Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Tiotropium inhibits methacholine-induced extracellular matrix production via β-catenin signaling in human airway smooth muscle cells
Authors Huo Y, Guan L, Xu J ![]() , Zhou L, Chen R
, Zhou L, Chen R
Received 30 November 2017
Accepted for publication 14 March 2018
Published 3 May 2018 Volume 2018:13 Pages 1469—1481
DOI https://doi.org/10.2147/COPD.S158552
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Yating Huo, Lili Guan, Jiawen Xu, Luqian Zhou, Rongchang Chen
State Key Laboratory of Respiratory Disease, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
Background: Airway remodeling is an important feature of chronic obstructive pulmonary disease (COPD) that is associated with disease severity and irreversible airflow limitation. An extensive alteration of the extracellular matrix (ECM) surrounding the airway smooth muscle (ASM) bundle is one of the pathological manifestations of airway remodeling, which contributes to the decline in lung function. Tiotropium, a long-acting inhaled muscarinic receptor antagonist, has been confirmed to play a role in preventing airway remodeling including ECM deposition beyond bronchodilation in vivo, but the relationship between ASM cell (ASMC) relaxation and ECM production remains unclear.
Purpose: In this study, we attempted to investigate the influence of tiotropium on ECM production by ASMCs and the underlying mechanism.
Methods: Tiotropium was added 30 minutes before the addition of methacholine to primary cultured human ASMCs. Protein expression was analylized by Western Blot and mRNA abundance was determined by real-time PCR.
Results: We found that tiotropium reduced collagen I protein expression, and the mRNA abundance of collagen I, fibronectin, and versican. β-catenin signaling was inactivated by inhibiting glycogen synthase kinase 3β (GSK3β) phosphorylation in this process. Tiotropum inhibited the amount of active β-catenin and its transcription activity. Furthermore, overexpression of active β-catenin by adenoviruses carrying the S33Y mutant resisted the suppressive effect of tiotropium on collagen I protein expression. However, silencing β-catenin by specific small interfering RNA enhanced the negative effect of tiotropium.
Conclusion: These findings suggest that relaxation of ASMCs by tiotropium can prevent ECM production through β-catenin signaling.
Keywords: tiotropium, collagen I, β-catenin, airway smooth muscle cell
Introduction
Airway remodeling is an important feature of chronic inflammatory airway diseases such as chronic obstructive pulmonary disease (COPD) and correlates with disease severity and irreversible airflow limitation. It is described as the structure changes in the airway wall caused by repeated injury and repair,1 including airway epithelial cell hyperplasia, squamous cell and goblet cell metaplasia, reticular basement membrane thickening, peribronchial fibrosis, and angiogenesis.2 Airway remodeling exists in small airways prior to the emphysematous destruction in COPD and induces clinical symptoms like shortness of breath at an early stage of COPD.3 However, currently there is no effective therapy for relieving or reversing airway remodeling.
Extracellular matrix (ECM) alteration plays a key role in the process of airway remodeling, which is likely to contribute to persistent tissue injury and airflow obstruction in COPD patients.4 ECM is a complicated network of macromolecules comprising collagens, elastic fibers, proteoglycans, fibronectin, and tenascin,5,6 and the contents of collagens,7 elastic fibers,8 and fibronectin4 are associated with the decline in the forced expiratory volume in 1 second (FEV1). It has been reported that there is an altered ECM composition in the airways of COPD patients who have mild airflow limitation,9 which suggests that it is necessary to prevent abnormal ECM deposition at the early stage of COPD. It has been suggested that ECM is mainly produced by fibroblasts, but many recent studies have shown that airway smooth muscle cells (ASMCs) may play a crucial role in producing ECM when exposed to allergens, cigarettes, and environmental pollution.10
ASMCs are the main structure cells in small airways, which are subjected to the vagus nerve and contribute to bronchoconstriction.11 The increased cholinergic tone is the primary reversible element of airflow limitation in COPD.12 Hence, tiotropium, the first long-acting inhaled muscarinic antagonist introduced to the market, has been used as a bronchodilator in COPD for >10 years. However, it has been suggested that mechanical forces caused by bronchoconstriction may play a crucial role in airway remodeling in asthma.13,14 Grainge et al have found that bronchoconstriction without inflammation induced airway remodeling in asthma patients.15 Furthermore, in recent years, several studies have indicated that anticholinergics may have effects beyond bronchodilation. For example, Pera et al have found that tiotropium inhibited airway remodeling including ECM deposition in a guinea pig model of COPD.16 These findings suggest that bronchodilators may prevent airway remodeling. However, there is not enough evidence for the relationship between ASMC relaxation and airway remodeling, and the mechanisms involved remain unclear.
The constriction and relaxation of ASMCs are associated with remodeling of the cytoskeleton. β-catenin is a protein that connects cadherins to the cytoskeleton at adherens junctions where it functions as a membrane-bound protein and influences cell stabilization and transmission of mechanical forces. However, cytosolic β-catenin plays a central role in the canonical Wnt/β-catenin signaling pathway. When it is rescued from GSK3β-mediated phosphorylation and proteasomal degradation, it translocates into the nucleus and regulates T-cell factor (TCF)/lymphoid enhancer factor (LEF)-mediated gene transcription.17 Recently, it has been suggested that mechanical forces within the airway wall can activate β-catenin signaling. Activation of β-catenin signaling (in WNT-dependent and -independent manners) has a regulatory function in airway remodeling involving ASMC proliferation, epithelial-to-mesenchymal transition, myofibroblast differentiation, and ECM production.18 Based on the aforementioned clues, we hypothesized that tiotropium inhibits ECM production through β-catenin signaling in ASMCs.
