Back to Journals » ImmunoTargets and Therapy » Volume 14
TLR9: A Double-Dealing Toll-Like Receptor
Authors Nielsen M ![]() , Nishizaki D, Kato S, Kurzrock R
, Nishizaki D, Kato S, Kurzrock R ![]()
Received 28 August 2025
Accepted for publication 9 December 2025
Published 30 December 2025 Volume 2025:14 Pages 1531—1554
DOI https://doi.org/10.2147/ITT.S563765
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Michael Shurin
Megan Nielsen,1 Daisuke Nishizaki,2,* Shumei Kato,2,* Razelle Kurzrock1
1Department of Medicine, Medical College of Wisconsin, Milwaukee, WI, USA; 2Moores Cancer Center, University of California San Diego, La Jolla, CA, USA
*These authors contributed equally to this work
Correspondence: Megan Nielsen, Medical College of Wisconsin, 8701 W Watertown Plank Road, Milwaukee, WI, 53226, USA, Email [email protected]
Abstract: In 2011, the Nobel Prize was awarded for the discovery of Toll-like receptors (TLRs) and their critical role in immunity. TLR9 is a key pattern recognition receptor that detects unmethylated cytosine-phosphate-guanine (CpG) DNA motifs, triggering innate and adaptive immune responses against pathogens and damaged host cells. Upon activation, TLR9 triggers signaling cascades that drive NF-κB, STAT3, and MAPK pathways, modulating inflammatory responses, cellular survival mechanisms, and immune regulation. While TLR9 activation is essential for immune defense, TLR9 acts as a double-dealing TLR in multiple pathologies, including cancer, autoimmunity, and chronic inflammatory disease. In cancer biology, TLR9 exhibits context-dependent roles, acting as a driver of tumorigenesis, a suppressor of tumor growth, and a regulator of immune responses, depending on the tumor type, signaling pathway, and microenvironment. It promotes tumorigenesis in leukemias, gliomas, and cancers of the prostate, bone, lung, and gastrointestinal tract, yet displays tumor-suppressive effects in triple-negative breast cancer, renal cell carcinoma, and virally-associated malignancies, through a variety of mechanisms. Clinically, synthetic CpG oligodeoxynucleotides (ODNs), which function as TLR9 agonists, have emerged as a promising approach in cancer immunotherapy, particularly in combination with other potent anticancer therapies. However, the dual nature of TLR9 signaling poses challenges for therapeutic applications. Its context-dependent effects contribute to inconsistent clinical outcomes and raise concerns about safety and toxicity. This review examines the immunologic function and signaling mechanisms of TLR9, with a focus on its complex, context-specific, and “double-dealing” roles in cancer pathogenesis and therapy.
Keywords: toll-like receptor 9, NF-κB, STAT3, TLR9 agonist, cancer, immunotherapy, inflammation
Introduction
The 2011 Nobel Prize in Physiology and Medicine was awarded to Bruce Beutler, Jules Hoffmann, and Ralph Steinman for their discoveries regarding immune system function, especially Toll-like receptors (TLRs).1 The TLR family of pattern recognition receptors (PRRs) identifies pathogen-associated molecular patterns (PAMPs) from invading microbes and damage-associated molecular patterns (DAMPs) from stressed or damaged host cells.2,3 TLRs can be broadly categorized by their cellular localization and ligand types. Cell-surface TLRs (TLR1, TLR2, TLR4, TLR5, TLR6) primarily sense lipid- and protein-based microbial components, whereas endosomal TLRs (TLR3, TLR7/8, TLR9) detect nucleic acids.4 Among the nucleic acid-sensing TLRs, TLR9 is unique in both its ligand specificity and signaling pathways. TLR9 is constitutively expressed in both plasmacytoid dendritic cells (pDCs) and B cells and primarily recognizes unmethylated cytosine-phosphate-guanosine (CpG) DNA–motifs that are common among bacteria and single-stranded DNA viruses, but rare, and usually methylated, in mammalian DNA.5 TLR9 signals through a myeloid differentiation 88 (MyD88)-dependent pathway to activate nuclear factor kappa B (NF-κB), activator protein 1 (AP-1), and a broad interferon regulatory factor (IRF) repertoire—including IRF-1, IRF-5, IRF-7, and IRF-8—stimulating both type I interferons (IFN) and pro-inflammatory cytokines.6 In contrast, TLR3 detects dsRNA and signals exclusively through the TIR-domain-containing adaptor-inducing beta interferon (TRIF) adaptor to activate a predominantly antiviral response.4,6 TLR7 and TLR8 signal through MyD88 and, like TLR9, can activate NF-κB and AP-1 pathways; however, they recognize ssRNA and are classically associated with an acute antiviral type I IFN response that differs from the broader range of signaling elicited by CpG DNA.6,7
The outcome of TLR9 activation depends on the nature and origin of its ligands, which include both exogenous microbial DNA and endogenous host-derived DNA. Exogenous CpG-rich DNA derived from bacteria and viruses are internalized by antigen presenting cells (APCs), including pDCs and B cells, and routed to endosomal compartments, where they drive a robust IRF-7-dependent type I IFN response to combat the pathogen.6 Exogenous TLR9 activation also plays a role in cancer, as several tumor types—including esophageal, gastric, colorectal, and pancreatic cancers—are exposed to bacterial CpG DNA that can activate TLR9 and contribute to chronic inflammation and tumorigenesis.8–10 In contrast, endogenous TLR9 ligands arise from nuclear and mitochondrial DNA (mtDNA) released during necrosis, apoptosis, hypoxia, oxidative stress, or cytotoxic therapy and preferentially activate tissue-resident cells, including epithelial, parenchymal, and stromal cells. Host DNA is typically methylated and therefore a poor activator of TLR9; however, many tumors exhibit profound genomic hypomethylation and release cell-free, unmethylated, CpG-rich DNA fragments that can activate TLR9 and drive pro-tumorigenic inflammation.11–14 Endogenous methylated CpG sequences may also activate TLR9 when delivered within extracellular vesicles or apoptotic bodies—structures that bypass the methylation barrier by physically trafficking DNA into endosomal compartments—although this delivery generally produces weaker, NF-κB-dominant signaling with minimal type I IFN production.6,15 Host mtDNA released during cell damage or stress is largely unmethylated and CpG-rich, capable of efficiently engaging TLR9 and contributing to tumorigenesis in many cancer types.16–19
Once CpG binding has occurred, TLR9 initiates a well-defined MyD88-dependent signaling cascade that stimulates both innate and adaptive immune responses.2 CpG binding recruits MyD88, MyD88 adaptor-like (MAL), and Src kinase-interacting membrane protein (SCIMP), forming a Myddosome complex with interleukin-1 receptor-associated kinase-1 and −4 (IRAK1, IRAK4).3 This complex recruits TNF receptor-associated factor-6 (TRAF6), which induces interferon regulatory factor 7 (IRF-7) activation and transforming growth factor-β-activated kinase 1 (TAK1) phosphorylation, leading to mitogen-activated protein kinase (MAPK)-dependent AP-1 activation and NF-κB translocation via inhibitor of NF-κB (IκB) phosphorylation (Figure 1).2,5 NF-κB, AP-1, and IRF-7 mediate TLR9-induced inflammation by promoting the transcription of proinflammatory cytokines, the production of costimulatory molecules, and the activation of adaptive immune cells.2,5 While TLR9 signaling is crucial for adequate immune responses to pathogen invasion, excessive TLR9 activation, resulting from TLR9 overexpression or CpG demethylation, can lead to inflammatory and neoplastic pathologies.3 This review will outline the involvement of TLR9 in tumor progression, immune modulation, novel cancer therapy strategies, and diseases beyond cancer.
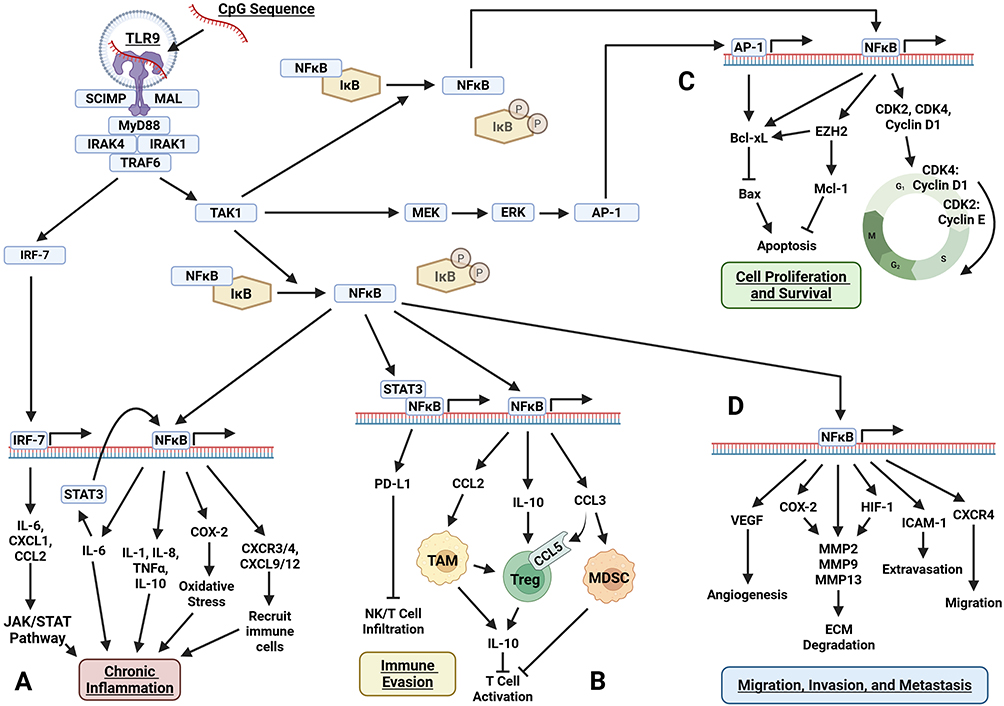 |
Figure 1 Pro-tumorigenic pathways activated by TLR9 in tumor cells. Upon CpG sequence recognition, TLR9–via MyD88 signaling–induces downstream activation of NFĸB, AP-1, and IRF-7. These transcription factors drive major tumor-promoting mechanisms including (A) chronic inflammation via induction of STAT3, IL-6, IL-8, IL-10, TNFα, COX-2, and chemokines; (B) immune evasion through STAT3/NFĸB-mediated PD-L1 expression and recruitment of TAMs, Tregs, and MDSCs; (C) cell proliferation and survival through upregulation of cyclins, CDKs, and anti-apoptotic proteins; and (D) migration, invasion, and metastasis through VEGF, HIF-1, ICAM-1, CXCR4, and MMPs. Figure created with BioRender. Abbreviations: AP-1, Activator Protein 1; Bax, BCL2-Associated X Protein; Bcl-xL, B-Cell Lymphoma Extra Large; CCL2, C-C Motif Chemokine Ligand 2; CCL3, C-C Motif Chemokine Ligand 3; CCL5, C-C Motif Chemokine Ligand 5; CDK2, Cyclin-Dependent Kinase 2; CDK4, Cyclin-Dependent Kinase 4; COX-2, Cyclooxygenase-2; CXCL1, C-X-C Motif Chemokine Ligand 1; CXCL9, C-X-C Motif Chemokine Ligand 9; CXCL12, C-X-C Motif Chemokine Ligand 12; CXCR3, C-X-C Chemokine Receptor Type 3; CXCR4, C-X-C Chemokine Receptor Type 4; ECM, Extracellular Matrix; ERK, Extracellular Signal-Regulated Kinase; EZH2, Enhancer of Zeste Homolog 2; HIF-1, Hypoxia-Inducible Factor 1; ICAM-1, Intercellular Adhesion Molecule 1; IκB, Inhibitor of Nuclear Factor Kappa B; IL-1, Interleukin-1; IL-6, Interleukin-6; IL-8, Interleukin-8; IL-10, Interleukin-10; IRAK1, Interleukin-1 Receptor-Associated Kinase 1; IRAK4, Interleukin-1 Receptor-Associated Kinase 4; IRF-7, Interferon Regulatory Factor 7; JAK/STAT, Janus Kinase/Signal Transducer and Activator of Transcription; MAL, MyD88-Adaptor-Like; MDSC, Myeloid-Derived Suppressor Cell; MEK, Mitogen-Activated Protein Kinase Kinase; Mcl-1, Myeloid Cell Leukemia 1; MMP2, Matrix Metalloproteinase 2; MMP9, Matrix Metalloproteinase 9; MMP13, Matrix Metalloproteinase 13; MyD88, Myeloid Differentiation Primary Response 88; NFκB, Nuclear Factor Kappa B; NK Cell, Natural Killer Cell; PD-L1, Programmed Death-Ligand 1; SCIMP, src Kinase-Interacting Membrane Protein; STAT3, Signal Transducer and Activator of Transcription 3; TAK1, Transforming Growth Factor-Beta-Activated Kinase 1; TAM, Tumor-Associated Macrophage; TNFα, Tumor Necrosis Factor Alpha; TRAF6, TNF Receptor-Associated Factor 6; Treg, Regulatory T Cell; TLR9, Toll-Like Receptor 9; VEGF, Vascular Endothelial Growth Factor. |
TLR9 Pathways in Cancer
TLR9 has a complex, context-dependent role in tumorigenesis, acting as both a stimulator of tumorigenesis and a promoter of antitumor immunity, depending on the cell type, tumor stage, and tumor microenvironment (TME).
Oncogenic Signaling and Tumor Progression
TLR9 signaling promotes tumorigenesis by driving proliferation, invasion, and immune modulation across multiple cancer types. TLR9 overexpression is correlated with poor outcomes in leukemia, glioma, osteosarcoma, and cancers of the stomach, colon, cervix, and prostate.20–26 Its downstream signaling influences tumor initiation, progression, and metastasis, with NF-κB acting as a central mediator of TLR9-induced oncogenesis.23,24 Upon activation, NF-κB regulates the transcription of proinflammatory cytokines, growth factors, and anti-apoptotic proteins.27 Cross-talk with signal transducer and activator of transcription 3 (STAT3) and AP-1 further amplifies these tumor-promoting effects. TLR9 also engages other central oncogenic pathways, including IRF-7 and MAPK. Collectively, these interconnected signaling cascades mediate TLR9’s tumor-promoting activity, driving several key hallmarks of cancer.
