Back to Journals » Journal of Inflammation Research » Volume 16
TMAO Promotes NLRP3 Inflammasome Activation of Microglia Aggravating Neurological Injury in Ischemic Stroke Through FTO/IGF2BP2
Authors Ge P, Duan H, Tao C, Niu S, Hu Y, Duan R, Shen A, Sun Y, Sun W ![]()
Received 29 November 2022
Accepted for publication 19 August 2023
Published 28 August 2023 Volume 2023:16 Pages 3699—3714
DOI https://doi.org/10.2147/JIR.S399480
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Pengxin Ge,1,* Huijie Duan,2,* Chunrong Tao,3,* Sensen Niu,4 Yiran Hu,5 Rui Duan,6 Aizong Shen,7 Yancai Sun,1 Wen Sun3
1Department of Pharmacy, Anhui Provincial Cancer Hospital, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230031, People’s Republic of China; 2School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing, 210009, People’s Republic of China; 3Stroke Center and Department of Neurology, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230001, People’s Republic of China; 4Digestive System Department, The Second Affiliated Hospital of Anhui Medical University, Hefei, 230601, People’s Republic of China; 5Department of Scientific Research, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230001, People’s Republic of China; 6Department of Neurology, Nanjing First Hospital, Nanjing Medical University, Nanjing, 210006, People’s Republic of China; 7Department of Pharmacy, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wen Sun; Aizong Shen, Email [email protected]; [email protected]
Objective: Stroke is a kind of cerebrovascular disease with high mortality. TMAO has been shown to aggravate stroke outcomes, but its mechanism remains unclear.
Materials and Methods: Mice were fed with 0.12% TMAO for 16 weeks. Then, mice were made into MCAO/R models. Neurological score, infarct volume, neuronal damage and markers associated with inflammation were assessed. Since microglia played a crucial role in ischemic stroke, microglia of MCAO/R mice were isolated for high-throughput sequencing to identify the most differentially expressed gene following TMAO treatment. Afterward, the downstream pathways of TMAO were investigated using primary microglia.
Results: TMAO promoted the release of inflammatory cytokines in the brain of MCAO/R mice and promoted the activation of OGD/R microglial inflammasome, thereby exacerbating ischemic stroke outcomes. FTO/IGF2BP2 inhibited NLRP3 inflammasome activation in OGD/R microglia by downregulating the m6A level of NLRP3. TMAO can inhibit the expression of FTO and IGF2BP2, thus promoting the activation of NLRP3 inflammasome in OGD/R microglia. In conclusion, these results demonstrated that TMAO promotes NLRP3 inflammasome activation of microglia aggravating neurological injury in ischemic stroke through FTO/IGF2BP2.
Conclusion: Our results demonstrated that TMAO promotes NLRP3 inflammasome activation of microglia aggravating neurological injury in ischemic stroke through FTO/IGF2BP2. These findings explained the molecular mechanism of TMAO aggravating ischemic stroke in detail and provided molecular mechanism for clinical treatment.
Keywords: stroke, TMAO, NLRP3 inflammasome, OGD/R, microglia, FTO, IGF2BP2
Introduction
Stroke, commonly known as cerebral apoplexy, is a group of diseases characterized by the loss of local neurological function caused by the disturbance of blood circulation in the brain. Ischemic stroke can be caused by hypertension, transient ischemic attack, diabetes, high choline, atrial fibrillation and so on. Ischemic stroke is one of the most common causes of death and disability worldwide.1 Stroke brings a huge burden to the family and society; therefore, humans are urgently looking for stroke prevention and treatment methods.
Trimethylamine N-oxide (TMAO) is formed by the oxidation of the choline metabolite trimethylamine (TMA) by monooxygenase 3 containing flavin in the liver.2 It can also be generated by the oxidation of TMA that takes place in the gut microbiota. TMAO is associated with varieties of neurological disorders, such as Alzheimer’s disease3,4 and Parkinson’s disease.5,6 Recently, TMAO has been reported to be indicative of ischemic stroke in patients.7 However, the mechanism by which TMAO aggravates stroke remains unclear.
Microglia is closely related to stroke. It has been reported that microglia promoted the release of inflammatory cytokines in stroke.8 In addition, NLRP3 inflammasome is activated in microglia under the context of stroke.9 NLRP3 is an inflammasome that impairs cellular function, and the association of NLRP3 with many diseases has been extensively studied, consisting of traumatic brain injury (TBI) and myocardial ischemia/reperfusion (I/R) injury.10,11 NLRP3 inflammasome is produced through stimulation by exogenous and endogenous harmful substances, initiating inflammatory and non-inflammatory responses that influence the stroke process.12 TMAO has been shown to promote NLRP3 inflammasome activation in a variety of diseases, such as cardiac fibrosis,13 vascular calcification.14 However, the molecular mechanism of TMAO on NLRP3 has been less studied in stroke microglial.
N6-methyladenosine (m6A) is the most abundant and reversible modification in mRNA. It can regulate the stability, export, splicing or translation of RNA, thus influencing disease processes such as obesity, cancer and viral infections.15–17 m6A modification consisted of installing by “writer”, demethylating by “eraser” and identifying by “reader”.18–20 FTO, which is a member of eraser, has been suggested to stimulate angiogenesis and anti-myocardial fibrosis after myocardial ischemia. Meanwhile, downregulation of FTO and ALKBH5 after stroke can exacerbate brain damage.21–23 IGF2BP2 is also reported to be one of the m6A “readers” and has been shown to have six RNA binding domains.24 FTO/IGF2BP2 plays an important role in a variety of diseases. There was a documentation indicated that MTA1 is the direct target gene of FTO and is regulated by an FTO/IGF2BP2 m6A-dependent mechanism in colorectal cancer.25 In addition, FTO/IGF2BP2 could be used as a diagnostic marker of type 2 diabetes.26,27 Nonetheless, FTO/IGF2BP2 has rarely been studied in stroke. It was reported that miR-1208 in EVs could target METTL3 to reduce NLRP3 mRNA methylation, suppress inflammatory factor release.28 Furthermore, total flavones of Abelmoschus manihot mediate NLRP3 inflammatory activation by targeting METTL3 to induce m6A modification on PTEN mRNA to improve podocyte pyroptosis in high glucose.29 However, there are a few studies on the effect of FTO/IGF2BP2 on NLRP3 in stroke.
Taking the above reports into consideration, we use MCAO/R mice and primary microglia to explore the molecular mechanism of NLRP3 inflammasome regulation and the effect of FTO/IGF2BP2 on NLRP3 in ischemic stroke, which provide diagnostic indicators and drug targets for the treatment of stroke.
