi-drugs and controlled drug release behaviors are still existed and need to be tackled urgently.
Enhanced permeability and retention (EPR) effects are the classic tumor targeting mechanism of typical nanodrugs such as Abraxane and Doxil. However, the accumulation of nanodrugs in tumors caused by the EPR effect is highly variable because of tumor heterogeneity.17 Active targeting, which makes use of ligands decorated on the nanoparticles (NPs) to specifically bind the receptors overexpressed on tumors, can achieve precise spatial localization, further trigger receptor-mediated endocytosis and promote targeted delivery of drugs.18 TMTP1, a 5-amino peptide (NVVRQ) screened through the FliTrx bacterial peptide display system in our laboratory, can specifically target highly metastatic tumors.19,20 Using TMTP1 as a tumor-targeting peptide, we have successfully constructed sTRAIL-TMTP1, TMTP1-TAT-NBD, TMTP1-GG-D(KLAKLAK)2 and DT390-TriTMTP1, which all exhibited excellent antitumor effects;21–24 the novel agents 99mTc-HYNIC-TMTP1 and TMTP1-PEG4-ICG also exhibited targeted imaging in breast and cervical cancers.25,26 Therefore, TMTP1-modified nanocarriers are promising for targeting solid tumors. In our preliminary research work, TMTP1-modified indocyanine green-loaded polymeric micelles exhibited targeted imaging of cervical cancer and metastatic sentinel lymph node.27
Tumor-targeted peptides can promote drug accumulation in tumors; however, targeted drug delivery is still challenging in part because of the biological complexity of tumors.28 In addition, the drug is effective only when it is released from the NDDSs and restors to its free form, which is also the key to design nanodrugs. Based on the tumor microenvironment (TME) characteristics such as low pH, hypoxia and high levels of glutathione (GSH), TME-responsive NDDSs can not only promote drug accumulation in the tumors but also trigger drug release.29–31 Recently, tetravalent platinum (PT(IV)) has been developed to alleviate the resistance and improve the biosafety of cisplatin. PT(IV) has better stability in the blood circulation, which can alleviate side effects caused by the premature drug leakage. PT(IV) also has a modifiable element for chemical linkage formation and avoids compromising drug activity. More importantly, PT(IV) is in the oxidized state and can be reduced to cisplatin.32,33 High levels of GSH in the TME can act as a reducing agents; GSH-responsive PT released NDDSs have been extensively researched.34,35 Hydrophobicity of PTX makes it difficult to be efficiently loaded, connecting paclitaxel with the polymer through chemical bonds can promote its loading. Sometimes, however, the connection is too tight to release the drug, hindering its antitumor effects. The tumor microenvironment is weakly acidic (pH 6.0–7.0) due to glycolysis, so pH-responsive NDDSs have been developed.36,37 Cis-aconitic anhydride (CA) is acid-sensitive and has been widely used to conjugate doxorubicin (DOX) and nanocarriers because the resultant chemical bond is unstable in an acidic environment.38–40 Therefore, it is expected to make use of CA to help achieve pH-dependent PTX release in the TME.
In this study, TMTP1-modified PEG-DSPE was used to deliver tetravalent platinum and paclitaxel prodrugs to treat cervical cancer. Cisplatin was oxidized and connected to lauric anhydride (LA) to form the prodrug PT (IV)-LA. Paclitaxel was linked to poly(ethylene glycol) (PEG) by acid-sensitive β carboxylate bond of CA to form the prodrug PTX-CA-PEG. TMTP1-modified PEG-DSPE was co-loaded with PT (IV)-LA and PTX-CA-PEG to form the nanodrug TMTP1-PT-PTX-NPs (TDNP). After intravenous administration, TDNP could be targeted delivered to cervical cancer through the passive and active targeting ability of EPR and TMTP1. When it reached the TME, high levels of GSH stimulated the reduction of tetravalent platinum to cisplatin, and the low pH triggered the cleavage of β carboxylate bond between PTX and CA to release paclitaxel. Further, cisplatin and paclitaxel could synergistically and effectively control the cervical tumor growth without life-threatening side effects.
Materials and Methods
Materials, Cell Lines and Animals
The cyclic polypeptide cTMTP1 (NVVRQC, end-collateral amide bond into a ring) was purchased in Wuhan Baiyixin Biotechnology Co., Ltd (Wuhan, China). Hydroxyl poly(ethylene glycol) (HO-PEG-OH)(Mw 2000 Da) and DSPE-PEG-Maleimide (DSPE-PEG-MAL) (Mw 3400 Da) were purchased from Aladdin Biochemical Technology Co., Ltd (Shanghai, China). Cis-aconitic anhydride (CA) and lauric anhydride (LA) were purchased from Alfa Aesar Chemical Co., Ltd (Beijing, China). Cisplatin (99.6%) was purchased from Kunming Guiyan Pharmaceutical Co. LTD (Yunnan, China). Paclitaxel was purchased from Shanghai Jinhe Pharmaceutical Co., Ltd. (Shanghai, China). Rhodamine B (RhoB), 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine (DIR), 4-dimethylaminopyridine (DMAP) and N,N’-dicyclohexylcarbodiimide (DCC) were purchased from Sigma-Aldrich (MO, USA). N,N-dimethyl formamide (DMF), dimethyl sulfoxide (DMSO), dichloromethane (DCM), triethylamine (TEA), chloroform (TCM), acetone, diethyl ether and methanol were of analytical grade and were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). 4,6-diamidino-2-phenylindoleD (DAPI) and immunohistochemistry (IHC) detection kit were purchased from Servicebio Biotechnology Co., Ltd (Wuhan, China).
Cervical cancer cell lines SiHa and HeLa were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin and 100μg/mL streptomycin (Invitrogen).
The female 4-week-old BALB/c-nude mice were purchased from Jicui Yaokang Biological Technology Co., Ltd (Nanjing, China). All animals were adapted to 1 week before the experiments under specific pathogen-free (SPF) conditions at the Experimental Animal Center of Tongji Hospital of Huazhong University of Science and Technology (HUST). All animal procedures were approved by the Ethics Committee for Animal Experiments of HUST.
Synthesis and Characterization of Prodrugs
Cisplatin (PT) was oxidized with 30% H2O2 solution at 70 ° C for 5 h. After cooling to room temperature, the oxidized tetravalent platinum (PT(IV)) was collected by centrifugation and dried. PT(IV) and LA at a molar ratio of 1:2 were dissolved in anhydrous DMSO, reacted for 24 h at room temperature, then precipitated twice with a mixed solution of acetone:ether at a volume ratio of 1:3, and dried to obtain the products PT(IV)-LA (yield: 73.4%).34
Paclitaxel (PTX) and CA at a molar ratio of 1:1.2 were reacted in anhydrous DCM for 12 h. After adding DCC and DMAP into the obtained solution for activation for 5 h, 0.8 times of HO-PEG-OH was added to continue the reaction at RT for 48 h. The obtained mixtures were centrifuged to remove the insoluble N,N′dicyclohexylurea (DCU). Then, the mixtures were dialyzed (MWCO: 3000 Da) against DMSO and water to remove the unreacted PTX, CA, DMAP and DMSO in dialysis bag for 48 h, respectively, and lyophilized to obtain products PTX-CA-PEG (yield: 67.8%).
Chemical structures of PT(IV)-LA and PTX-CA-PEG were characterized by 1H-NMR spectra (Bruker AVANCE III 400 MHz NMR spectrometer, Switzerland) and Fourier transform infrared spectrometer (FTIR, PerkinElmer, USA).
Preparation of Nanoparticles
NPs were prepared by ultrasonic emulsification. One milligram TMTP1 and 6 mg DSPE-PEG-MAL were dissolved in DMF and reacted for 8 h to obtain the TMTP1-PEG-DSPE.27 Then, 3 mg TMTP1-PEG-DSPE, 3 mg NH2-PEG-DSPE, 6 mg PTX-CA-PEG and 2.5 mg PT(IV)-LA were dissolved in 300 µL TCM and 50 µL DMSO. The mixture was dropped into ultrapure water and immediately emulsified with an ultrasonicator (Scientz, JY92-IIDN) for 5 min in an ice bath. The suspension was stirred to evaporate TCM at room temperature overnight and dialyzed (MWCO: 3000 Da) with water to remove excess DMSO. After centrifugation at 3000 rpm for 5 min to remove unloaded drugs, the suspension was concentrated to obtain cisplatin and paclitaxel co-loaded nanodrug TMTP1-PT/PTX-NP (TDNP). Cisplatin and paclitaxel co-loaded nanodrug PT/PTX-NPs (DNP) without TMTP1 modification was prepared as the same as the TDNP, except for replacing TMTP1-PEG-DSPE with NH2-PEG-DSPE. TMTP1-modified single-drug-loaded nanodrug TMTP1-PTX-NP (T-PTX-NP) and TMTP1-PT-NP (T-PT-NP) were also prepared using the same method as for TDNP. RhoB-labelled nanoparticles including RhoB-NP and TMTP1-RhoB-NP were prepared to replace the prodrugs with 200 µg of RhoB.