Materials and methods
Cell culture
Human ASMCs (HASMCs) for all experiments were donated by Dr Cheng Yuanxiong (The Third Affiliated Hospital of Southern Medical University, Guangzhou, China). HASMCs were isolated from healthy segments of the lobar or main bronchus obtained from three different lung resection donors after giving informed consents, which were approved by the Ethics Committee of Nanfang Hospital, Southern Medical University (NFEC-201109-K1). Table 1 summarizes the clinical conditions of the tissue donors. Cell type identification was carried out by immunofluorescence detection of smooth muscle α-actin, a contractile phenotype marker protein. Primary cultured HASMCs from passages 2 to 10 were used for the experiments, and the experiments were in triplicate in the three cell lines. Cells were incubated in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum (FBS) (Gibco, Life Technologies, Australia) and antibiotics (100 U/mL penicillin and 100 U/mL streptomycin). Cells were washed twice with phosphate-buffered saline (PBS), and then serum starved in DMEM with antibiotics for 12 hours. Subsequently, the cells were stimulated with methacholine in serum-free medium supplemented with antibiotics for 24 hours. Tiotropium (10 μM) was added 30 minutes before the addition of methacholine.
 | Table 1 Clinical conditions for the tissue donors |
Immunofluorescence
HASMCs were cultured in 48-well plates for 24 hours at 50% confluence. Next, cells were fixed with 4% paraformaldehyde for 15 minutes, washed with PBS 3×5 minutes, and then permeabilized with 0.1% Triton X-100 for 7 minutes. After three washes with PBS, the cells were blocked with 5% bovine serum albumin (BSA) for 2 h at room temperature. They were then incubated with specific primary antibodies at 4°C for at least 12 hours, followed by goat anti-mouse IgG/FITC-antibody for 1 hour at room temperature. Finally, the nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI; Beyotime, Shanghai, China) for 15 minutes. Images were obtained using an inverted fluorescence microscope (Olympus-FL 500; Olympus Corporation, Tokyo, Japan).
Isolation of mRNA and real-time PCR analysis
Total mRNA was extracted by using MiniBEST Universal RNA Extraction Kit (TaKaRa Bio, Tokyo, Japan) according to the manufacturer’s protocol. The mRNA was quantified by a NanoDrop Spectrophotometer (NanoDrop Tech, Rockland, DE, USA). For reverse transcription, 1 μg of total mRNA per sample was used with PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa Bio) and cDNA was stored at −20°C until further use. The gene-specific primers (listed in Table 2) were obtained from Sangon Biotech (Shanghai, China). Quantitative real-time PCR was performed with SYBR Premix Ex Taq II (TaKaRa Bio) and Roche LightCycler 480 instrument II (Roche Diagnostics, Basel, Switzerland). The target gene levels were quantified with the 2(−ΔΔct) relative quantification method, which was normalized to GAPDH.
 | Table 2 Primers used for the determination of genes’ expression |
Western blot analysis
Whole cell lysates were extracted by using the radioimmunoprecipitation assay lysis buffer containing protease inhibitors and phosphatase inhibitors. Nucleoproteins were extracted by using Nuclear-Cytosol Extraction Kit (FDbio science, Hangzhou, China) according to the manufacturer’s instructions. Protein concentration was detected by the BCA Protein Assay Kit (FDbio science). All the lysates were stored at −80°C until further use.
Western blotting was conducted following standard procedures. Equal amounts of protein (15–25 μg/lane) were electrophoresed in 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% BSA and then incubated with specific primary antibodies at 4°C for at least 12 h. Subsequently, the blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 hour at room temperature. The proteins of interest were detected by enhanced chemiluminescence reagents (FDbio science), and the band intensities were quantified by using the ImageJ software (National Institute of Health, Bethesda, MD, USA). The expression of the target proteins was normalized against the loading control, β-actin or LaminB1.
β-catenin small interfering RNA (siRNA) transfection
A specific double-stranded siRNA against the β-catenin transcript or a negative control (Shanghai Genechem Co., Ltd., Shanghai, China) was transfected into HASMCs at a final concentration of 90 nM when cells were 50% confluent in six-well cluster plates using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were cultured in serum-free DMEM without any supplements for 6 hours. Next, cells were washed once with PBS and then incubated in DMEM with 10% FBS for another 42 hours. Subsequently, the medium was replaced with serum-free DMEM and stimulated with tiotropium, which was added 30 minutes before the addition of methacholine. Transfected cells were harvested for the extraction of total protein or mRNA after 24 hours.
β-catenin S33Y mutant transfection
The active β-catenin mutant, S33Y-β-catenin, is resistant to GSK3-mediated phosphorylation and proteasomal degradation because of a serine-to-tyrosine substitution at position 33. The adenovirus packaging was conducted by a professional company (Shanghai Genechem Co., Ltd.), and the transduction efficiency was measured by green fluorescent protein (GFP) fluorescence using a fluorescence microscope. Cells were incubated in DMEM with 10% FBS. The recombinant adenovirus was directly transfected into HASMCs at 50% confluence in six-well cluster plates for 48 hours (multiplicity of infection [MOI] = 100). A GFP expression vector was used as a negative control. Consecutively, the medium was replaced with serum-free DMEM, followed by tiotropium stimulation, which was added 30 minutes before the addition of methacholine. Transfected cells were harvested for total protein or mRNA extraction after 24 hours.
Statistical analyses
All quantitative data are presented as mean ± SD and analyzed using SPSS v.16.0 (SPSS Inc., Chicago, IL, USA). Multiple comparisons were analyzed by one-way analysis of variance (ANOVA), followed by Student–Neuman–Keuls test with equal variances determined by the homogeneity of variance test. Differences were considered to be statistically significant when P<0.05.
Antibodies and reagents
HRP-conjugated goat anti-rabbit antibody and HRP-conjugated goat anti-mouse antibody were purchased from FDbio science. Mouse anti-alpha smooth muscle actin antibody was from Abcam (Cambridge, MA, USA). Rabbit anti-phospho-GSK3β antibody, rabbit anti-total β-catenin antibody, rabbit anti-nonphosphorylated-β-catenin antibody were from Cell Signaling Technology (Danvers, MA, USA). Mouse anti-GSK3(α+β) antibody was from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Rabbit anti-collagen I antibody was from Bioss (Beijing, China). Methacholine and tiotropium were from Sigma-Aldrich Co. (St Louis, MO, USA).
Ethics
According to the ethics committee at our own institution (the Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University), there is no need to have the approval as the research used donated isolated human cells. All the patients whose cells were used provided written informed consent.
Results
Cell culture and identification
Primary cultured HASMCs were fusiform when observed under an inverted light microscope and showed the typical “hill and valley” pattern when confluent (Figure 1A). We used the second generation of cells for identification. Cells expressed smooth muscle α-actin, a contractile phenotype marker protein that appears green under immunofluorescence microscopy and the contractile fibrils were visible when magnified at 400 times (Figure 1B).