Materials and Methods
Animal and Treatments
C57BL/6J mice (8 weeks, male, 25–30g) were purchased from Model Animal Research Center of Nanjing University (Nanjing, China); meanwhile, all animal studies were conducted with approval from the Animal Research Ethics Committee of University of Science and Technology of China (2022-N(A)-089) and conducted in the Experimental Animal Centre. The animals used in this study were handled according to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publications No. 8023, revised 1978). In the experiments, all mice were fed with or without 0.12% TMAO (Sigma-Aldrich, USA) for 16 weeks.30,31
HPLC-MS/MS Detection of TMAO Level
TMAO plasma and brain levels were measured using liquid chromatography mass spectrometry. Briefly, 60 μL of samples were aliquoted into 1.5 mL Axygen tubes and mixed with 10 μL of a 1ug/mL internal standard consisting of d9-TMAO in methanol. Proteins in the samples were precipitated and supernatants (5 μL) were analyzed by injection into a Waters BEH C18 column (2.1 × 50 mm, 1.7 μm; Cat. No. 03433919615148, Massachusetts, USA) at a flow rate of 0.4 mL/min using an LC‐20AD Shimadzu pump system and SIL‐20AXR autosampler interfaced with a Triple Quad 4500MD mass spectrometry (AB SCIEX, Framingham, MA). By mixing solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in methanol) in different ratios, starting at 10% B, a discontinuous gradient was generated to separate the analytes and then linearly increased to 80% B over 1.0 min, then hold for 1.8 min, then return to 10% B. Quantification of TMAO was performed using multiple reaction monitoring (MRM) transitions at m/z 76.1→59, d9‐TMAO at m/z 85.1→68.1.
Animal Model of MCAO/R
The MCAO/R surgical was performed in mice using a modified endovascular suture method described by Longa et al.32 Mice were anesthetized with isoflurane (Abbott Park, USA). Briefly, after exposure of the right common carotid artery, external carotid artery (ECA), and internal carotid artery (ICA), a suture was introduced into the ECA and extended from the ICA to the opening of the middle cerebral artery (MCA). The MCA was blocked by inserting a silk thread from the carotid bifurcation. Sutures were removed after 60 minutes of occlusion and then reperfused. Finally, the neck wound was sutured with surgical thread. The Sham group underwent the same surgery, except that no wires were inserted.
Neurological Score Determination
At 24 h after MCAO/R, neurological deficit scores were evaluated according to the 5-point scoring system described by Longa et al by an examiner blinded to the experiment.32 The higher the neurological deficit score, the more severe the motor injury.
TTC Staining
Mice brain collection was performed 24 hours after MCAO/R. The mice brains were flash-frozen in a −20 °C refrigerator for about 20 minutes to facilitate slicing. The mice brains were sliced and placed in 2% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich, USA). Covered with tin foil and placed in a temperature box at 37°C for 15 ~ 30 min. Turned the brain tablet from time to time to make even contact with the staining solution. Infarct size was determined using ImageJ analysis software (Image-Pro plus, USA).
Hematoxylin and Eosin Staining
After the neurological score determination, the mice were anesthetized with isoflurane and perfused with normal saline and fixed with paraformaldehyde in phosphate buffered saline (PBS). Then, the mice brain was sliced. Histological changes were analyzed after hematoxylin and eosin staining.
Real-Time Reverse Transcription-Quantitative PCR (RT-qPCR) for mRNA Expression
At 24 h after MCAO/R, mice were anesthetized with isoflurane and the brain tissue was dissected. Total RNA was extracted from the brain using an RNA extraction kit and reverse transcribed into cDNA using a commercial kit (TAKARA, Japan). Real-time PCR was performed using an ABI 7500 sequence detection system (Applied Biosystems, Foster City, CA) mixed with SYBR Green 2 × PCR master Mix (TAKARA, Japan), cDNA template, forward and reverse primers. Thirty-five cycles: 95°C for 15s, 60°C for 60s. The following primers were used: IL-1β: forward TGACCTGGGCTGTCCTGATG, reverse GGTGCTCATGTCCTCATCCTG; IL-6: forward CCACTTCACAAGTCGGAGGC, reverse GCAAGTGCATCATCGTTGTTCAT; TNF-α: forward GACCCTCACACTCAGATCATCTTCT, reverse CCTCCACTTGGTGGTTTGCT. β-actin: forward TGAGCTGCGTTTTACACCCT, reverse GCCTTCACCGTTCCAGTTTT. Data were analyzed using the ABI 7500 sequence detection system software. The quantity of TNF-α, IL-1β, and IL-6 mRNA was normalized to that of β-actin using the comparative (2−ΔΔCt) method.
ELISA for IL-1β, IL-6 and TNF-α Serum Concentrations
The brain homogenates or the collected supernatant medium of primary microglial was centrifuged at 12,000 rpm for 10 min. The IL-1β (#MLB00C, R&D Systems, Inc. USA), IL-6 (#M6000B, R&D Systems, Inc. USA) and TNF-α (#MTA00B, R&D Systems, Inc., USA) levels were determined using a corresponding ELISA kit.
Immunofluorescence
Microglia were fixed with paraformaldehyde, permeabilized with Triton X-100, washed and then blocked by BSA. After washing, the cells were stained with anti-IBA1 (1:100, GT10312, ThermoFisher, USA) for overnight at 4°C. After washing, the cells were incubated with anti-mouse secondary antibodies (1:200, A-11001, Invitrogen, USA) for 1 h at 37°C. After washing, the cells were stained with DAPI (Sigma, USA) for 15 min. Samples were observed and photographed with a fluorescence microscope (ZEISS, Germany) and the number of Iba1+ cells was counted from three randomly selected microscope fields.
Western Blot Analysis
Total protein in the ischemic brain tissue and cell lysis buffer were extracted with the protein extraction kit (Key GEN Biotech, China), and the protein quantity was assessed with the BCA protein assay kit (Key GEN Biotech, China). The protein samples were separated electrophoretically and transferred to polyvinylidene difluoride membranes (Millipore, USA). After the membranes were blocked with 5% fat-free milk powder in TBST buffer, they were incubated overnight at 4 °C with anti-NLRP3 (1:1000; #15101, Cell Signaling Technology, USA), anti-IGF2BP2 (1:1000, #ab124930, Abcam Biotechnology, UK), anti-FTO (1:1000, #ab280081, Abcam Biotechnology, UK), anti-β-actin (1:1000; #4970, Cell Signaling Technology, USA). Next, the membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody at room temperature for 2 h and washed again. Immune complexes were assessed with peroxidase and an enhanced chemiluminescence system (Thermo Fisher, USA). The Western blot band densities were quantified by Quantity one software (BioRad, USA).