Inflammatory Pathways and Tumor Initiation
Early in tumorigenesis, TLR9-induced activation of NF-κB promotes chronic inflammation and supports cell proliferation and survival, particularly in inflammatory-driven malignancies such as gastric, pancreatic, and colorectal cancer (CRC) (Figure 1A).3 Major inflammatory mediators stimulated by NF-κB, including interleukin 1 (IL-1), interleukin 6 (IL-6), interleukin 8 (IL-8), and tumor necrosis factor-alpha (TNF-α), amplify its signaling through positive feedback, thereby sustaining a proinflammatory TME.28 In gastric cancer, high TLR9 expression correlates with inflammation severity and marks progression from H. pylori gastritis to gastric adenocarcinoma.26 In addition to stimulating proinflammatory cytokines IL-6, IL-8, and TNFɑ, TLR9-induced NF-κB activation also upregulates cyclooxygenase-2 (COX-2), facilitating the release of reactive oxygen species (ROS) from malignant adenocarcinoma cells and tumor-associated macrophages (TAMs).29 This process induces oxidative stress and DNA damage, creating an inflammatory TME that accelerates tumor progression. A similar TLR9/NF-κB-driven COX-2 pathway occurs in CRC, accompanied by the release of chemokine receptors 3 and 4 (CXCR3, CXCR4) and chemokine ligands 9 and 12 (CXCL9, CXCL12) which recruit additional immune cells.24,30 This effect is mediated by NF-κB’s interaction with STAT3—a protein that regulates many aspects of cell growth, survival, and differentiation—and is frequently dysregulated in CRC and other tumors.30 NF-κB cooperatively binds with STAT3 to activate genes that drive tumor-promoting inflammation and cancer cell proliferation, invasion, angiogenesis, and metastasis.28 IL-6, an essential downstream effector of NF-κB:STAT3 signaling, regulates proliferation, survival, and inflammation and is critical in early CRC tumorigenesis.30 Similarly, in pancreatic cancer, TLR9 activation in tumor cells induces nearby stellate cells to secrete inflammatory cytokines (IL-1, IL-10, TNF-α) and chemokines (CCL3, CCL11, CCL12), contributing to a proinflammatory TME that promotes tumorigenesis.8
TLR9 activation is associated with pro-tumorigenic inflammation in many other cancer types as well; however, the exact mechanisms underlying this association are not fully understood. In chronic lymphocytic leukemia (CLL), high TLR9 expression is correlated with increased production of proinflammatory cytokines and chemokines, as well as elevated NF-κB and STAT3 activity.25 Similarly, TLR9 is upregulated in glioma, where it is associated with advanced histopathological grades, stem cell-like phenotypes, and tumor progression.21,31 In these tumors, IRF-7 is a key regulator of type I interferons and a major mediator of pro-tumorigenic inflammation.31 Its activation in glioma promotes secretion of IL-6 and chemokine ligands 1 and 2 (CXCL1, CXCL2), activating the Janus kinase/STAT3 pathway and promoting stem cell-like phenotypes and angiogenesis.31 Although no studies have specifically examined the effects of TLR9 on IRF-7 signaling in glioma, the known activation of IRF-7 downstream of TLR9 suggests a potential connection between TLR9 expression and IRF-7-mediated inflammation in this context.
Regulation of Cell Proliferation and Survival
TLR9 signaling through NF-κB, STAT3, and AP-1 promotes cell cycle progression and inhibits apoptosis across many tumor types by inducing expression of cell cycle regulators and upregulating survival genes (Figure 1C). B-cell lymphoma-extra large (Bcl-xL), a central mediator of cell survival, is upregulated in CLL, liver, pancreatic, and colorectal neoplasms.24,32–34 Bcl-xL promotes survival by inhibiting the pro-apoptotic factor Bcl-2-associated X protein (Bax), which prevents mitochondrial pore formation and blocks cytochrome c release.27 In CRC, Bcl-xL expression is regulated by NF-κB, whereas other tumors rely on alternative mechanisms.24 TLR9 signaling in CLL activates enhancer of zeste homolog 2 (EZH2), an epigenetic regulator that upregulates Bcl-xL and myeloid cell leukemia 1 (Mcl-1), likely by transcriptionally silencing tumor suppressors that normally inhibit their expression.32 This decreases the cleavage of pro-apoptotic factors, including poly (ADP-ribose) polymerase (PARP) and caspase-3, suppressing apoptosis and enhancing cell proliferation.32 In pancreatic cancer, TLR9 regulates Bcl-xL through the MAPK pathway, as evidenced by increased phosphorylation of the MAPK protein extracellular signal-regulated kinase (ERK).34 Similar to pancreatic cancer, TLR9-driven Bcl-xL expression in hepatocellular carcinoma (HCC) occurs independently of NF-κB and IRF-7, suggesting an alternative mechanism potentially involving MAPK/AP-1 signaling.33 AP-1 is a downstream effector of TLR9 signaling and is implicated in other tumors, such as oral squamous cell carcinoma (OSCC).35 Given that AP-1 can directly regulate Bcl-xL expression, it is plausible that TLR9 activation increases Bcl-xL activity via AP-1, thereby suppressing apoptosis and promoting tumorigenesis in HCC.
TLR9 signaling promotes cell cycle progression through NF-κB-mediated activation of key cell cycle proteins. In HR+ breast cancer, osteosarcoma, and cervical cancer, TLR9/NF-κB signaling upregulates cyclin D1, facilitating the transition from G1 to S phase and promoting cell proliferation.23,36,37 TLR9 also increases the expression of cyclin-dependent kinase (CDK) proteins, including CDK4 in cervical cancer and CDK2 in osteosarcoma and lung cancer.23,37,38 In OSCC, TLR9 activates AP-1, which binds to the cyclin D1 promoter, enhancing its transcription and promoting cell cycle progression.35
Tumor Migration, Invasion, and Metastasis
Invasion, angiogenesis, and intravasation/extravasation are critical processes driving tumor progression and metastasis.39 Elevated TLR9 expression is linked to enhanced invasion, migration, angiogenesis, and metastasis across various cancers through specific signaling pathways (Figure 1D). In prostate and pancreatic cancer, TLR9 upregulates vascular endothelial growth factor (VEGF), a key regulator of angiogenesis, increasing tumor cell invasion and migration in both cell lines34 and patients.20 NF-κB mediates this effect by binding to the VEGF promoter and initiating its transcription.20,27 VEGF then promotes endothelial cell proliferation, vascular permeability, and vasculogenesis, increasing nutrient delivery to tumor cells.20
Extracellular matrix (ECM) degradation is crucial to tumor invasion and is primarily driven by matrix metalloproteinases (MMPs).39 These proteolytic enzymes are frequently overexpressed in cancer and positively correlated with TLR9 signaling in many tumor types. TLR9 activation induces the release of MMP-2 and MMP-9 in osteosarcoma, glioma, glioblastoma, lung, gastric, and prostate cancer, while MMP-13 contributes to ECM degradation in prostate cancer.23,29,40–43 NF-κB can directly upregulate MMPs27 or act indirectly via COX-2–a proinflammatory mediator that increases MMP expression.44 In brain tumors, hypoxia amplifies TLR9-induced MMP expression.40 A common hallmark of solid tumors, hypoxia promotes angiogenesis and invasion by activating HIF-1, a transcription factor that upregulates MMPs and further enhances TLR9 signaling in a positive feedback loop that perpetuates tumor invasion.40
TLR9 activity in lung and prostate cancer is associated with increased CXCR4 expression, which drives tumor cell migration.41,42 To facilitate metastasis, chemokines secreted in blood vessels and distant organs bind to chemokine receptors on tumor cells, directing their migration to distant sites. At these sites, cell adhesion molecules facilitate the attachment and extravasation of tumor cells into new tissues. This occurs in lung cancer, where TLR9 activity increases the expression of intercellular adhesion molecule 1 (ICAM-1).41 By regulating critical molecules such as VEGF, MMPs, CXCR4, and ICAM-1, TLR9 facilitates tumor invasion, migration, and metastasis. As a result, it is a promising target for therapies aimed at limiting tumor spread in aggressive cancers.
There is evidence to suggest that TLR9 may facilitate tumor progression in part by promoting or sustaining the epithelial-mesenchymal transition (EMT), a key mechanism that enables malignant cells to acquire migratory and invasive phenotypes. In lung adenocarcinoma, EMT induction enhances sensitivity to CpG stimulation, leading to increased cytokine production—an effect that is lost when TLR9 is inhibited.45 In breast cancer, high TLR9 expression correlates with more aggressive, invasive subtypes, and EMT-like transcriptional profiles.46 For example, TLR9 expression is positively associated with expression of zinc finger E-box-binding homeobox 2 (ZEB2), Snail family transcriptional repressor 1 (SNAIL), twist family bHLH transcription factor 1 (TWIST1), and vimentin.46 Additional studies in cancer models are needed to fully characterize this mechanism; however, TLR9 activation is known to induce EMT in non-malignant inflammatory settings, supporting the plausibility of a similar pathway in tumors.47
Immune Modulation and Tumor Immune Evasion
The immune system plays a dual role in tumorigenesis, with distinct immune cell populations either suppressing or supporting cancer development. TAMs, myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs) promote tumor progression by producing growth factors, pro-angiogenic molecules, and matrix-degrading enzymes (Figure 1B).39 In CLL, TLR9 increases the secretion of the anti-inflammatory cytokine IL-10 and recruits Tregs, which suppress the proliferation of cytotoxic CD4+ T cells and enable tumor immune escape.48 In pancreatic cancer, TLR9-dependent signaling directly recruits Tregs and MDSCs to the TME via secretion of CCL3, which binds to chemokine receptor 5 (CCR5) on Tregs.8 Within the TME, Tregs release IL-10, suppressing major histocompatibility complex class II (MHC II) expression and costimulatory signals in antigen-presenting cells, while MDSCs inhibit cytotoxic T cell activation, collectively enhancing immunosuppression and promoting tumor progression.8 MDSCs similarly accelerate tumor growth in prostate cancer. In this context, TLR9-induced upregulation of leukemia inhibitory factor, an IL-6 family cytokine, activates and recruits a specific subpopulation of MDSCs, namely polymorphonuclear (PMN)-MDSCs, to the TME.49 These PMN-MDSCs trigger STAT3 activation, driving the expression of IL-10 and arginase-1 to sustain an immunosuppressive TME.49
Mitochondrial DNA (mtDNA) stress is characterized by the release of mtDNA into the cytosol and is often indicative of oncogenesis.3 In HCC, TLR9 detects cytosolic mtDNA and activates the NF-κB pathway, leading tumor cells to secrete CCL2, which recruits TAMs.50 These TAMs then release IL-10 and chemokine ligands 7 and 22 (CCL17, CCL22) to attract additional immunosuppressive cells and support HCC progression.50 Notably, TLR9 activation also promotes immune escape in both HCC and OSCC by STAT3-mediated upregulation of programmed cell death ligand 1 (PD-L1) on tumor cells.51,52 This upregulation reduces the infiltration and activation of NK cells and CD4+/CD8+ T cells within the TME while increasing Tregs, collectively contributing to an immunosuppressive TME that supports cancer progression.51,52
Antitumorigenic Pathways
TLR9 signaling can protect against cancer development in a few specific contexts. TLR9 overexpression is associated with improved outcomes in triple-negative breast cancer (TNBC), renal cell carcinoma (RCC), neuroblastoma, and cancers of the cervix, head and neck, oropharynx, and pancreas.46,53–58 In this protective role, TLR9 interferes with several key hallmarks of oncogenesis, including tumor initiation, evasion, and immune system dysregulation (Figure 2). In these pathways, TLR9 appears to promote a more balanced and stable inflammatory response, which may help limit tumor growth and progression. However, these antitumorigenic mechanisms may differ based on tumor types and microenvironments.
