Back to Journals » Journal of Inflammation Research » Volume 18
TIMP4 as a Potential Complementary Biomarker and Therapeutic Target in Membranous Nephropathy: A Multi-Omics Investigation with Clinical Validation
Authors Chen Q, Wang G, Zhao R, Hu Q, Li J, Wang Y, Ran J, Huang Q, Yu G, Luo Y, Liao X ![]()
Received 7 June 2025
Accepted for publication 15 October 2025
Published 25 October 2025 Volume 2025:18 Pages 14755—14769
DOI https://doi.org/10.2147/JIR.S545422
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wenjian Li
Qingsong Chen, Gang Wang, Ruo Zhao, Qiyuan Hu, Jiayun Li, Yuting Wang, Jingyang Ran, Qi Huang, Guiquan Yu, Yanjia Luo, Xiaohui Liao
Department of Nephrology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China
Correspondence: Yanjia Luo, Email [email protected] Xiaohui Liao, Email [email protected]
Background: Despite genome-wide association studies having revealed risk loci for membranous nephropathy (MN), the functional mechanisms linking genetic variants to disease pathogenesis remain poorly understood.
Methods: We implemented a multi-stage analytical framework, integrating proteome-wide association studies (PWAS) and transcriptome-wide association studies (TWAS) to systematically prioritize MN-associated biomarkers. Genetic causality was rigorously established through summary data-based Mendelian randomization (SMR) and Bayesian colocalization analysis. Clinical validation was performed in a MN cohort. Additionally, single-cell transcriptomic profiling resolved cell-type-specific expression patterns of candidate targets. The therapeutic potential was further evaluated through drug target interaction and molecular docking.
Results: PWAS identified three proteins associated with MN (PLA2R1, LY75, TIMP4), while TWAS of whole blood and kidney cortex tissues implicated significant associations for LY75, POLR2I, and NFKB1 in MN. TIMP4 was uniquely prioritized as a high-confidence candidate through concordant evidence from PWAS significance (P = 2.67 × 10− 2), causal association by SMR (PSMR = 3.61 × 10− 4, PHEIDI = 3.49 × 10− 1), and genetic colocalization (PP.H4 = 8.01 × 10− 1). Clinically, serum TIMP4 levels were significantly elevated in MN patients versus controls (2267.1 vs 1581.4 pg/mL, P < 0.001). Single-cell analysis revealed TIMP4 expression was predominant in endothelial cells, mesangial cells, fibroblasts and proximal tubule cells. Molecular docking showed the binding between TIMP4 and potential therapeutic drugs.
Conclusion: This study suggests TIMP4 may serve as a novel complementary biomarker and potential therapeutic target in MN. Systematic integration of multi-omics and clinical data provides promising insights for future drug development and mechanistic exploration.
Keywords: membranous nephropathy, multi-omics, proteome-wide association study, transcriptome-wide association study, genome-wide association study
Introduction
Membranous nephropathy (MN), an immune-mediated glomerulopathy characterized by subepithelial immune deposits and glomerular basement membrane thickening, is the leading cause of nephrotic syndrome in adults and a major contributor of chronic kidney disease (CKD) globally.1 Among primary glomerular diseases, MN ranks second in prevalence following IgA nephropathy,2 yet epidemiological studies reveal its disproportionately rapid growth in incidence and disease burden compared to other glomerulopathies, highlighting its increasing clinical and public health urgency.3 Contemporary management of MN employs risk-stratified regimens integrating supportive care, targeted B-cell depletion and immunosuppressive therapies.4,5 Although these strategies achieve remission in 60–80% of patients, persistent challenges include treatment refractoriness and drug-related side effects.5 These limitations mainly originate from unresolved mechanistic questions. Thus, providing new insights into MN pathogenesis and identifying novel druggable targets represent critical research priorities.
MN exhibits a pronounced genetic predisposition, with genome-wide association studies (GWAS) successfully delineating multiple risk loci and advancing our understanding of its genetic architecture.6–8 Despite these breakthroughs, the causal genes and functional mechanisms underlying GWAS-identified loci remain largely unresolved, and translating genetic associations into actionable therapeutic targets or clinically relevant biomarkers has encountered limited success.9 Recent advances in high-throughput transcriptomic and proteomic profiling now enable systematic characterization of tissue-specific and circulating molecular signatures, presenting a robust foundation for linking genetic associations to mechanistic insights into MN pathogenesis. Capitalizing on these technological advancements, integrative multi-omics frameworks have been developed, combining quantitative trait locus (QTL) mapping, particularly expression QTLs (eQTLs) and protein QTLs (pQTLs), with GWAS to prioritize causal mediators.10 Two pivotal computational strategies exemplify this approach: transcriptome-wide association studies (TWAS) and proteome-wide association studies (PWAS).11,12 TWAS infers the regulatory effects of cis-acting genetic variants on gene expression, linking these effects to disease-associated GWAS signals, while PWAS extends similar principles to proteomic data, identifying proteins whose abundance is genetically correlated with MN risk. Through parallel interrogation of transcriptional and proteomic layers, TWAS and PWAS collectively illuminate how genetic variants mechanistically converge on disease-driving pathways. Although these approaches provide critical steps in candidate gene prioritization, their findings require further validation to distinguish causal effectors from coincidental associations in some instances. These methods have been complemented by summary data-based Mendelian Randomization (SMR) and Bayesian colocalization analysis, which statistically interrogate shared causal mechanisms between molecular QTLs and GWAS signals.13,14 SMR leverages genetic instruments to test causal relationships between molecular traits (eg, gene expression or protein levels) and disease risk.13 Bayesian colocalization quantifies the probability that overlapping causal variants drive both QTL and GWAS associations.14 Collectively, a multi-omics paradigm, that integrates GWAS, QTL mapping, TWAS/PWAS, and causal inference approaches, holds transformative potential to dissect the mechanistic hierarchy of genetic contributions to MN and unveil novel biomarkers and therapeutic targets.