Characteristics of NPs and in vitro Drug Release
The particle sizes and zeta potentials of the nanoparticles were determined by dynamic light scattering (DLS, Zeta Plus, Brookhaven Instruments, USA). The morphologies were imaged by transmission electron microscopy (TEM, JEM-1230, Japan). High-performance liquid chromatography (HPLC, Shimadzu, LC-20A, Japan) and atomic absorption spectroscopy (AAS, VARIAN, USA) were used to detect the content of paclitaxel and cisplatin in NPs to obtain the loading efficiency (LE) and loading capacity (LC). The calculated formulas for LC and LE were as follows:

In the formula, Mdrug represents the drug content in NPs, MNP represents the all content in the NPs, and Mdrug added represents the total drug content when preparing the NPs.
The stability of NPs at 4°C was determined by detecting the changes in particle sizes within 2 weeks.
In vitro drug release behavior was determined as the published researches.35,41 In details, 3 mL TDNP suspension under different pH (5.5, 6.5, 7.4) was infused into a dialysis bag (MWCO: 3500 Da) and dialyzed against 25mL phosphate-buffered saline (PBS) at the same pH containing 10% FBS and 0.5% (v/v) Tween 80 at 37°C. Then, the system was gently shaken at a speed of 100 rpm in the dark. At scheduled time intervals, 3 mL PBS was withdrawn and then replaced with 3 mL fresh PBS. The content of PTX and PT was measured by HPLC and AAS. The PT release was detected in PBS of pH 7.4 with different GSH levels (0 mM, 1 mM, 10 mM). The detail was as the same with the PTX.
The methods for detection of PTX12 and PT35 were as follows. For the analysis of the PTX content, the samples were dissolved in methanol and centrifuged to remove the precipitate. A 20µL sample was measured on a HPLC instrument equipped with a C18 column (Welch, 4.6×250 mm, 5 µm). The mobile phase was acetonitrile/water (50/50, v/v) at a flow rate of 1.0 mL/min; and UV absorbance was detected at a wavelength of 227 nm at 25 °C. The elute time was 14.5 min. For the analysis of the PT content, the sample was dissolved in HNO3 and diluted HNO3 with water to a concentration of 3%. A 20µL sample was measured on AAS at a wavelength of 265.9 nm.
Cellular Uptake in vitro
Cellular uptake was investigated using RhoB as a fluorescence probe. Cervical cancer cell lines SiHa and HeLa cells were cultured in 12-well plates at a density of 1.0×105/well, then different NPs (RhoB-NP, T-RhoB-NP) loaded with RhoB (3 µg/mL) were added and further incubated for 1h, 2h and 4h. Free RhoB was used as a control. Flow cytometry analysis was used to detect the fluorescence intensity which indicated the cellular uptake of NPs. Similarly, SiHa and HeLa cells were seeded onto the sterilized glass slides in 12-well plates at a density of 5×104/well. NPs containing RhoB (5 µg/mL) were added and incubated for 2 h. Then, the cells were fixed with 4% paraformaldehyde solution for 10 min, and the cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, 1 µg/mL). Cellular uptake images of NPs were obtained under a fluorescence microscope (Zeiss LSM510, Goettingen, Germany). To further identify the endocytic pathways of the NPs, SiHa and HeLa cells were incubated with endocytic inhibitors for 0.5 h prior to the T-RhoB-NPs, including methyl-β-cyclodextrin (Mβ-CD, 5 mM), chlorpromazine (CHOL, 30 µM), dynasore (DYN, 20 µM) and nocodazole (NOC, 20 µM). Then, cells were washed with PBS to remove the inhibitors and incubated with T-RhoB-NPs (5 µg/mL) for 2 h. Flow cytometry analysis was used to detect the fluorescence intensity which indicated the relative cellular uptake of NPs.
Cytotoxicity of NPs in vitro
Cytotoxicity of NPs was evaluated via the Cell Counting Kit-8 (CCK8, Dojindo) assay and flow cytometry analysis. SiHa and HeLa cells were cultured in 96-well plates at a density of 1.0×104/well. After incubation overnight, PT, PTX, Free (free PT+PTX), T-PTX-NP, T-PT-NP, DNP and TDNP with different drug concentration of 0–50 µmol/L (50 µM, 25 µM, 12.5 µM, 4.17 µM, 1.39 µM, 0.46 µM, 0 µM) were added to the cells and incubated for 24 h or 48 h. The drug concentration of cells treated with FREE, DNP and TDNP was defined by the content of PT. Cells without any drug treatment were used as the control group. The cells were subjected to the CCK8 assay and absorbance at 450nm was measured on a microplate reader (BioTek, ELx808, USA). The cell viability and drug combination index (CI) were calculated according to the formulas:
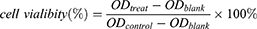
In the formula, Dm1 and Dm2 were the concentrations of each drug alone resulting in fixed ratio growth inhibition, and D1 and D2 were the concentrations of each drug in the combination leading to the same ratio of growth inhibition. Generally, CI values below 1 or above 1 suggested drug synergy and antagonism, respectively. CI values below 0.3 was considered strong synergy.42
In addition, SiHa and HeLa cells were cultured in 12-well plates at a density of 2.0×105/well. FREE, T-PTX-NP, T-PT-NP, DNP and TDNP with PTX at 2.11 µmol/L and PT at 3 µmol/L were added and incubated for 24 h. Then, the cells were collected for flow cytometry analysis according to the procedures of the Annexin V-FITC/PI Apoptosis Detection Kit (Becton Dickinson, USA).
In vivo Tumor Distribution and Pharmacokinetic Study
Near-infrared fluorescence imaging (NIRF) with NPs labeled with DIR was used to track the tissue distribution and tumor accumulation of NPs. Tumor-bearing BALB/c-nude mice were randomly divided into three groups (n=3) and were administered separately via tail vein injection with DIR, DIR-DNP-NP (DIR-NP), DIR-TDNP-NP (T-DIR-NP) (at an equivalent dose of DIR, 0.5 mg/kg, 100 µL/mouse). At the designated time points (0.5h, 2h, 6h, 12h and 24h), mice were anesthetized for NIRF using an IVIS Spectrum Imaging System (Caliper, USA) with an excitation wavelength of 780 nm and an emission wavelength of 845 nm. After imaging, the main organs (heart, liver, spleen, lung and kidney) and tumors were collected at 24 h for NIR imaging ex vivo.
In vivo Antitumor Efficacy
Cervical cancer cells (1.25×107) of SiHa were inoculated subcutaneously onto the hips of 6-week-old female BALB/c-nude mice. When the tumor size reached 30–100mm3, mice were randomly divided into six groups (n=6). Then PBS, FREE, T-PTX-NP, T-PT-NP, DNP and TDNP were administered intravenously at an equivalent dose of 8 mg/kg PTX and 4 mg/kg PT at 2-day intervals for 4 times. The body weight and tumor size were monitored every other day until the mice died or tumor diameter reached 1.5 cm. Tumor volume was calculated by the formula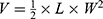 , where V, L and W represented the volume, length and width of the tumor, respectively. Tumors and the main organs (heart, liver, spleen, lung and kidney) were obtained and stained with hematoxylin and eosin (H&E) for histopathological analysis. At the same time, tumor sections were obtained for immunohistochemistry staining to detect the expression of apoptosis-related proteins.
, where V, L and W represented the volume, length and width of the tumor, respectively. Tumors and the main organs (heart, liver, spleen, lung and kidney) were obtained and stained with hematoxylin and eosin (H&E) for histopathological analysis. At the same time, tumor sections were obtained for immunohistochemistry staining to detect the expression of apoptosis-related proteins.
In vivo Safety Analysis
Acute and repeated toxicity studies were performed on the 6-week-old female BALB/c mice according to the research.14 For the acute toxicity study, mice were administrated with TDNP or combination of FREE once at an equivalent dose of 20 mg/kg PTX and 10 mg/kg PT. The survival state and body weight of the mice were monitored every day for 14 days. For the repeated toxicity study, mice were randomly divided into two groups including FREE and TDNP (n=5). Then, different drugs were administrated at an equivalent dose of 8mg/kg PTX and 4 mg/kg PT at 2-day intervals for four times in total. Similarly, the survival state and body weight of the mice were monitored every other day for 1 month. At the endpoint, all mice were sacrificed. Blood samples were collected for blood cell counting and biochemical indicator detection including alanine aminotransferase (ALT), aspartate aminotransferase (AST), urea (UREA), creatinine (CR). Main organs (heart, liver, spleen, lung and kidney) were stained with H&E for histopathological analysis.
Statistical Analysis
The results were analyzed by using unpaired one-way analysis of variance and unpaired t-test with GraphPad Prism 6.0 (GraphPad Software, CA, USA). Values of p < 0.05 were considered significant (ns: no significance, *p < 0.05, *p < 0.01, ***p < 0.001, ****p < 0.0001). Each experiment was repeated at least three times.