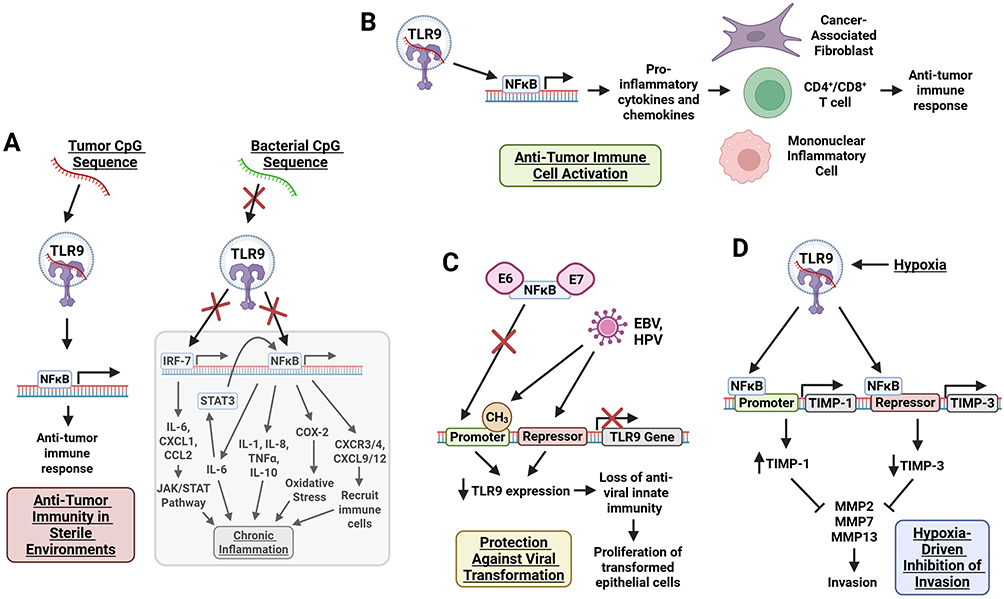 |
Figure 2 Anti-tumorigenic roles of TLR9 in pre-cancerous cells. Upon CpG sequence recognition, TLR9 activates NFĸB through MyD88-dependent signaling, driving transcriptional regulation that (A) supports anti-tumor immunity in sterile environments; (B) sustains innate immune defenses to protect against viral transformation; (C) modulates the expression of hypoxia-sensitive invasion suppressors, TIMP-1 and TIMP-3; and (D) promotes anti-tumor immune cell activation through induction of pro-inflammatory cytokines and chemokines. Figure created with BioRender. Abbreviations: CCL2, C-C Motif Chemokine Ligand 2; CD4+/CD8+, Cluster of Differentiation 4+/8+ T Cells; COX-2, Cyclooxygenase-2; CpG, Cytosine-phosphate-Guanine; CXCL1, C-X-C Motif Chemokine Ligand 1; CXCL9, C-X-C Motif Chemokine Ligand 9; CXCL12, C-X-C Motif Chemokine Ligand 12; CXCR3/4, C-X-C Chemokine Receptor Type 3/4; EBV, Epstein-Barr Virus; E6/E7, Oncoproteins of Human Papillomavirus (HPV); HPV, Human Papillomavirus; IL-1, Interleukin-1; IL-6, Interleukin-6; IL-8, Interleukin-8; IL-10, Interleukin-10; IRAK1, Interleukin-1 Receptor-Associated Kinase 1; IRAK4, Interleukin-1 Receptor-Associated Kinase 4; IRF-7, Interferon Regulatory Factor 7; iNOS, Inducible Nitric Oxide Synthase; JAK/STAT, Janus Kinase/Signal Transducer and Activator of Transcription; MAL, MyD88-Adaptor-Like; MMP2, Matrix Metalloproteinase 2; MMP7, Matrix Metalloproteinase 7; MMP13, Matrix Metalloproteinase 13; MyD88, Myeloid Differentiation Primary Response 88; NFκB, Nuclear Factor Kappa B; SCIMP, src Kinase-Interacting Membrane Protein; STAT3, Signal Transducer and Activator of Transcription 3; TIMP-1, Tissue Inhibitor of Metalloproteinases 1; TIMP-3, Tissue Inhibitor of Metalloproteinases 3; TLR9, Toll-Like Receptor 9; TNFα, Tumor Necrosis Factor Alpha; TRAF6, TNF Receptor-Associated Factor 6. |
TLR9 and Viral Oncology
Oncoviruses contribute to the malignant transformation of many tumors, including gynecological, hematological, head and neck, and liver cancers. Effective viral immune responses are crucial in preventing tumorigenesis and are mediated, in part, by PRRs, like TLR9, in organ tissues and nearby immune cells. TLR9 recognizes unmethylated CpG motifs present in viral DNA; however, oncogenic viruses frequently suppress its activity to evade immune detection. For example, TLR9 is downregulated by Epstein-Barr virus (EBV) in oropharyngeal cancer and by E6 and E7 from human papillomavirus 16 (HPV16) in head and neck and cervical neoplasms.54,55 This impairs antiviral innate immunity and promotes the growth of transformed epithelial cells (Figure 2C). In head and neck cancer, restoring TLR9 expression stabilized p16, a CDK4 inhibitor, which prolonged the S phase and activated cell cycle checkpoints, ultimately reducing cell proliferation and colony formation.54 This may explain why low TLR9 expression is associated with higher tumor grades in cervical cancer.59 In contrast, other research has instead linked high TLR9 expression with more advanced cervical cancer, so further research is needed to understand TLR9’s role in this malignancy.22 In early-stage ovarian tumors, high TLR9 expression is associated with extended patient survival; however, the underlying mechanism for this observation has not been investigated.56 The presence of DNA from HPV, EBV, and cytomegalovirus has been detected in ovarian malignancies, suggesting a potential viral contribution to ovarian carcinogenesis.60 Given this, TLR9 may play a similar antitumor role in ovarian cancer as it does in cervical and head and neck cancers.
TLR9 in Tumors with Minimal Microbial Exposure
Upregulation of TLR9 is associated with improved survival in patients with pancreatic cancer.57 This finding aligns with in vitro studies demonstrating that TLR9 signaling suppresses pancreatic cancer migration, invasion, and metastasis.61 Although the underlying mechanism is unclear, one hypothesis suggests that the relatively sterile environment of the pancreas–compared to bacterial-rich organs such as the oral cavity, stomach, and colon–may allow TLR9 to promote antitumor immune responses without triggering the proinflammatory signaling pathways that drive tumorigenesis in tissues more frequently exposed to microbiota (Figure 2A).57 Similar associations between TLR9 expression and favorable prognostic factors in tumors from other relatively sterile organs, such as the kidney and brain, support this hypothesis. In RCC, high TLR9 expression correlates with improved survival, and in neuroblastoma, it inversely correlates with disease stage.53,58 However, other tumor-intrinsic mechanisms may be involved as this process cannot fully explain the observed decrease in pancreatic tumor cell proliferation, invasion, and metastasis in tumor cell cultures that lack immune cells. Furthermore, in mucoepidermoid salivary gland carcinoma, high TLR9 expression is associated with less invasion and better survival despite this tumor developing in the oral cavity, an area rich in bacteria.62 These observations suggest that TLR9 exerts context-dependent antitumor effects through both immune-mediated and tumor-intrinsic mechanisms.
Hypoxia-Driven Modulation of Invasion Pathways
The antitumor effects of TLR9 are context-dependent, especially in TNBC, where hypoxic conditions significantly influence TLR9’s activity. Although TLR9 is generally upregulated in TNBC, a subset of TNBC patients with low TLR9 expression exhibit significantly decreased survival, suggesting a protective role for TLR9 in this setting.46 The ability of hypoxia to promote tumor cell invasion in TNBC depends on TLR9 expression. When TLR9 expression is low, hypoxia enhances invasion through transcriptional inactivation of tissue inhibitor of metalloproteinase 3 (TIMP-3) and downregulation of TIMP-1 and TIMP-2.63 This leads to increased activity of MMP-2, MMP-7, and MMP-13, resulting in unchecked proteolytic activity and increasing tumor cell invasion (Figure 2D). In contrast, TNBC cells with normal or high TLR9 expression respond differently: hypoxia downregulates TIMP-3 without inactivating it, upregulates TIMP-1, and has no effect on TIMP-2.63 This balance in MMP activity restricts tumor cell invasion and may explain the increased survival observed in TNBCs with higher TLR9 expression. Notably, less aggressive subtypes, such as ER+ breast cancer, do not exhibit this trend, likely due to tumor-specific differences in TLR9 activity and downstream signaling under hypoxic conditions.63
Although differences in TLR9 expression correlate with invasion in TNBC, the minimal or absent expression of TLR9 in the low-TLR9 subtype of TNBC makes it unlikely that TLR9 itself directly mediates the hypoxia-induced loss of TIMP-3. Instead, the absence of TLR9 signaling may permit alternative pathways to drive invasion. TLR9 is known to inhibit the signaling of other endosomal TLRs. Chloroquine–an inhibitor of endosomal acidification and TLR activation–reduces invasion in TNBC cells, suggesting that other TLRs on the endosomal membrane may induce pro-invasive phenotypes.63 Therefore, loss of TLR9 in TNBC may allow other endosomal TLRs to promote invasion through TIMP-3 inactivation.63 However, this hypothesis requires experimental validation.
Immune Activation and Tumor Suppression
Although TLR9 expression in tumor cells appears to induce tumor-promoting immune responses, it may also facilitate antitumor immunity in specific contexts and cancer types. For example, TNBC tumors with high TLR9 expression exhibit an inflammatory TME that correlates with improved prognosis.46 TLR9-induced activation of NF-κB drives the expression of proinflammatory cytokines and chemokines, which recruit cancer-associated fibroblasts, mononuclear inflammatory cells, and CD4+/CD8+ T cells (Figure 2B). Importantly, in this context, immune cells are directed against the tumor, thereby strengthening antitumor immunity and contributing to improved outcomes.46 Interestingly, TLR9’s antitumor immune effects may be particularly relevant following cancer treatment, as in vivo responses to doxorubicin chemotherapy in TNBC depend on TLR9 expression status.14,64
High TLR9 expression also correlates with increased survival in RCC. Given the immunogenic nature of RCC, TLR9’s role in antitumor immunity may explain this observation, but this mechanism has not been investigated.58 The specific immune populations mediating TLR9’s effects in TNBC and RCC require further exploration. However, CD8+ T cells are unlikely to be the primary immune cells involved, as TLR9 expression does not correlate with tumor-infiltrating CD8+ T cell count in either TNBC or RCC.64
Immune Cell TLR9 Expression in Tumor Progression and Defense
Given TLR9’s involvement in both innate and adaptive immunity, its signaling in immune cells can exert either tumor-promoting or tumor-suppressive effects, depending on the context. Evidence largely supports TLR9’s anticancer effects, primarily mediated by pDC and B cells–the only two cell types that constitutively express TLR9. TLR9 signaling in pDCs triggers IFN secretion, which upregulates costimulatory cluster of differentiation 80/86 (CD80/86) molecules on APCs—including pDCs and macrophages—thereby promoting activation of CD4+ T cells (Th1), CD8+ T cells, and NK cells to support antitumor immunity (Figure 3A).65 Additionally, pDCs secrete TNF-related apoptosis-inducing ligand (TRAIL), which binds to death receptors on tumor cells and initiates apoptosis (Figure 3C).66 TLR9 signaling enhances humoral immunity by driving B cell differentiation into plasma cells, increasing antibody secretion, and enabling antibody-dependent cellular cytotoxicity (Figure 3D).65 Immune cell TLR9 signaling exerts antitumor effects across multiple tumor types. For example, TLR9 activation enhances NK and CD8+ T cell cytotoxicity in cutaneous T-cell lymphoma (CTCL), drives DC-dependent CD4+ T cell responses in melanoma, and promotes DC expansion, CD8+ T cell infiltration, and antibody production in lung cancer.65,67,68
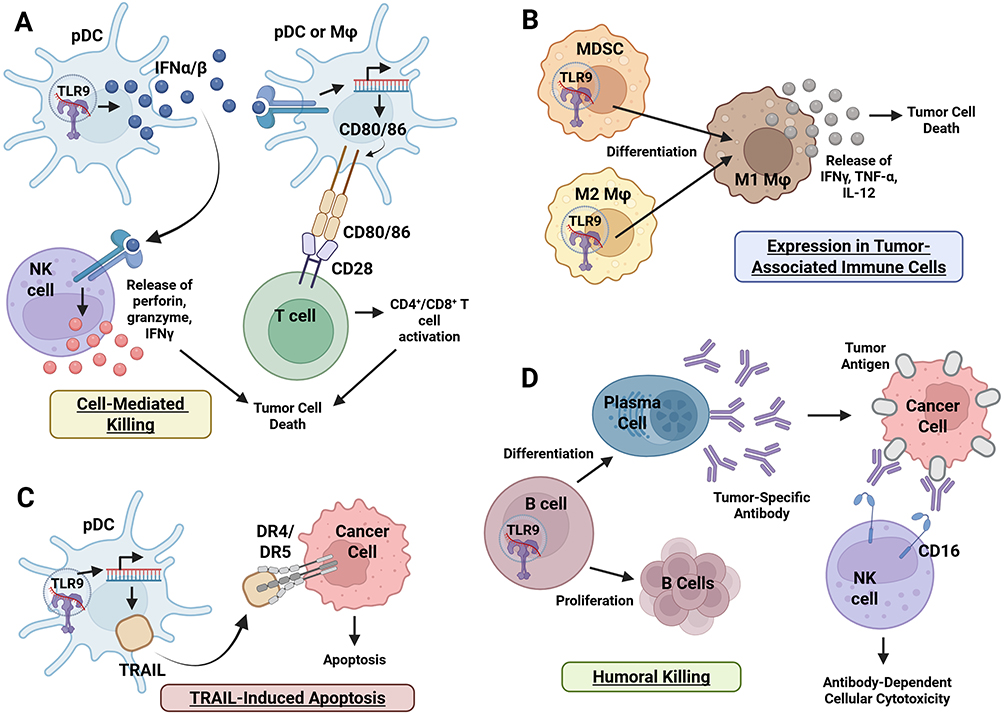 |
Figure 3 TLR9 expression in immune cells. Upon CpG sequence recognition, TLR9 induces tumor clearance primarily via pDCs, macrophages, and B cells. TLR9 signaling promotes (A) cell-mediated killing through type I IFN-mediated activation of NK- and T-cell cytotoxicity; (B) differentiation of tumor associated immune cells toward a tumoricidal M1 phenotype; (C) TRAIL-induced apoptosis via death ligand expression; and (D) humoral killing driven by B cell proliferation, plasma cell differentiation, and ADCC. Collectively, these responses enable tumor cell apoptosis through innate, cell-mediated, and humoral pathways. Figure created with BioRender. Abbreviations: ADCC, Antibody-Dependent Cellular Cytotoxicity; CD16, Cluster of Differentiation 16; CD28, Cluster of Differentiation 28; CD4+/CD8+, Cluster of Differentiation 4+/8+ T Cells; CD80/86, Cluster of Differentiation 80/86; CpG, Cytosine-phosphate-Guanine; DR4/DR5, Death Receptor 4/5; IFNα/β, Interferon Alpha/Beta; IFNγ, Interferon Gamma; IL-12, Interleukin-12; M1 Mφ, M1 Macrophage (Proinflammatory); M2 Mφ, M2 Macrophage (Anti-inflammatory); MDSC, Myeloid-Derived Suppressor Cell; NK cell, Natural Killer Cell; pDC, Plasmacytoid Dendritic Cell; TLR9, Toll-Like Receptor 9; TNF-α, Tumor Necrosis Factor Alpha; TRAIL, TNF-Related Apoptosis-Inducing Ligand. |
Alternatively, because tumor-promoting immune cells (eg, TAMs, MDSCs, and Tregs) contribute to cancer progression, TLR9 signaling within these populations may paradoxically enhance tumor growth. In prostate cancer, TLR9 activation in MDSCs drives STAT3 activation and immunosuppressive effects, inhibiting CD8+ T cell proliferation and the secretion of IFNγ- and graγnzyme-B.69 TLR9 expression is elevated in Tregs from patients with head and neck squamous cell carcinoma compared to healthy controls, suggesting that TLR9 may enhance Treg-mediated immunosuppression and tumor immune escape.70 Similarly, high TLR9 expression in TAMs correlates with increased tumor recurrence in murine models of melanoma, bladder, and colon cancer.71
In contrast, other studies suggest that TLR9 expression in tumor-associated immune cells can promote antitumor immunity. In gastric, liver, and colon cancers, TLR9 signaling blocks the immunosuppressive function of MDSCs, restores T cell activity and promotes MDSC differentiation into antitumor macrophages (Figure 3B).72–74 TLR9 activation in liver cancer also increases the ratio of M1 (antitumor) to M2 (pro-tumor) TAMs. In melanoma and neuroblastoma models, TLR9 activates macrophages to induce tumor cell apoptosis via IFN-γ, IL-12, and TNF-ɑ secretion.75 Overall, the role of immune TLR9 signaling in cancer is complex and largely dependent on the surrounding TME and immune cells involved.