Our study employed a multi-tiered analytical framework to systematically investigate the pathogenic mechanisms and therapeutic targets in MN. The workflow of this study is presented in Figure 1. Initially, we integrated proteomic and transcriptomic association analyses through parallel PWAS and TWAS, enabling multi-omics screening of MN-associated candidate biomarkers. Subsequently, a dual causal inference approach combining SMR and Bayesian colocalization analysis was applied to validate genetic causality of prioritized targets. The predictive findings were then rigorously validated in a MN cohort, single-cell transcriptomic profiling for cellular-resolution localization, and structure-based virtual drug screening. This comprehensive strategy established a complete evidence chain, ultimately constructing a multidimensional validation system that bridges computational insights with experimental confirmation in MN research.
 |
Figure 1 Flowchart of the study. Abbreviations: PWAS, proteome-wide association study; TWAS, transcriptome-wide association study; GWAS, genome-wide association study; MN, membranous nephropathy; SMR, summary data-based Mendelian randomization. |
Methods
MN GWAS Data
The GWAS summary statistics for MN (ebi-a-GCST010005) were obtained from the IEU Open GWAS project (https://opengwas.io/datasets/ebi-a-GCST010005), comprising 7979 individuals of European ancestry (2150 cases and 5829 controls).
Human Plasma Proteome Data
We leveraged two independent plasma proteomic datasets for cis-pQTL analyses. For PWAS, data from the Atherosclerosis Risk in Communities (ARIC) study were used, comprising 4657 plasma proteins measured in 7213 European American participants.12 For subsequent SMR and colocalization analyses, we incorporated plasma pQTL data from the deCODE Health study, which performed large-scale proteomic profiling of 4907 distinct plasma proteins in 35,559 Icelandic participants.15
Human Whole Blood and Kidney Cortex Transcriptomic Data
Whole blood and kidney cortex eQTL data were derived from the Genotype-Tissue Expression (GTEx) V8 database16 and their corresponding mRNA weights can be accessed at http://gusevlab.org/projects/fusion. Further details are publicly available on the official GTEx website (https://gtexportal.org/home/).
Proteome-Wide and Transcriptome-Wide Association Analyses
PWAS and TWAS represented advanced integrative frameworks that synergized molecular QTL data with GWAS to elucidate protein-level and gene-level associations with disease susceptibility. Both approaches followed a standardized pipeline implemented in FUSION.11,12 In the TWAS analysis, we first trained gene expression prediction models using GTEx V8 reference panels from whole blood and kidney cortex tissues, selecting optimal weights via cross-validation across multiple algorithms. We then applied these eQTL weights to impute transcriptomic profiles in the MN GWAS cohort and calculated disease-associated z-scores by integrative analysis of genetic effects and expression weights. For PWAS, an analogous workflow was executed using plasma pQTL weights derived from the ARIC cohort proteomic dataset, where genotype-protein abundance correlations were modeled to predict disease-relevant proteomic signatures. The two analytical pipelines shared core computational parameters: (1) linkage disequilibrium reference from the 1000 Genomes European population; (2) a significance threshold of false discovery rate (FDR) < 0.05; (3) default FUSION configurations for association testing.
SMR and Bayesian Colocalization Analysis
We performed SMR to test causal relationships between molecular traits and MN risk. The analysis leveraged cis-QTL as instrumental variables, with effect estimates harmonized across QTL and MN GWAS datasets. The Heterogeneity in Dependent Instruments (HEIDI) test was implemented within the SMR framework to distinguish horizontal pleiotropy from linkage disequilibrium confounding.17 A genome-wide significance threshold of SMR P < 0.05 was applied for primary inference, complemented by HEIDI P > 0.05 to exclude pleiotropy.13 To assess the probability of shared causal variations, we performed Bayesian colocalization analysis using the coloc R package. We evaluated shared causal signals by examining the posterior probabilities (PP) for five predefined hypotheses (H0-H4).14 The threshold for significance was set at PP.H4 greater than 0.8.18
Candidate Prioritization Framework
We established a three-tiered evidence framework to evaluate targets identified through PWAS and TWAS, integrating causal validation via SMR and Bayesian colocalization analysis. High-confidence targets (Tier 1) required genome-wide significance in PWAS/TWAS with dual validation through both SMR and colocalization analysis. Moderate-confidence targets (Tier 2) met PWAS/TWAS thresholds with validation by either SMR or colocalization analysis. Low-confidence targets (Tier 3) exhibited only PWAS/TWAS significance without evidence from SMR or colocalization analysis.
Clinical Validation Cohort
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of the Second Affiliated Hospital of Chongqing Medical University (approval number: 2023166). All participants provided written informed consent prior to enrollment. Between September 2023 and December 2024, we recruited 42 adult patients (age ≥ 18 years) with primary MN from the Department of Nephrology. Concurrently, 42 age- and sex-matched healthy controls were selected from individuals undergoing routine health screenings at the same institution. The diagnosis of primary MN was made in accordance with the Kidney Disease Improving Global Outcomes (KDIGO) guidelines,4 based on either kidney biopsy or serological testing for anti-phospholipase A2 receptor (PLA2R) antibody in patients with compatible clinical presentations. All participants underwent comprehensive clinical evaluation to screen for associated conditions, including autoimmune diseases, chronic infections, malignancies, and medication exposures, to rigorously exclude secondary forms of MN. To evaluate potential heterogeneity in biomarker associations, all MN patients were further stratified by serum PLA2R antibody status (positive or negative) for subgroup analyses. Exclusion criteria comprised the following conditions: (1) diabetic nephropathy, hypertensive nephrosclerosis, or other glomerular disorders; (2) systemic comorbidities such as active infections, malignancies, cardiac or hepatic insufficiency, or pulmonary hypertension; (3) neuropsychiatric disorders; (4) pregnancy or lactation status; (5) recent use of glucocorticoids or immunosuppressive agents; and (6) incomplete essential medical documentation. Patient-level demographic and clinical data were summarized if available.
Blood Sample Analysis
Peripheral blood samples were collected from all 84 participants using standard venipuncture procedures. Serum isolation was performed by clotting blood in serum separator tubes for 30 minutes at room temperature, followed by centrifugation at 1500 g for 15 min at 4 °C. The serum was aliquoted into polypropylene microcentrifuge tubes and stored at −80 °C. TIMP4 concentrations were quantified using a commercial enzyme-linked immunosorbent assay (ELISA) system (KE00198; Proteintech) according to the manufacturer’s protocols.