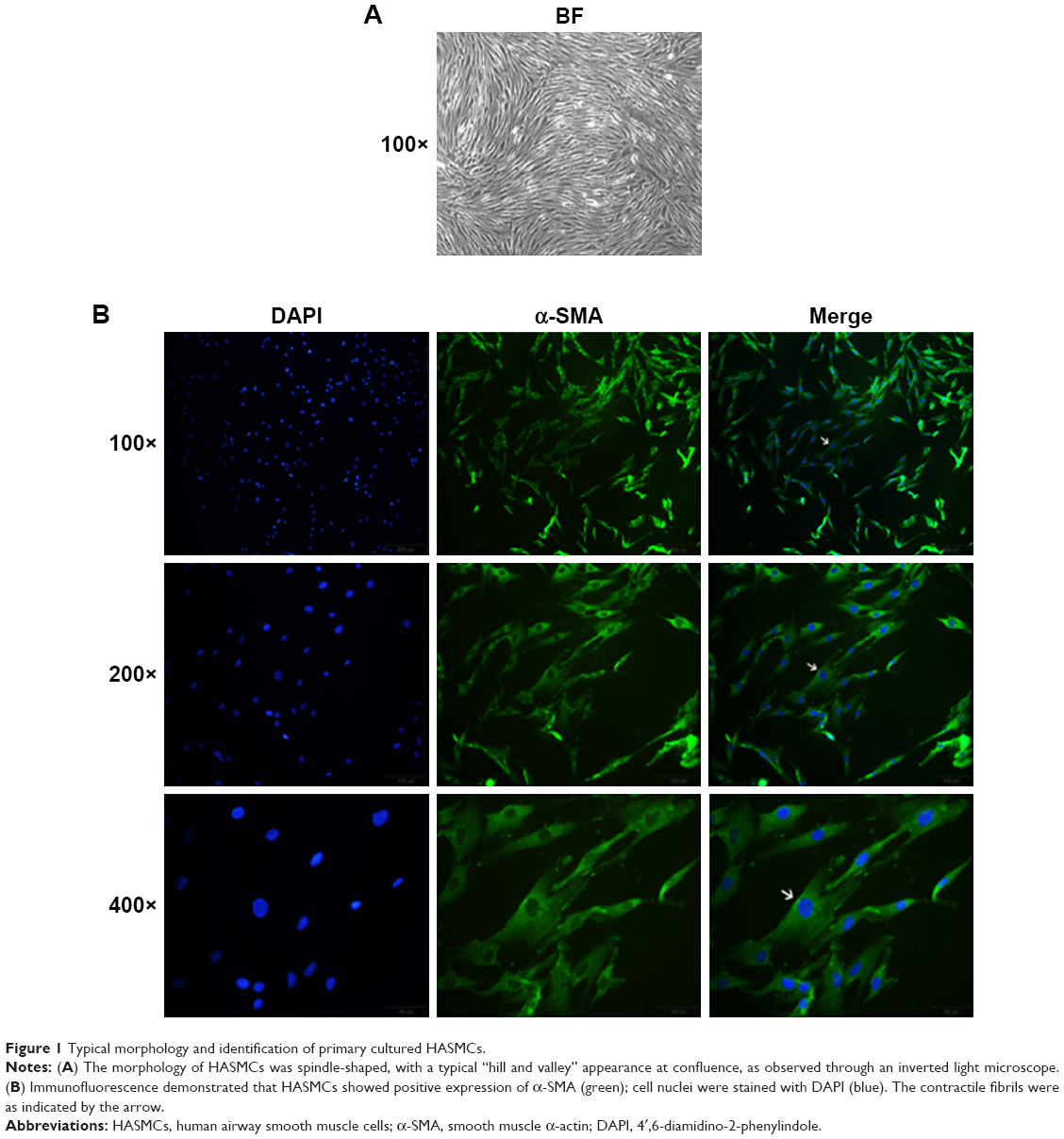 | Figure 1 Typical morphology and identification of primary cultured HASMCs. |
Tiotropium inhibits methacholine-induced ECM expression
First, we investigated whether tiotropium can inhibit methacholine-induced ECM production by HASMCs. We stimulated cells with an increasing concentration of methacholine from 0.1 to 10 μM for 24 hours. The expression of collagen I reached the maximum at 10 μM (Figure 2A). Thus, we performed the following experiments with 10 μM methacholine. Then, we examined the optimal concentration of tiotropium for inhibiting methacholine-induced ECM production by stimulating HASMCs with increasing concentrations of tiotropium (0.1–100 μM). Western blot analysis revealed that tiotropium decreased collagen I protein expression from 10 μM (Figure 2B). Accordingly, we used tiotropium at 10 μM for the following experiments. Furthermore, real-time PCR showed that the gene expression of collagen I, fibronectin, and versican was decreased in HASMCs exposed to 10 μM tiotropium (Figure 2C). These data revealed that tiotropium inhibited ECM production by ASMCs.
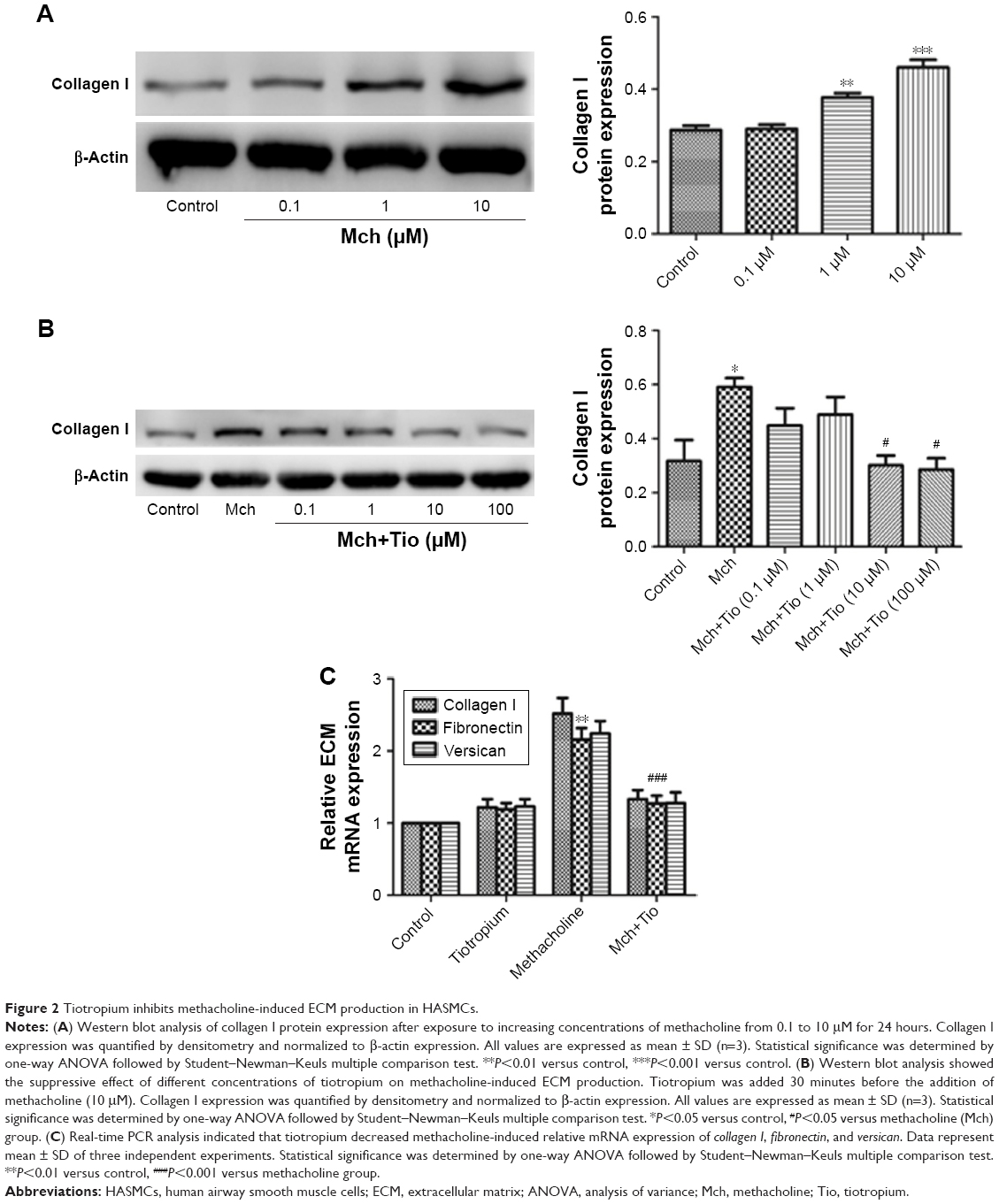 | Figure 2 Tiotropium inhibits methacholine-induced ECM production in HASMCs. |
Tiotropium inhibits β-catenin signaling in ASMCs