Results
Synthesis of Cisplatin and Paclitaxel Prodrugs
The synthetic pathways of cisplatin and paclitaxel prodrugs are shown in Supplementary Figure 1. Their structures were characterized by 1H-NMR and FTIR (Figure 1). For the NMR analysis (Figure 1A), the peak of –NH3 of PT appeared at 5.9ppm (a) in PT(IV)-LA; the peak of –CH2- and –CH3 of LA appeared at 0.8ppm (b) and 1.4ppm (c) in PT(IV)-LA. In the spectrum of PTX-CA-PEG, the peak of –HC=C< and HOOC–CH2– of CA appeared at 6.9ppm (d) and 3.7 ppm (e) in PTX-CA-PEG spectrum. Also, the chemical shifts of 3.6ppm (f), typically belonged to protons in repeated units of -CH2CH2-O in PEG. Meanwhile, the rest peak could correspond to the peaks of PTX one by one, which demonstrated the successful synthesis of PTX-CA-PEG. And the grafting degree of PTX could be calculated by the ratio of peak areas of -CH2CH2-O in PEG and methyl groups (0.8ppm, g) in PTX in PTX-CA-PEG spectrum. For the FT-IR analysis (Figure 1B), the peaks at 3242 cm−1 attributed to stretching vibration of −NH3(νN−H), and the range of 2956~2852 cm−1, 1375 cm−1 and 720 cm−1 belonged to the stretching and bending vibration of –CH3(νC−H) and –CH2–(νC−H) in carbon chain of LA in PT(IV)-LA spectrum. The peak at 1655cm−1 attributed to the stretching vibration of carbonyl group (δC=O) generated by ester linkers. In the spectrum of PTX-CA-PEG, the appearance of strong peak at 842 cm−1 typically belonged to bending vibration of O-CH2(δC-O) in part of PEG, and broad peak around 3423 cm−1 attributed to –COOH group generated by alcoholysis of anhydride in CA. These results indicated the successful synthesis of cisplatin and paclitaxel prodrugs.
 |
Figure 1 Synthesis and molecular characteristics of cisplatin and paclitaxel prodrugs. (A) 1H-NMR spectra of PT(IV)-LA and PTX-CA-PEG. (B) FTIR spectra of PT(IV)-LA and PTX-CA-PEG. |
Preparation and Characterization of Drug-Loaded NPs
A series of NPs loaded with chemotherapeutics were prepared by the ultrasonic emulsification method. The DLS images (Figure 2A and Table 1) show that the average diameters of NPs were between 100 and 250 nm with a low polydispersity index (PDI) <0.3 and negative or near-neutral surface zeta potential. TDNP and DNP with average diameters of 233.5±8.6nm and 242±8.1nm, respectively, were larger than T-PTX-NP with average diameters of approximately 216.3±4.7nm, and T-PT-NP with average diameters of approximately 159.2±3.8nm. Except T-PTX-NP with a negative zeta potential of −38.69±0.197mV, all other NPs exhibited near-neutral surface charges. The PDI of NPs were 0.140±0.020 for TDNP, 0.153±0.016 for DNP, 0.232±0.026 for T-PTX-NP and 0.242±0.042 for T-PT-NP, which all were <0.3, indicating the homogeneity of NPs. The morphology determined by transmission electron microscopy (TEM) (Figure 2B) was a spherical shape with smaller size distribution than the diameter measured by DLS. High loading of drug is a necessary prerequisite for their application. As shown in Table 1, the loading efficiency (LE) and loading capacity (LC) of PTX for TDNP were 89.17% and 5.05%, respectively, which were slightly lower than that for DNP and T-PTX-NP. LE and LC of PTX were 89.45% and 5.07% for DNP, and 93.29% and 5.29% for T-PTX-NP, respectively. The LE and LC of PT for TDNP was also high to 85.98% and 2.5%, which was slightly lower than that for T-PT-NP. LE and LC of PT were 86.24% and 2.5% for DNP, and 89.98% and 2.6% for T-PT-NP, respectively. EDX pattern of TDNP showed the existence of Pt and Cl peaks (Figure 2C and D and Supplementary Figure 2A and B), indicating the presence of cisplatin in the nanoparticles. Moreover, Pt4f XPs spectrum of TDNP (Figure 2E and Supplementary Figure 2C) showed an increased peak, which attributed to PT(IV)-LA in TDNP (Pt4f=78.4 eV), indicating the presence of oxidized PT in TDNP. The stability of the NPs was evaluated by monitoring the changes of the diameter in PBS. There were slight changes in 2 weeks (Figure 2F).
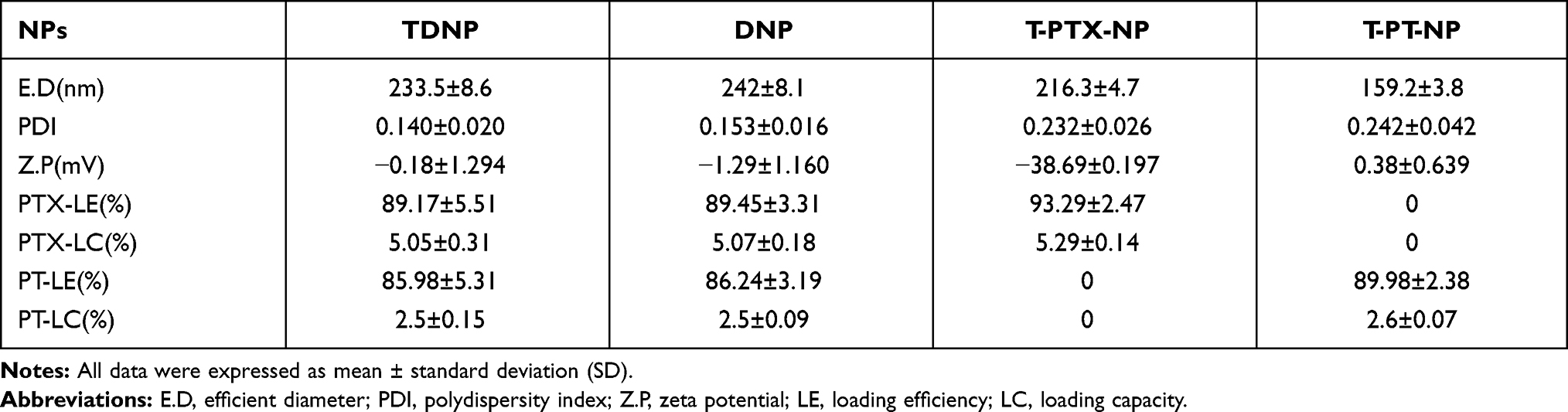 |
Table 1 The Size Distribution and Zeta Potential of NPs and the Content of PT and PTX in NPs |
 |
Figure 2 Preparation and characteristics of NPs and in vitro drug release. (A) The size distribution of T-PTX-NP, T-PT-NP, DNP and TDNP detected by DLS. (B) The TEM images of T-PTX-NP, T-PT-NP, DNP and TDNP. (C and D) The distribution of Pt elements in TDNP detected by EDS. (E) Pt4f XPS spectrum of TDNP and PT. (F) The diameter stability of different nanoparticles after 2 week storage at 4°C in PBS. (G) The pH-dependent release of PTX in PBS with different pH (7.4, 6.5 and 5.5). (H) The redox-dependent release of PT in PBS of pH 7.4 with different GSH levels (0 mM, 1 mM and 10 mM). |
TDNP Exhibited Stimuli-Dependent Drug Release in vitro
The release profiles of PTX and PT of TDNP were evaluated in the low pH or high GSH environment. As shown in Figure 2G, PTX was released quickly within 4 h, and reached the plateau at almost 12 h. PTX release in PBS at pH 5.5 was significantly faster than that in PBS at pH 7.4, while the drug release rate in PBS at pH 6.5 was midway between the two. To release 50% of PTX would spend 2 h and 24 h in PBS at pH 5.5 and pH 6.5, respectively, while it only reached approximately 30% in PBS at pH 7.4 up to 72 h. The accumulative release proportions of PTX at 72 h were 89.1% at pH 5.5, 62.3% at pH 6.5 and 32.1% at pH 7.4. The oxidized PT(IV) can be reduced to cisplatin with a high level of a reducing agent such as GSH. After immersing the TDNP with 10 mM GSH at 72 h, rapid PT release was achieved within 12 h, and the accumulative release rate reached approximately 77% at 72 h. By contrast, TDNP was stable in PBS with 0 or 1 mM GSH, and the PT accumulative release rate at 72 h was approximately 35% which is less than half of that in PBS with 10 mM GSH (Figure 2H).