Exploring Target Signals at the Single-Cell Level
To investigate cell-type specificity, we analyzed single-cell RNA sequencing (scRNA-seq) data from three public datasets (GSE241302, GSE131685 and GSE171458) comprising renal specimens of 9 MN patients and 6 living donor controls.19,20 Data were processed using Seurat v5.1.0 through the following workflow: initial quality control filtered cells using thresholds of 200–6000 detected genes per cell, < 20,000 total RNA counts, and < 30% mitochondrial gene content; log-normalized counts were then computed alongside identification of 2000 highly variable genes; principal component analysis dimensionality reduction was subsequently conducted and harmonized across batches using the Harmony algorithm; shared nearest neighbor graphs were constructed from the top 20 principal components, followed by clustering at a resolution of 0.5 to delineate cell states; finally, cell-type identities were annotated based on marker genes reported in the literature.19,20
Potential Therapeutic Drugs Prediction
We performed compound enrichment analysis using the Enrichr platform (https://maayanlab.cloud/Enrichr/) to identify chemical compounds associated with the candidate targets. The platform generated drug-target association profiles containing three key parameters: enrichment scores to quantify interaction strength, statistically significant P values, and false discovery rates adjusted for multiple testing.
Molecular Docking
Compound structures retrieved from PubChem (https://pubchem.ncbi.nlm.nih.gov/) in SDF format were converted to PDB using Open Babel (v2.3.2). The receptor protein acquired from UniProt (https://www.uniprot.org/) underwent hydrogenation, charge neutralization, and conversion to PDBQT format via AutoDockTools. Molecular docking was executed in AutoDock Vina (v1.1.2). The resulting complexes were analyzed for interaction profiling using PLIP and visualized with PyMOL.
Statistical Analysis
Analyses were conducted using R (v4.4.1) and GraphPad Prism (v10.0). Continuous variables were analyzed using parametric (Student’s t-test) or non-parametric (Mann–Whitney U) methods based on normal distribution. Categorical variables were compared by χ²-tests or Fisher’s exact tests. Correlation analysis employed Pearson (normal distribution) or Spearman (non-normal distribution) coefficients. For multiple comparisons, the ANOVA or Kruskal–Wallis test with the post hoc Bonferroni test was applied. Statistical significance was defined as a two-tailed P < 0.05.
Results
Plasma PWAS Results for MN
We performed a PWAS analysis by integrating MN GWAS results with human plasma proteomes data to identify genetically predicted protein biomarkers for MN. This approach identified three genes, including PLA2R1 (P = 5.19 × 10−40), LY75 (P = 2.47 × 10−2) and TIMP4 (P = 2.67 × 10−2), whose plasma protein abundances were associated with MN (Figure 2 and Table S1).
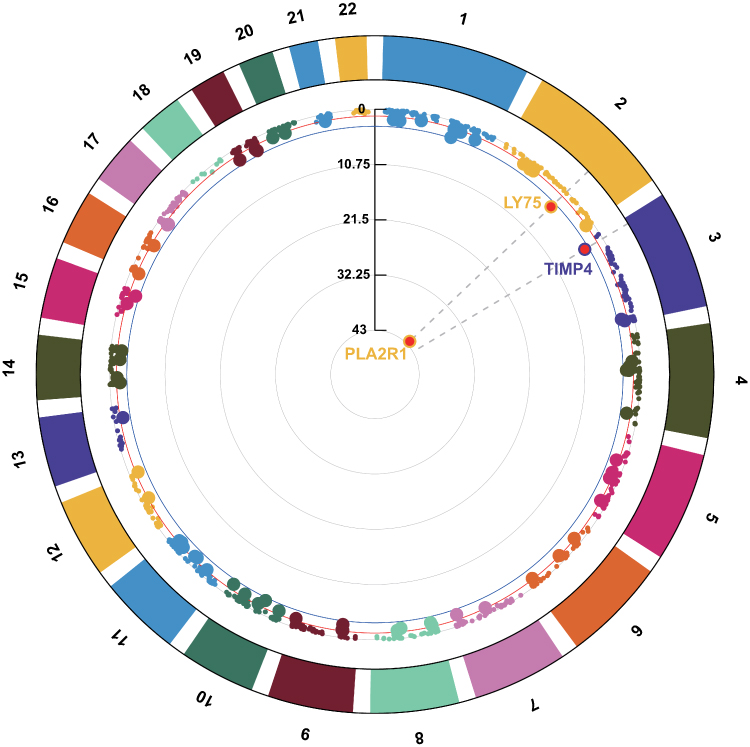 |
Figure 2 The circular Manhattan plot of PWAS results for MN. Each segment on the circular axis represents a chromosome. Dots represent individual genes, with radial position indicating -log10(p) of PWAS. The blue circle denotes FDR = 0.05. |
TWAS Results for MN
We conducted tissue-specific TWAS using GTEx V8 reference panels from whole blood and kidney cortex. Blood-derived TWAS revealed three genes showing expression-phenotype associations with MN (Table S2), including LY75 (P = 8.60×10−16), POLR2I (P = 6.81×10−4), and NFKB1 (P = 3.49×10−2). Kidney cortex analysis demonstrated strong association at LY75 (P = 1.14 × 10−21; Table S3). Interestingly, LY75 exhibited shared associations across kidney cortex and whole blood. The genomic architecture of these genes was visualized through circular Manhattan plots (Figure 3).