Next, we investigated whether tiotropium affects β-catenin signaling. β-catenin is the central mediator in the canonical Wnt/β-catenin signaling pathway, and its expression and localization are determined by the phosphorylation of GSK3β. To test our hypothesis, ASMCs were stimulated with different tiotropium concentrations (0.1–100 μM). Both total β-catenin and active β-catenin protein expression were significantly decreased from 10 μM, which is in accordance with the effect on ECM production (Figure 3A and B). Moreover, tiotropium decreased β-catenin mRNA abundance in HASMCs (Figure 3C). GSK3β phosphorylation is necessary for the activation of β-catenin signaling. Western blot analysis demonstrated that tiotropium markedly inhibited GSK3β phosphorylation in HASMCs (Figure 3D). These data indicated that tiotropium suppressed β-catenin signaling by preventing GSK3β phosphorylation.
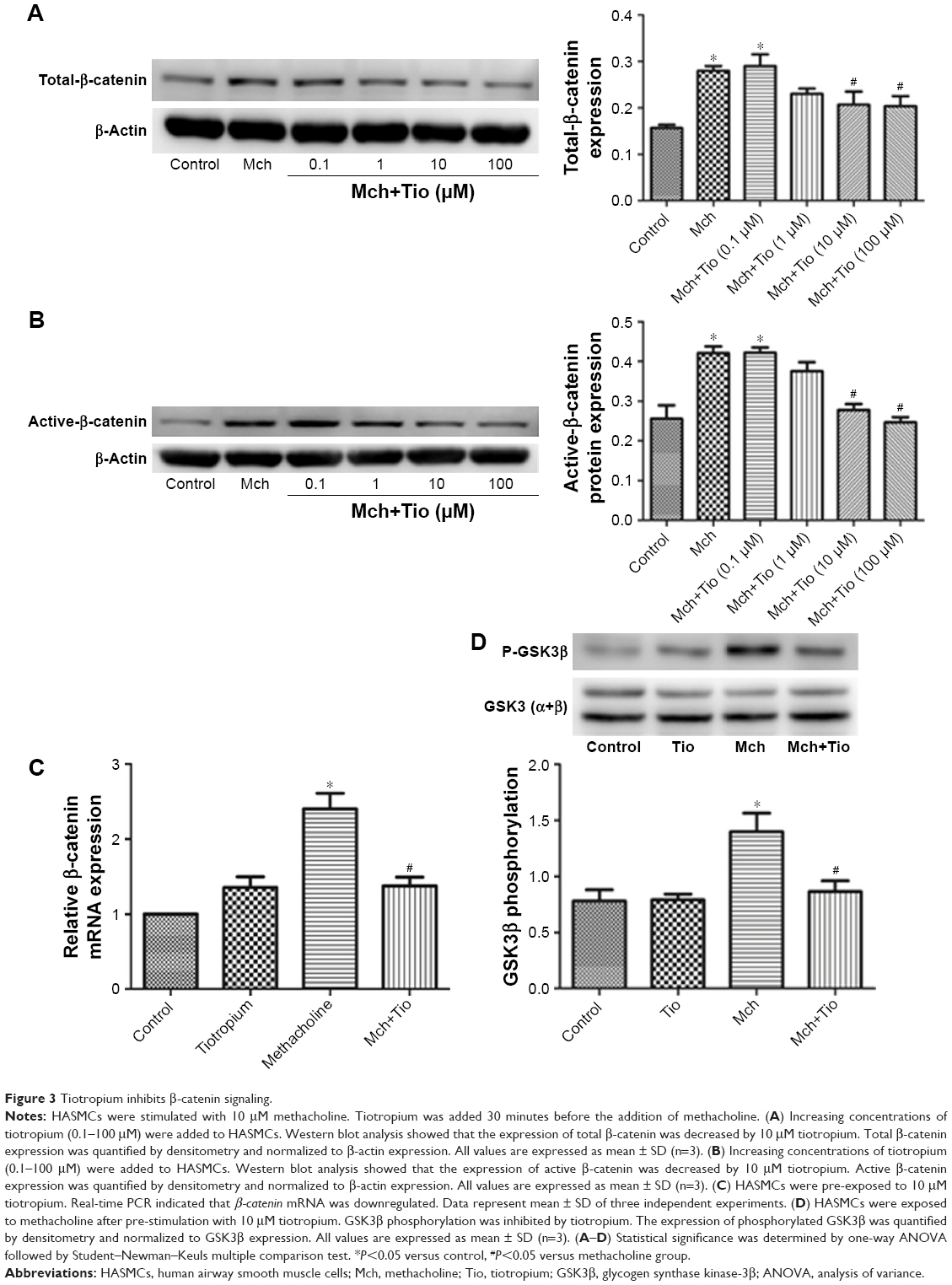 | Figure 3 Tiotropium inhibits β-catenin signaling. |
Tiotropium inhibits the transcription activity of active β-catenin
Then, we investigated the effect of tiotropium on the transcription activity of active β-catenin. Western blot analysis showed that tiotropium reduced the expression of active β-catenin (Figure 4A). Non-phosphorylated β-catenin translocates from the cytoplasm to nucleus and interacts with TCF/LEF to start target gene transcription. Hence, we examined the amount of active β-catenin in the nucleus. With tiotropium pre-incubation, the abundance of active β-catenin that translocated into the nucleus was also decreased (Figure 4B). These data showed that tiotropium decreased the amount of active β-catenin and suppressed its transcription activity.