TMTP1 Modification Improved the Endocytosis of NPs
The endocytosis of NPs by cervical cancer cells was investigated by using RhoB as a tracer. Flow cytometry analysis (Figures 3A and B and Supplementary Figure 3) confirmed that the RhoB-loaded NPs entered tumor cells in a time-dependent manner, which was most prominent within cells treated with T-RhoB-NP. With the extension of the incubation time, the fluorescence intensity of cells treated with T-RhoB-NP increased significantly in contrast with other groups. In addition, the increase of fluorescence intensity in HeLa was more apparent than that in SiHa cells. Statistically, the fluorescence intensity of T-RhoB-NP was enhanced 2.51-fold in SiHa cells and 3.11-fold in HeLa cells when compared with free RhoB, and 1.31-fold in SiHa cells and 1.99-fold in HeLa cells when compared with RhoB-NP at 2h. After 4h incubation, the fluorescence intensity of HeLa cells treated with T-RhoB-NP was further elevated to 3.69-fold and 2.54-fold in HeLa when compared with RhoB and RhoB-NP, respectively. While the increased fluorescence intensity in SiHa cells treated with T-RhoB-NP was decreased, which was 2.33-fold and 1.25-fold when compared with RhoB and RhoB-NP, respectively. Microscopic fluorescence images (Figure 3C) also confirmed that RhoB-labeled NPs displayed stronger fluorescence intensity than free RhoB. More importantly, TMTP1 modification further strengthened the fluorescence intensity of the cells. As the result of flow cytometry analysis, HeLa also exhibited stronger fluorescence intensity than SiHa cells when treated with same NPs, which was more obvious in the T-RhoB-NP group.
 |
Figure 3 In vitro cellular uptake of RhoB-labelled NPs in SiHa and HeLa cells and endocytic mechanisms. (A and B) Statistical analysis of mean fluorescence intensity (MFI) of cervical cancer cells treated with RhoB and RhoB-labelled NPs (RhoB: 3 µg/mL) within 4 h by flow cytometry analysis. Data were expressed as mean ± standard deviation(SD). (*p<0.05, **p<0.01). (C) Fluorescence images of cervical cancer cells treated with RhoB and RhoB-labelled NPs (RhoB: 5 µg/mL) for 2 h. The red was for RhoB and blue was for DAPI. (D and E) The cellular uptake analysis of cells by flow cytometry after pretreatment with methyl-β-cyclodextrin (Mβ-CD, 5 mM), chlorpromazine (CHOL, 30 µM), dynasore (DYN, 20 µM) and nocodazole (NOC, 20 µM) in HeLa and SiHa cells. *p < 0.05, **p < 0.01, and ****p < 0.0001. Abbreviation: ns, no significance. |
TDNP Entered Cancer Cells by Caveolae and Clathrin-Dependent Pathways
We further explored the specific cellular uptake pathways of TMTP1-modified nanoparticles. Different endocytic inhibitors were incubate with SiHa and HeLa cells, including methyl-β-cyclodextrin (Mβ-CD) that inhibited caveolae-dependent endocytosis, chlorpromazine (CHOL) that inhibited clathrin-dependent endocytosis, dynasore (DYN) that inhibited caveolae and clathrin-dependent endocytosis, and nocodazole (NOC) that inhibited micropinocytosis.43–46 As shown in Figure 3D and E, CHOL and DYN could remarkably inhibited the endocytosis, while Mβ-CD and NOC only inhibited the endocytosis slightly. These results indicated that T-RhoB-NPs entered cells through caveolae and clathrin-dependent pathways; the major endocytic pathways for T-RhoB-NPs was the caveolae-dependent endocytosis. The relative cellular uptake of T-RhoB-NPs in HeLa cells decrease to 0.750±0.044 in the Mβ-CD-treated group, 0.717±0.001 in the CHOL-treated group, 0.654±0.099 in the DYN-treated group, and 0.953±0.059 in the NOC-treated group. The cellular uptake of T-RhoB-NPs in SiHa cells decrease to 0.757±0.133 in the Mβ-CD-treated group, 0.571±0.036 in the CHOL-treated group, 0.529±0.006 in the DYN-treated group, and 0.849±0.035 in the NOC-treated group.
TDNP Exhibited Strong Cytotoxicity in vitro
The cytotoxicity of TDNP was investigated in cervical cancer cells of SiHa and HeLa. As shown in Figure 4A, cell survival was inhibited in a concentration-dependent and time-dependent manner. PTX or PT exhibited limited cytotoxicity, and nanoformulation modified with TMTP1 could increase the cytotoxicity. Cells treated with TDNP displayed the lowest survival rate, indicating the strongest cytotoxicity. The IC50 values of HeLa cells treated with TDNP at 24 h and 48 h were 2.72 µM and 1.03 μM, respectively. The IC50 values of SiHa cells treated with TDNP at 24 and 48 h were 3.01 µM and 1.59 μM, respectively (Table 2). The CI50 of TDNP in cervical cells was <1 at 24h and 48h, indicating the strong synergistic cytotoxicity. The CI50 of TDNP were 0.53 and 0.38 in HeLa cells, and 0.40 and 0.31 in SiHa cells (Table 3). DNP also had a CI50 <1, but it was larger than that of TDNP, indicating the weaker synergistic cytotoxicity. The IC50 and CI50 of FREE were similar with TDNP.
 |
Table 2 The IC50 of Different Drugs to the Cervical Cancer Cells at 24h and 48h |
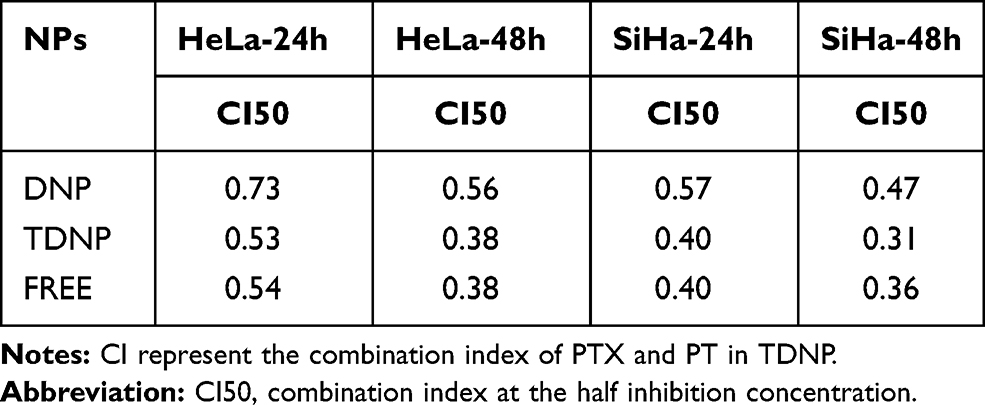 |
Table 3 The CI50 of TDNP to the Cervical Cancer Cells at 24h and 48h |
 |
Figure 4 Cytotoxicity of nanodrugs in SiHa and HeLa cells. (A) The survival rates of HeLa and SiHa cells treated with different drugs (PT, PTX, FREE, T-PTX-NP, T-PT-NP, DNP and TDNP) for 24 h and 48 h. The drug concentration of FREE, DNP and TDNP was defined by the content of PT. Data were expressed as mean ± standard deviation (SD). (B) The apoptosis of cells treated with different drugs investigated by flow cytometry analysis. The drug concentrations of PT and PTX were 3.00 µM and 2.11 µM, respectively. The data in the lower left, upper left, upper right, and lower right corner represented living cells, fragmented cells, late apoptotic cells and early apoptotic cells, respectively. |
The effect of combination therapy was also investigated in vitro by subjecting the SiHa and HeLa cells to different NPs and analyzing them with the Annexin V-FITC/PI Apoptosis Detection Kit. As shown in Figure 4B, HeLa and SiHa cells treated with TDNP had a survival rate of 56.72% and 83.82%, respectively, which was higher than that in the group of DNP, with cell survival rates of 66.63% and 88.01%, respectively, indicating the stronger cytotoxicity of TDNP than DNP. In addition, TDNP showed a better antitumor effect than single-drug loaded nanodrugs, with cell survival rates of 70.33% of T-PT-NP and 73.29% of T-PTX-NP in HeLa cells. The apoptosis rates in cells treated with T-PTX-NP or T-PT-NP in SiHa cells were also far lower than that for TDNP, which was 89.64% and 90.57%, respectively. Regarding the FREE group, the survival rates were 55.68% in HeLa cells and 81.45% in SiHa cells, which was consistent with the results of CCK8 assay. There was no significant difference in the survival rate between TDNP and FREE.