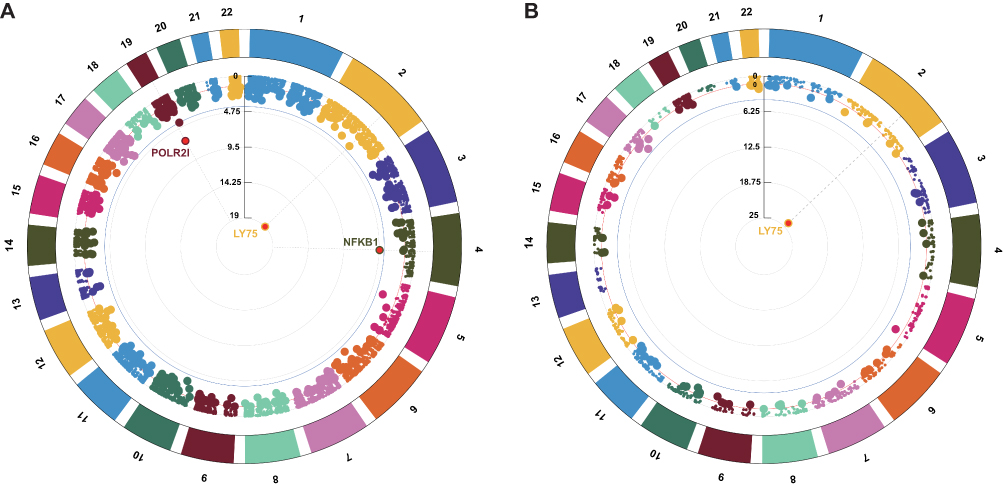 |
Figure 3 Circular Manhattan plots of TWAS results for MN. (A) Results using whole blood eQTL data. (B) Results using kidney cortex eQTL data. Each segment on the circular axis represents a chromosome. Dots represent individual genes, with radial position indicating -log10(p) of TWAS. The blue circle denotes FDR = 0.05. |
SMR Analyses for MN
To evaluate putative causal genes for MN at both transcriptional and protein levels, we performed SMR and HEIDI tests. Plasma proteome analysis using pQTL data identified TIMP4 as the only protein with consistent evidence of causality, showing significant SMR association (PSMR = 3.61 × 10−4) and no heterogeneity (PHEIDI = 3.49 × 10−1). Transcriptome-wide analyses using whole blood and kidney cortex eQTL datasets yielded divergent results. NFKB1 was not included in the SMR analysis due to the lack of genome-wide significant eQTLs in whole blood data. None of the blood-derived TWAS candidates (LY75 and POLR2I) passed both SMR significance (PSMR < 0.05) and HEIDI threshold (PHEIDI > 0.05). Although kidney cortex LY75 showed nominal SMR association (PSMR = 6.68 × 10−7), significant heterogeneity (PHEIDI = 1.80 × 10−3) suggested pleiotropy, precluding causal interpretation. Complete results are provided in Tables S4–S6.
Bayesian Colocalization Evidence
Colocalization analysis of three candidate proteins revealed shared genetic influences for two targets, TIMP4 (PP.H4 = 8.01 × 10−1) and LY75 (PP.H4 = 8.43 × 10−1), both of which exhibited concordant variant-phenotype associations between MN susceptibility and plasma protein levels (Figure 4). Transcriptome-wide colocalization analysis did not identify any genes showing evidence of shared causality at the specified threshold. Detailed colocalization probabilities for all candidates are provided in Tables S7 and S8.
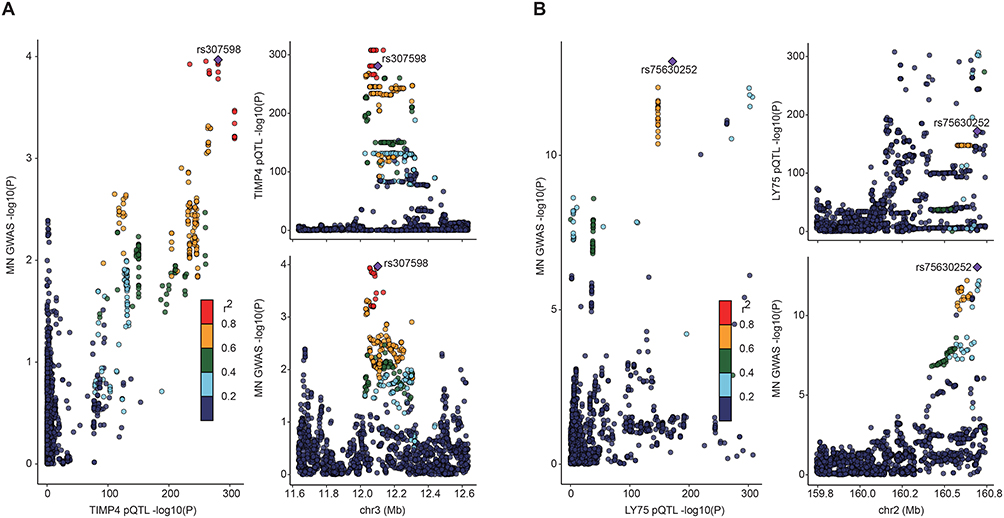 |
Figure 4 The results of Bayesian colocalization analysis. (A) TIMP4; (B) LY75. The figure illustrates meaningful colocalization results based on plasma proteins, with the instrumental variables and their chromosomal positions visualized. |
Prioritization of TIMP4 as a Causal Candidate for MN
Based on the results of our candidate prioritization framework, we identified plasma protein TIMP4 as a key signal that passed both association and causal tests and was categorized as Tier 1. The remaining proteins or genes associated with MN were classified as Tier 2 or 3 (Table S9). As a Tier 1 candidate supported by both proteomic and causal evidence, TIMP4 was prioritized for clinical validation, single-cell analysis, and molecular docking to elucidate its pathogenic role in MN.
Clinical Validation of TIMP4 in MN
Among 84 participants (median age 57.5 years; 57.1% male), demographic and clinical characteristics were balanced between 42 MN patients and 42 matched controls (Table 1). Serum TIMP4 concentrations were significantly elevated in MN patients [2267.1 (2001.0–2807.0) pg/mL] compared to controls [1581.4 (1427.8–1714.4) pg/mL] (P < 0.001; Figure 5A). TIMP4 levels showed no significant correlation with age, sex, body mass index, comorbidities (hypertension, diabetes or coronary artery disease), or disease severity markers including urine albumin-to-creatinine ratio, serum albumin, and estimated glomerular filtration rate (eGFR) in MN patients (all P > 0.05; Table 2). Subgroup analysis stratified by PLA2R antibody status revealed no significant difference in TIMP4 levels between PLA2R-positive and PLA2R-negative MN patients [2352.0 (1990.8–2901.7) vs 2246.6 (2041.9–2604.9) pg/mL; adjusted P > 0.99], and their baseline characteristics are detailed in Table S10. Notably, in the three-group comparison, both PLA2R-positive and PLA2R-negative MN patients exhibited significantly elevated TIMP4 concentrations compared to healthy controls (Figure 5B).