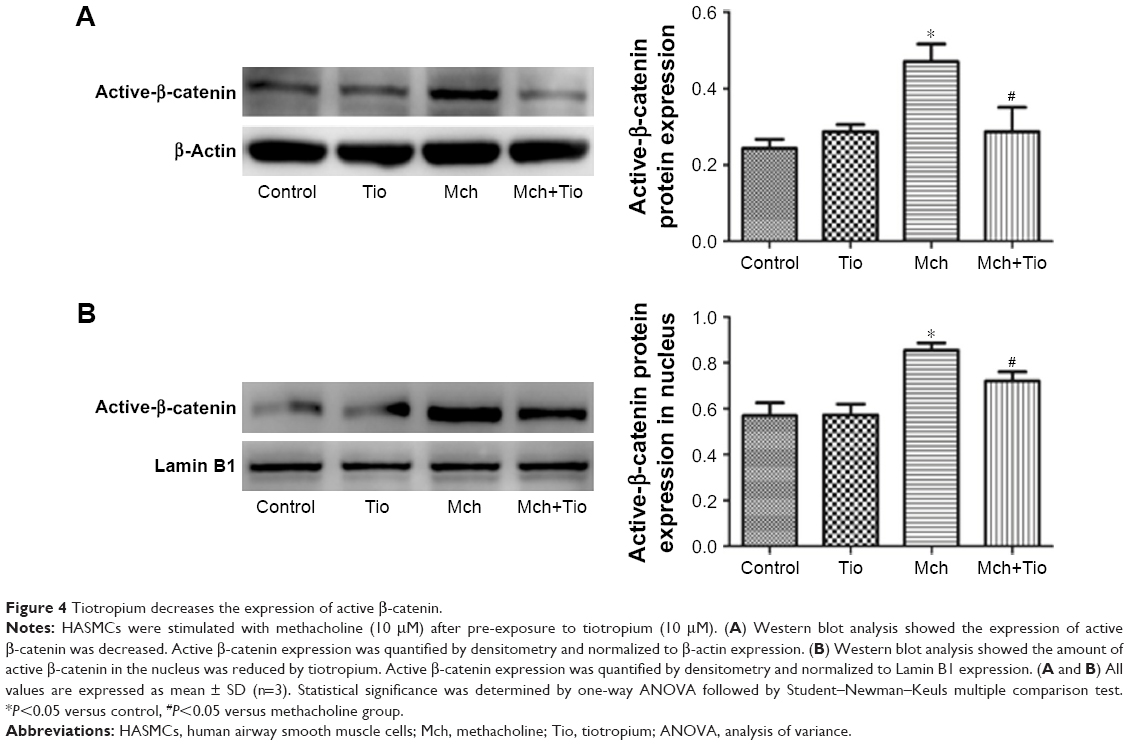 | Figure 4 Tiotropium decreases the expression of active β-catenin. |
Overexpression of β-catenin suppresses the effect of tiotropium on ECM production
To further investigate the role of β-catenin in the tiotropium-induced inhibition of ECM expression in HASMCs, we examined the negative effect of tiotropium on the expression of collagen I protein when β-catenin is overexpressed. To this end, we used adenoviruses carrying a constitutively active β-catenin mutant (S33Y-β-catenin). The mutant has a serine-to-tyrosine substitution at position 33, rescuing β-catenin from GSK3β-mediated phosphorylation and proteasomal degradation. Primary cultured HASMCs were transfected with the S33Y-β-catenin mutant, while control cultures were transfected with GFP. A satisfactory infection efficiency was obtained at MOI =100, which was measured by green fluorescence under fluorescence microscopy (Figure 5A). The abundance of β-catenin mRNA was elevated as evidenced by real-time PCR (Figure 5B), and the expression of total β-catenin was markedly increased (Figure 5C). Furthermore, transfection with the S33Y-β-catenin mutant increased the basal level of active β-catenin (Figure 5D). In the control cells, tiotropium reduced the methacholine-induced expression of active β-catenin and collagen I, as described earlier. However, tiotropium did not decrease the methacholine-induced production of active β-catenin and collagen I when cells were transfected with the S33Y-β-catenin mutant. In fact, the expression level was higher than it was upon stimulation with methacholine alone (Figure 5D and E). These data revealed that β-catenin played a crucial role in the suppression of methacholine-induced collagen I expression by tiotropium in HASMCs.