TDNP Had a Prolonged Blood Circulation and Favorable Tumor Accumulation in vivo
The distribution and tumor accumulation of NPs in vivo were investigated using DIR as a tracker for NIRF. Compared with the free DIR group in which the fluorescent signal accumulated mainly in the liver, the DIR-labeled NP group also exhibited strong fluorescence accumulation in tumors (Figure 5A). Among them, TMTP1-modified NPs showed the strongest fluorescent signal in tumors. The distinguishable fluorescent signal in tumors was visible at 30min after tail vein injection, and gradually increased follow the time and reached its maximum at 24h. In addition, the attenuation of fluorescent intensity in the DIR group was most significant, and was much slower in the DIR-labeled NP group. TMTP1-modified NPs exhibited the slowest attenuation of fluorescent intensity, indicating the prolonged blood circulation of TDNP. The ex vivo NIR images (Figure 5B) of the main organs and tumors harvested after 24h post-injection also showed that the strongest fluorescent signal in tumors was observed in the T-DIR-NP group. In addition to the tumor, the fluorescent signal was also observed in liver and kidney, but the fluorescent signal in liver of the T-DIR-NP group was weaker than that of the DIR-NP group, indicating the tumor targeting effect of TMTP1. Quantitatively, T-DIR-NP accumulation in SiHa tumors was 1.70-fold higher compared with DIR-NP, and 9.55-fold higher compared with free DIR at 24h (Figure 5C).
 |
Figure 5 The distribution of NPs in SiHa tumor bearing mice. (A) NIRF imaging of SiHa tumor bearing mice injected with free DIR and DIR-labelled NPs with DIR concentration of 0.5mg/kg during 24h in vivo. Ex=780nm, Em=845nm. The black circle indicated the location of the tumor. (B) NIRF imaging of ex vivo organs and tumors at 24h. Ex=780nm, Em=845nm. (C) Fluorescent intensity in the main organs and tumors were calculated by the software of the IVIS imaging system. ***p<0.001. |
TDNP Exhibited Effective Antitumor Effect without Life-Threatening Side Effects in vivo
Combination antitumor effects were evaluated using the subcutaneous model with SiHa cells inoculated on the hip. As shown in Figure 6C–I, all mice treated with TDNP showed obvious tumor growth inhibition and delayed tumor growth. Compared with the PBS group, the TDNP group demonstrated the strongest antitumor effect; other groups including T-PTX-NP, T-PT-NP and DNP also showed statistically significant tumor inhibition. In addition, the TDNP group also showed a stronger antitumor effect than the T-PTX-NP, T-PT-NP and DNP groups. The endpoint volume of tumor treated with TDNP showed a significant statistical difference compared with other groups, and the tumor suppressive effect was elevated 3.98, 3.63, 2.81 and 2.21 times higher than that in the FREE, T-PTX-NP, T-PT-NP and DNP groups (Figure 6A). However, even with TDNP it is difficult to eradicate the tumor, especially, when the tumor volume exceeds 500mm3, the tumor growth was uncontrolled. There was no significant prolongation of survival in mice except for those in the TDNP group (Figure 6J). During the treatment, no obvious behavioral and mental abnormalities were observed. Importantly, no body weight loss was observed while a slight weight increase was found (Figure 6B). Histopathological analysis of tumors (Figure S4) showed different levels of tissue necrosis and apoptotic cells, especially in the TDNP group. The expression of apoptosis protein caspase-3 was also most significant in the TDNP group (Supplementary Figure 4). H&E images (Supplementary Figure 4) showed that there was no obvious tissue abnormality or damage.
 |
Figure 6 The antitumor efficacy of combination therapy on the SiHa subcutaneous tumor model. (A) The volume of the tumor at the endpoint of treatment calculated by the formula: tumor volume=1/2×length×width×width. The “*” on the column and line represent the statistical significance between the each group and PBS or TDNP group respectively. (*p<0.05, **p<0.01). (B) The changes of the body weight during the treatment process. (C) The changes of the tumor volume during the treatment process. (D–I) The changes of the tumor volume in each group during the treatment process. (J) Survival analysis. Abbreviations: ns, no significance. |
TDNP Exhibited Better Biosafety
In the acute toxicity study of TDNP (Figure 7A–C), there was a slight weight loss of about 4.3% at 4 days after drug administration but was regained in subsequent observations, and no obvious changes on appearance and behavior were observed. In contrast, mice in the FREE group became gradually emaciated, hypopraxia, and apathetic with messy, dull hair and low response to stimuli. As shown in Figure 7C, body weight in the FREE group dropped sharply and all mice died within one week after drug administration. In the repeat toxicity study (Figure 7D–F), both groups lost weight, but the body weight in the FREE group dropped more significantly. Two mice died during the process of free drug administration and one mouse in the TDNP group died at the end of the observation period. In all other mice, weight was regained at the late period of the observation. As shown in Figure 7G–I, the blood cell counts of a few mice had slightly increased or decreased, but there were no statistically significant differences among the groups. UREA values of mice in the repeated toxicity study were significantly increased, but other biochemical indicators were not significantly abnormal (Figure 7J–M). All mice that died before the observation endpoint were not subjected to blood cell analysis and blood biochemical analysis because blood samples were not obtained. H&E staining analysis showed all mice treated with TDNP had clear tissue structure, and no obvious tissue abnormalities or damage were observed, while mice treated in the FREE-A group had obvious bleeding features (Figures 7N and Supplementary Figure 5).
 |
Figure 7 Safety analysis in vivo. (A–F) The body weight changes of the FREE and TDNP group in the acute and repeated toxicity study. (G–I) Blood cell analysis at the endpoint of the observation, including erythrocyte (RBC), platelet (PLT) and leukocytes (WBC). Abnormal high and low values were marked with red and blue frame respectively. (J–M) Biomedical indicators including ALT, AST, UREA and CR at the endpoint of the observation. Abnormal high values were marked with red frame respectively (**p<0.01). (N) H&E staining images of the kidney. |
Discussion
Effective delivery of anticancer agents to tumors is one of the clinical goals in cancer therapy. However, chemotherapeutics for cervical cancer such as carboplatin, oxaliplatin, Lipusu and Abraxane cannot meet the goal because of the tumor heterogeneity, poor bioavailability and side effects resulting from nonspecific drug distribution.47,48 Traditional nanocarriers for drug delivery are challenged by double drug co-delivery, low loading capacity, insufficient tumor accumulation and uncontrollable drug release. In our study, we designed the NDDS with EPR, active targeting and TME-responsive characteristics to co-delivery PT and PTX prodrugs for effective cervical cancer therapy. TDNP exhibited excellent advantages as follows: (a) active and passive targeting of TMTP1 and EPR effect of nanoparticles, (b) co-delivery of PT and PTX and TME-responsive drug release, (c) prolonged blood circulation and increased accumulation in tumors, and (d) synergistic antitumor effects of PTX and PT and reduced side effects.
Almost NDDSs approved by the US FDA load a single drug; however, combination therapy is imperative for cancer because of its cellular complexity and heterogeneity.49 Co-delivery of different drugs is one of the major advantages of nanocarriers; however, its application has been hindered by poor miscibility among drugs and low loading capacity.50,51 In general, chemotherapeutics can be loaded into nanocarriers through physical packaging, chemical linkage or electrostatic adsorption. However, physical packaging and electrostatic adsorption are associated with premature drug release, resulting in undesirable side effects. Linking drugs with a polymer has improved aqueous solubility of hydrophobic drugs, elevated loading efficiency, better stability during blood circulation and improved pharmacokinetics.52 Stimuli-dependent linkage is especially preferable because of its capability for tumor-specific drug release.52 The acid anhydride bond in cis-aconitic anhydride can react with the hydroxyl or amino group to form a β-carboxylate bond or amide bond. In a weakly acidic environment, the ester bond or amide bond can be hydrolyzed to release the connected drug.53–55 In our study, PTX was coupled with PEG by the pH-sensitive β-carboxylic amide linkage, which could improve the hydrophilicity of PTX and could be further self-assembled to nanoparticles. Similarly, PT with metallic properties linked with LA increased load efficiency in NPs.35 Therefore, TDNP was responsible for successful co-delivery of cisplatin and paclitaxel prodrugs, and the loading efficiencies were high up to 80% (Table 1). Moreover, PEGylation, which can provide the hydrophilic protective shell to prevent adsorption of plasma protein and subsequent elimination of mononuclear phagocytic system (MPS), has been approved for clinical application by the US FDA.56 Prodrugs coated with PEG-DSPE improved the stability of NPs (Figure 2F), which assists the accumulation of NPs in tumor. Controlled drug release aimed at site-specially targeting release can enhance drug concentration in tumors and reduce off-target side effects, which is an important feature of nanoparticles used for cancer therapy. As the pH-sensitive linker, CA had the similar acid-responsive mechanism with 2.3-dimethylmaleic anhydride (DMA).57,58 Conjugated double bond in CA was destroyed by linking PTX with CA, which resulted to the instability of PTX-CA-PEG. β-carboxylate bond in PTX-CA-PEG tended to be broken to restore the stable structure of CA, resulting in the release of PTX.55 In our study, sustained release of PT and PTX of TDNP were observed (Figure 2G and H). PTX and PT exhibited a faster and more effective release under the conditions of low pH and high level of GSH condition, indicating that TDNP could release PT and PTX in TME for subsequent cancer therapy.