Primary Microglia Cell Culture and Oxygen-Glucose Deprivation/Reoxygenation (OGD/R) Treatment
Newborn mice for 1 to 2 days were taken. After disinfection with 75% alcohol, the brain tissues were removed by craniotomy under sterile conditions and placed in pre-cooled D-Hanks solution, and the surface blood was washed off with cold D-Hanks solution. The blood vessels and meninges were carefully dissected under the microscope and all bleeding spots were removed. The cortex and hippocampus were transferred to a 15 mL centrifuge tube containing D-Hanks solution. Add 0.25% trypsin with 1 mL pipette and gently blow and mix well. Then, the centrifuge tube was placed in a constant temperature incubator for 3 min. The digestion was terminated with DMEM/F12 medium containing 10% FBS when the liquid was flowing. The terminated liquid was filtered through a 70 μm cell sieve, and the filtered liquid was collected and centrifuged (1000 r/min, 5min). After precipitation, the cells were suspended, inoculated in 2×106 into 75 cm2 culture bottles, and cultured in an incubator. The culture was incubated with DMEM/F12 (Gibco, USA) containing 10% FBS (Gibco, USA) based on 24 h total liquid exchange. On the 5th day, the culture was changed again and continued to the 8th day. On the 8th day of culture, the cells were obviously stratified. DMEM/F12 (Gibco, USA) medium with 10% FBS (Gibco, USA) was used for liquid exchange. On the 9th day, the culture medium was removed from the culture bottle to the centrifuge tube and placed into the culture box for cultivation. Then, the new DMEM/F12 medium of 10% FBS was injected into the culture flask, and the flask was sealed with sealing glue, and the flask was shaken for 2 h with a 37 °C constant temperature shaker (200 r/min). The scattered cells were collected and seeded directly into culture bottles or well plates and cultured in a constant temperature incubator. At the same time, DMEM/F12 (Gibco, USA) medium containing 10% FBS (FBS, Gibco, USA) was added to the original mixed culture flask. After 5 days, the second batch of microglia could be harvested. The culture medium in the new culture flask/well plate was decanted after 25 min by differential adhesion method. The culture medium prepared in advance on the 8th day was centrifuged and added to the culture flask or well plate. After 24 h, the culture medium was completely changed and then used for the test.
The establishment of OGD/R model: oxygen–glucose deprivation stage: the original cell medium was aspirated, rinsed once with phosphate buffer, cultured with sugar-free Kreb’s solution instead of complete medium, and placed into a modular hypoxic culture device. Mixed gas (95% N2+5% CO2) was injected into the device, and the air was fully replaced for 10 min. The inlet and outlet were clamped, and the device was incubated with cells in an incubator for 6 h. Reoxygenation stage: At the end of anoxic culture, sugar-free Kreb’s solution was aspirated, rinsed with phosphate buffer once, and then incubated with complete medium under normal conditions for different times. To determine the effects of TMAO on OGD/R primary microglia cell, we added 200 μM TMAO (Sigma-Aldrich, USA) to the OGD/R primary microglia cell.
Isolation of Adult Microglia
Adult mice were anesthetized and brains were harvested after heart perfusion with ice-cold Duchenne phosphate buffer. This was followed by tissue isolation, debris removal, and red blood cell removal, followed by single-cell suspension using an adult brain isolation kit (Miltenyi Biotec, Germany). Microglia were further isolated from single-cell suspensions using MACS separation columns (Miltenyi Biotec, Germany) and magnetic CD11b microbeads (Miltenyi Biotec, Germany).
Lentivirus Infection
The lentivirus system (OEC, OE-IGF2BP2, OE-FTO, shC, sh-IGF2BP2, sh-FTO) was used to infect microglia. These productions are purchased from Shanghai Genechem. These lentivirus infected microglia were harvested after incubating with 1 mg/mL puromycin at 37°C for 72 h. Their infection efficiency was verified by Western blot.
RNA Extraction Library Construction and Sequencing
Adult microglia cells from MCAO/R mice were sequenced. Total RNA was extracted by Trizol reagent (Thermo Fisher, USA). mRNA was purified from total RNA (5ug) using Dynabeads Oligo (dT) (Thermo Fisher, USA), and then was fragmented by divalent cations under elevated temperature (Magnesium RNA Fragmentation Module (NEB, USA) under 94°C 5–7min). The cleaved RNA fragments were reverse-transcribed to cDNA by SuperScript™ II Reverse Transcriptase (Invitrogen, USA), which were next used to synthesise U-labeled second-stranded DNAs with E. coli DNA polymerase I (NEB, USA), RNase H (NEB, USA) and dUTP Solution (Thermo Fisher, USA). An A-base was added to the blunt ends of each strand to ligate to indexed adapters, each of which contained a T-base overhang. After the heat-labile UDG enzyme (NEB, USA) treatment of the U-labeled second-stranded DNAs, the products were amplified as following: 95°C for 3 min; 8 cycles of at 98°C for 15 sec; 60°C for 15 sec; 72°C for 30 sec; 72°C for 5 min. The average insert size for the final cDNA libraries were 300±50 bp. Lastly, we performed the 2×150bp paired-end sequencing (PE150) on an Illumina Novaseq™ 6000 (LC-Bio Technology Co., Ltd., Hangzhou, China). The sequencing data have been deposited in GenBank (GSE216349).
Bioinformatics Analysis
The potential m6A modification site on NLRP3 mRNA was analyzed by m6Avar (http://www.cuilab.cn/sramp/).
RNA Immunoprecipitation (RIP)
m6A modifications on NLRP3 mRNA were detected using the Magna RIP™Quad RNA-binding Protein immunoprecipitation Kit (17–704, Millipore, USA). Briefly, 200 mg of total RNA was enriched with anti-M6A antibody (ABE572, Merck, USA), anti-IGF2BP2 antibody (ab128175, Abcam, USA) or IgG-coupled protein A/G magnetic beads in 500 mL of 1×IP buffer supplemented with RNase inhibitors. Overnight at 4 ° C. The RNA of interest was immunoprecipitated with beads. One-tenth of each RNA fragment was stored as an input control for qPCR analysis with primers: NLRP3, 5′-CCATCAATGCTGCTTCGACA-3′ and 5′-GAGCTCAGAACCAATGCGAG-3′.
Statistical Analyses
All data were expressed as Mean ± Standard deviation (SD). GraphPad prism 8 (GraphPad Software, Inc., La Jolla, USA) was used for statistical analysis of experimental data in this chapter. Unpaired t-test was used for comparison between two groups, and one-way ANOVA test was used for comparison between multiple groups Dunnett method was used to analyze the data. When P < 0.05, significant differences were considered.
Results
TMAO Aggravated Neuronal Injury in the Brain of MCAO/R Mice
In order to investigate the effect of TMAO, mice were treated with TMAO. As shown in Figure 1A and B, given TMAO increased plasma and brain TMAO concentrations in the Sham group, the plasma and brain TMAO levels in MCAO/R+Vehicle group are higher than Sham group; meanwhile, the plasma and brain TMAO levels were further elevated in MCAO/R+TMAO group. As shown in Figure 1C–F, TMAO did not cause cerebral infarction in Sham mice. After 24 h of MCAO/R, neurological deficits were examined. The scores of the MCAO/R+Vehicle group were significantly higher than the Sham group, but the scores of the MCAO/R+Vehicle group was lower than MCAO/R+TMAO group (Figure 1C). Cerebral infarction was detected by TTC staining, which is indicated in Figure 1D and E. There was no cerebral infarction in the Sham group, but there were extensive lesions in the brain in the MCAO/R+Vehicle group. In the MCAO/R+TMAO group, infarct size was significantly increased compared with that in the MCAO/R+Vehicle group. HE staining showed histological changes in the MCAO/R+TMAO and MCAO/R+Vehicle group compared with that of mice in the Sham group. The Sham group showed normal round intact brain neurons with normal nuclei and intact tissue structure. HE staining revealed the mice brain in MCAO/R group exists with nucleus pyknosis, vacuolation, and shrunken neuronal bodies in the focal point of the infarcted region. After pretreatment with TMAO, the focal point of the infarction was significantly expanded when compared with that in the MCAO/R+vehicle group (Figure 1F). In conclusion, these results showed that TMAO aggravated neuronal injury in the brain of MCAO/R mice.