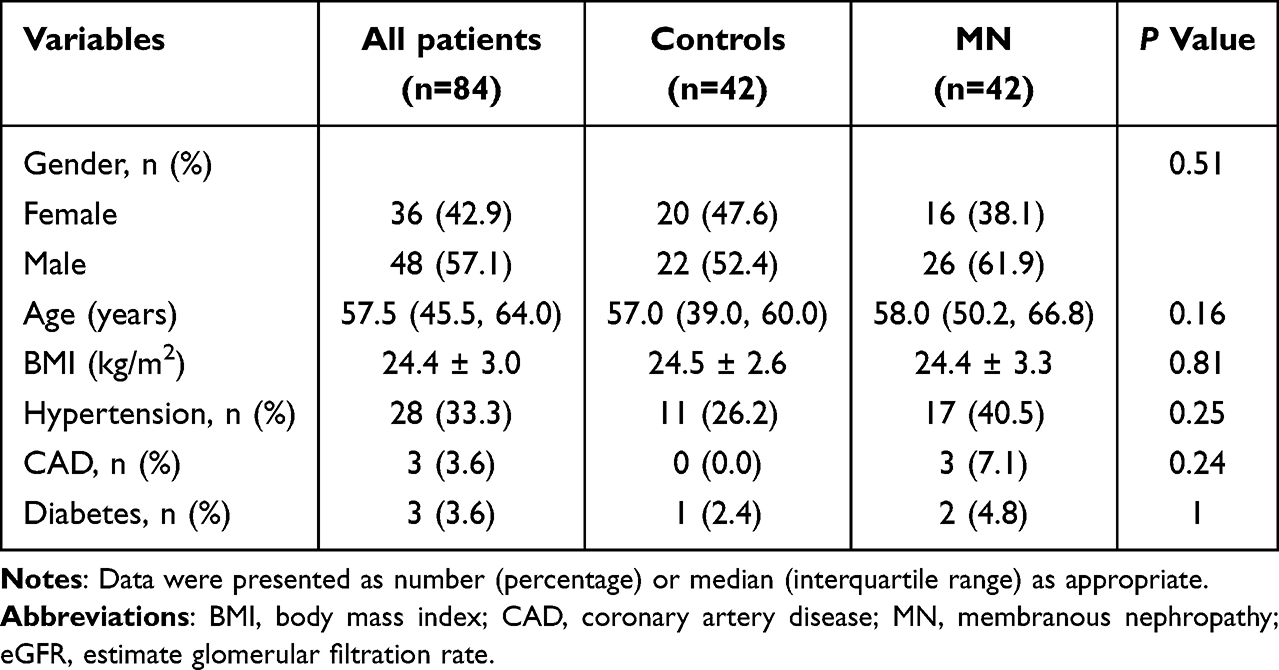 |
Table 1 Characteristics of Patients with MN and Healthy Controls |
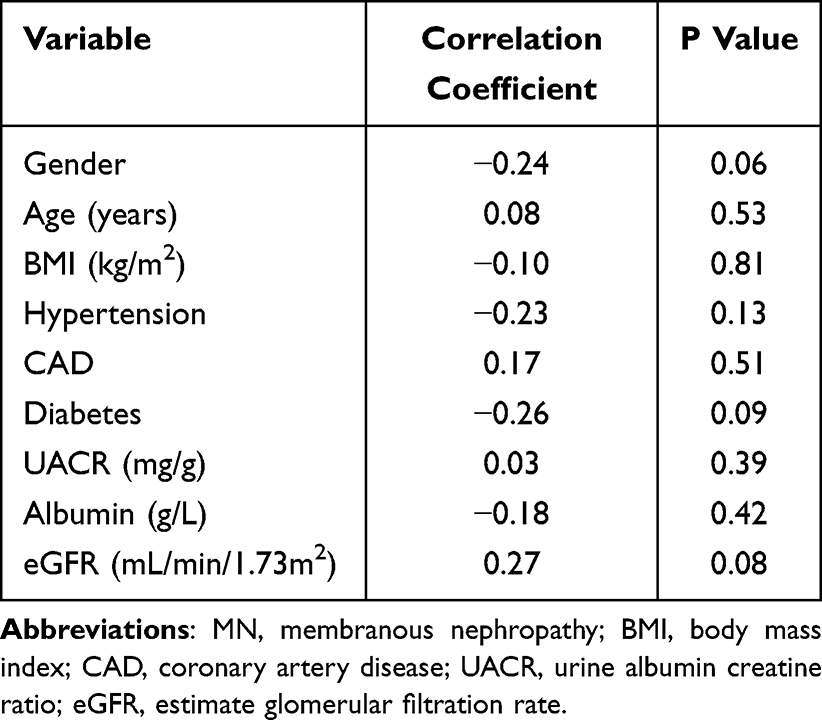 |
Table 2 Correlation Analysis of Clinical Indicators with Serum Levels of TIMP4 in MN |
 |
Figure 5 Validation of TIMP4 protein expression. (A) TIMP4 levels in MN patients vs controls. (B) TIMP4 expression across PLA2R-positive MN, PLA2R-negative MN, and control groups. ***P < 0.001; ns, not significant (P > 0.05). Abbreviation: MN, membranous nephropathy. |
Expression Pattern of TIMP4 at the Single-Cell Level
To investigate cell-type-specific enrichment of the TIMP4 gene in human kidney tissue, we performed integrated analysis of scRNA-seq datasets from 9 MN patients and 6 controls using Harmony batch correction (Figure 6A). Following integration, cellular clustering based on established marker gene expression profiles identified 18 transcriptionally distinct clusters corresponding to 13 canonical renal cell types (Figure 6B-D). Subsequent analysis revealed sparsely restricted distribution of TIMP4 expression across clusters (Figure 6E). Detectable TIMP4 transcripts were predominantly observed in endothelial cells (EC), mesangial cells (MC), fibroblasts (Fib) and proximal tubule cells (PT), with minimal detection in other lineages. This expression pattern indicates predominant localization of TIMP4 to epithelial-stromal niches rather than broad renal distribution.