Tumors allows particles with a diameter of less than 1 µm to pass through the leaky vasculature and remain at the tumor site due to poor lymphatic system drainage, which is called the EPR effect, and it has been widely employed for passive targeting of nanoparticles.17 However, EPR-mediated tumor targeting is closely related to the physicochemical properties of the nanoparticles such as size, shape and surface charge.59–62 Researchers have studied the size-dependent tumor targeting of different nanoparticles.59,63,64 Collectively, nanoparticles with diameter 5–400nm are more preferable because the larger nanoparticles are subjected to uptake by MPS and the smaller nanoparticles are subjected to rapid renal clearance. Negative surface charge is associated with better blood circulation stability but limited cellular uptake, while positive surface charge produces the opposite effects.65,66 It is difficult to have a trade-off between “positive surface charge/size increase/greater tumor retention/high background” versus “negative surface charge/size decrease/lower tumor retention/less background.” In our study, the hydrophobic size diameter of less than 300nm and near-neutral surface charge (Figure 2 and Table 1) endowed nanoparticles with the better blood circulation stability and passive tumor targeted capacity via the EPR effect. Once nanodrug reaches the tumor zone via the EPR effect, it needs to specifically recognize the tumor and be taken up by cancer cells to take an action. Targeting moieties have been applied for specially binding to the cancer cells, which included antibodies, peptides and aptamers.38,67–69 Among these, peptides has been extensively studied for nanoformulation because of their advantages of simple structure, easy preparation, versatility and low immunogenicity.67,70 TMTP1, a tumor-targeted peptide, can identify highly metastatic tumors by binding to a possible receptor, XPNPEP2,27 which is overexpressed on highly metastatic cancer cells and closely related to the tumor metastasis.71,72 In our study, TDNP modified with TMTP1 exhibited increased cellular uptake (Figure 3) in vitro and enhanced tumor accumulation in tumors (Figure 5), which may be the result of the interaction of TMTP1 and XPNPEP2. In addition, the more obvious fluorescent signal in HeLa than in SiHa cells may be attributed to the greater expression of XPNPEP2 in HeLa cells.27
Nano-drugs are evaluated on the basis of two aspects: efficiency and toxicity. The synergistic antitumor effect of TDNP was demonstrated. In vitro (Figure 4A), TMTP1-modified NPs elevated the cytotoxicity of PTX and PT, which was a result of the protective effect of the nanoformulation and TMTP1-mediated improved cellular uptake. The combination index <1 indicated a synergism of TDNP on the cervical cells.14 The apoptosis results from flow cytometry (Figure 4B) showed a strong apoptosis rate for TDNP in comparison with that in other groups, which may attribute to the enhanced cellular uptake and sustained drug release. However, TDNP did not show better antitumor effect than FREE in vitro (Figure 4) because they had similar cytotoxicity, IC50, CI50 and apoptosis rate. The reason may partly be that FREE could entered cancer cells by passive diffusion in cell culture media, resulting in faster cytotoxicity.73,74 By contrast, TDNP entered the cells by the caveolae and clathrin-dependent pathways (Figure 3D and E) and elicited cytotoxicity after releasing from the NPs. In addition, cell culture media which lack of low pH and high redox may also result in the slow release of PT and PTX.73 In vivo, DIR-labeled NPs had prolonged blood circulation and increased accumulation in tumors, indicating the protective effect of the NPs as compared with the free drugs (Figure 5). Moreover, modification of TMTP1 further achieved better tumor localization, suggesting more chemotherapeutics was deliver to the tumor for a better antitumor effect. In SiHa-bearing mice, TDNP exhibited an improved therapeutic index through suppressing tumor growth and prolonging the survival (Figure 6). Toxicity profiles are a major concern in translating nanomedicine into clinical application. How to alleviate the toxicities of NPs represents a novel research direction for the future. Nephrotoxicity and allergic reactions were the common side effects of cisplatin and paclitaxel. In the toxicity studies, obvious bleeding and acute tubular injury was observed in the FREE group, and high dose of the chemotherapeutics caused the death of mice in one week. In comparison, mice tolerated TDNP well, and no obvious abnormalities in histopathology, blood count analysis and biochemical indicators were observed (Figure 7). These results suggested that TDNP has a better antitumor effect and biosafety for further clinical translation.
Collectively, we have developed a novel tumor-target platform to co-deliver cisplatin and paclitaxel for cervical cancer therapy. First, TNDP with a diameter of less than 300nm and negative zeta potential guaranteed the stability and the EPR effect, which could translate to a long life in the blood circulation. Second, TMTP1 modification endowed the TDNP with active targeting capability, further enhancing drug accumulation in tumors and cancer cell endocytosis, which ensured an adequate amount of drugs in tumor sites for effective therapy. Third, TME with low pH and high level of GSH could trigger drug fast release and restore to the active style, which also promotes drug accumulation in tumors and is the prerequisite for drugs to become active. These above characteristics ensured that TDNP had a better therapeutic index by improving efficacy and alleviating the off-target toxicity, which is an effective strategy to improve the dilemma of TP chemotherapy. Moreover, TDNP has potential applications in other cancers.
Conclusion
In our study, we designed the NDDS with a high drug loading efficiency of over 80%, passive EPR, active TMTP1-targeting and TME-responsive drug release to co-deliver PT and PTX prodrugs for effective cervical cancer therapy. TNDP with a diameter of less than 300nm and negative zeta potential showed excellent stability and passive tumor-targeting via the EPR effect. TMTP1 modification further endowed the TDNP with active targeting capability, further enhancing drug accumulation in tumors and cancer cell endocytosis, which ensured adequate amount of drugs in the tumor site for effective therapy. Moreover, PTX and PT in TDNP exhibited the faster and more effective release under conditions of low pH and high level of GSH, which indicated that TDNP could release PT and PTX in TME for subsequent cancer therapy. Based on above results, TDNP had cytotoxicity in vitro and the better antitumor effect in vivo by improving efficacy and alleviating off-target toxicity. In summary, we have successfully provided an effective strategy to elevate the therapeutic index of TP in cervical cancer therapy and this method may also be promising for treating other tumors.
Abbreviations
TP, cisplatin-taxane; TME, tumor microenvironment; CC, cervical cancer; PT, cisplatin; PTX, paclitaxel; NDDS, nano-based drug delivery systems; NP, nanoparticle; GSH, glutathione; PT(IV), tetravalent platinum; CA, cis-aconitic anhydride; DOX, doxorubicin; LA, lauric anhydride; PEG, polyethylene glycol; TDNP, TMTP1-PT-PTX-NPs; HO-PEG-OH, hydroxyl poly(ethylene glycol); DSPE-PEG-MAL, DSPE-PEG-maleimide; RhoB, rhodamine B; DIR, 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine; DMAP, 4-dimethylaminopyridine; DCC, N,N’-dicyclohexylcarbodiimide; DMF, N,N-dimethyl formamide; DMSO, dimethyl sulfoxide; DCM, dichloromethane; TEA, trimethylamine; TCM, chloroform; DAPI, 4,6-diamidino-2-phenylindole; IHC, immunohistochemistry; ATCC, American Type Culture Collection; DMEM, Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; SPF, specific pathogen-free; HUST, Huazhong University of Science and Technology; RT, room temperature; DCU, N,N′dicyclohexylurea; DNP, PT/T-PTX-NPs; T-PTX-NP, TMTP1-T-PTX-NPs; T-PT-NP, TMTP1-T-PT-NPs; DLS, dynamic light scattering; TEM, transmission electron microscopy; HPLC, high-performance liquid chromatography; UV-Vis-NIR, ultraviolet - visible light - near infrared; AAS, spectrophotometer and atomic absorption spectroscopy; LE, loading efficiency; LC, loading capacity; PBS, phosphate-buffered saline; CCK8, Cell Counting Kit-8; NIRF, near-infrared fluorescence imaging; HE, hematoxylin and eosin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; UREA, urea; CR, creatinine; ANOVA, unpaired one-way analysis of variance.
Ethics Approval and Informed Consent
All animals used in our study were breed in a pathogen-free environment. All animal procedures were approved by the Ethics Committee for Animal Experiments of Tongji hospital in Hubei province with the approved number of TJH-201,903,005. The guidelines for the welfare and treatment of the laboratory animals included “Guidelines for Ethical Review of Laboratory Animals”, “Regulations of Hubei Province on the Administration of Laboratory Animals“,”Regulations of the Laboratory Animal Ethics Committee of Huazhong University of Science and Technology” and “Regulations of the Experimental Animal Welfare Ethics Committee of Tongji Hospital”
Consent for Publication
All authors have read this manuscript and would like to have it considered exclusively for publication in International Journal of Nanomedicine. None of the material related to this manuscript has been published or is under consideration for publication elsewhere, including the internet.
Acknowledgments
We thank Analytical and Testing Center of HUST for NMR and TEM measurements, the Environment and Health Research Platform of School of Public Health of HUST for AAS analysis and Department of Pharmacy of Tongji Hospital for HPLC analysis.