TLR9 Therapeutics in Cancer
TLR9-based cancer therapies take advantage of the receptor’s ability to both activate and regulate the immune system (Tables 1 and 2). The clinical translation of TLR9 modulators has yielded inconsistent results that reflect TLR9’s complex, context-dependent biology. Given TLR9’s dual role in tumorigenesis, the efficacy of TLR9 agonists or antagonists may depend more on tumor type and microenvironment than on the specific modulator itself. This section examines the clinical applications of TLR9 modulators and analyzes their performance through the lens of TLR9 biology and the molecular pathways discussed earlier.
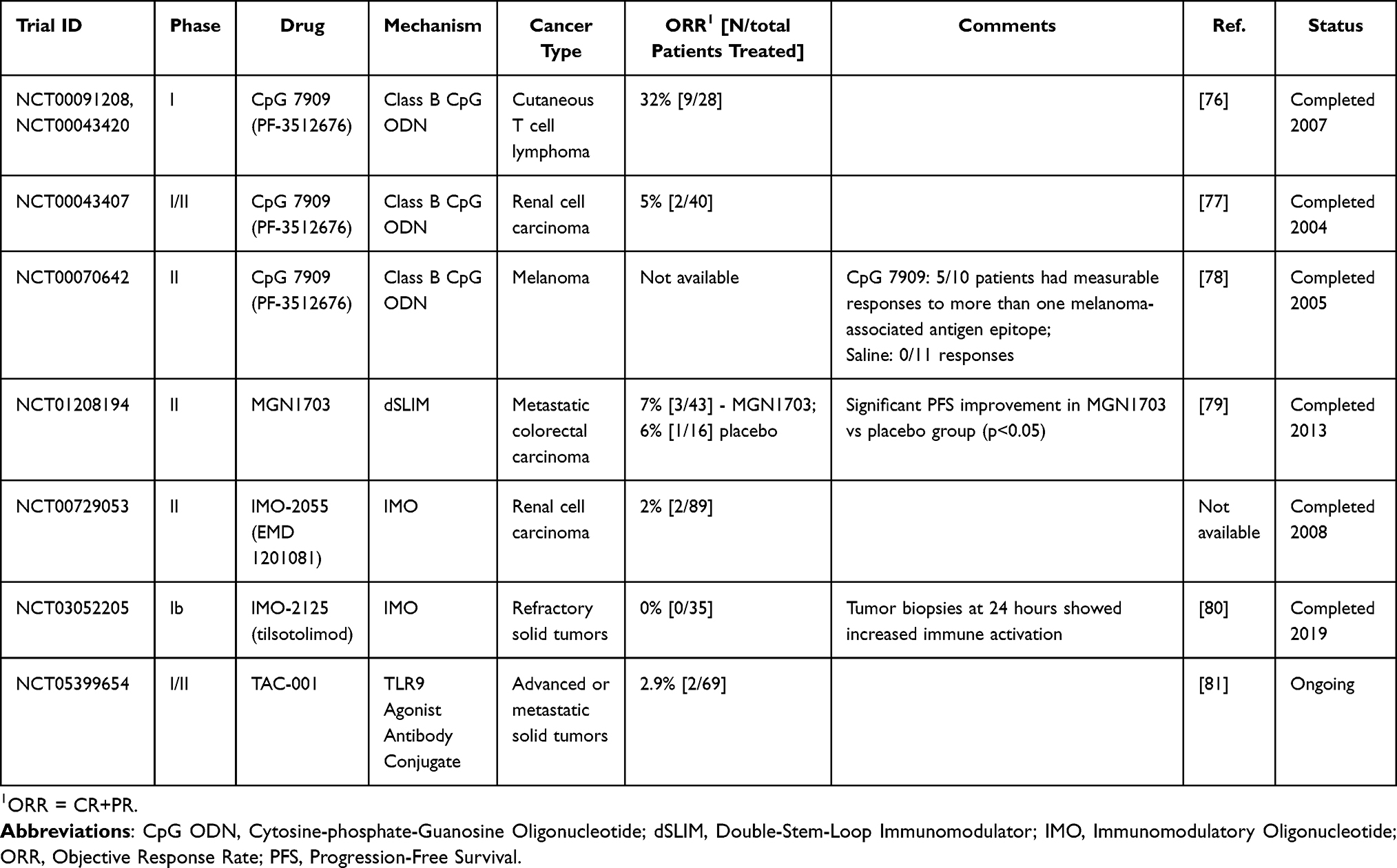 |
Table 1 Examples of Clinical Trials Using TLR9 Agonists as Single Agents |
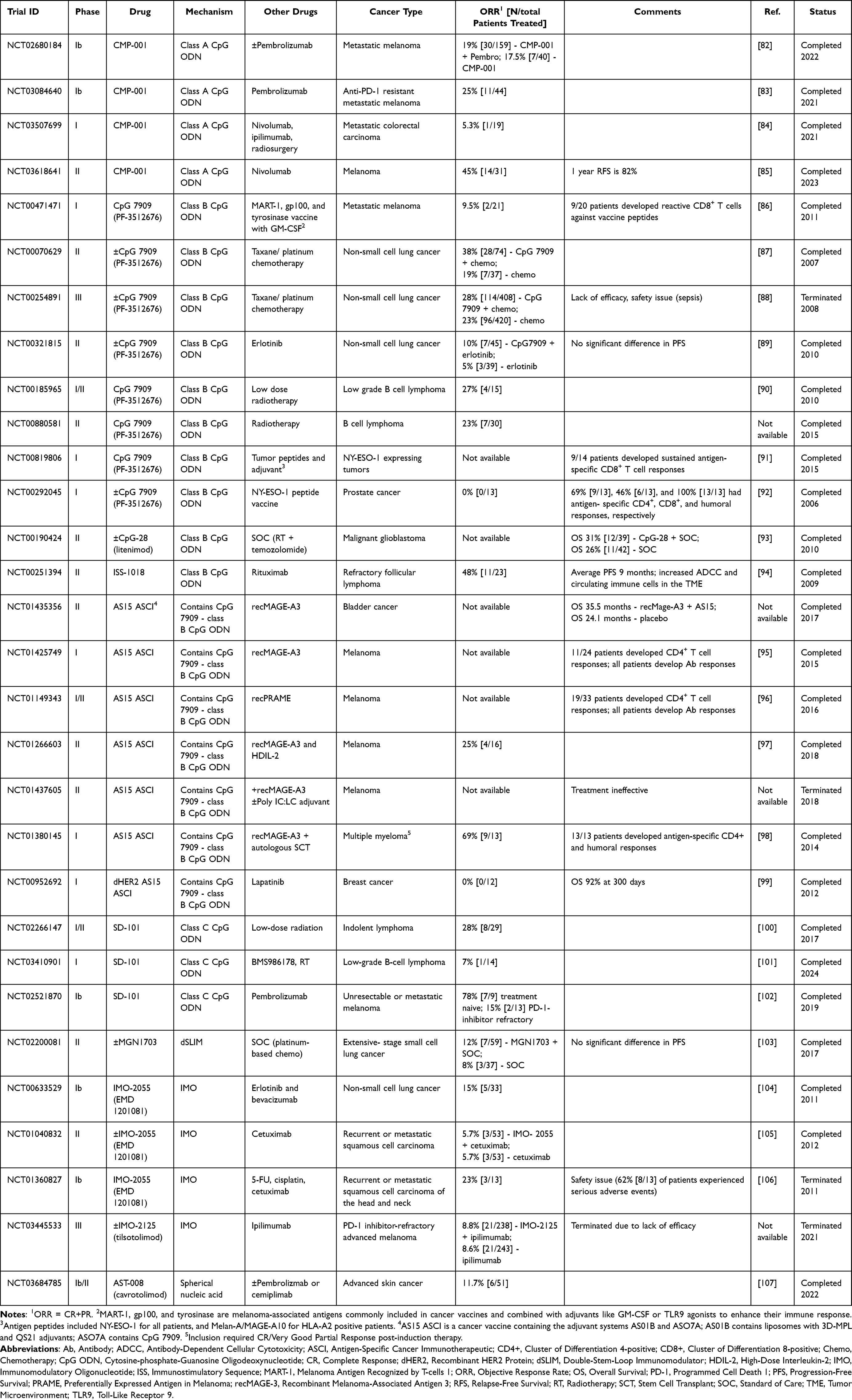 |
Table 2 Examples of Clinical Trials Using TLR9 Agonists in Combination with Other Therapies |
TLR9 Agonists
Synthetic oligodeoxynucleotides (ODNs) mimic endogenous CpG DNA motifs and induce TLR9 signaling. As monotherapies, CpG ODNs are insufficient to reverse the immunosuppressive state of most cancers; however, many TLR9 agonists are being explored in combination with other therapies. TLR9 agonists consist of synthetic, single-stranded DNA sequences containing unmethylated cytosine-guanine dinucleotides and are divided into three classes–A, B, and C–based on their structure and immunostimulatory effects.108 Class A ODNs, including CMP-001, consist of a central palindromic phosphodiester CpG motif and a 3’ poly-G-tail containing a phosphorothioate backbone.108 These CpG agonists predominantly activate pDCs and are selectively retained with MyD88-IRF-7 complexes in pDC endosomes, leading to sustained IRF-7 activation, robust IFN-ɑ secretion, and indirect activation of T and NK cells.6,108,109 In melanoma, CMP-001 has been combined with immune checkpoint inhibitors (ICI), pembrolizumab, and nivolumab (Table 2). A Phase II trial combining CMP-001 with nivolumab demonstrated a promising ORR of 45% (14/31 patients); however, response rates vary across other melanoma trials.85 Notably, the combination of CMP-001 and pembrolizumab achieved a 25% ORR (11/44 patients) in patients resistant to prior anti-PD-1 therapy, suggesting that TLR9 agonists may help overcome resistance to PD-1 blockade.83 The promising, yet variable efficacy of Class A ODNs in melanoma likely reflects their reliance on type I IFN-mediated immune activation, a mechanism that is particularly effective in immunologically “cold” tumors with limited immune infiltration and intrinsic resistance to PD-1 blockade.83 Consistent with this, CMP-001 appears to be more effective in noninflamed tumors.110 However, in tumors with highly immunosuppressive TMEs, including some melanoma subtypes, TLR9-driven antitumor immunity may be attenuated or suppressed. pDCs in these settings may be scarce, dysfunctional, or overwhelmed by competing immunosuppressive signaling, diminishing the type I IFN antitumor response.83 This likely underlies the variable outcomes observed across trials. Overall, the Class A CpG agonists appear to depend on a TME that retains a functional pDC compartment lacking significant competing immunosuppression.
Class B CpG ODNs are characterized by a fully phosphorothioate backbone and incorporate one or more 6-mer CpG motifs.108 CpG-7909 is the most extensively studied TLR9 agonist and has been investigated both as a monotherapy and in combination with various other cancer therapies, including chemotherapy, radiotherapy, peptide vaccines, and targeted therapies, across multiple tumor types (Tables 1 and 2). In pDCs, Class B ODNs are rapidly routed to lysosomes and therefore induce only weak type I IFN responses.6,109 Instead, they act predominantly on B cells to promote Th1-type immune responses and enhance MHC II expression on APCs.5 These mechanisms are particularly relevant in hematologic malignancies, which often contain a TME enriched with TLR9-expressing immune cells. Although Class B ODNs have demonstrated limited efficacy as monotherapy in solid tumors (Table 1), CpG-7909 achieved an objective response rate (ORR) of 32% (9/28 patients) in a Phase I study for CTCL, a disease driven by immature dendritic cells that promote Th2 polarized T cell proliferation and immune dysregulation.76 In this setting, Class B TLR9 agonists act directly on malignant and infiltrating immune cells, enhancing antigen presentation and restoring a Th1-dominant antitumor response.76 In solid tumors, however, Class B CpGs have demonstrated limited benefit even when used in combination regimens (Table 2). Non-small cell lung cancer (NSCLC) initially appeared to be a promising target for TLR9 modulators due to its immunosuppressive TME and expression of TLR9 in tumor cells and surrounding myeloid cells.87 While preclinical studies and phase II trials suggested that CpG-7909-induced APC activation could synergize with cytotoxic taxane/platinum chemotherapy, this combination therapy failed to enhance clinical responses or extend progression free survival (PFS) in a Phase III NSCLS trial and was associated with significant adverse effects, including sepsis, despite improving objective responses in a phase II trial.87,88 These findings suggest that Class B ODN-driven pathways are insufficient to overcome the profound immunosuppressive TME typical of NSCLC. Additionally, systemic CpG agonist delivery may have primarily engaged the pro-tumorigenic NF-κB/STAT3 axis of TLR9 signaling within NSCLC cells, effectively reinforcing the tumor’s growth pathways and negating any beneficial immune effects.38,41,87 It is possible that NSCLC may be better suited for treatment with Class A CpG ODNs, which induce robust type I IFN production and pDC activation that more directly counteract the myeloid suppression, defective antigen presentation, and impaired innate signaling characteristic of NSCLC. Similar patterns have been observed across other solid tumors. While Class B CpGs often elicit CD4+, CD8+, and/or humoral responses, these rarely translate into meaningful tumor regression.86,91,92
In hematologic malignancies, Class B ODNs are most effective when paired with therapies that complement their immune-activating mechanisms. A phase II trial combining ISS-1018–a Class B CpG ODN primarily utilized as a vaccine adjuvant–with rituximab, reported an ORR of 48% (11/23) in patients with refractory follicular lymphoma.94 In this context, Class B ODNs enhance rituximab-mediated antibody-dependent cellular cytotoxicity (ADCC) by triggering TLR9-dependent activation of APCs and subsequent cytokine-mediated activation NK cells, increasing antigen presentation and Fc-mediated cytotoxicity.94 Class B ODNs can also synergize with radiotherapy which produces highly immunogenic tumor antigens, that, when coupled with TLR9-driven APC activation, enables APC cross-presentation and drives expansion of tumor-reactive CD8+ T cells.90 In a phase I/II trail of low-grade B-cell lymphoma, the combined regimen of radiotherapy and CpG-7909 resulted in an ORR of 27% and increased tumor-reactive memory CD8+ T cells.90 However, clinical responses were diminished in patients with Treg-inducing tumors, further reinforcing that the efficacy of Class B ODNs is contingent upon a permissive TME and suggesting that co-administration with Treg-modulating agents like CTLA-4 inhibitors may be critical for success in immunosuppressive contexts.90
Class C ODNs incorporate characteristics of both preceding classes and feature a fully phosphorothioate backbone and palindromic CpG motifs.108 They elicit broader immune responses by inducing robust type I IFN responses while also promoting B cell, APC, and NK call activation.108 This may allow Class C ODNs to overcome the limitations of Class A and B ODNs. SD-101, a Class C ODN, has been combined with radiotherapy in lymphomas and pembrolizumab in metastatic melanoma, with promising early results (Table 2). In combination with pembrolizumab, SD-101 achieved clinical responses in 78% (7/9) of patients with PD-1 inhibitor-naive tumors.83 This exceeds the typical 35–40% clinical response rate typically observed with pembrolizumab monotherapy.83 In this context, TLR9 agonism triggers robust innate activation that enhances T cell recruitment and priming, transforming the TME into a state that is conducive to checkpoint blockade.83 However, it is unclear whether the moderate IFN-α induction of Class C ODNs will be sufficient to overcome the immunosuppressive TME of less immunogenic solid tumors. Several phase I/II trials are currently investigating SD-101 combination therapies in pancreatic (NCT04050085), prostate (NCT03007732), liver (NCT05220722), and breast (NCT01042379) cancers.