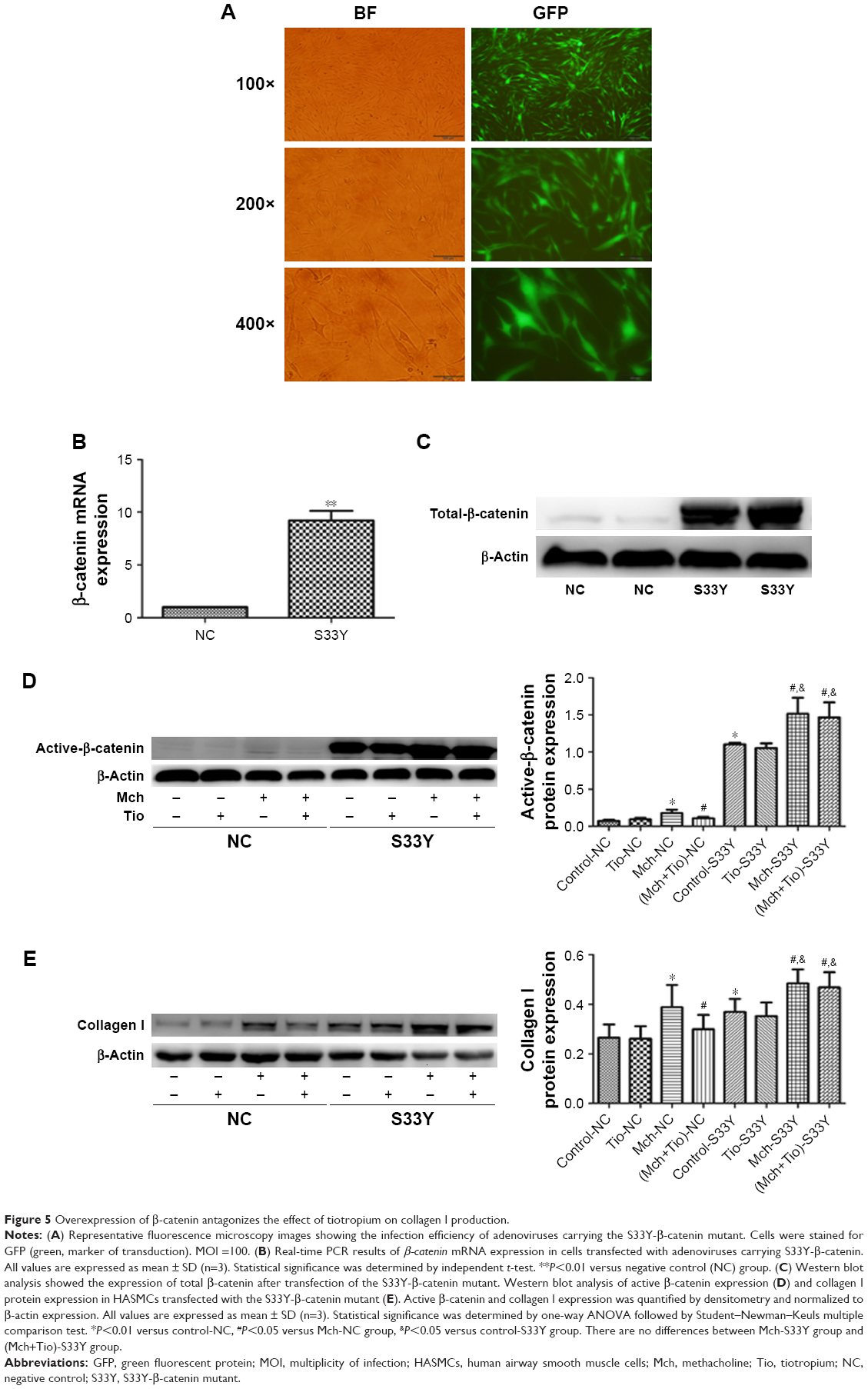 | Figure 5 Overexpression of β-catenin antagonizes the effect of tiotropium on collagen I production. |
Functional role of β-catenin in the tiotropium-induced inhibition of ECM production
Finally, we investigated the effect of β-catenin silencing on the suppressive effect of tiotropium on collagen I expression in ASMCs. Cells were transfected with specific β-catenin siRNA, and the optimal reduction in β-catenin expression was achieved with 90 nM β-catenin siRNA (Figure 6A). Silencing of β-catenin reduced the basal level of total β-catenin (Figure 6B). As mentioned previously, tiotropium inhibited the methacholine-induced expression of total β-catenin and collagen I. When transfected with β-catenin siRNA, the methacholine-induced total β-catenin and collagen I protein expression were downregulated. Furthermore, the expression levels of total β-catenin and collagen I were much lower when cells were treated with tiotropium together with β-catenin silencing compared with the effect of tiotropium alone (Figure 6B and C). These data indicated that β-catenin was sufficient for the tiotropium-induced inhibition of collagen I production.
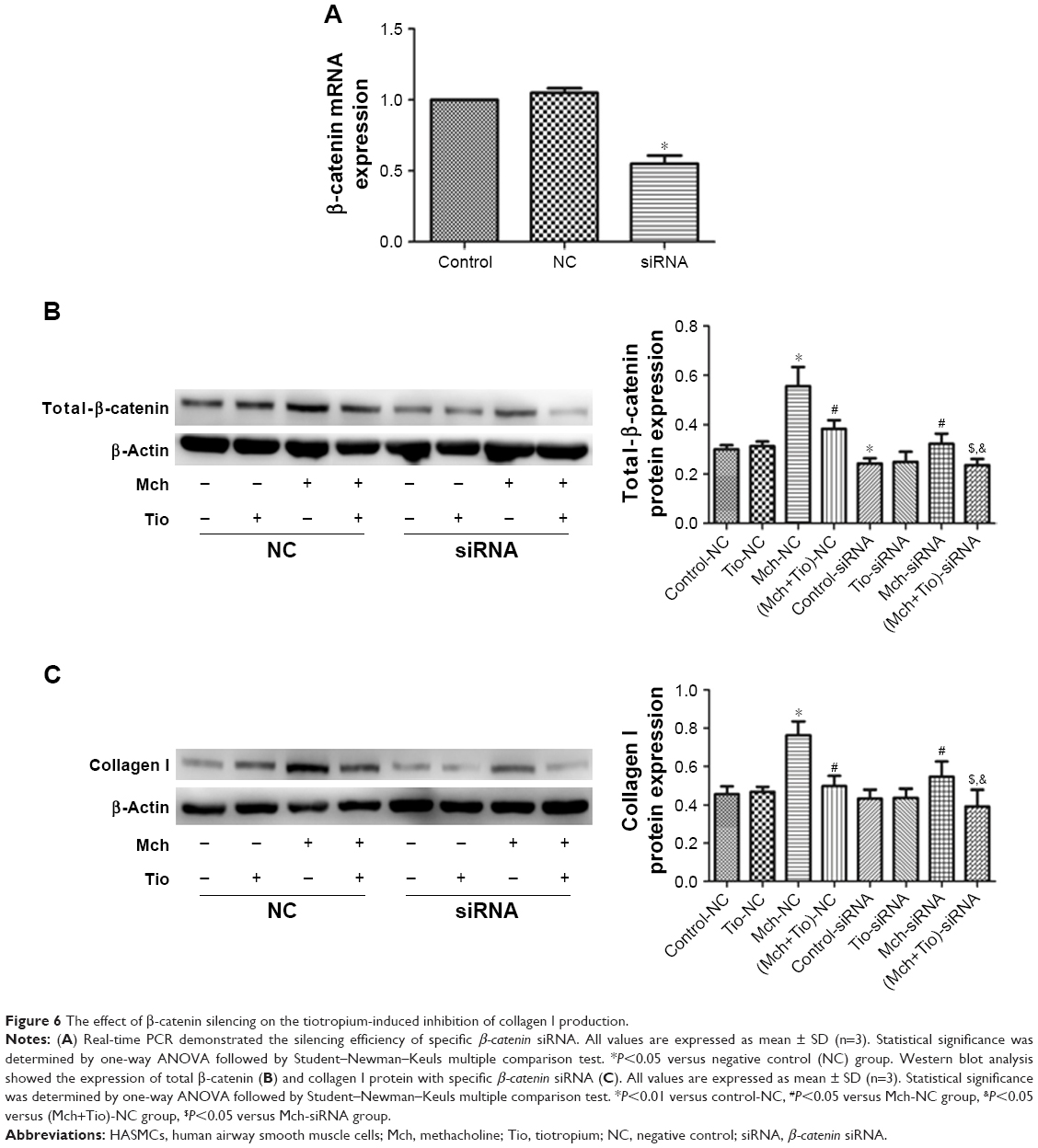 | Figure 6 The effect of β-catenin silencing on the tiotropium-induced inhibition of collagen I production. |
Discussion
In this study, we investigated the relationship between the relaxation of ASMCs and ECM production. Tiotropium, a common muscarinic receptor antagonist that is used as a bronchodilator in COPD, can reduce methacholine-induced ECM production through β-catenin signaling. It decreased the expression of collagen I protein and various ECM genes including collagen I, fibronectin, and versican. The suppressive effect of tiotropium on ECM production in ASMCs was exerted by preventing β-catenin signaling. Tiotropium decreased the expression of total β-catenin and the abundance of β-catenin mRNA. It decreased the transcription activity of non-phosphorylated β-catenin by inhibiting GSK3β phosphorylation. Furthermore, overexpression of active β-catenin suppressed the tiotropium-induced inhibition of collagen I production, while β-catenin silencing enhanced the suppressive influence of tiotropium. Taken together, these data indicated that relaxation of ASMCs by tiotropium was beneficial for controlling methacholine-induced ECM production in ASMCs.