 |
Figure 1 TMAO aggravated neuronal injury in the brain of MCAO/R mice. (A) Plasma TMAO levels were measured by HPLC/MS (n=6). (B) Brain TMAO levels were measured by HPLC/MS (n=6). (C) Neurological deficit scores evaluated 24 h after reperfusion in C57BL/6J mice after MCAO/R injury (n=18). (D) The infarct volume assessed 24 h after reperfusion in C57BL/6J mice after MCAO/R injury by TTC staining (n=6). (E) Infarction volume as a percentage of the Sham value (n=6). (F) Representative images of ischemic brain tissue with hematoxylin and eosin staining (n=6). Scale bar, 20 µm. Magnification of the microphotograph, × 400. Results were shown as mean ± SD. *P <0.05, ***P <0.001, ****P <0.0001 versus the Sham+Vehicle group; ####P <0.0001 versus the MCAO/R+Vehicle group. |
TMAO Promoted the Release of Inflammatory Cytokines in the Brain of MCAO/R Mice
Neuroinflammation is an important factor in neuronal apoptosis, so we next examined the expression of inflammatory factors in the brain of MCAO/R mice. As shown in Figure 2A–C, IL-1β, IL-6 and TNF-α mRNA levels were increased slightly in the Sham+TMAO group compared with the Sham+Vehicle group, but there was no significant difference. IL-1β, IL-6 and TNF-α mRNA levels were notably upregulated in the MCAO/R+Vehicle group compared with Sham group and were enlarged by TMAO administration. As shown in Figure 2D–F, brain protein levels of IL-1β and IL-6 were increased in the Sham+TMAO group compared with the Sham+Vehicle group, but TNF-α has no significant change. Meanwhile, the brain concentrations of the inflammatory cytokines IL-1β, IL-6 and TNF-α were significantly increased in the MCAO/R+Vehicle group relative to the Sham+Vehicle group at 24 h after the induction of ischemia-reperfusion injury. Compared with the MCAO/R+Vehicle group, the IL-1β, IL-6 and TNF-α concentrations were significantly upregulated in the MCAO/R+TMAO groups. To sum up, these suggest that TMAO promoted the release of inflammatory cytokines in the brain of MCAO/R mice.
 |
Figure 2 TMAO promoted the release of inflammatory cytokines in the brain of MCAO/R mice. (A–C) The levels of IL-1β, IL-6, TNF-α mRNA in the mice brain were detected by qRT-PCR (n=6). (D–F) The levels of IL-1β, IL-6 and TNF-α in the mice brain were detected by ELISA (n=6). Results were shown as mean ± SD. *P <0.005, **P <0.01, ****P <0.0001 versus the Sham+Vehicle group; ##P <0.01, ####P <0.0001 versus the MCAO/R+Vehicle group. |
TMAO Promoted NLRP3 Inflammasome Activation in OGD/R Microglia
Next, we investigated the molecular mechanism of TMAO affecting neuroinflammation in vitro, so primary microglia were extracted. Above all, we verified the purity of primary microglia by immunofluorescence with Iba-1 antibody. The results showed that microglia accounted for more than 90% of the extracted cells (Figure 3A). We also detected the expression of NLRP3 in the primary microglia. Adding TMAO to control primary microglia increased NLRP3 expression and IL-1β protein levels in the brain (Figure S1A–C). Compared with the Control+Vehicle group, OGD/R+Vehicle and OGD/R+TMAO group significantly upregulated the levels of NLRP3, especially in OGD/R+TMAO group (Figure 3B and C). As shown in Figure 3D, the supernatant concentrations of the IL-1β was significantly increased in the OGD/R+Vehicle. Compared with the OGD/R+Vehicle group, the supernatant IL-1β concentrations was further upregulated in the OGD/R+TMAO groups. In general, our research indicated that TMAO promoted NLRP3 inflammasome activation in OGD/R microglia.
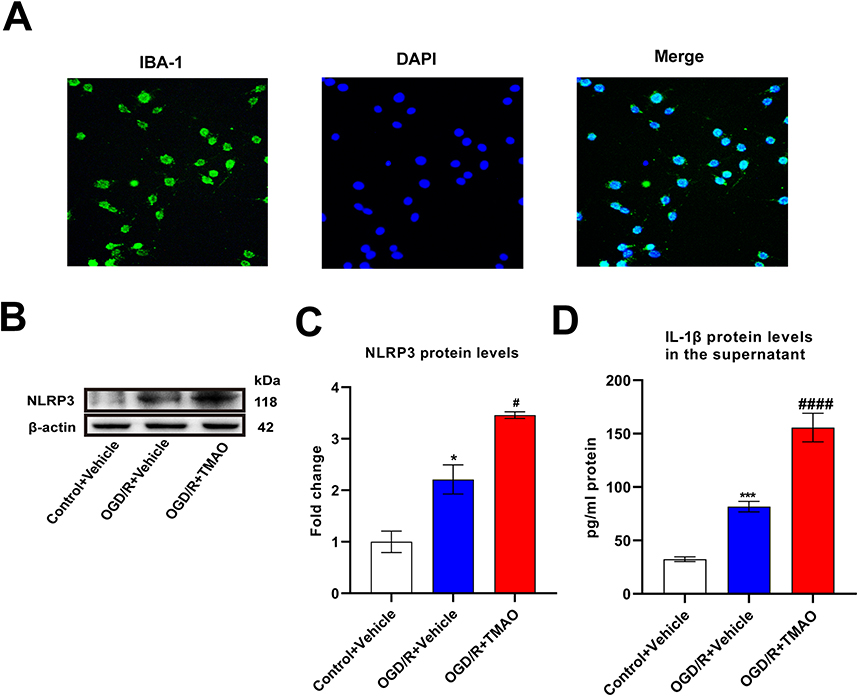 |
Figure 3 TMAO promoted NLRP3 inflammasome activation in OGD/R microglia. (A) Immunofluorescence staining to determine the expression and distribution of IBA-1 in the primary microglia, Scale bar = 20 μm (n=3). (B and C) The expression of NLRP3 in primary microglia was detected by Western blot assay. β-actin was used as an internal control (n=3). (D) The content of IL-1β in the supernatant of primary microglia was detected by ELISA assay (n=3). Results were shown as mean ± SD. *P <0.05, ***P <0.001 versus the Control+Vehicle group; #P <0.05, ####P <0.0001 versus the OGD/R+Vehicle group. |
TMAO Promoted NLRP3 Inflammasome Activation in OGD/R Microglia by Down-Regulating IGF2BP2
To further investigate the mechanisms of TMAO affects microglia, we explored gene expression changes affected by TMAO in adult microglia by high-throughput sequencing. The results showed a significant change in gene expression caused by TMAO, with 210 genes downregulated and 5 upregulated at a cut-off of 6 folds and P<0.05 (Figure 4A). Among them, only IGF2BP2 was associated with the m6A-related genes, thereby we chose IGF2BP2 as the research object. IGF2BP2 expression was reduced by TMAO administration in both Sham mice and Control primary microglia cells (Figure S2A–D). Compared with Sham+Vehicle group, IGF2BP2 level in MCAO/R+Vehicle group decreased. Compared with the MCAO/R+Vehicle group, MCAO/R+TMAO group significantly downregulated the levels of IGF2BP2 (Figure 4B and C). As shown in Figure 4D and E, the overexpression of IGF2BP2 was induced by IGF2BP2 overexpressing lentivirus, while the expression of IGF2BP2 was reduced after IGF2BP2 knockdown lentivirus. However, NLRP3 expression showed the opposite result, NLRP3 expression was increased by TMAO administration in Sham mice (Figure S1D and E). NLRP3 expression decreased when IGF2BP2 was overexpressed, whereas NLRP3 expression increased when IGF2BP2 expression decreased (Figure 4F and G). The supernatant of IL-β decreased in OGD/R+OE-IGF2BP2 group, nonetheless the supernatant of IL-β could increase in OGD/R+sh-IGF2BP2 group (Figure 4H). In summary, these results demonstrated that TMAO promoted NLRP3 inflammasome activation in OGD/R microglia by down-regulating IGF2BP2.