Funding
This work was supported by the National Science Foundation of China (81472444, 82002764 and 81902661).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Cohen PA, Jhingran A, Oaknin A, Denny L. Cervical cancer. Lancet. 2019;393(10167):169–182. doi:10.1016/s0140-6736(18)32470-x
3. Oberoi HS, Nukolova NV, Kabanov AV, Bronich TK. Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Deliv Rev. 2013;65(13–14):1667–1685. doi:10.1016/j.addr.2013.09.014
4. Khalifa AM, Elsheikh MA, Khalifa AM, Elnaggar YSR. Current strategies for different paclitaxel-loaded nano-delivery systems towards therapeutic applications for ovarian carcinoma: a review article. J Control Release. 2019;311–312:125–137. doi:10.1016/j.jconrel.2019.08.034
5. Patra JK, Das G, Fraceto LF, et al. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnology. 2018;16(1):71. doi:10.1186/s12951-018-0392-8
6. Wang X, Guo Z. Targeting and delivery of platinum-based anticancer drugs. Chem Soc Rev. 2013;42(1):202–224. doi:10.1039/c2cs35259a
7. Kim J, Pramanick S, Lee D, Park H, Kim WJ. Polymeric biomaterials for the delivery of platinum-based anticancer drugs. Biomater Sci. 2015;3(7):1002–1017. doi:10.1039/c5bm00039d
8. Browning RJ, Reardon PJT, Parhizkar M, et al. Drug delivery strategies for platinum-based chemotherapy. ACS Nano. 2017;11(9):8560–8578. doi:10.1021/acsnano.7b04092
9. Wan X, Beaudoin JJ, Vinod N, et al. Co-delivery of paclitaxel and cisplatin in poly(2-oxazoline) polymeric micelles: implications for drug loading, release, pharmacokinetics and outcome of ovarian and breast cancer treatments. Biomaterials. 2019;192:1–14. doi:10.1016/j.biomaterials.2018.10.032
10. Wang G, Wang Z, Li C, et al. RGD peptide-modified, paclitaxel prodrug-based, dual-drugs loaded, and redox-sensitive lipid-polymer nanoparticles for the enhanced lung cancer therapy. Biomed Pharmacother. 2018;106:275–284. doi:10.1016/j.biopha.2018.06.137
11. Liu B, Han L, Liu J, Han S, Chen Z, Jiang L. Co-delivery of paclitaxel and TOS-cisplatin via TAT-targeted solid lipid nanoparticles with synergistic antitumor activity against cervical cancer. Int J Nanomedicine. 2017;12:955–968. doi:10.2147/IJN.S115136
12. Shen W, Chen X, Luan J, Wang D, Yu L, Ding J. Sustained codelivery of cisplatin and paclitaxel via an injectable prodrug hydrogel for ovarian cancer treatment. ACS Appl Mater Interfaces. 2017;9(46):40031–40046. doi:10.1021/acsami.7b11998
13. Tian J, Min Y, Rodgers Z, et al. Co-delivery of paclitaxel and cisplatin with biocompatible PLGA-PEG nanoparticles enhances chemoradiotherapy in non-small cell lung cancer models. J Mater Chem B. 2017;5(30):6049–6057. doi:10.1039/C7TB01370A
14. Cai L, Xu G, Shi C, Guo D, Wang X, Luo J. Telodendrimer nanocarrier for co-delivery of paclitaxel and cisplatin: a synergistic combination nanotherapy for ovarian cancer treatment. Biomaterials. 2015;37:456–468. doi:10.1016/j.biomaterials.2014.10.044
15. Desale SS, Soni KS, Romanova S, Cohen SM, Bronich TK. Targeted delivery of platinum-taxane combination therapy in ovarian cancer. J Control Release. 2015;220(Pt B):651–659. doi:10.1016/j.jconrel.2015.09.007
16. Xiao H, Song H, Yang Q, et al. A prodrug strategy to deliver cisplatin(IV) and paclitaxel in nanomicelles to improve efficacy and tolerance. Biomaterials. 2012;33(27):6507–6519. doi:10.1016/j.biomaterials.2012.05.049
17. Kalyane D, Raval N, Maheshwari R, Tambe V, Kalia K, Tekade RK. Employment of enhanced permeability and retention effect (EPR): nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater Sci Eng C Mater Biol Appl. 2019;98:1252–1276. doi:10.1016/j.msec.2019.01.066
18. He J, Li C, Ding L, et al. Tumor targeting strategies of smart fluorescent nanoparticles and their applications in cancer diagnosis and treatment. Adv Mater. 2019;31(40):e1902409. doi:10.1002/adma.201902409
19. Yang W, Luo D, Wang S, et al. TMTP1, a novel tumor-homing peptide specifically targeting metastasis. Clin Cancer Res. 2008;14(17):5494–5502. doi:10.1158/1078-0432.CCR-08-0233
20. Xiao M, Hong Z, Sun L, et al. TMTP1, a novel tumor-homing peptide, specifically targets hematological malignancies and their metastases. J Huazhong Univ Sci Technolog Med Sci. 2011;31(5):608. doi:10.1007/s11596-011-0569-y
21. Liu R, Ma X, Wang H, et al. The novel fusion protein sTRAIL-TMTP1 exhibits a targeted inhibition of primary tumors and metastases. J Mol Med. 2014;92(2):165–175. doi:10.1007/s00109-013-1093-2
22. Liu R, Xi L, Luo D, et al. Enhanced targeted anticancer effects and inhibition of tumor metastasis by the TMTP1 compound peptide TMTP1-TAT-NBD. J Control Release. 2012;161(3):893–902. doi:10.1016/j.jconrel.2012.05.002
23. Ma X, Xi L, Luo D, et al. Anti-tumor effects of the peptide TMTP1-GG-D(KLAKLAK)(2) on highly metastatic cancers. PLoS One. 2012;7(9):e42685. doi:10.1371/journal.pone.0042685
24. Ma X, Lv P, Ye S, et al. DT390-triTMTP1, a novel fusion protein of diphtheria toxin with tandem repeat TMTP1 peptide, preferentially targets metastatic tumors. Mol Pharm. 2013;10(1):115–126. doi:10.1021/mp300125k
25. Li F, Cheng T, Dong Q, et al. Evaluation of (99m)Tc-HYNIC-TMTP1 as a tumor-homing imaging agent targeting metastasis with SPECT. Nucl Med Biol. 2015;42(3):256–262. doi:10.1016/j.nucmedbio.2014.11.001
26. Zhou Y, Jiang G, Wang W, et al. A novel near-infrared fluorescent probe TMTP1-PEG4-ICG for in vivo tumor imaging. Bioconjug Chem. 2018;29(12):4119–4126. doi:10.1021/acs.bioconjchem.8b00756
27. Wei R, Jiang G, Lv M, et al. TMTP1-modified indocyanine green-loaded polymeric micelles for targeted imaging of cervical cancer and metastasis sentinel lymph node in vivo. Theranostics. 2019;9(24):7325–7344. doi:10.7150/thno.35346
28. Dai Q, Wilhelm S, Ding D, et al. Quantifying the ligand-coated nanoparticle delivery to cancer cells in solid tumors. ACS Nano. 2018;12(8):8423–8435. doi:10.1021/acsnano.8b03900
29. Shah VM, Nguyen DX, Al Fatease A, et al. Liposomal formulation of hypoxia activated prodrug for the treatment of ovarian cancer. J Control Release. 2018;291:169–183. doi:10.1016/j.jconrel.2018.10.021
30. Hao Y, Zheng C, Wang L, et al. Covalent self-assembled nanoparticles with pH-dependent enhanced tumor retention and drug release for improving tumor therapeutic efficiency. J Mater Chem B. 2017;5(11):2133–2144. doi:10.1039/c6tb02833k
31. El-Sawy HS, Al-Abd AM, Ahmed TA, El-Say KM, Torchilin VP. Stimuli-responsive nano-architecture drug-delivery systems to solid tumor micromilieu: past, present, and future perspectives. ACS Nano. 2018;12(11):10636–10664. doi:10.1021/acsnano.8b06104
32. Kaludjerović GN, Miljković D, Momcilović M, et al. Novel platinum(IV) complexes induce rapid tumor cell death in vitro. Int J Cancer. 2005;116(3):479–486. doi:10.1002/ijc.21080
33. Wilson JJ, Lippard SJ. Synthesis, characterization, and cytotoxicity of platinum(IV) carbamate complexes. Inorg Chem. 2011;50(7):3103–3115. doi:10.1021/ic2000816
34. Ling X, Tu J, Wang J, et al. Glutathione-responsive prodrug nanoparticles for effective drug delivery and cancer therapy. ACS Nano. 2019;13(1):357–370. doi:10.1021/acsnano.8b06400
35. Ling X, Chen X, Riddell IA, et al. Glutathione-scavenging poly(disulfide amide) nanoparticles for the effective delivery of Pt(IV) prodrugs and reversal of cisplatin resistance. Nano Lett. 2018;18(7):4618–4625. doi:10.1021/acs.nanolett.8b01924