Small airway remodeling is a crucial pathology in COPD and impairs lung function via changes in the ECM.19 As reported, the expression of collagen I, III, and IV, fibronectin, and lamin is increased in COPD patients and is associated with the decline in lung function.20 Furthermore, collagen I and III were increased in the small airways of COPD patients with GOLD stage 1 and 2.21 This means that the alteration of ECM exists in the early stages of COPD and that early intervention in abnormal ECM deposition may be helpful. An important observation of airway wall remodeling is the increased ECM deposition surrounding ASM bundles.22 The traditional view of ASMCs is that they mainly contribute to bronchoconstriction via muscarinic M3 receptors under the control of the parasympathetic nerve; however, it overlooked the synthetic capabilities of ASMCs. Actually, they serve as a biological factory that produces many cytokines, chemokines, growth factors, adhesion molecules, and bronchoprotective factors and promote the formation of interstitial ECM.23 Furthermore, it is suggested that mechanical forces within the airway walls, due to bronchoconstriction, may be the initial cause of airway remodeling.24 Hence, it would be useful to find a way to prevent ECM production by ASMCs.
The parasympathetic nerve is the major nerve distributed in airways, and its neurotransmitter induces bronchoconstriction and mucus secretion via muscarinic M2 and M3 receptors.25 In this regard, anticholinergics such as tiotropium are used as bronchodilator therapy in COPD patients. However, increasing attention has been paid to the function of muscarinic agonists in airway remodeling. It has been reported that stimulation of muscarinic receptors leads to the proliferation and mutation of ASMCs,26 human lung fibroblast proliferation,27 collagen synthesis,28 and matrix metalloproteinase (MMP) expression.29 Interestingly, recent studies have indicated that mechanical changes due to muscarinic agonist-induced bronchoconstriction may also contribute to airway remodeling. Accordingly, there is an increasing number of studies focused on the anti-remodeling effect of muscarinic receptor antagonists. For example, tiotropium treatment prevented the increased peribronchial collagen deposition in a guinea pig COPD model16 and attenuated MMP-2 expression in lung fibroblasts after TNF-α stimulation.30 A large clinical trial announced that tiotropium improved the annual decline in the FEV1 in COPD patients of GOLD stage 1 or 2,31 but the mechanism remains unclear. Based on the relationship between ECM deposition and FEV1 decline, we hypothesized that tiotropium affects FEV1 decline via inhibition of ECM production. In this study, we demonstrated that tiotropium inhibited methacholine-induced ECM production in HASMCs. It reduced the collagen I protein expression and the mRNA abundance of collagen I, fibronectin, and versican. Our data showed direct evidence of the influence of tiotropium on methacholine-induced ECM production in HASMCs, which expanded the understanding of the effect of muscarinic receptor antagonists on airway remodeling.
Methacholine-induced ASMCs constriction is associated with the actin cytoskeletal remodeling. In cells, β-catenin functions as a membrane-bound protein connecting E-cadherin and the cytoskeleton at the adherens junctions to stabilize cell–cell contacts and transmit mechanical forces between cells and tissues.32 In the cytoplasm, the amount and localization of β-catenin are mainly regulated by a multiprotein destruction complex containing GSK3β, adenomatous polyposis coli, axin, and casein kinase 1, which constitutively phosphorylates β-catenin, leading to subsequent proteasomal degradation.33 On the contrary, phosphorylation of GSK3β rescues β-catenin from this destructive phosphorylation, leading to cytosolic accumulation of non-phosphorylated β-catenin. Then, cytosolic non-phosphorylated β-catenin translocates into the nucleus and activates β-catenin-dependent gene transcription, including ECM genes.34 β-catenin signaling is important in repair and regeneration process and plays a crucial role in a variety of pathologies from fibrosis to cancer.31 Interestingly, there is growing evidence that mechanical forces, like stretch and compression, can activate β-catenin signaling in cells and may play an important role in airway remodeling.18 Herein, we confirmed that tiotropium reduced total β-catenin expression by inhibiting GSK3β phosphorylation. Furthermore, the expression of active β-catenin was decreased, and the translocation into the nucleus was prevented by tiotropium treatment. The effect of tiotropium on ECM production was weakened when active β-catenin was overexpressed. This means that at least tiotropium can inhibit ECM production by inactivating β-catenin signaling in ASMCs. However, considering the regulation of the membrane-bound pool of β-catenin is associated with the cytosolic pool of β-catenin, further investigation is required to elucidate whether the anti-remodeling effect of tiotropium is mediated by other biochemical processes or airway mechanics, such as mechanotransduction.35
Conclusion
Airway remodeling is a serious problem in COPD therapy because of its various manifestations and multiple mechanisms. ECM deposition in airways is one of the characteristics of airway remodeling and correlates with disease severity of COPD. Our study provided new evidence for bronchodilators in preventing airway remodeling. As airway remodeling exists at the early stage of COPD and bronchoconstriction is a common and faster process, it is necessary to consider the appropriate time to use bronchodilators in COPD treatment.