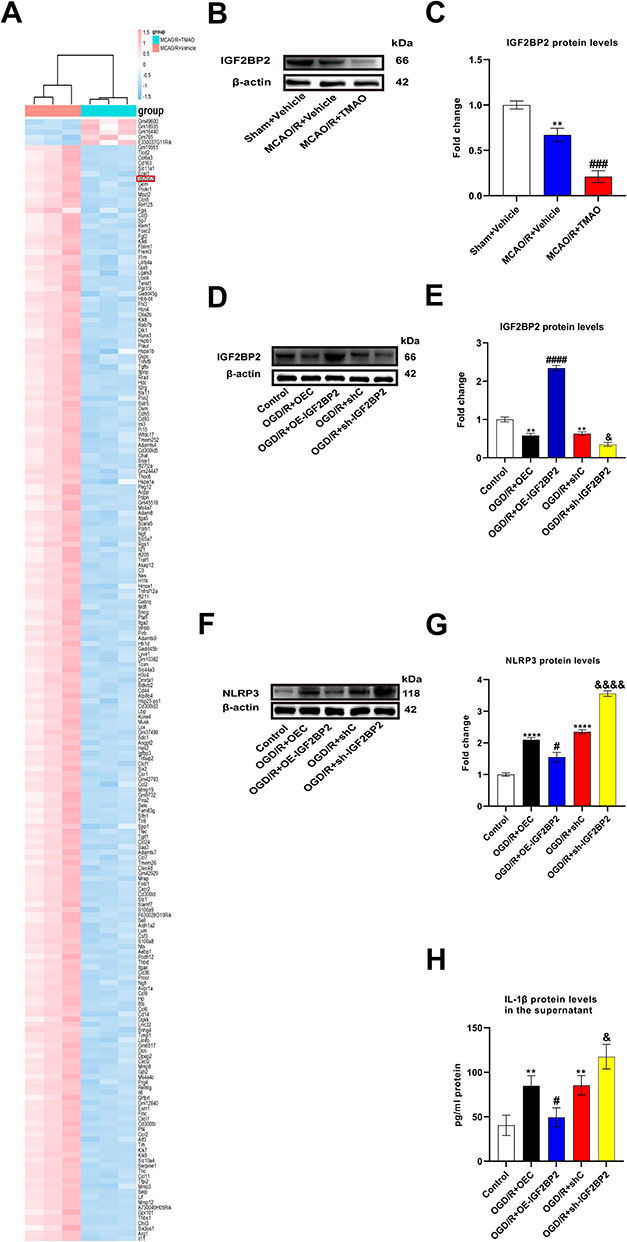 |
Figure 4 TMAO promoted NLRP3 inflammasome activation in OGD/R microglia by down-regulating IGF2BP2. (A) Top 215 differentially expressed gene were detected through gene microarray analysis. Genes in red indicates overexpression; those in blue indicate reduced expression. A total of 5 genes were up-regulated and 210 genes were down-regulated (n=3). (B and C) The expression of IGF2BP2 in C57BL/6J mice brain was detected by Western blot assay. β-actin was used as an internal control (n=6). (D and E) The expression of IGF2BP2 in primary microglia was detected by Western blot assay. β-actin was used as an internal control (n=3). (F and G) The expression of NLRP3 in primary microglia was detected by Western blot assay. β-actin was used as an internal control (n=3). (H) The content of IL-1β in the supernatant of primary microglia was detected by ELISA assay (n=3). Results were shown as mean ± SD. **P <0.01 versus the Sham+Vehicle group; ###P <0.001 versus the MCAO/R+Vehicle group; **P <0.01, ****P <0.0001 versus the Control group; #P <0.05, ####P <0.0001 versus the OGD/R+OEC group; &P <0.05, &&&&P <0.0001 versus the OGD/R+shC group. |
FTO/IGF2BP2 Inhibited NLRP3 Inflammasome Activation in OGD/R Microglia by Downregulating the m6A Level of NLRP3
It has been reported that IGF2BP2 can recognize m6A-modified mRNA. We used m6Avar to analyze the potential m6A sites on NLRP3 mRNA, and the m6A site of NLRP3 mRNA position has high confidence (Figure 5A and Table S1). Additionally, the result of RNA immunoprecipitation (RIP) analysis showed that NLRP3 mRNA indeed occurred m6A modification (Figure 5B), and IGF2BP2 interacted with NLRP3 mRNA in primary microglia cells (Figure 5C). After FTO was knocked down, the expression of FTO decreased significantly (Figure 5D and E). RIP analysis showed that knockdown of FTO significantly increased m6A modification on NLRP3 mRNA (Figure 5F), and inhibited IGF2BP2 binding NLRP3 mRNA (Figure 5G). In Figure 5H and I, NLRP3 expression in OGD/R+shC group was higher than that in Control group. NLRP3 expression decreased when FTO was overexpressed; meanwhile, NLRP3 expression increased when FTO expression decreased. Compared with OGD/R+shC group, the supernatant of IL-β increased in OGD/R+sh-FTO group (Figure 5J). In short, these results indicated that FTO/IGF2BP2 inhibited NLRP3 inflammasome activation in OGD/R microglia by downregulating the m6A level of NLRP3.