 |
Figure 6 Single-cell RNA sequencing profile of kidney tissues. (A) UMAP projection after Harmony batch correction, integrating datasets across samples. (B) Eighteen distinct clusters visualized by UMAP plotting. (C) Dot plot illustrating the expression of marker genes in different cell types. (D) Annotation of 13 major cell types based on marker genes. (E) Violin plot depicting TIMP4 expression levels across different cell populations. Abbreviations: MN, membranous nephropathy; PT, proximal tubule cells; Epi, epithelial cells; PC, principal cells; LOH, loop of Henle cells; EC, endothelial cells; IC, intercalated cells; MC, mesangial cells; Per, pericyte; Pod, podocytes; Fib, fibroblasts; Myeloid, myeloid cells; NK/T, natural killer cells and T cells; BC, B cells. |
Identification of TIMP4-Targeting Compounds
Given the critical role of human proteins as potential therapeutic targets, we assessed whether TIMP4 could be a druggable target. Our computational screening pipeline identified seven compounds with significant interaction potential (P < 0.05; Table S11) through the Enrichr platform. Following acute toxicity evaluation via PubChem chemical safety profiles, four compounds were excluded due to acute toxicity. The three remaining candidates, specifically the oxidized phospholipid 1-palmitoyl-2-oxovaleroyl-sn-glycero-3-phosphorylcholine (POVPC), the histone deacetylase inhibitor scriptaid, and the β-adrenergic agonist isoproterenol, were selected for molecular docking studies. Subsequent molecular docking unveiled distinct binding patterns with TIMP4. Scriptaid exhibited the strongest interaction, with a binding energy of −5.8 kcal/mol, followed by isoproterenol (−4.4 kcal/mol) and POVPC (−4.1 kcal/mol). Detailed binding conformations are illustrated in Figure 7.
 |
Figure 7 Molecular docking results of three candidates. (A) Scriptaid; (B) Isoproterenol; (C) POVPC. |
Discussion
The development of mechanism-based therapies for MN has been limited by an incomplete understanding of disease pathogenesis. Our multi-omics framework represents the first systematic integration of proteomic, transcriptomic, scRNA-seq, and clinical evidence to explore potential molecular alterations and therapeutic targets in MN. Through PWAS, we identified PLA2R1, LY75, and TIMP4 as significantly associated with MN, while TWAS highlighted LY75, POLR2I, and NFKB1 as candidates associated with MN in both blood and kidney cortex. Crucially, SMR and Bayesian colocalization analyses provided robust evidence for a causal relationship between TIMP4 and MN, further supported by elevated serum TIMP4 levels in MN patients. Furthermore, drug prediction and molecular docking identified scriptaid, isoproterenol, and POVPC as potential compounds exhibiting stable binding configurations with TIMP4.
TIMP4, a member of the TIMP family, exhibited a heterogeneous expression pattern, with prominent enrichment in heart, kidney, pancreas, colon, brain and adipose tissue.21 Emerging evidence implicated TIMP4 in diverse pathological processes including cancer metastasis, pulmonary hypertension, and neuropsychiatric disorders. Nevertheless, its role in kidney homeostasis and diseases, particularly in humans, remained relatively unexplored. Although animal studies suggested a potential link, such as progressive elevation of renal TIMP4 in streptozotocin-induced diabetic rats at 2 and 4 months postinduction22 and increased TIMP4 levels in the renal medulla of hypertensive nephropathy models,23 direct evidence in human kidney diseases is limited. In this study, we observed elevated circulating TIMP4 levels in MN patients. Notably, no significant correlation was found between TIMP4 levels and eGFR, implying that this increase is unlikely to be a secondary effect of renal functional impairment. Given the preserved mean eGFR (85.5 mL/min/1.73m²) in our cohort, these data suggest that TIMP4 elevation may occur during early phases of kidney injury, potentially preceding significant eGFR decline. Regrettably, the present study lacks direct evidence of TIMP4 expression levels in renal tissues. Further investigations are warranted to determine the tissue origin of serum TIMP4 and its correlation with renal expression patterns. Our scRNA-seq analysis delineated a cell-type-specific expression landscape of TIMP4 in the kidney, with prominent signals in EC, MC, Fib and PT, suggesting its potential role in extracellular matrix (ECM) remodeling and fibrogenesis. This finding aligned with a prior report that localized TIMP4 predominantly to endothelial cells,24 and expanded its cellular sources to tubular epithelial cells and fibroblasts. Notably, Camp et al confirmed the presence of TIMP4 in the tubular epithelium by in situ immunolabeling in hypertensive nephropathy rats,23 suggesting that tubular TIMP4 expression may be a conserved feature across species and injury models. Future studies should isolate these cell types from disease contexts to quantify TIMP4 dynamics under stress conditions.
The molecular mechanisms of TIMP4 in MN pathogenesis are still unclear. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) are pivotal regulators of ECM homeostasis, inflammation, and cellular remodeling in kidney diseases. Among MMPs, MMP-2 and MMP-9 (collectively MMP-2/9) have been extensively investigated in kidney diseases. Accumulating evidence demonstrated the pathogenic effects of MMP-2/9 overexpression in both acute kidney injury (AKI) and CKD. In AKI models, MMP-2/9 expression was significantly upregulated and knockdown of their expression attenuated kidney injury.25,26 Similarly, in diabetic nephropathy, elevated MMP-2/9 exacerbated proteinuria, and their suppression was associated with reduced histological injury.27,28 Notably, clinical and preclinical studies consistently reported MMP-9 elevation in MN.29,30 As endogenous MMP inhibitors, TIMPs often increased concomitantly with MMPs in some kidney diseases,23,31 suggesting a potential feedback loop. This pattern was also observed in MN, where a study of 26 patients demonstrated elevated plasma TIMP-1 and MMP-2 levels.32 Our finding of elevated TIMP4 in MN aligned with this paradigm. Therefore, we hypothesize that TIMP4 upregulation may reflect a homeostatic response to MMP-driven injury, analogous to TIMP4 counteracting MMP hyperactivity in hypertensive nephropathy.23 However, determining whether the upregulation of TIMP4 is adaptive or maladaptive requires functional validation. Given its role in cellular remodeling and ECM homeostasis, elucidating the relationship between TIMP4 and established histopathological indicators of renal fibrosis, particularly interstitial fibrosis and tubular atrophy, could provide valuable insights into its mechanistic influence on MN progression. Future studies should therefore prioritize this association by targeting enrollment of patients across a broad spectrum of fibrotic severity.