Modifications to the sequence length, shape, and delivery mechanism of TLR9 agonists have been pursued to potentially enhance their antitumor immune activity.108 MGN1703, a TLR9 agonist with a double-stem loop structure, significantly improved PFS in metastatic CRC but did not improve clinical responses or PFS when combined with standard-of-care therapies in extensive-stage small-cell lung cancer (Tables 1 and 2). Immunomodulatory oligonucleotides (IMOs) contain CpR dinucleotides, which substitute the standard guanine nucleotide in CpG motifs with 7-deazaguanosine to increase IFN-ɑ secretion and B cell activation.108 However, in clinical trials, neither IMO-2055 nor IMO-2125 improved clinical outcomes when combined with various targeted agents or immunotherapies. Additionally, one study evaluating IMO-2055 alongside 5-FU, cisplatin, and cetuximab (NCT01360827) was terminated due to serious adverse events (Table 2). These findings suggest that while modified TLR9 agonists may amplify innate immune signaling in vitro, this does not reliably translate into effective antitumor immunity in patients, potentially due to the constraints of an immunosuppressive TME. Collectively, the disappointing clinical performance of modified TLR9 agonists suggests that structural optimization alone is unlikely to improve therapeutic efficacy of TLR9 agonists, highlighting the need for combination strategies that directly address these immune constraints.
More recently, next-generation TLR9-modulating platforms have sought to overcome these limitations through precision delivery rather than sequence optimization. AST-008 (cavrotolimod) is a spherical nucleic acid-based TLR9 agonist, whose structure enhances cellular uptake and endosomal trafficking.107 In combination with anti-PD-1 therapy, AST-008 led to an ORR of 11.7% (6/51) in patients with advanced skin cancers refractory to prior PD-1 blockade.107 Similarly, Toll-like Receptor Agonist Antibody Conjugates (TRAACs), developed by Tallac Therapeutics, deliver highly potent CpG agonists directly to specific immune cell subsets. TAC-001, which conjugates a CpG agonist to an anti-CD22 antibody, selectively targets and activates CD22+ B cells to promote antitumor immunity while minimizing off-target systemic TLR9 activation.81 Early results from the ongoing phase I/II INCLINE-101 trial (NCT05399654) show that TAC-001 monotherapy achieved an ORR of 2.9% [2/69] in patients with advanced solid tumors refractory to immune checkpoint inhibitors.81 Another TRAAC, ALTA-002, targets SIRPα on myeloid cells and has demonstrated robust antitumor activity across multiple preclinical tumor models.111 Collectively, these novel mechanisms of CpG delivery illustrate a shift towards targeted, cell-specific TLR9 activation, a strategy that may help overcome the intrinsic limitations of earlier TLR9 agonists while reducing systemic toxicity.
Tumor vaccines targeting cancer-associated peptides such as MAGE, PRAME, and NY-ESO-1 have incorporated TLR9 agonists as adjuvants to enhance antigen-specific immune responses. In this setting, TLR9 agonism provides a strong innate activation signal that supports the development of robust adaptive immune responses which are critical for effective vaccination.95 The AS15 antigen-specific cancer immunotherapeutic (ASCI) adjuvant system—a liposomal formulation containing CpG-7909, 3D-MPL, and QS-21—induced durable antigen-specific CD4+ T cell and antibody responses in melanoma and multiple myeloma patients.95,96,98 However, most studies report limited expansion of cytotoxic CD8+ T cells, consistent with the signaling profile of Class B ODNs, which predominantly activate B cells and conventional APCs to generate a CD4+ Th1 response.95,96,98,99 These limitations suggest that Class A ODN which trigger robust type I IFN signaling and pDC-mediated cross-priming, or Class C ODNs, which combine Class A and Class B mechanisms, may serve as more effective adjuvants when CD8+ T cell responses are required. Additionally, several studies highlight regulatory T cells as a barrier to vaccine efficacy, indicating that TLR9-based vaccine adjuvants may benefit from combination strategies that modulate Treg activity, such as immune checkpoint blockade.95,97,98
Overall, TLR9 agonists show promise in cancer therapy due to their ability to activate both innate and adaptive immune responses, particularly when combined with treatments such as ICIs, chemotherapy, radiation, and tumor vaccines. It appears that the immunologic context in which TLR9 agonists are applied significantly influence their efficacy, potentially to a greater extent than the CpG chemistry itself. TLR9 signaling can generate potent innate and adaptive immune activation, but its antitumor effects depend on the presence of functional pDCs, effective antigen presentation, and a TME capable of supporting T cell recruitment and priming. Across studies, TLR9 agonists show the greatest activity when paired with therapies that relieve immunosuppressive pathways or provide complementary sources of tumor antigens. However, challenges remain due to safety concerns, variable clinical outcomes, and underwhelming results in phase III clinical trials. A clearer understanding of which TLR9-dependent pathways dominate in individual tumors and how each tumor’s immune composition shapes those pathways will be important for refining the use of TLR9 agonists. As such, future progress will likely depend on developing more effective combination therapies and identifying biomarkers that can help guide patient selection and predict treatment response.
TLR9 Antagonists
Inhibiting TLR9 is a rational therapeutic approach in cancers where its activation drives tumor progression through chronic inflammation, immune evasion, and metastasis. In preclinical studies, chloroquine—a TLR9 inhibitor—reduces tumor size in mice with intrahepatic cholangiocarcinoma and suppresses proliferation and invasion in esophageal cancer models.3,112 Additionally, TLR9 antagonist ODN INH18 significantly reduces in vitro cervical cancer proliferation and induces apoptosis by decreasing Bcl-2 expression and increasing Bax.37 Despite demonstrating some efficacy in preclinical models, TLR9 antagonists lack sufficient evidence to progress to clinical trials.
While most molecular studies highlight TLR9’s role in tumor growth and progression, nearly all current treatment approaches use TLR9 agonists instead of antagonists. This discrepancy suggests that TLR9’s ability to activate the immune system outweighs its tumor-promoting effects, especially when paired with therapies that reduce immunosuppression. Additional research is necessary to refine the design, delivery, and implementation of TLR9 modulators.
Role of TLR9 Beyond Cancer
Infection
Upon bacterial or viral infection, TLR9 detects unmethylated CpG motifs in pathogenic DNA sequences and triggers a strong type I IFN response; however, TLR9 overactivation is observed in multiple viral infections.17 TLR9 contributes to symptom severity in COVID-19 patients and is central to immune dysregulation in HIV infection.113,114 Despite this, Class C CpG ODNs are effective at decreasing viral load and activating B cells, pDCs, and NK cells in chronic viral infections, such as hepatitis C.115,116 Additionally, as vaccine adjuvants, Class B CpG ODNs boost both humoral and cell-mediated immunity. Within two weeks, most recipients given CpG-7909 alongside the hepatitis B vaccine achieved protective anti-hepatitis B IgG levels, compared to none who received the vaccine alone.117 The TLR9 adjuvant group also developed a higher proportion of high-avidity antigen-specific antibodies.118 These improvements are particularly beneficial for immunocompromised patients, such as HIV+ individuals. When used as an adjuvant, CpG-7909 significantly boosted antibody titers, T cell responses, and long-term protection against hepatitis B in HIV+ recipients compared to the standard vaccine alone.119
Autoimmune Disease
In autoimmunity, TLR9 acts as both a promoter and suppressor of disease, depending on the context. In systemic lupus erythematosus (SLE), TLR9 directly recognizes self-DNA-containing immune complexes, leading to the release of IFN-α from pDCs and the activation of B cells, which perpetuates autoantibody production.120 SLE patients exhibit increased TLR9 expression, and targeted disruption of TLR9 signaling in B cells reduces inflammatory cytokines and alleviates symptoms.121 Conversely, TLR9-deficient lupus-prone mice exhibit increased pDC activation, IFN-ɑ production, and disease severity despite reduced high-affinity autoantibodies.122,123 This suggests that TLR9’s net effect is protective despite enhancing antibody affinity. In rheumatoid arthritis (RA) patients, TLR9 overexpression in monocytes and pDCs worsens symptoms, whereas TLR9 antagonism maintains remission in those at high risk for disease recurrence.124,125 TLR9 is also linked with the progression of both type I and type II diabetes; however, the exact mechanism is unclear and needs additional investigation.126,127
Inflammatory Conditions
TLR9 plays a context-dependent role in inflammatory disorders such as inflammatory bowel disease (IBD) and allergies. In acute colitis, apical TLR9 suppresses gut inflammation via Treg activation, whereas in chronic IBD, high TLR9 expression correlates with disease severity and production of proinflammatory cytokines.128,129 Clinically, prophylactic CpG-ODN administration prevents intestinal inflammation in murine IBD, while ODN antagonists decrease gut inflammation and cytokine secretion.129,130 In clinical trials, DIMS0150, a TLR9 agonist, led to higher remission rates and increased mucosal healing in patients with ulcerative colitis.131 In allergies, including asthma and rhinitis, TLR9 ligands suppress inflammation and reprogram the allergic Th2-dominant immune response toward a Th1 profile.132,133 Using this approach, a TLR9-agonist-based vaccine containing a CpG-ODN conjugated to the ragweed allergen significantly improved symptoms in allergic rhinitis patients.132
Conclusion and Future Directions
TLR9 exhibits multifaceted, context-dependent roles in humans, acting as a promoter of tumorigenesis, mediator of antitumor immunity, and regulator of immune responses in infection, autoimmunity, and inflammatory diseases. In this way, it is similar to other immunoregulatory agents—such as TLR3, inducible T-cell co-stimulator (ICOS), V-domain immunoglobulin suppressor of T cell activation (VISTA), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3)—that have both stimulatory and inhibitory effects in a context-dependent manner, and whose expression varies across cell types and between patients.134–140 While TLR9’s immunomodulatory nature enables the recognition and targeting of pathogens and cancer cells, its overexpression in tumors contributes to chronic inflammation, cell survival, metastatic spread, and immune evasion. The pathways and consequences of TLR9 signaling in cancer vary by tumor type, stage, and microenvironment, with NF-κB, STAT3, and COX-2 serving as key mediators. Despite this, therapeutic strategies to inhibit TLR9 are extremely limited, as TLR9 antagonists have shown minimal success in translational studies. The primary therapeutic interest in TLR9 centers on its ability to activate antitumor immunity, yet the immunologic context of the tumor greatly influences efficacy. Classic synthetic CpG ODNs activate pDCs, NK cells, and T cells, although their clinical efficacy is highly variable and frequently limited by an immunosuppressive TME. Consistent with these observations, Class A and C ODNs appear most effective in combination with ICIs, and Class B ODNs demonstrate potential alongside tumor vaccines. Importantly, differences in CpG ligand class, backbone chemistry, and structural configuration also determine whether TLR9 signaling is biased toward potent type I IFN production in pDCs or toward B cell, APC, and Th1-type activation, further contributing to heterogeneity of clinical responses observed in trials. Across cancer types, successful responses consistently correlate with TMEs that retain functional pDCs, intact antigen presentation, and the capacity to support type I IFN-driven immune priming. These insights have shifted TLR9 modulators away from simple sequence modification and toward strategies that more directly address TME constraints, including novel precision delivery technologies and synergistic immune combination regimens.
Future research should prioritize mapping TLR9-dominant signaling pathways within specific tumor contexts and identifying predictive biomarkers to stratify patients that are most likely to benefit from TLR-9-based therapies. Given TLR9’s dependence on intact pDC function, type I IFN signaling, and myeloid cells capable of supporting innate activation, biomarkers such as pDC abundance, IFN- transcriptional signatures, and NF-B/STAT3 expression patterns may help distinguish tumors with sensitivity versus resistance to TLR9 agonists. The development of circulating biomarkers—particularly cell-free DNA (cfDNA)-based assays—offers an additional opportunity to integrate TLR9 therapeutics with continuous tumor monitoring.141,142 Because cfDNA reflects tumor burden, mutational landscape, and immune activity, pairing TLR9 agonists with cfDNA-guided diagnostics could enable adaptive treatment strategies, earlier identification of immune activation or resistance, and more precise patient selection.143,144 In parallel, recent advances in methylation-based liquid biopsy assays demonstrate how CpG modification patterns can be leveraged in clinical diagnostics. For example, the Belay Vantage™ assay uses enzymatic DNA conversion to evaluate MGMT promoter methylation in low input cerebrospinal fluid-derived cfDNA from patients with nervous system tumors, highlighting how CpG methylation profiling is increasingly being integrated into translational oncology workflows.145 Because TLR9 activation is highly sensitive to CpG methylation status, these developments reinforce the potential for methylation-informed biomarkers to guide patient selection for TLR9-targeted therapies. Ultimately, the integration of biomarker profiling, cfDNA-guided monitoring, and optimized combination regimens has the potential to transform TLR9 modulators into more precise and reliable immunotherapeutic strategies.