 |
Figure 5 FTO/IGF2BP2 inhibited NLRP3 inflammasome activation in OGD/R microglia by downregulating the m6A level of NLRP3. (A) The potential m6A sites on NLRP3 mRNA were analyzed by m6Avar. (B and C) RIP assays were used to detect the interaction between m6A/IGF2BP2 and NLRP3 mRNA (n=3). (D and E) The expression of FTO in primary microglia was detected by Western blot assay. β-actin was used as an internal control (n=3). (F and G) RIP assays were used to detect the interaction between m6A/IGF2BP2 and NLRP3 mRNA in primary microglia infected with shRNA-FTO for 48 h (n=3). (H and I) The expression of NLRP3 in primary microglia was detected by Western blot assay. β-actin was used as an internal control (n=3). (J) The content of IL-1β in the supernatant of primary microglia was detected by ELISA assay (n=3). Results were shown as mean ± SD. **P <0.01, ****P <0.0001 versus the IgG group, *P <0.05 versus the shC group; ***P <0.001, ****P <0.0001 versus the IgG group in sh-C; ###P <0.001, ####P <0.0001 versus the IgG group in sh-FTO; *P <0.05, ***P <0.001 versus the Control group; ###P <0.001 versus the OGD/R+sh-C group. |
TMAO Promoted NLRP3 Inflammasome Activation in OGD/R Microglia by Inhibiting FTO Expression
To explore whether TMAO promotes NLRP3 inflammasome activation in OGD/R microglia via FTO, both TMAO and FTO overexpressing lentiviruses were added to primary OGD/R microglia. The result demonstrated that the expression of FTO increased in OE-FTO group (Figure 6A and B). The NLRP3 protein expression in OGD/R+OEC+Vehicle group was slightly higher than that in Control group. The TMAO-induced upregulation of NLRP3 protein levels was reversed by FTO overexpression (Figure 6C and D). Equally, the TMAO-induced upregulation of IL-β protein levels in the supernatant was reversed by FTO overexpression (Figure 6E). Overall, these results indicated that TMAO promoted NLRP3 inflammasome activation in OGD/R microglia by inhibiting FTO expression.
 |
Figure 6 TMAO promoted NLRP3 inflammasome activation in OGD/R microglia by inhibiting FTO expression. (A and B) The expression of FTO in primary microglia was detected by Western blot assay. β-actin was used as an internal control (n=3). (C and D) The expression of NLRP3 in primary microglia was detected by Western blot assay. β-actin was used as an internal control (n=3). (E) The content of IL-1β in the supernatant of primary microglia was detected by ELISA assay (n=3). Results were shown as mean ± SD. **P <0.01 versus the OEC group; *P <0.05, ***P <0.001 versus the Control group; ###P <0.001, ####P <0.0001 versus OGD/R+Vehicle+OEC group; &&P <0.01, &&&P <0.001 versus the OGD/R+TMAO+OEC group. |
TMAO Inhibited FTO Expression but Promoted NLRP3 Expression in MCAO/R Mice
To verify effects of TMAO on expression of FTO and NLRP3 in MCAO/R mice, we administered TMAO to MCAO/R mice and measured the changes of NLRP3 and FTO gene expression by WB. FTO expression was slightly inhibited by TMAO administration in Sham mice, but there was no significant difference (Figure S3A and B). NLRP3 expression was elevated after ischemic stroke in mice and higher after TMAO administration (Figure 7A and B). In contrast, FTO expression was reduced after ischemic stroke and even lower after TMAO administration (Figure 7C and D). In summary, these results showed that TMAO inhibited FTO expression but promoted NLRP3 expression in MCAO/R mice.
 |
Figure 7 TMAO inhibited FTO expression but promoted NLRP3 expression in MCAO/R mice. (A and B) The expression of NLRP3 in MCAO/R mice brain was detected by Western blot assay. β-actin was used as an internal control (n=6). (C and D) The expression of FTO in MCAO/R mice brain was detected by Western blot assay. β-actin was used as an internal control (n=6). Results were shown as mean ± SD. *P <0.05, **P <0.01 versus the Sham+Vehicle group; #P <0.05, ###P <0.001, versus MCAO/R+Vehicle group. |
Discussion
TMAO can be used as a marker of stroke.7,33–35 Plasma TMAO levels were associated with adverse functional outcomes and mortality 3 months after stroke in a dose-dependency.7 TMAO levels in brain tissue are associated with neuronal oxidative stress and mitochondrial dysfunction, and may further cause neurological dysfunction.35 Recent studies have shown that TMAO exacerbates neurological damage in ischemic stroke by promoting astrocyte polarization and glial scar formation.36 At the same time, our research shows that TMAO aggravated neuronal injury in the brain of MCAO/R mice.
Increasingly, evidence supports the fact that the inflammatory response is related to infarct severity and infarct volume.37–39 In the hyperacute phase after stroke, the peripheral immune system rapidly activates and inflammatory cytokines are released in large quantities as a response to the stroke-induced brain injury. This first acute systemic response is followed by a state of immunosuppression, which is characterized by loss and unresponsiveness of immune cells. Later, in the chronic phase after stroke, a third and less well-understood phase is characterized by a low-grade sustained residual inflammation that might potentially impact on the long-term outcome of stroke patients.40 Our research shows that TMAO aggravated inflammatory response in stroke.
The recruitment of microglia in brain parenchymal tissue after ischemic stroke can increase the infarct volume. It was reported that microglia activation could induce various inflammatory mediators that lead to cellular damage and death, thus further aggravating brain damage in stroke.41,42 Minocycline and Fluoxetine has a neuroprotective effect on brain injury after intracerebral hemorrhage by inhibiting microglial activation.43,44 In our study, we found that TMAO promotes microglial activation to worsen stroke.
NLRP3 is involved in microglia-induced inflammation. NLRP3 levels in microglia correlate with brain injury and the pathology of central nervous system diseases.45,46 Some studies have shown that reducing NLRP3 inflammasome activation in microglia cells could inhibit neuroinflammation and ameliorates ischemic stroke injury.47 Targeting the microglial NLRP3 inflammasome may provide a new way for treatment of PD.48 Triggering microglial NLRP3 inflammasome activation is critical contributor to AD pathology.49 Furthermore, our research demonstrated that TMAO promotes NLRP3 inflammasome activation of microglia aggravating neurological injury in stroke.
M6A is involved in the development of stroke. It was reported that YTHDC1, one of m6A readers, can alleviate ischemic stroke by destabilizing PTEN mRNA.50 METTL3 mediated miRNA m6A modification promotes stress granule generation in the early stage of acute ischemic stroke.51 Obesity gene FTO is a risk factor for large atherosclerotic stroke in Chinese.52 In thyroid papillary carcinoma, FTO inhibits APOE expression and thus tumor growth through IGF2BP2-mediated m6A modification53. In epithelial ovarian cancer, FTO inhibits SNAI2 expression and attenuates cancer cell growth and metastasis in an M6a-IGF2BP2-dependent manner.54 However, the FTO/IGF2BP2 pathway has been poorly studied in ischemic stroke. Finally, we found that FTO/IGF2BP2 is a TMAO pathway that promotes NLRP3 inflammasome in ischemic stroke, so FTO/IGF2BP2 pathway has important significance for stroke.
It should be noted that this study has a limitation. DMB, a TMAO formation inhibitor, was not used in vivo studies. We will conduct relevant studies in the future.
In conclusion, these results demonstrated that TMAO promotes NLRP3 inflammasome activation of microglia aggravating neurological injury in stroke through FTO/IGF2BP2. Our study explained the molecular mechanism of TMAO aggravating stroke in detail and provided molecular mechanism for clinical treatment.