Another important finding in our study was the significant elevation of TIMP4 in MN patients, regardless of PLA2R antibody status. Currently, the PLA2R antibody test serves as a valuable first-line diagnostic tool for primary MN, enabling the identification of PLA2R-related cases and reducing the need for kidney biopsies in some patients. However, approximately 20–30% of MN patients are PLA2R antibody-negative, leaving a significant diagnostic gap.33 For these individuals, kidney biopsy remains the gold standard for diagnosis, underscoring the unmet need for reliable non-invasive biomarkers. The discovery of elevated TIMP4 levels across all MN patients, independent of PLA2R antibody status, carries important clinical implications. TIMP4 may serve as a universal biomarker for MN, potentially bridging this critical diagnostic gap for PLA2R antibody-negative cases, but its clinical utility needs to be validated in larger prospective studies.
Through a three-tiered evidence framework, our results showed the genetic association of PLA2R1, LY75, POLR2I, and NFKB1 with MN. Consistent with this research, previous genomic studies have revealed risk alleles within the PLA2R1 and NFKB1 locus.6,7 As a major target antigen in MN, PLA2R1 gene variations and susceptibility to MN have been well established. Importantly, the detection of PLA2R antibody has been widely used in clinical practice for the diagnosis and prognostic assessment of MN. Nuclear Factor Kappa-B (NFKB) is a ubiquitously expressed transcription factor that orchestrates immune responses and inflammatory processes through transcriptional regulation of target genes.34 Xie et al reported NFKB1 (rs230540) as a novel genome-wide significant risk locus for MN,7 highlighting the potential role of the NFKB pathway in primary MN pathogenesis. Our identification of LY75 and POLR2I as MN-associated loci represents a novel genetic architecture, which needs further investigation.
The strengths of our work lie in several aspects. Methodologically, we established a vertically integrated analysis pipeline from discovery to validation to elucidate the pathogenic mechanisms of complex diseases and identify potential drug targets. Specifically, this study pioneered PWAS and TWAS approaches integrated with multi-omics data to investigate MN multidimensionally. The three-tiered evidence framework screening approach facilitated robust target identification and minimized false-positive associations. In addition, scRNA-seq analysis uncovered cell type-specific gene expression, providing valuable insights into the cellular mechanisms of MN. From a clinical perspective, the validation in MN cohorts not only confirmed the robustness of multi-omics screening but also highlighted the clinical translational value of the research.
However, several limitations should be acknowledged. First, the pooled GWAS data derived from European ancestry may limit the broader applicability of these results to other populations, though employing a validation cohort of Chinese participants partially addressed this limitation. Second, the single-center, cross-sectional design lacking external validation and with a limited sample size not only hampers our ability to analyze temporal dynamics and the impact of TIMP4 on MN progression but also limits the generalizability of our findings. Future multi-center, large-scale prospective studies with longitudinal data are needed to establish the dynamics and prognostic value of serum TIMP4 in MN. Third, due to the limited number of cases available, analyses of the PLA2R antibody-negative subgroup should be considered exploratory and their findings warrant cautious interpretation. Fourth, since antibodies against other antigens (THSD7A, NELL1, etc.) were not routinely tested, whether additional subtypes exist among PLA2R-negative patients remains unclear. Further research incorporating expanded antibody panels is warranted to improve the subclassification of MN. Fifth, our exclusive focus on MN precludes comparisons with other glomerulopathies or across disease phases. Future work should therefore characterize TIMP4 expression across diverse glomerular diseases, including proliferative and nonproliferative subtypes at various stages, to evaluate its potential clinical utility for differential diagnosis. Finally, the computational prediction of scriptaid, isoproterenol, and POVPC as TIMP4-targeting compounds remains limited by the lack of experimental validation.
Conclusion
The lack of correlation between TIMP4 and anti-PLA2R titers, together with the known role of TIMP4 in ECM regulation, supports the interpretation of TIMP4 as a likely downstream marker of renal injury and remodeling rather than a primary immunologic driver of MN. Accordingly, TIMP4 may provide complementary clinical value, particularly in PLA2R-negative disease for risk stratification and monitoring, while its diagnostic specificity requires multicenter validation against other glomerular diseases.
Data Sharing Statement
The research data are available from the corresponding author upon reasonable request.
Research Ethics and Consent
The Institutional Review Board of the Second Affiliated Hospital of Chongqing Medical University reviewed and approved the study. All participants provided written informed consent prior to enrollment.
Acknowledgments
We are grateful to the researchers who provided the original data. Figure 1 was created by Figdraw (www.figdraw.com).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81873604), Chongqing Medical Scientific Research project (Joint project of Chongqing Health Commission and Science and Technology Bureau) (2022GDRC005), Chongqing Natural Science Foundation (CSTB2022NSCQ-MSX0984) and the CQMU Program for Youth lnnovation in Future Medicine (W0173).
Disclosure
The authors declare no competing financial or non-financial interests.