36. Du JZ, Mao CQ, Yuan YY, Yang XZ, Wang J. Tumor extracellular acidity-activated nanoparticles as drug delivery systems for enhanced cancer therapy. Biotechnol Adv. 2014;32(4):789–803. doi:10.1016/j.biotechadv.2013.08.002
37. Deirram N, Zhang C, Kermaniyan SS, Johnston APR, Such GK. pH-responsive polymer nanoparticles for drug delivery. Macromol Rapid Commun. 2019;40(10):e1800917. doi:10.1002/marc.201800917
38. Du C, Deng D, Shan L, et al. A pH-sensitive doxorubicin prodrug based on folate-conjugated BSA for tumor-targeted drug delivery. Biomaterials. 2013;34(12):3087–3097. doi:10.1016/j.biomaterials.2013.01.041
39. Sun D, Ding J, Xiao C, Chen J, Zhuang X, Chen X. Preclinical evaluation of antitumor activity of acid-sensitive PEGylated doxorubicin. ACS Appl Mater Interfaces. 2014;6(23):21202–21214. doi:10.1021/am506178c
40. Sun D, Ding J, Xiao C, Chen J, Zhuang X, Chen X. pH-responsive reversible PEGylation improves performance of antineoplastic agent. Adv Healthcare Mater. 2015;4(6):844–855. doi:10.1002/adhm.201400736
41. Sohn JS, Yoon DS, Sohn JY, Park JS, Choi JS. Development and evaluation of targeting ligands surface modified paclitaxel nanocrystals. Mater Sci Eng C Mater Biol Appl. 2017;72:228–237. doi:10.1016/j.msec.2016.11.065
42. Li J, Li Y, Wang Y, et al. Polymer prodrug-based nanoreactors activated by tumor acidity for orchestrated oxidation/chemotherapy. Nano Lett. 2017;17(11):6983–6990. doi:10.1021/acs.nanolett.7b03531
43. Wu M, Guo H, Liu L, Liu Y, Xie L. Size-dependent cellular uptake and localization profiles of silver nanoparticles. Int J Nanomedicine. 2019;14:4247–4259. doi:10.2147/IJN.S201107
44. Wei X, Wei R, Jiang G, et al. Mechanical cues modulate cellular uptake of nanoparticles in cancer via clathrin-mediated and caveolae-mediated endocytosis pathways. Nanomedicine. 2019;14(5):613–626. doi:10.2217/nnm-2018-0334
45. Li L, Xi WS, Su Q, et al. Unexpected size effect: the interplay between different-sized nanoparticles in their cellular uptake. Small. 2019;15(38):e1901687. doi:10.1002/smll.201901687
46. Ding L, Yao C, Yin X, et al. Size, shape, and protein corona determine cellular uptake and removal mechanisms of gold nanoparticles. Small. 2018;14(42):e1801451. doi:10.1002/smll.201801451
47. Lajous H, Lelievre B, Vauleon E, Lecomte P, Garcion E. Rethinking alkylating(-like) agents for solid tumor management. Trends Pharmacol Sci. 2019;40(5):342–357. doi:10.1016/j.tips.2019.03.003
48. Sofias AM, Dunne M, Storm G, Allen C. The battle of “nano” paclitaxel. Adv Drug Deliv Rev. 2017;122:20–30. doi:10.1016/j.addr.2017.02.003
49. Xu X, Ho W, Zhang X, Bertrand N, Farokhzad O. Cancer nanomedicine: from targeted delivery to combination therapy. Trends Mol Med. 2015;21(4):223–232. doi:10.1016/j.molmed.2015.01.001
50. Hu CM, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Deliv. 2010;1(2):323–334. doi:10.4155/tde.10.13
51. Parhi P, Mohanty C, Sahoo SK. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discov Today. 2012;17(17–18):1044–1052. doi:10.1016/j.drudis.2012.05.010
52. Xu H, Ma H, Yang P, et al. Targeted polymer-drug conjugates: current progress and future perspective. Colloids Surf B Biointerfaces. 2015;136:729–734. doi:10.1016/j.colsurfb.2015.10.001
53. Zhuo S, Zhang F, Yu J, Zhang X, Yang G, Liu X. pH-sensitive biomaterials for drug delivery. Molecules. 2020;25(23). doi:10.3390/molecules25235649
54. Cheng X, He L, Xu J, et al. Oxygen-producing catalase-based prodrug nanoparticles overcoming resistance in hypoxia-mediated chemo-photodynamic therapy. Acta Biomater. 2020;112:234–249. doi:10.1016/j.actbio.2020.05.035
55. Cong Y, Ji L, Gao Y-J, et al. Microenvironment-induced in situ self-assembly of polymer–peptide conjugates that attack solid tumors deeply. Angew Chem Int Ed Engl. 2019;58(14):4632–4637. doi:10.1002/anie.201900135
56. Hussain Z, Khan S, Imran M, Sohail M, Shah SWA, de Matas M. PEGylation: a promising strategy to overcome challenges to cancer-targeted nanomedicines: a review of challenges to clinical transition and promising resolution. Drug Deliv Transl Res. 2019;9(3):721–734. doi:10.1007/s13346-019-00631-4
57. Li J, Ke W, Li H, Zha Z, Han Y, Ge Z. Endogenous stimuli-sensitive multistage polymeric micelleplex anticancer drug delivery system for efficient tumor penetration and cellular internalization. Adv Healthc Mater. 2015;4(15):2206–2219. doi:10.1002/adhm.201500379
58. Li J, Han Y, Chen Q, et al. Dual endogenous stimuli-responsive polyplex micelles as smart two-step delivery nanocarriers for deep tumor tissue penetration and combating drug resistance of cisplatin. J Mater Chem B. 2014;2(13):1813–1824. doi:10.1039/c3tb21383h
59. Kang H, Rho S, Stiles WR, et al. Size-dependent EPR effect of polymeric nanoparticles on tumor targeting. Adv Healthc Mater. 2020;9(1):e1901223. doi:10.1002/adhm.201901223
60. Kang H, Hu S, Cho MH, Hong SH, Choi Y, Choi HS. Theranostic nanosystems for targeted cancer therapy. Nano Today. 2018;23:59–72. doi:10.1016/j.nantod.2018.11.001
61. Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134(4):2139–2147. doi:10.1021/ja2084338
62. Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9(9–10):1521–1532. doi:10.1002/smll.201201390
63. Tong X, Wang Z, Sun X, et al. Size dependent kinetics of gold nanorods in EPR mediated tumor delivery. Theranostics. 2016;6(12):2039–2051. doi:10.7150/thno.17098
64. Sykes EA, Chen J, Zheng G, Chan WC. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8(6):5696–5706. doi:10.1021/nn500299p
65. Li Y, Yang HY, Thambi T, Park JH, Lee DS. Charge-convertible polymers for improved tumor targeting and enhanced therapy. Biomaterials. 2019;217:119299. doi:10.1016/j.biomaterials.2019.119299
66. Han SS, Li ZY, Zhu JY, et al. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small. 2015;11(21):2543–2554. doi:10.1002/smll.201402865
67. Hu JJ, Xiao D, Zhang XZ. Advances in peptide functionalization on mesoporous silica nanoparticles for controlled drug release. Small. 2016;12(25):3344–3359. doi:10.1002/smll.201600325
68. Zhu G, Chen X. Aptamer-based targeted therapy. Adv Drug Deliv Rev. 2018;134:65–78. doi:10.1016/j.addr.2018.08.005
69. Ahmad A, Khan F, Mishra RK, Khan R. Precision cancer nanotherapy: evolving role of multifunctional nanoparticles for cancer active targeting. J Med Chem. 2019;62(23):10475–10496. doi:10.1021/acs.jmedchem.9b00511
70. Field LD, Delehanty JB, Chen Y, Medintz IL. Peptides for specifically targeting nanoparticles to cellular organelles: quo vadis? Acc Chem Res. 2015;48(5):1380–1390. doi:10.1021/ar500449v
71. Cheng T, Wei R, Jiang G, et al. XPNPEP2 is overexpressed in cervical cancer and promotes cervical cancer metastasis. Tumour Biol. 2017;39(7):1010428317717122. doi:10.1177/1010428317717122
72. Li F, Dai Y, Xu H, et al. XPNPEP2 is associated with lymph node metastasis in prostate cancer patients. Sci Rep. 2019;9(1):10078. doi:10.1038/s41598-019-45245-5
73. Yi X, Lian X, Dong J, et al. Co-delivery of pirarubicin and paclitaxel by human serum albumin nanoparticles to enhance antitumor effect and reduce systemic toxicity in breast cancers. Mol Pharm. 2015;12(11):4085–4098. doi:10.1021/acs.molpharmaceut.5b00536
74. Riddell IA. Cisplatin and oxaliplatin: our current understanding of their actions. Met Ions Life Sci. 2018;18. doi:10.1515/9783110470734-007
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.