Funding
This work was supported by the Natural Science Foundation of Anhui Province (No. 2108085MH271) and the Fundamental Research Funds for the Central Universities (YD9110002028).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Faralli A, Bigoni M, Mauro A, Rossi F, Carulli D. Noninvasive strategies to promote functional recovery after stroke. Neural Plast. 2013;2013:854597. doi:10.1155/2013/854597
2. Janeiro MH, Ramírez MJ, Milagro FI, Martínez JA, Solas M. Implication of trimethylamine N-Oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients. 2018;10(10):1398. doi:10.3390/nu10101398
3. Vogt NM, Romano KA, Darst BF, et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res Ther. 2018;10(1):124. doi:10.1186/s13195-018-0451-2
4. Wang QJ, Shen YE, Wang X, et al. Concomitant memantine and Lactobacillus plantarum treatment attenuates cognitive impairments in APP/PS1 mice. Aging. 2020;12(1):628–649. doi:10.18632/aging.102645
5. Chen SJ, Kuo CH, Kuo HC, et al. The gut metabolite trimethylamine N-oxide is associated with parkinson’s disease severity and progression. Mov Disord. 2020;35(11):2115–2116. doi:10.1002/mds.28246
6. Chung SJ, Rim JH, Ji D, et al. Gut microbiota-derived metabolite trimethylamine N-oxide as a biomarker in early Parkinson’s disease. Nutrition. 2021;83:111090. doi:10.1016/j.nut.2020.111090
7. Zhai Q, Wang X, Chen C, et al. Prognostic value of plasma trimethylamine N-oxide levels in patients with acute ischemic stroke. Cell Mol Neurobiol. 2019;39(8):1201–1206. doi:10.1007/s10571-019-00714-3
8. Borozdenko DA, Shmigol TA, Ezdoglian AA, et al. The effect of a new N-hetero cycle derivative on behavior and inflammation against the background of ischemic stroke. Molecules. 2022;27(17):5488. doi:10.3390/molecules27175488
9. Ding R, Li H, Liu Y, et al. Activating cGAS-STING axis contributes to neuroinflammation in CVST mouse model and induces inflammasome activation and microglia pyroptosis. J Neuroinflammation. 2022;19(1):137. doi:10.1186/s12974-022-02511-0
10. Liu HD, Li W, Chen ZR, et al. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res. 2013;38(10):2072–2083. doi:10.1007/s11064-013-1115-z
11. Guo Z, Yu S, Chen X, Ye R, Zhu W, Liu X. NLRP3 is involved in ischemia/reperfusion injury. CNS Neurol Disord Drug Targets. 2016;15(6):699–712. doi:10.2174/1871527315666160321111829
12. Yang KL, Li WH, Liu YJ, et al. Hydrogen sulfide attenuates neuroinflammation by inhibiting the NLRP3/Caspase-1/GSDMD pathway in retina or brain neuron following rat ischemia/reperfusion. Brain Sci. 2022;12(9):1245. doi:10.3390/brainsci12091245
13. Zhang X, Fu Y, Li H, et al. H3 relaxin inhibits the collagen synthesis via ROS- and P2X7R-mediated NLRP3 inflammasome activation in cardiac fibroblasts under high glucose. J Cell Mol Med. 2018;22(3):1816–1825. doi:10.1111/jcmm.13464
14. Zhang X, Li Y, Yang P, et al. Trimethylamine-N-oxide promotes vascular calcification through activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) inflammasome and NF-κB (Nuclear Factor κB) signals. Arterioscler Thromb Vasc Biol. 2020;40(3):751–765. doi:10.1161/ATVBAHA.119.313414
15. Zhang C, Fu J, Zhou Y. A review in research progress concerning m6A methylation and immunoregulation. Front Immunol. 2019;10:922. doi:10.3389/fimmu.2019.00922
16. Hong K. Emerging function of N6-methyladenosine in cancer. Oncol Lett. 2018;16(5):5519–5524. doi:10.3892/ol.2018.9395
17. Wu F, Cheng W, Zhao F, Tang M, Diao Y, Xu R. Association of N6-methyladenosine with viruses and related diseases. Virol J. 2019;16(1):133. doi:10.1186/s12985-019-1236-3
18. Chen B, Li Y, Song R, Xue C, Xu F. Functions of RNA N6-methyladenosine modification in cancer progression. Mol Biol Rep. 2019;46(2):2567–2575. doi:10.1007/s11033-019-04655-4
19. Tong J, Flavell RA, Li HB. RNA m(6)A modification and its function in diseases. Front Med. 2018;12(4):481–489. doi:10.1007/s11684-018-0654-8
20. Liu J, Harada BT, He C. Regulation of Gene Expression by N(6)-methyladenosine in Cancer. Trends Cell Biol. 2019;29(6):487–499. doi:10.1016/j.tcb.2019.02.008
21. Chokkalla AK, Mehta SL, Kim T, Chelluboina B, Kim J, Vemuganti R. Transient focal ischemia significantly alters the m(6)A epitranscriptomic tagging of RNAs in the brain. Stroke. 2019;50(10):2912–2921. doi:10.1161/STROKEAHA.119.026433
22. Mathiyalagan P, Adamiak M, Mayourian J, et al. FTO-Dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation. 2019;139(4):518–532. doi:10.1161/CIRCULATIONAHA.118.033794
23. Xu K, Mo Y, Li D, et al. N(6)-methyladenosine demethylases Alkbh5/Fto regulate cerebral ischemia-reperfusion injury. Ther Adv Chronic Dis. 2020;11:2040622320916024. doi:10.1177/2040622320916024
24. Dai N, Rapley J, Angel M, Yanik MF, Blower MD, Avruch J. mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 2011;25(11):1159–1172. doi:10.1101/gad.2042311
25. Ruan DY, Li T, Wang YN, et al. FTO downregulation mediated by hypoxia facilitates colorectal cancer metastasis. Oncogene. 2021;40(33):5168–5181. doi:10.1038/s41388-021-01916-0
26. Lee YH, Kang ES, Kim SH, et al. Association between polymorphisms in SLC30A8, HHEX, CDKN2A/B, IGF2BP2, FTO, WFS1, CDKAL1, KCNQ1 and type 2 diabetes in the Korean population. J Hum Genet. 2008;53(11–12):991–998. doi:10.1007/s10038-008-0341-8
27. Gamboa-Meléndez MA, Huerta-Chagoya A, Moreno-Macías H, et al. Contribution of common genetic variation to the risk of type 2 diabetes in the Mexican Mestizo population. Diabetes. 2012;61(12):3314–3321. doi:10.2337/db11-0550
28. Zhou H, Shen X, Yan C, et al. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate osteoarthritis of the knee in mice model by interacting with METTL3 to reduce m6A of NLRP3 in macrophage. Stem Cell Res Ther. 2022;13(1):322. doi:10.1186/s13287-022-03005-9
29. Liu BH, Tu Y, Ni GX, et al. Total flavones of abelmoschus manihot ameliorates podocyte pyroptosis and injury in high glucose conditions by targeting METTL3-Dependent m(6)A modification-mediated NLRP3-inflammasome activation and PTEN/PI3K/Akt signaling. Front Pharmacol. 2021;12:667644. doi:10.3389/fphar.2021.667644
30. Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi:10.1038/nature09922
31. Zhu W, Gregory JC, Org E, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165(1):111–124. doi:10.1016/j.cell.2016.02.011
32. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi:10.1161/01.STR.20.1.84
33. Xu D, Zhao W, Song J, et al. The relationship of large-artery atherothrombotic stroke with plasma trimethylamine N-Oxide level and blood lipid-related indices: a cross-sectional comparative study. Biomed Res Int. 2021;2021:5549796. doi:10.1155/2021/5549796
34. Gong L, Wang H, Zhu X, et al. Nomogram to predict cognitive dysfunction after a minor ischemic stroke in hospitalized-population. Front Aging Neurosci. 2021;13:637363. doi:10.3389/fnagi.2021.637363
35. Li D, Ke Y, Zhan R, et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell. 2018;17(4):e12768. doi:10.1111/acel.12768
36. Su H, Fan S, Zhang L, Qi H. TMAO aggregates neurological damage following ischemic stroke by promoting reactive astrocytosis and glial scar formation via the Smurf2/ALK5 axis. Front Cell Neurosci. 2021;15:569424. doi:10.3389/fncel.2021.569424
37. Pawluk H, Kołodziejska R, Grześk G, et al. Selected mediators of inflammation in patients with acute ischemic stroke. Int J Mol Sci. 2022;23(18):10614. doi:10.3390/ijms231810614
38. Wang N, Yang Y, Qiu B, et al. Correlation of the systemic immune-inflammation index with short- and long-term prognosis after acute ischemic stroke. Aging. 2022;14(16):6567–6578. doi:10.18632/aging.204228
39. Simats A, Liesz A. Systemic inflammation after stroke: implications for post-stroke comorbidities. EMBO Mol Med. 2022;14(9):e16269. doi:10.15252/emmm.202216269
40. Kim JY, Park J, Chang JY, Kim SH, Lee JE. Inflammation after ischemic stroke: the role of leukocytes and glial cells. Exp Neurobiol. 2016;25(5):241–251. doi:10.5607/en.2016.25.5.241
41. Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84(1):49–59. doi:10.1139/Y05-143
42. Wood PL. Microglia as a unique cellular target in the treatment of stroke: potential neurotoxic mediators produced by activated microglia. Neurol Res. 1995;17(4):242–248. doi:10.1080/01616412.1995.11740321
43. Xue M, Mikliaeva EI, Casha S, Zygun D, Demchuk A, Yong VW. Improving outcomes of neuroprotection by minocycline: guides from cell culture and intracerebral hemorrhage in mice. Am J Pathol. 2010;176(3):1193–1202. doi:10.2353/ajpath.2010.090361
44. Park SH, Lee YS, Yang HJ, Song GJ. Fluoxetine potentiates phagocytosis and autophagy in microglia. Front Pharmacol. 2021;12:770610. doi:10.3389/fphar.2021.770610
45. de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab. 2014;34(3):369–375. doi:10.1038/jcbfm.2013.227
46. de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28(13):3404–3414. doi:10.1523/JNEUROSCI.0157-08.2008
47. Zhu H, Jian Z, Zhong Y, et al. Janus kinase inhibition ameliorates ischemic stroke injury and neuroinflammation through reducing NLRP3 inflammasome activation via JAK2/STAT3 pathway inhibition. Front Immunol. 2021;12:714943. doi:10.3389/fimmu.2021.714943
48. Haque ME, Akther M, Jakaria M, Kim IS, Azam S, Choi DK. Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov Disord. 2020;35(1):20–33. doi:10.1002/mds.27874
49. Hanslik KL, Ulland TK. The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front Neurol. 2020;11:570711. doi:10.3389/fneur.2020.570711
50. Zhang Z, Wang Q, Zhao X, et al. YTHDC1 mitigates ischemic stroke by promoting Akt phosphorylation through destabilizing PTEN mRNA. Cell Death Dis. 2020;11(11):977. doi:10.1038/s41419-020-03186-2
51. Si W, Li Y, Ye S, et al. Methyltransferase 3 mediated miRNA m6A methylation promotes stress granule formation in the early stage of acute ischemic stroke. Front Mol Neurosci. 2020;13:103. doi:10.3389/fnmol.2020.00103
52. Song Z, Qiu L, Hu Z, Liu J, Liu D, Hou D. Evaluation of the obesity genes FTO and MC4R for contribution to the risk of large artery atherosclerotic stroke in a Chinese Population. Obes Facts. 2016;9(5):353–362. doi:10.1159/000448588
53. Huang J, Sun W, Wang Z, et al. FTO suppresses glycolysis and growth of papillary thyroid cancer via decreasing stability of APOE mRNA in an N6-methyladenosine-dependent manner. J Exp Clin Cancer Res. 2022;41(1):42. doi:10.1186/s13046-022-02254-z
54. Sun M, Zhang X, Bi F, et al. FTO inhibits epithelial ovarian cancer progression by destabilising SNAI1 mRNA through IGF2BP2. Cancers. 2022;14(21):5218. doi:10.3390/cancers14215218
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Modified Citrus Pectin Alleviates Cerebral Ischemia/Reperfusion Injury by Inhibiting NLRP3 Inflammasome Activation via TLR4/NF-ĸB Signaling Pathway in Microglia
Cui Y, Zhang NN, Wang D, Meng WH, Chen HS
Journal of Inflammation Research 2022, 15:3369-3385
Published Date: 9 June 2022
Punicalagin Attenuates LPS-Induced Inflammation and ROS Production in Microglia by Inhibiting the MAPK/NF-κB Signaling Pathway and NLRP3 Inflammasome Activation

Lo J, Liu CC, Li YS, Lee PY, Liu PL, Wu PC, Lin TC, Chen CS, Chiu CC, Lai YH, Chang YC, Wu HE, Chen YR, Huang YK, Huang SP, Wang SC, Li CY
Journal of Inflammation Research 2022, 15:5347-5359
Published Date: 15 September 2022
Inhibition of Dectin-1 Alleviates Neuroinflammatory Injury by Attenuating NLRP3 Inflammasome-Mediated Pyroptosis After Intracerebral Hemorrhage in Mice: Preliminary Study Results
Ding Z, Zhong Z, Wang J, Zhang R, Shao J, Li Y, Wu G, Tu H, Yuan W, Sun H, Wang Q
Journal of Inflammation Research 2022, 15:5917-5933
Published Date: 19 October 2022
Nrf2 Deficiency Exacerbates Parkinson’s Disease by Aggravating NLRP3 Inflammasome Activation in MPTP-Induced Mouse Models and LPS-Induced BV2 Cells
Lu R, Zhou X, Zhang L, Hao M, Yang X
Journal of Inflammation Research 2024, 17:6277-6295
Published Date: 10 September 2024
Low-Frequency (1 Hz) Repetitive Trans-Spinal Magnetic Stimulation Attenuates NLRP3-Driven Neuroinflammation and Enhances Motor Recovery in Spinal Cord-Injured Mice
Song H, Yang Y, Huang L, Li Y, Sun B, Yu Z, Zhang M, Yang D
Journal of Inflammation Research 2025, 18:11263-11284
Published Date: 18 August 2025