Highlights
● TLR9 promotes and suppresses tumorigenesis via tumor- and context-specific pathways
● NF-κB/STAT3 signaling is central to TLR9-driven inflammation and tumor survival
● TLR9 activation in plasmacytoid dendritic cells and NK cells drives tumor apoptosis
● TLR9 agonist combinations activate antitumor responses in select patients/tumors
● Biomarkers are needed to stratify patients and predict responses to TLR9 modulators
Abbreviations
AP-1, Activator Protein 1; ADCC, Antibody-Dependent Cellular Cytotoxicity; APCs, Antigen-Presenting Cells; ASCI, Antigen-Specific Cancer Immunotherapeutic; Bax, Bcl-2 associated x protein; Bcl-xL, B-cell lymphoma-extra Large; CCL, C-C Motif Chemokine Ligand; CCR, C-C Motif Chemokine Receptor; CDK, Cyclin-Dependent Kinase; CLL, Chronic Lymphocytic Leukemia; COX-2, Cyclooxygenase-2; CpG, Cytosine-Phosphate-Guanosine; CRC, Colorectal Cancer; CTCL, Cutaneous T-Cell Lymphoma; CTLA-4, Cytotoxic T-Lymphocyte-Associated Protein 4; CXCL, C-X-C Motif Chemokine Ligand; CXCR, C-X-C Motif Chemokine Receptor; DAMPs, Damage-Associated Molecular Patterns; EBV, Epstein-Barr Virus; ECM, Extracellular Matrix; EMT, Epithelial-Mesenchymal Transition; ERK, Extracellular Signal-Regulated Kinase; EZH2, Enhancer of Zeste Homolog 2; GBM, Glioblastoma Multiforme; HCC, Hepatocellular Carcinoma; HIF-1, Hypoxia-Inducible Factor 1; HPV16, Human Papillomavirus 16; IBD, Inflammatory Bowel Disease; ICAM-1, Intercellular Adhesion Molecule 1; ICIs, Immune Checkpoint Inhibitors; ICOS, Inducible T-Cell Co-Stimulator; IFN, Interferon; IκB, Inhibitor of NF-κB; IL, Interleukin; IMOs, Immunomodulatory Oligonucleotides; IRAK, Interleukin-1 Receptor-Associated Kinase; IRF, Interferon Regulatory Factor; IRF-7, Interferon Regulatory Factor 7; MAL, MyD88 Adaptor-Like; MAPK, Mitogen-Activated Protein Kinase; Mcl-1, Myeloid Cell Leukemia 1; MDSCs, Myeloid-Derived Suppressor Cells; MHC II, Major Histocompatibility Complex Class II; MMP, Matrix Metalloproteinase; mtDNA, Mitochondrial DNA; MyD88, Myeloid Differentiation 88; NF-κB, Nuclear Factor-Kappa B; NK, Natural Killer; NSCLC, Non-Small Cell Lung Cancer; ODNs, Oligodeoxynucleotides; OSCC, Oral Squamous Cell Carcinoma; PAMPs, Pathogen-Associated Molecular Patterns; PARP, Poly(ADP-Ribose) Polymerase; PD-1, Programmed Cell Death Protein 1; PD-L1, Programmed Death-Ligand 1; pDCs, Plasmacytoid Dendritic Cells; PFS, Progression-Free Survival; PMN-MDSCs, Polymorphonuclear Myeloid-Derived Suppressor Cells; PRR, Pattern Recognition Receptor; RA, Rheumatoid Arthritis; RCC, Renal Cell Carcinoma; ROS, Reactive Oxygen Species; SCIMP, Src Kinase-Interacting Membrane Protein; SLE, Systemic Lupus Erythematosus; SNAIL, Snail Family Transcriptional Repressor 1; STAT3, Signal Transducer and Activator of Transcription 3; TAMs, Tumor-Associated Macrophages; Th1, T Helper 1; TIM-3, T Cell Immunoglobulin and Mucin Domain-Containing Protein 3; TIMP, Tissue Inhibitor of Metalloproteinase; TLR9, Toll-Like Receptor 9; TME, Tumor Microenvironment; TNF, Tumor Necrosis Factor; TRAAC, Toll-Like Receptor Agonist Antibody Conjugate; TRAF6, TNF Receptor-Associated Factor 6; TRAIL, TNF-Related Apoptosis-Inducing Ligand; Tregs, Regulatory T Cells; TRIF, TIR-domain-containing adaptor protein-inducing beta interferon; TWIST1, Twist Family bHLH Transcription Factor 1; VEGF, Vascular Endothelial Growth Factor; VISTA, V-domain Immunoglobulin Suppressor of T Cell Activation; ZEB2, Zinc Finger E-box-Binding Homeobox 2.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
RK is funded in part by 5U01CA180888-08 and 5UG1CA233198-05.
Disclosure
Dr. Kurzrock has received research funding from Boehringer Ingelheim, Debiopharm, Foundation Medicine, Genentech, Grifols, Guardant, Incyte, Konica Minolta, Medimmune, Merck Serono, Omniseq, Pfizer, Sequenom, Sysmex, Takeda, and TopAlliance and from the NCI; as well as consultant and/or speaker fees and/or advisory board/consultant for Actuate Therapeutics, AstraZeneca, Bicara Therapeutics, Inc., Biological Dynamics, Caris, Daiichi, Datar Cancer Genetics, EISAI, EMD Serono, EOM Pharmaceuticals, Iylon, Jackson Laboratories, LabCorp, Lanauria Therapeutics, Merck, NeoGenomics, Neomed, OneCell, Pfizer, Precirix, Prosperdtx, Quanta Therapeutics, Recordati, Regeneron, Roche, TD2/Volastra, Turning Point Therapeutics, X-Biotech; has an equity interest in CureMatch Inc.; serves on the Board of CureMatch and CureMetrix and XZOM, and is a co-founder of CureMatch. The authors report no other conflicts of interest in this work.
References
1. Nobel Prize to immunology. Nat Rev Immunol. 2011;11(11):714. doi:10.1038/nri3103
2. Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev. 2008;60(7):795–804. doi:10.1016/j.addr.2007.12.004
3. Alzahrani B. The biology of toll-like receptor 9 and its role in cancer. Crit Rev Eukaryot Gene Expr. 2020;30(5):457–474. doi:10.1615/CritRevEukaryotGeneExpr.2020036214
4. El-Zayat SR, Sibaii H, Mannaa FA. Toll-like receptors activation, signaling, and targeting: an overview. Bull Natl Res Cent. 2019;43(1):187. doi:10.1186/s42269-019-0227-2
5. Vollmer J. TLR9 in health and disease. Int Rev Immunol. 2006;25(3–4):155–181. doi:10.1080/08830180600743107
6. Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19(1):24–32. doi:10.1016/j.smim.2006.12.004
7. Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol. 2015;109(1):14.12.1–14.12.10. doi:10.1002/0471142735.im1412s109
8. Zambirinis CP, Levie E, Nguy S, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med. 2015;212(12):2077–2094. doi:10.1084/jem.20142162
9. Varga MG, Shaffer CL, Sierra JC, et al. Pathogenic H elicobacter pylori strains translocate DNA and activate TLR9 via the cancer-associated cag type IV secretion system. Oncogene. 2016;35(48):6262–6269. doi:10.1038/onc.2016.158
10. Kauppila JH, Karttunen TJ, Saarnio J, et al. Short DNA sequences and bacterial DNA induce esophageal, gastric, and colorectal cancer cell invasion. APMIS Acta Pathol Microbiol Immunol Scand. 2013;121(6):511–522. doi:10.1111/apm.12016
11. Niu Z, Tang W, Liu T, et al. Cell-free DNA derived from cancer cells facilitates tumor malignancy through Toll-like receptor 9 signaling-triggered interleukin-8 secretion in colorectal cancer. Acta Biochim Biophys Sin. 2018;50(10):1007–1017. doi:10.1093/abbs/gmy104
12. Fűri I, Kalmár A, Wichmann B, et al. Cell free DNA of tumor origin induces a ‘metastatic’ expression profile in HT-29 cancer cell line. PLoS One. 2015;10(7):e0131699. doi:10.1371/journal.pone.0131699
13. Kang TH, Mao C-P, Kim YS, et al. TLR9 acts as a sensor for tumor-released DNA to modulate anti-tumor immunity after chemotherapy. J Immunother Cancer. 2019;7(1):260. doi:10.1186/s40425-019-0738-2
14. Tuomela J, Sandholm J, Kaakinen M, et al. DNA from dead cancer cells induces TLR9-mediated invasion and inflammation in living cancer cells. Breast Cancer Res Treat. 2013;142(3):477–487. doi:10.1007/s10549-013-2762-0
15. de Jong SD, Basha G, Wilson KD, et al. The immunostimulatory activity of unmethylated and methylated CpG oligodeoxynucleotide is dependent on their ability to colocalize with TLR9 in late endosomes. J Immunol Baltim Md. 2010;184(11):6092–6102. doi:10.4049/jimmunol.0802442
16. Lai Y-H, Liu H-Y, Huang C-Y, Chau Y-P, Wu S. Mitochondrial‑DNA‑associated TLR9 signalling is a potential serological biomarker for non‑small cell lung cancer. Oncol Rep. 2019;41(2):999–1006. doi:10.3892/or.2018.6855
17. Saber MM, Monir N, Awad AS, Elsherbiny ME, Zaki HF. TLR9: a friend or a foe. Life Sci. 2022;307:120874. doi:10.1016/j.lfs.2022.120874
18. Ward GA, Dalton RP, Meyer BS, et al. Oxidized mitochondrial DNA engages TLR9 to activate the NLRP3 inflammasome in myelodysplastic syndromes. Int J Mol Sci. 2023;24(4):3896. doi:10.3390/ijms24043896
19. Borah S, Mishra R, Dey S, et al. Prognostic value of circulating mitochondrial DNA in prostate cancer and underlying mechanism. Mitochondrion. 2023;71:40–49. doi:10.1016/j.mito.2023.05.005
20. Zeng X-Z, Huang Z-S, Fang H-P, et al. Coexpression of TLR9 and VEGF-C is associated with lymphatic metastasis in prostate cancer. Asian J Androl. 2022;24(4):380–385. doi:10.4103/aja202167
21. Wang C, Cao S, Yan Y, et al. TLR9 expression in glioma tissues correlated to glioma progression and the prognosis of GBM patients. BMC Cancer. 2010;10(1):415. doi:10.1186/1471-2407-10-415
22. Lee J-W, Choi -J-J, Seo ES, et al. Increased toll-like receptor 9 expression in cervical neoplasia. Mol Carcinog. 2007;46(11):941–947. doi:10.1002/mc.20325
23. Jing Y, Jia M, Zhuang J, Han D, Zhou C, Yan J. TLR9 exerts an oncogenic role in promoting osteosarcoma progression depending on the regulation of NF-κB signaling pathway. Biol Pharm Bull. 2022;45(12):1733–1742. doi:10.1248/bpb.b22-00295
24. Luo Q, Zeng L, Tang C, Zhang Z, Chen Y, Zeng C. TLR9 induces colitis‑associated colorectal carcinogenesis by regulating NF‑κB expression levels. Oncol Lett. 2020;20(4):1. doi:10.3892/ol.2020.11971
25. Kennedy E, Coulter E, Halliwell E, et al. TLR9 expression in chronic lymphocytic leukemia identifies a promigratory subpopulation and novel therapeutic target. Blood. 2021;137(22):3064–3078. doi:10.1182/blood.2020005964
26. Wang TR, Peng JC, Qiao YQ, et al. Helicobacter pylori regulates TLR4 and TLR9 during gastric carcinogenesis. Int J Clin Exp Pathol. 2014;7(10):6950–6955.
27. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446(5):475–482. doi:10.1007/s00428-005-1264-9
28. Fan Y, Mao R, Yang J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013;4(3):176–185. doi:10.1007/s13238-013-2084-3
29. Sokolova O, Naumann M. NF-κB Signaling in Gastric Cancer. Toxins. 2017;9(4):119. doi:10.3390/toxins9040119
30. Vaiopoulos AG, Athanasoula K, Papavassiliou AG. NF-κB in colorectal cancer. J Mol Med. 2013;91(9):1029–1037. doi:10.1007/s00109-013-1045-x
31. Jin X, Kim S-H, Jeon H-M, et al. Interferon regulatory factor 7 regulates glioma stem cells via interleukin-6 and Notch signalling. Brain. 2012;135(4):1055–1069. doi:10.1093/brain/aws028
32. Chartomatsidou E, Ntoufa S, Kotta K, et al. Inhibition of EZH2 and immune signaling exerts synergistic antitumor effects in chronic lymphocytic leukemia. Blood Adv. 2019;3(12):1891–1896. doi:10.1182/bloodadvances.2018030262
33. Tanaka J, Sugimoto K, Shiraki K, et al. Functional cell surface expression of toll-like receptor 9 promotes cell proliferation and survival in human hepatocellular carcinomas. Int J Oncol. 2010;37(4):805–814. doi:10.3892/ijo_00000730
34. Grimmig T, Moench R, Kreckel J, et al. Toll like receptor 2, 4, and 9 signaling promotes autoregulative tumor cell growth and VEGF/PDGF expression in human pancreatic cancer. Int J Mol Sci. 2016;17(12):2060. doi:10.3390/ijms17122060
35. Min R, Siyi L, Wenjun Y, et al. Toll-like receptor-9 agonists increase cyclin D 1 expression partly through activation of activator protein-1 in human oral squamous cell carcinoma cells. Cancer Sci. 2012;103(11):1938–1945. doi:10.1111/j.1349-7006.2012.02394.x
36. Wang W, Kong P, Ma G, et al. Characterization of the release and biological significance of cell-free DNA from breast cancer cell lines. Oncotarget. 2017;8(26):43180–43191. doi:10.18632/oncotarget.17858
37. Wang L, Zhang S, Cai H, et al. Inhibiting TLR9 signaling stimulates apoptosis and cell cycle arrest, and alleviates angiogenic property in human cervical cancer cells. Endocr Metab Immune Disord Drug Targets. 2022;22(5):510–517. doi:10.2174/1871530321666210622112753
38. Xu L, Wang C, Wen Z, et al. Selective up-regulation of CDK2 is critical for TLR9 signaling stimulated proliferation of human lung cancer cell. Immunol Lett. 2010;127(2):93–99. doi:10.1016/j.imlet.2009.10.002
39. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med. 2016;94(5):509–522. doi:10.1007/s00109-015-1376-x
40. Sandholm J, Tuomela J, Kauppila JH, Harris KW, Graves D, Selander KS. Hypoxia regulates Toll-like receptor-9 expression and invasive function in human brain cancer cells in vitro. Oncol Lett. 2014;8(1):266–274. doi:10.3892/ol.2014.2095
41. Ren T, Wen Z-K, Liu Z-M, Liang Y-J, Guo Z-L, Xu L. Functional expression of TLR9 is associated to the metastatic potential of human lung cancer cell. Cancer Biol Ther. 2007;6(11):1704–1709. doi:10.4161/cbt.6.11.4826
42. Luo Y, Jiang Q-W, Wu J-Y, et al. Regulation of migration and invasion by toll-like receptor-9 signaling network in prostate cancer. Oncotarget. 2015;6(26):22564–22574. doi:10.18632/oncotarget.4197
43. Ilvesaro JM, Merrell MA, Swain TM, et al. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate. 2007;67(7):774–781. doi:10.1002/pros.20562
44. Di JM, Pang J, Sun QP, et al. Toll-like receptor 9 agonists up-regulates the expression of cyclooxygenase-2 via activation of NF-κB in prostate cancer cells. Mol Biol Rep. 2010;37(4):1849–1855. doi:10.1007/s11033-009-9620-5
45. Kobayashi K, Koyama K, Suzukawa M, et al. Epithelial-mesenchymal transition promotes reactivity of human lung adenocarcinoma A549 cells to CpG ODN. Allergology International. 2016;65:S45–52. doi:10.1016/j.alit.2016.06.010
46. Meseure D, Vacher S, Drak Alsibai K, et al. Biopathological significance of TLR9 expression in cancer cells and tumor microenvironment across invasive breast carcinomas subtypes. Cancer Microenviron. 2016;9(2–3):107–118. doi:10.1007/s12307-016-0186-1
47. Planté-Bordeneuve T, Pilette C, Froidure A. The epithelial-immune crosstalk in pulmonary fibrosis. Front Immunol. 2021;12:631235. doi:10.3389/fimmu.2021.631235
48. Ringelstein-Harlev S, Avivi I, Fanadka M, Horowitz NA, Katz T. Chronic lymphocytic leukemia cells acquire regulatory B-cell properties in response to TLR9 and CD40 activation. Cancer Immunol Immunother. 2018;67(5):739–748. doi:10.1007/s00262-018-2128-x
49. Won H, Moreira D, Gao C, et al. TLR9 expression and secretion of LIF by prostate cancer cells stimulates accumulation and activity of polymorphonuclear MDSCs. J Leukoc Biol. 2017;102(2):423–436. doi:10.1189/jlb.3MA1016-451RR
50. Bao D, Zhao J, Zhou X, et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene. 2019;38(25):5007–5020. doi:10.1038/s41388-019-0772-z
51. Zhou B, Yan J, Guo L, et al. Hepatoma cell-intrinsic TLR9 activation induces immune escape through PD-L1 upregulation in hepatocellular carcinoma. Theranostics. 2020;10(14):6530–6543. doi:10.7150/thno.44417
52. Ma L, Qin N, Wan W, et al. TLR9 activation induces immunosuppression and tumorigenesis via PARP1/PD-L1 signaling pathway in oral squamous cell carcinoma. Am J Physiol-Cell Physiol. 2024;326(2):C362–C381. doi:10.1152/ajpcell.00061.2023
53. Brignole C, Marimpietri D, Paolo DD, et al. Therapeutic targeting of TLR9 inhibits cell growth and induces apoptosis in neuroblastoma. Cancer Res. 2010;70(23):9816–9826. doi:10.1158/0008-5472.CAN-10-1251
54. Parroche P, Roblot G, Le Calvez-Kelm F, et al. TLR9 re-expression in cancer cells extends the S-phase and stabilizes p16INK4a protein expression. Oncogenesis. 2016;5(7):e244–e244. doi:10.1038/oncsis.2016.49
55. Stępień E, Strycharz-Dudziak M, Malm M, Drop B, Polz-Dacewicz M. Serum and tissue level of TLR9 in EBV-associated oropharyngeal cancer. Cancers. 2021;13(16):3981. doi:10.3390/cancers13163981
56. Vlad C, Dina C, Kubelac P, Vlad D, Pop B, Achimas Cadariu P. Expression of toll-like receptors in ovarian cancer. J BUON off J Balk Union Oncol. 2018;23(6):1725–1731.
57. Leppänen J, Helminen O, Huhta H, et al. High toll-like receptor (TLR) 9 expression is associated with better prognosis in surgically treated pancreatic cancer patients. Virchows Arch. 2017;470(4):401–410. doi:10.1007/s00428-017-2087-1
58. Ronkainen H, Hirvikoski P, Kauppila S, et al. Absent Toll-like receptor-9 expression predicts poor prognosis in renal cell carcinoma. J Exp Clin Cancer Res. 2011;30(1):84. doi:10.1186/1756-9966-30-84
59. Moreno-Eutimio MA, Acosta-Altamirano G, Vargas-Hernández VM. Expression of toll-like receptor 9 in cervical intraepithelial neoplasia from Mexican women. J Cytol Histol. 2013;4(4):1–3. doi:10.4172/2157-7099.1000181
60. Pathak S, Wilczyński JR, Paradowska E. Factors in oncogenesis: viral infections in ovarian cancer. Cancers. 2020;12(3):561. doi:10.3390/cancers12030561
61. Wu H-Q, Wang B, Zhu S-K, Tian Y, Zhang J-H, Wu H-S. Effects of CPG ODN on biological behavior of PANC-1 and expression of TLR9 in pancreatic cancer. World J Gastroenterol. 2011;17(8):996–1003. doi:10.3748/wjg.v17.i8.996
62. Korvala J, Harjula T, Siirilä K, et al. Toll-like receptor 9 expression in mucoepidermoid salivary gland carcinoma may associate with good prognosis. J Oral Pathol Med. 2014;43(7):530–537. doi:10.1111/jop.12160
63. Tuomela J, Sandholm J, Karihtala P, et al. Low TLR9 expression defines an aggressive subtype of triple-negative breast cancer. Breast Cancer Res Treat. 2012;135(2):481–493. doi:10.1007/s10549-012-2181-7
64. Mella M, Kauppila JH, Karihtala P, et al. Tumor infiltrating CD8 + T lymphocyte count is independent of tumor TLR9 status in treatment naïve triple negative breast cancer and renal cell carcinoma. OncoImmunology. 2015;4(6):e1002726. doi:10.1080/2162402X.2014.1002726
65. Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27(2):161–167. doi:10.1038/sj.onc.1210911
66. Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117(5):1184–1194. doi:10.1172/JCI31414
67. Zhang X, Munegowda MA, Yuan J, Wei Y, Xiang J. Optimal TLR9 signal converts tolerogenic CD4–8– dCs into immunogenic ones capable of stimulating antitumor immunity via activating CD4+ Th1/Th17 and NK cell responses. J Leukoc Biol. 2010;88(2):393–403. doi:10.1189/jlb.0909633
68. Gallotta M, Assi H, Degagné É, Kannan SK, Coffman RL, Guiducci C. Inhaled TLR9 agonist renders lung tumors permissive to PD-1 blockade by promoting optimal CD4+ and CD8+ T-cell Interplay. Cancer Res. 2018;78(17):4943–4956. doi:10.1158/0008-5472.CAN-18-0729
69. Hossain DMS, Pal SK, Moreira D, et al. TLR9-targeted STAT3 silencing abrogates immunosuppressive activity of myeloid-derived suppressor cells from prostate cancer patients. Clin Cancer Res. 2015;21(16):3771–3782. doi:10.1158/1078-0432.CCR-14-3145
70. Wild CA, Brandau S, Lindemann M, et al. Toll-like receptors in regulatory T cells of patients with head and neck cancer TLRs in T-reg cells of HNSCC patients. Arch Otolaryngol Neck Surg. 2010;136(12):1253–1259. doi:10.1001/archoto.2010.195
71. Gao C, Kozlowska A, Nechaev S, et al. TLR9 signaling in the tumor microenvironment initiates cancer recurrence after radiotherapy. Cancer Res. 2013;73(24):7211–7221. doi:10.1158/0008-5472.CAN-13-1314
72. Zoglmeier C, Bauer H, Nörenberg D, et al. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res. 2011;17(7):1765–1775. doi:10.1158/1078-0432.CCR-10-2672
73. Ghosh CC, Heatherton KR, Connell KPO, et al. Regional infusion of a class C TLR9 agonist enhances liver tumor microenvironment reprogramming and MDSC reduction to improve responsiveness to systemic checkpoint inhibition. Cancer Gene Ther. 2022;29(12):1854–1865. doi:10.1038/s41417-022-00484-z
74. Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol. 2012;188(4):1592–1599. doi:10.4049/jimmunol.1101304
75. Buhtoiarov IN, Lum HD, Berke G, Sondel PM, Rakhmilevich AL. Synergistic activation of macrophages via CD40 and TLR9 results in T cell independent antitumor effects1. J Immunol. 2006;176(1):309–318. doi:10.4049/jimmunol.176.1.309
76. Kim YH, Girardi M, Duvic M, et al. Phase I trial of a Toll-like receptor 9 agonist, PF-3512676 (CPG 7909), in patients with treatment-refractory, cutaneous T-cell lymphoma. J Am Acad Dermatol. 2010;63(6):975–983. doi:10.1016/j.jaad.2009.12.052
77. Thompson JA, Kuzel T, Drucker BJ, Urba WJ, Bukowski RM. Safety and efficacy of PF-3512676 for the treatment of stage IV renal cell carcinoma: an open-label, multicenter phase I/II study. Clin Genitourin Cancer. 2009;7(3):E58–E65. doi:10.3816/CGC.2009.n.025
78. Molenkamp BG, Sluijter BJR, Van Leeuwen PAM, et al. Local administration of PF-3512676 CpG-B instigates tumor-specific CD8+ T-cell reactivity in melanoma patients. Clin Cancer Res. 2008;14(14):4532–4542. doi:10.1158/1078-0432.CCR-07-4711
79. Schmoll H-J, Wittig B, Arnold D, et al. Maintenance treatment with the immunomodulator MGN1703, a toll-like receptor 9 (TLR9) agonist, in patients with metastatic colorectal carcinoma and disease control after chemotherapy: a randomised, double-blind, placebo-controlled trial. J Cancer Res Clin Oncol. 2014;140(9):1615–1624. doi:10.1007/s00432-014-1682-7
80. Babiker H, Borazanci E, Subbiah V, et al. 1031P tilsotolimod engages the TLR9 pathway to promote antigen presentation and type I IFN signaling in solid tumours. Ann Oncol. 2020;31:S711–S712. doi:10.1016/j.annonc.2020.08.1151
81. Henry JT, Pavlick AC, Humphries T, et al. Abstract CT045: a Phase 1/2 trial of TAC-001 (TLR9 agonist-CD22 mAb) in advanced or metastatic solid tumors. Cancer Res. 2025;85(8_Supplement_2):CT045. doi:10.1158/1538-7445.AM2025-CT045
82. Milhem M, Zakharia Y, Davar D, et al. 304 Intratumoral injection of CMP-001, a toll-like receptor 9 (TLR9) agonist, in combination with pembrolizumab reversed programmed death receptor 1 (PD-1) blockade resistance in advanced melanoma. J Immunother Cancer. 2020;8(Suppl 3). doi:10.1136/jitc-2020-SITC2020.0304
83. Ribas A, Medina T, Kirkwood JM, et al. Overcoming PD-1 blockade resistance with CpG-A toll-like receptor 9 agonist vidutolimod in patients with metastatic melanoma. Cancer Discov. 2021;11(12):2998–3007. doi:10.1158/2159-8290.CD-21-0425
84. Lawrence YR, Lieberman S, Redinsky I, et al. Combination treatment of intratumoral vidutolimod (CMP-001), radiosurgery, nivolumab, and ipilimumab for metastatic colorectal carcinoma. J Clin Oncol. 2023;41(4_suppl):123. doi:10.1200/JCO.2023.41.4_suppl.123
85. Davar D, Karunamurthy A, Hartman D, et al. 303 phase II trial of neoadjuvant nivolumab (Nivo) and intra-tumoral (IT) CMP-001 in high-risk resectable melanoma (Neo-C-Nivo): final results. J Immunother Cancer. 2020;8(Suppl 3). doi:10.1136/jitc-2020-SITC2020.0303
86. Tarhini AA, Leng S, Moschos SJ, et al. Safety and immunogenicity of vaccination with MART-1 (26–35, 27L), gp100 (209–217, 210M), and tyrosinase (368–376, 370D) in adjuvant with PF-3512676 and GM-CSF in metastatic melanoma. J Immunother. 2012;35(4):359–366. doi:10.1097/CJI.0b013e31825481fe
87. Manegold C, Gravenor D, Woytowitz D, et al. Randomized phase II trial of a toll-like receptor 9 agonist oligodeoxynucleotide, PF-3512676, in combination with first-line taxane plus platinum chemotherapy for advanced-stage non–small-cell lung cancer. J Clin Oncol off J Am Soc Clin Oncol. 2008;26(24):3979–3986. doi:10.1200/JCO.2007.12.5807