Acknowledgments
We thank Dr Yuanxiong Cheng for donating human ASMC. The study was supported by the Guangzhou Healthcare collaborative innovation major project (201604020012) and the State’s Key Project of Research and Development Plan (2017YFC1310601).
Author contributions
All authors contributed toward data analysis, drafting, and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Hirota N, Martin JG. Mechanisms of airway remodeling. Chest. 2013;144(3):1026–1032. | ||
Mauad T, Dolhnikoff M. Pathologic similarities and differences between asthma and chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2008;14(1):31–38. | ||
Hogg JC, McDonough JE, Gosselink JV, Hayashi S. What drives the peripheral lung–remodeling process in chronic obstructive pulmonary disease? Proc Am Thorac Soc. 2009;6(8):668–672. | ||
Annoni R, Lanças T, Yukimatsu Tanigawa R, et al. Extracellular matrix composition in COPD. Eur Respir J. 2012;40(6):1362–1373. | ||
van der Geld YM, van Straaten JFM, Postma DS, Timens W, Lenfant C. Role of proteoglycans in development and pathogenesis of emphysema. In: Garg HG, Roughley PJ, Hales CA, editors. Proteoglycans in Lung Disease. New York: Marcel Dekker; 2002:241–267. | ||
Jones FS, Jones PL. The tenascin family of ECM glycoproteins: structure, function, and regulation during embryonic development and tissue remodelling. Dev Dyn. 2000;218(2):235–259. | ||
Gosselink JV, Hayashi S, Elliott WM, et al. Differential expression of tissue repair genes in the pathogenesis of COPD. Am J Respir Crit Care Med. 2010;181(12):1329–1335. | ||
Deslee G, Woods JC, Moore CM, et al. Elastin expression in very severe human COPD. Eur Respir J. 2009;34(2):324–331. | ||
McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–1575. | ||
Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am J Physiol Lung Cell Mol Physiol. 2013;305(12):L912–L933. | ||
Meurs H, Dekkers BG, Maarsingh H, Halayko AJ, Zaagsma J, Gosens R. Muscarinic receptors on airway mesenchymal cells: novel findings for an ancient target. Pulm Pharmacol Ther. 2013;26(1):145–155. | ||
Kistemaker LE, Gosens R. Acetylcholine beyond bronchoconstriction roles in inflammation and remodeling. Trends Pharmacol Sci. 2015;36(3):164–171. | ||
Tschumperlin DJ, Liu F, Tager AM. Biomechanical regulation of mesenchymal cell function. Curr Opin Rheumatol. 2013;25(1):92–100. | ||
Tschumperlin DJ, Drazen JM. Chronic effects of mechanical force on airways. Annu Rev Physiol. 2006;68:563–583. | ||
Grainge CL, Lau LC, Ward JA, et al. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. 2011;364(21):2006–2015. | ||
Pera T, Zuidhof A, Valadas J, et al. Tiotropium inhibits pulmonary inflammation and remodelling in a guinea pig model of COPD. Eur Respir J. 2011;38(4):789–796. | ||
Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31(12):2714–2736. | ||
Kumawat K, Koopmans T, Gosens R. β-Catenin as a regulator and therapeutic target for asthmatic airway remodeling. Expert Opin Ther Targets. 2014;18(9):1023–1034. | ||
Karakiulakis G, Roth M. Muscarinic receptors and their antagonists in COPD: anti-inflammatory and anti remodeling effects. Mediators Inflamm. 2012;2012(8):409580. | ||
Parameswaran K, Willems-Widyastuti A, Alagappan VK, Radford K, Kranenburg AR, Sharma HS. Role of extracellular matrix and its regulators in human airway smooth muscle biology. Cell Biochem Biophys. 2006;44(1):139–146. | ||
Lambers C, Qi Y, Eleni P, et al. Extracellular matrix composition is modified by β2-agonists through cAMP in COPD. Biochem Pharmacol. 2014;91(3):400–408. | ||
Pera T, Gosens R, Lesterhuis AH, et al. Cigarette smoke and lipopolysaccharide induce a proliferative airway smooth muscle phenotype. Respir Res. 2010;11:48. | ||
Johnson SR, Knox AJ. Synthetic functions of airway smooth muscle in asthma. Trends Pharmacol Sci. 1997;18(8):288–292. | ||
Gosens R, Grainge C. Bronchoconstriction and airway biology: potential impact and therapeutic opportunities. Chest. 2015;147(3):798–803. | ||
Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res. 2006;7:73. | ||
Kistemaker LE, Oenema TA, Meurs H, Gosens R. Regulation of airway inflammation and remodeling by muscarinic receptors: perspectives on anticholinergic therapy in asthma and COPD. Life Sci. 2012;91(21–22):1126–1133. | ||
Matthiesen S, Bahulayan A, Kempkens S, et al. Muscarinic receptors mediate stimulation of human lung fibroblast proliferation. Am J Respir Cell Mol Biol. 2006;35(6):621–627. | ||
Haag S, Matthiesen S, Juergens UR, Racke K. Muscarinic receptors mediate stimulation of collagen synthesis in human lung fibroblasts. Eur Respir J. 2008;32(3):555–562. | ||
Asano K, Shikama Y, Shoji N, Hirano K, Suzaki H, Nakajima H. Tiotropium bromide inhibits TGF-β-induced MMP production from lung fibroblasts by interfering with Smad and MAPK pathways in vitro. Int J Chron Obstruct Pulmon Dis. 2010;5:277–286. | ||
Asano K, Shikama Y, Shibuy Y, et al. Suppressive activity of tiotropium bromide on matrix metalloproteinase production from lung fibroblasts in vitro. Int J Chron Obstruct Pulmon Dis. 2008;3(4):781–789. | ||
Zhou Y, Zhong NS, Li X, et al. Tiotropium in early-stage chronic obstructive pulmonary disease. N Engl J Med. 2017;377(10):923–935. | ||
Gough NR. Wnt/beta-catenin signaling in development and disease. Sci Signal. 2012;5(206):eg2. | ||
Forde JE, Dale TC. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci. 2007;64:1930–1944. | ||
Baarsma HA, Menzen MH, Halayko AJ, Meurs H, Kerstjens HA, Gosens R. β-Catenin signaling is required for TGF-β1-induced extracellular matrix production by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2011;301:L956–L965. | ||
Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316–323. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.