References
1. Ronco P, Beck L, Debiec H, et al. Membranous nephropathy. Nat Rev Dis Primers. 2021;7(1):69. doi:10.1038/s41572-021-00303-z
2. Nieto-Gañán I, Iturrieta-Zuazo I, Rita C, Carrasco-Sayalero Á. Revisiting immunological and clinical aspects of membranous nephropathy. Clin Immunol. 2022;237:108976. doi:10.1016/j.clim.2022.108976
3. Hu R, Quan S, Wang Y, et al. Spectrum of biopsy proven renal diseases in Central China: a 10-year retrospective study based on 34,630 cases. Sci Rep. 2020;10(1):10994. doi:10.1038/s41598-020-67910-w
4. Rovin BH, Adler SG, Barratt J, et al. Executive summary of the KDIGO 2021 guideline for the management of glomerular diseases. Kidney Int. 2021;100(4):753–779. doi:10.1016/j.kint.2021.05.015
5. Deng L, Xu G. Update on the application of monoclonal antibody therapy in primary membranous nephropathy. Drugs. 2023;83(6):507–530. doi:10.1007/s40265-023-01855-y
6. Stanescu HC, Arcos-Burgos M, Medlar A, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med. 2011;364(7):616–626. doi:10.1056/NEJMoa1009742
7. Xie J, Liu L, Mladkova N, et al. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat Commun. 2020;11(1):1600. doi:10.1038/s41467-020-15383-w
8. Berchtold L, Letouzé E, Alexander MP, et al. HLA-D and PLA2R1 risk alleles associate with recurrent primary membranous nephropathy in kidney transplant recipients. Kidney Int. 2021;99(3):671–685. doi:10.1016/j.kint.2020.08.007
9. Wang M, Yang J, Fang X, Lin W, Yang Y. Membranous nephropathy: pathogenesis and treatments. MedComm. 2024;5(7):e614. doi:10.1002/mco2.614
10. Battle A, Khan Z, Wang SH, et al. Genomic variation. Impact of regulatory variation from RNA to protein. Science. 2015;347(6222):664–667. doi:10.1126/science.1260793
11. Gusev A, Ko A, Shi H, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245–252. doi:10.1038/ng.3506
12. Zhang J, Dutta D, Köttgen A, et al. Plasma proteome analyses in individuals of European and African ancestry identify cis-pQTLs and models for proteome-wide association studies. Nat Genet. 2022;54(5):593–602. doi:10.1038/s41588-022-01051-w
13. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi:10.1038/ng.3538
14. Giambartolomei C, Zhenli liu J, Zhang W, et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics. 2018;34(15):2538–2545. doi:10.1093/bioinformatics/bty147
15. Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712–1721. doi:10.1038/s41588-021-00978-w
16. GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–1330. doi:10.1126/science.aaz1776
17. Wu Y, Zeng J, Zhang F, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun. 2018;9(1):918. doi:10.1038/s41467-018-03371-0
18. Sun Z, Yun Z, Lin J, et al. Comprehensive mendelian randomization analysis of plasma proteomics to identify new therapeutic targets for the treatment of coronary heart disease and myocardial infarction. J Transl Med. 2024;22(1):404. doi:10.1186/s12967-024-05178-8
19. Xu J, Shen C, Lin W, et al. Single-cell profiling reveals transcriptional signatures and cell-cell crosstalk in anti-PLA2R positive idiopathic membranous nephropathy patients. Front Immunol. 2021;12:683330. doi:10.3389/fimmu.2021.683330
20. Shi M, Wang Y, Zhang H, et al. Single-cell RNA sequencing shows the immune cell landscape in the kidneys of patients with idiopathic membranous nephropathy. Front Immunol. 2023;14:1203062. doi:10.3389/fimmu.2023.1203062
21. Melendez-Zajgla J, Del Pozo L, Ceballos G, Maldonado V. Tissue inhibitor of metalloproteinases-4. The road less traveled. Mol Cancer. 2008;7:85. doi:10.1186/1476-4598-7-85
22. Singh RM, Howarth FC, Adeghate E, Bidasee K, Singh J, Waqar T. Type 1 diabetes mellitus induces structural changes and molecular remodelling in the rat kidney. Mol Cell Biochem. 2018;449(1–2):9–25. doi:10.1007/s11010-018-3338-4
23. Camp TM, Smiley LM, Hayden MR, Tyagi SC. Mechanism of matrix accumulation and glomerulosclerosis in spontaneously hypertensive rats. J Hypertens. 2003;21(9):1719–1727. doi:10.1097/00004872-200309000-00022
24. Peeney D, Fan Y, Gurung S, et al. Whole organism profiling of the Timp gene family. Matrix Biol Plus. 2023;18:100132. doi:10.1016/j.mbplus.2023.100132
25. Kunugi S, Shimizu A, Kuwahara N, et al. Inhibition of matrix metalloproteinases reduces ischemia-reperfusion acute kidney injury. Lab Invest. 2011;91(2):170–180. doi:10.1038/labinvest.2010.174
26. Lee SY, Hörbelt M, Mang HE, et al. MMP-9 gene deletion mitigates microvascular loss in a model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2011;301(1):F101–109. doi:10.1152/ajprenal.00445.2010
27. Ramnath RD, Butler MJ, Newman G, et al. Blocking matrix metalloproteinase-mediated syndecan-4 shedding restores the endothelial glycocalyx and glomerular filtration barrier function in early diabetic kidney disease. Kidney Int. 2020;97(5):951–965. doi:10.1016/j.kint.2019.09.035
28. Hirata T, Fan F, Fan L, et al. Knockout of matrix metalloproteinase 2 opposes hypertension- and diabetes-induced nephropathy. J Cardiovasc Pharmacol. 2023;82(6):445–457. doi:10.1097/fjc.0000000000001473
29. McMillan JI, Riordan JW, Couser WG, Pollock AS, Lovett DH. Characterization of a glomerular epithelial cell metalloproteinase as matrix metalloproteinase-9 with enhanced expression in a model of membranous nephropathy. J Clin Invest. 1996;97(4):1094–1101. doi:10.1172/jci118502
30. Zakiyanov O, Kalousová M, Kratochvilová M, Kríha V, Zima T, Tesar V. Changes in levels of matrix metalloproteinase-2 and −9, pregnancy-associated plasma protein-A in patients with various nephropathies. J Nephrol. 2013;26(3):502–509. doi:10.5301/jn.5000136
31. Kassiri Z, Oudit GY, Kandalam V, et al. Loss of TIMP3 enhances interstitial nephritis and fibrosis. J Am Soc Nephrol. 2009;20(6):1223–1235. doi:10.1681/asn.2008050492
32. Bauvois B, Mothu N, Nguyen J, Nguyen-Khoa T, Nöel LH, Jungers P. Specific changes in plasma concentrations of matrix metalloproteinase-2 and −9, TIMP-1 and TGF-beta1 in patients with distinct types of primary glomerulonephritis. Nephrol Dial Transplant. 2007;22(4):1115–1122. doi:10.1093/ndt/gfl743
33. Ragy O, Abass W, Kanigicherla DAK, et al. PLA2R autoantibodies, a multifaceted biomarker in nephrotic syndrome and membranous nephropathy. Nephrol Dial Transplant. 2025;40(8):1458–1469. doi:10.1093/ndt/gfaf012
34. Guo Q, Jin Y, Chen X, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther. 2024;9(1):53. doi:10.1038/s41392-024-01757-9
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.