88. Hirsh V, Paz-Ares L, Boyer M, et al. Randomized phase III trial of paclitaxel/carboplatin with or without PF-3512676 (toll-like receptor 9 agonist) as first-line treatment for advanced non–small-cell lung cancer. J Clin Oncol off J Am Soc Clin Oncol. 2011;29(19):2667–2674. doi:10.1200/JCO.2010.32.8971
89. Belani CP, Nemunaitis JJ, Chachoua A, et al. Phase 2 trial of erlotinib with or without PF-3512676 (CPG 7909, a Toll-like receptor 9 agonist) in patients with advanced recurrent EGFR-positive non-small cell lung cancer. Cancer Biol Ther. 2013;14(7):557–563. doi:10.4161/cbt.24598
90. Brody JD, Ai WZ, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28(28):4324–4332. doi:10.1200/JCO.2010.28.9793
91. Karbach J, Gnjatic S, Bender A, et al. Tumor-reactive CD8 + T-cell responses after vaccination with NY-ESO-1 peptide, CpG 7909 and Montanide® ISA-51: association with survival. Int, J, Cancer. 2010;126(4):909–918. doi:10.1002/ijc.24850
92. Karbach J, Neumann A, Atmaca A, et al. Efficient in vivo priming by vaccination with recombinant NY-ESO-1 protein and CpG in antigen Naïve prostate cancer patients. Clin Cancer Res off J Am Assoc Cancer Res. 2011;17(4):861–870. doi:10.1158/1078-0432.CCR-10-1811
93. Ursu R, Carpentier A, Metellus P, et al. Intracerebral injection of CpG oligonucleotide for patients with de novo glioblastoma—A phase II multicentric, randomised study. Eur J Cancer. 2017;73:30–37. doi:10.1016/j.ejca.2016.12.003
94. Friedberg JW, Kelly JL, Neuberg D, et al. Phase II study of a TLR-9 agonist (1018 ISS) with rituximab in patients with relapsed or refractory follicular lymphoma. Br J Haematol. 2009;146(3):282–291. doi:10.1111/j.1365-2141.2009.07773.x
95. Slingluff CL, Petroni GR, Olson WC, et al. A randomized pilot trial testing the safety and immunologic effects of a MAGE-A3 protein plus AS15 immunostimulant administered into muscle or into dermal/subcutaneous sites. Cancer Immunol Immunother. 2016;65(1):25–36. doi:10.1007/s00262-015-1770-9
96. Gutzmer R, Rivoltini L, Levchenko E, et al. Safety and immunogenicity of the PRAME cancer immunotherapeutic in metastatic melanoma: results of a phase I dose escalation study. ESMO Open. 2016;1(4):e000068. doi:10.1136/esmoopen-2016-000068
97. McQuade JL, Homsi J, Torres-Cabala CA, et al. A phase II trial of recombinant MAGE-A3 protein with immunostimulant AS15 in combination with high-dose Interleukin-2 (HDIL2) induction therapy in metastatic melanoma. BMC Cancer. 2018;18(1):1274. doi:10.1186/s12885-018-5193-9
98. Cohen AD, Lendvai N, Nataraj S, et al. Autologous lymphocyte infusion supports tumor antigen vaccine–induced immunity in autologous stem cell transplant for multiple myeloma. Cancer Immunol Res. 2019;7(4):658–669. doi:10.1158/2326-6066.CIR-18-0198
99. Hamilton E, Blackwell K, Hobeika AC, et al. Phase I clinical trial of HER2-specific immunotherapy with concomitant HER2 kinase inhibtion. J Transl Med. 2012;10(1):28. doi:10.1186/1479-5876-10-28
100. Frank MJ, Reagan PM, Bartlett NL, et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. 2018;8(10):1258–1269. doi:10.1158/2159-8290.CD-18-0743
101. Shree T, Czerwinski D, Haebe S, et al. A phase I clinical trial adding OX40 agonism to in situ therapeutic cancer vaccination in patients with low-grade b-cell lymphoma highlights challenges in translation from mouse to human studies. Clin Cancer Res. 2025;31(5):868–880. doi:10.1158/1078-0432.CCR-24-2770
102. Ribas A, Medina T, Kummar S, et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov. 2018;8(10):1250–1257. doi:10.1158/2159-8290.CD-18-0280
103. Thomas M, Ponce-Aix S, Navarro A, et al. Immunotherapeutic maintenance treatment with toll-like receptor 9 agonist lefitolimod in patients with extensive-stage small-cell lung cancer: results from the exploratory, controlled, randomized, international phase II IMPULSE study. Ann Oncol. 2018;29(10):2076–2084. doi:10.1093/annonc/mdy326
104. Smith DA, Conkling P, Richards DA, et al. Antitumor activity and safety of combination therapy with the toll-like receptor 9 agonist IMO-2055, erlotinib, and bevacizumab in advanced or metastatic non-small cell lung cancer patients who have progressed following chemotherapy. Cancer Immunol Immunother. 2014;63(8):787–796. doi:10.1007/s00262-014-1547-6
105. Ruzsa A, Sen M, Evans M, et al. Phase 2, open-label, 1:1 randomized controlled trial exploring the efficacy of EMD 1201081 in combination with cetuximab in second-line cetuximab-naïve patients with recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN). Invest New Drugs. 2014;32(6):1278–1284. doi:10.1007/s10637-014-0117-2
106. Machiels J-P, Kaminsky M-C, Keller U, et al. Phase Ib trial of the toll-like receptor 9 agonist IMO-2055 in combination with 5-fluorouracil, cisplatin, and cetuximab as first-line palliative treatment in patients with recurrent/metastatic squamous cell carcinoma of the head and neck. Invest New Drugs. 2013;31(5):1207–1216. doi:10.1007/s10637-013-9933-z
107. Milhem MM, Wise-Draper TM, Chandra S, et al. Phase 1b/2 study evaluating safety, efficacy and immune effects of TLR9 agonist cavrotolimod with anti-PD-1 antibodies among patients with advanced solid tumors. J Immunother Cancer. 2025;13(7):e011651. doi:10.1136/jitc-2025-011651
108. Zhang Z, Kuo JC-T, Yao S, Zhang C, Khan H, Lee RJ. CpG oligodeoxynucleotides for anticancer monotherapy from preclinical stages to clinical trials. Pharmaceutics. 2021;14(1):73. doi:10.3390/pharmaceutics14010073
109. Honda K, Ohba Y, Yanai H, et al. Spatiotemporal regulation of MyD88–IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434(7036):1035–1040. doi:10.1038/nature03547
110. Luke JJ, Bao R, Kirkwood JM, et al. Abstract CT032: CMP-001 demonstrates improved response in noninflamed anti-PD-1 refractory melanoma and response is associated with serum CXCL10. Cancer Res. 2021;81(13_Supplement):CT032. doi:10.1158/1538-7445.AM2021-CT032
111. Harrabi O, Chen A, Sangalang E, et al. 780 ALTA-002, a SIRPα-directed TLR9 agonist antibody conjugate activates myeloid cells and promotes anti-tumor immunity. J Immunother Cancer. 2021;9(Suppl 2). doi:10.1136/jitc-2021-SITC2021.780
112. Mohamed FEZ, Jalan R, Minogue S, et al. Inhibition of TLR7 and TLR9 reduces human cholangiocarcinoma cell proliferation and tumor development. Dig Dis Sci. 2022;67(5):1806–1821. doi:10.1007/s10620-021-06973-9
113. Costa TJ, Potje SR, Fraga-Silva TFC, et al. Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vascul Pharmacol. 2022;142:106946. doi:10.1016/j.vph.2021.106946
114. O’Brien M, Manches O, Wilen C, et al. CD4 receptor is a key determinant of divergent HIV-1 sensing by plasmacytoid dendritic cells. PLoS Pathog. 2016;12(4):e1005553. doi:10.1371/journal.ppat.1005553
115. McHutchison JG, Bacon BR, Gordon SC, et al. Phase 1B, randomized, double-blind, dose-escalation trial of CPG 10101 in patients with chronic hepatitis C virus. Hepatology. 2007;46(5):1341–1349. doi:10.1002/hep.21773
116. Libri NA, Barker SJ, Rosenberg WMC, Semper AE. A class C CpG toll-like receptor 9 agonist successfully induces robust interferon-alpha production by plasmacytoid dendritic cells from patients chronically infected with hepatitis C. J Viral Hepat. 2009;16(5):315–324. doi:10.1111/j.1365-2893.2008.01011.x
117. Cooper CL, Davis HL, Morris ML, et al. CPG 7909, an immunostimulatory TLR9 agonist oligodeoxynucleotide, as adjuvant to Engerix-B HBV vaccine in healthy adults: a double-blind phase I/II study. J Clin Immunol. 2004;24(6):693–701. doi:10.1007/s10875-004-6244-3
118. Siegrist C-A, Pihlgren M, Tougne C, et al. Co-administration of CpG oligonucleotides enhances the late affinity maturation process of human anti-hepatitis B vaccine response. Vaccine. 2004;23(5):615–622. doi:10.1016/j.vaccine.2004.07.014
119. Cooper CL, Davis HL, Angel JB, et al. CPG 7909 adjuvant improves hepatitis B virus vaccine seroprotection in antiretroviral-treated HIV-infected adults. AIDS Lond Engl. 2005;19(14):1473–1479. doi:10.1097/01.aids.0000183514.37513.d2
120. Lövgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, Rönnblom L. Induction of interferon-α by immune complexes or liposomes containing systemic lupus erythematosus autoantigen– and Sjögren’s syndrome autoantigen–associated RNA. Arthritis Rheum. 2006;54(6):1917–1927. doi:10.1002/art.21893
121. Rao H, Zeng Q, Liang Y, Xiao C, Xie S, Xu X. Correlation between TLR9 expression and cytokine secretion in the clinical diagnosis of systemic lupus erythematosus. Mediators Inflamm. 2015;2015(1):710720. doi:10.1155/2015/710720
122. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202(2):321–331. doi:10.1084/jem.20050338
123. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–428. doi:10.1016/j.immuni.2006.07.013
124. Lacerte P, Brunet A, Egarnes B, Duchêne B, Brown JP, Gosselin J. Overexpression of TLR2 and TLR9 on monocyte subsets of active rheumatoid arthritis patients contributes to enhance responsiveness to TLR agonists. Arthritis Res Ther. 2016;18(1):10. doi:10.1186/s13075-015-0901-1
125. Han J, Li X, Luo X, et al. The mechanisms of hydroxychloroquine in rheumatoid arthritis treatment: inhibition of dendritic cell functions via Toll like receptor 9 signaling. Biomed Pharmacother Biomedecine Pharmacother. 2020;132:110848. doi:10.1016/j.biopha.2020.110848
126. Liu M, Peng J, Tai N, et al. Toll-like receptor 9 negatively regulates pancreatic islet beta cell growth and function in a mouse model of type 1 diabetes. Diabetologia. 2018;61(11):2333–2343. doi:10.1007/s00125-018-4705-0
127. Zhou J, Zhang X, Ji L, Jiang G, Sánchez García FJ. Identification of potential biomarkers of type 2 diabetes mellitus-related immune infiltration using weighted gene coexpression network analysis. BioMed Res Int. 2022;2022(1):9920744. doi:10.1155/2022/9920744
128. Lee J, Mo J-H, Katakura K, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8(12):1327–1336. doi:10.1038/ncb1500
129. Obermeier F, Dunger N, Strauch UG, et al. CpG motifs of bacterial DNA essentially contribute to the perpetuation of chronic intestinal inflammation. Gastroenterology. 2005;129(3):913–927. doi:10.1053/j.gastro.2005.06.061
130. Rachmilewitz D, Karmeli F, Takabayashi K, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology. 2002;122(5):1428–1441. doi:10.1053/gast.2002.32994
131. Atreya R, Bloom S, Scaldaferri F, et al. Clinical effects of a topically applied toll-like receptor 9 agonist in active moderate-to-severe ulcerative colitis. J Crohns Colitis. 2016;10(11):1294–1302. doi:10.1093/ecco-jcc/jjw103
132. Simons FER, Shikishima Y, Van Nest G, Eiden JJ, HayGlass KT. Selective immune redirection in humans with ragweed allergy by injecting Amb a 1 linked to immunostimulatory DNA. J Allergy Clin Immunol. 2004;113(6):1144–1151. doi:10.1016/j.jaci.2004.03.003
133. Santeliz JV, Nest GV, Traquina P, Larsen E, Wills-Karp M. Amb a 1–linked CpG oligodeoxynucleotides reverse established airway hyperresponsiveness in a murine model of asthma. J Allergy Clin Immunol. 2002;109(3):455–462. doi:10.1067/mai.2002.122156
134. Hsieh ML, Nishizaki D, Adashek JJ, Kato S, Kurzrock R. Toll-like receptor 3: a double-edged sword. Biomark Res. 2025;13(1):32. doi:10.1186/s40364-025-00739-5
135. Nikanjam M, Kato S, Nishizaki D, et al. ICOS and ICOS ligand: expression patterns and outcomes in oncology patients. Ther Adv Med Oncol. 2025;17:17588359251330514. doi:10.1177/17588359251330514
136. Nishizaki D, Kurzrock R, Miyashita H, et al. Viewing the immune checkpoint Vista: landscape and outcomes across cancers. ESMO Open. 2024;9(4):102942. doi:10.1016/j.esmoop.2024.102942
137. Krishnamurthy N, Nishizaki D, Lippman SM, et al. High CTLA-4 transcriptomic expression correlates with high expression of other checkpoints and with immunotherapy outcome. Ther Adv Med Oncol. 2024;16:17588359231220510. doi:10.1177/17588359231220510
138. Lim J, Kurzrock R, Nishizaki D, et al. Pan-cancer analysis of TIM −3 transcriptomic expression reveals high levels in pancreatic cancer and interpatient heterogeneity. Cancer Med. 2024;13(1):e6844. doi:10.1002/cam4.6844
139. Ahmed J, Nishizaki D, Miyashita H, et al. TIM-3 transcriptomic landscape with clinical and immunomic correlates in cancer. Am J Cancer Res. 2024;14(5):2493–2506. doi:10.62347/MQFF6404
140. Chen H-Z, Kim NH, Nishizaki D, et al. PD-1 transcriptomic landscape across cancers and implications for immune checkpoint blockade outcome. Npj Genomic Medicine. 2025;10(1):21. doi:10.1038/s41525-025-00465-9
141. Miyar A, Habibi I, Ebrahimi A, et al. Predictive and prognostic value of TLR9 and NFKBIA gene expression as potential biomarkers for human glioma diagnosis. J Neurol Sci. 2016;368:314–317. doi:10.1016/j.jns.2016.07.046
142. Powles T, Assaf ZJ, Davarpanah N, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595(7867):432–437. doi:10.1038/s41586-021-03642-9
143. Cisneros-Villanueva M, Hidalgo-Pérez L, Rios-Romero M, et al. Cell-free DNA analysis in current cancer clinical trials: a review. Br J Cancer. 2022;126(3):391–400. doi:10.1038/s41416-021-01696-0
144. Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–238. doi:10.1038/nrc.2017.7
145. Schilter KF, Nie Q, Adams JN, et al. Analytical validation of the Belay Vantage™ assay for evaluation of MGMT promoter methylation using enzymatically converted tumorDNA from cerebrospinal fluid. Cancer Genetics. 2025;294-295:94–98. doi:10.1016/j.cancergen.2025.04.001
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.