Back to Journals » Drug Design, Development and Therapy » Volume 10
Ticagrelor promotes atherosclerotic plaque stability in a mouse model of advanced atherosclerosis
Authors Preusch MR, Rusnak J, Staudacher K, Mogler C ![]() , Uhlmann L, Sievers P, Bea F, Katus HA, Blessing E, Staudacher I
, Uhlmann L, Sievers P, Bea F, Katus HA, Blessing E, Staudacher I
Received 3 February 2016
Accepted for publication 20 April 2016
Published 26 August 2016 Volume 2016:10 Pages 2691—2699
DOI https://doi.org/10.2147/DDDT.S105718
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Michael R Preusch,1 Jonas Rusnak,1 Kathrin Staudacher,2 Carolin Mogler,3 Lorenz Uhlmann,4 Philipp Sievers,1 Florian Bea,1 Hugo A Katus,1 Erwin Blessing,1 Ingo Staudacher1
1Department of Internal Medicine III, 2Department of Neonatology, 3Department of Pathology, 4Institute of Medical Biometry and Informatics, University of Heidelberg, Heidelberg, Germany
Objective: There is increasing evidence supporting the role of platelets in atherosclerotic vascular disease. The G-protein-coupled receptor P2Y12 is a central mediator of platelet activation and aggregation but has also been linked to platelet-independent vascular disease. Ticagrelor is an oral P2Y12 antagonist that is used as a standard treatment in patients after acute myocardial infarction. However, the effects of ticagrelor on advanced atherosclerosis have not been investigated.
Materials and methods: Twenty-week-old apolipoprotein-E-deficient mice received standard chow or standard chow supplemented with 0.15% ticagrelor (approximately 270 mg/kg/day) for 25 weeks. The lesion area was evaluated in the aortic sinus by Movat’s pentachrome staining and lesion composition, thickness of the fibrous cap, and size of the necrotic core evaluated by morphometry. RAW 264.7 macrophages were serum starved and treated with ticagrelor in vitro for the detection and quantification of apoptosis. In addition, oxLDL uptake in RAW 264.7 macrophages was evaluated.
Results: A trend toward the reduction of total lesion size was detected. However, data did not reach the levels of significance (control, n=11, 565,881 µm2 [interquartile range {IQR} 454,778–603,925 µm2] versus ticagrelor, n=13, 462,595 µm2 [IQR 379,740–546,037 µm2]; P=0.1). A significant reduction in the relative area of the necrotic core (control, n=11, 0.46 [IQR 0.4–0.51] versus ticagrelor, n=13, 0.34 [IQR 0.31–0.39]; P=0.008), and a significant increase in fibrous caps thickness (control, n=11, 3.7 µm [IQR 3.4–4.2 µm] versus ticagrelor, n=13, 4.7 [IQR 4.3–5.5 µm], P=0.04) were seen in ticagrelor-treated mice. In vitro studies demonstrated a reduction in apoptotic RAW 264.7 macrophages (control 0.07±0.03 versus ticagrelor 0.03±0.03; P=0.0002) when incubated with ticagrelor. Uptake of oxLDL in RAW 264.7 was significantly reduced when treated with ticagrelor (control 9.2 [IQR 5.3–12.9] versus ticagrelor 6.4 [IQR 2.5–9.5], P=0.02).
Conclusion: The present study demonstrates for the first time a plaque-stabilizing effect of ticagrelor in a model of advanced vascular disease, potentially induced by a reduction of oxLDL uptake or an inhibition of apoptosis as seen in vitro.
Keywords: atherosclerosis, inflammation, apoptosis, ticagrelor, platelets, P2Y12, oxLDL, apoE mouse
Introduction
Beyond hemostasis, platelets are critically involved in the initiation of atherosclerosis, but the effects of platelet activation throughout the atherosclerotic process within the already established lesions are unclear. In vitro studies demonstrate that platelets adhere to intact but activated endothelium as an initial step in atherogenesis.1 Platelet-induced alterations of the endothelium support chemotaxis, adhesion, and transmigration of monocytes into vessel walls, a key step in atherosclerosis.2–4 Animal studies further confirm the critical role of platelet adhesion in atherosclerotic lesion formation and reveal anti-atherosclerotic properties, for example, of acetylsalicylic acid.5,6 Recently, the junctional adhesion molecule A has been identified to increase platelet inflammation leading to an increase in atherosclerotic lesion development.7 The G-protein-coupled receptor, P2Y12, on platelets is implicated in cardiovascular disease, and inhibition of the P2Y12 receptor is a well-validated and effective therapeutic target for the prevention of myocardial infarction and ischemic stroke.8–12 Therefore, European Society of Cardiology (ESC) guidelines recommend ticagrelor, in addition to acetylsalicylic acid, to treat acute coronary syndrome.10,11 Evidence further supports an anti-inflammatory role for P2Y12 receptor inhibition.13–15 However, whether potential anti-atherosclerotic effects are mediated due to an inhibition of platelet activation or anti-inflammatory effects elsewhere in the vascular wall is not clear.
It has recently been demonstrated that inhibition of P2Y12 on vessel walls attenuates early atherosclerotic lesion progression, whereas platelet P2Y12 inhibition had no effect.16 In line with these findings, Li et al17 documented a reduction in atherosclerotic lesion size in apoE−/−/P2Y12−/− double knockout mice.17
Collectively, these data show that P2Y12 inhibition may exert further beneficial effects in addition to inhibition of platelet aggregation.
The aim of this study is to evaluate the effectiveness of P2Y12 inhibition by ticagrelor treatment in a clinically relevant model of atherosclerosis. Clinically, atherosclerosis is detected and treated in late stages. To mirror this phenotype, we set up an animal model that displays advanced atherosclerosis. This setup is supported by clinical data showing beneficial effects of long-term treatment with ticagrelor in patients with a prior myocardial infarction.12
Materials and methods
Animals and drug administration
Dose finding
Female apoE−/− mice with a C57BL/6 background were housed in the animal care facility at the University of Heidelberg in a temperature-controlled room on a 12-hour light cycle. The mice were randomized, at 8 weeks of age, to receive standard chow supplemented with ticagrelor (AstraZeneca, Gothenburg, Sweden), 0.05% (90 mg/kg/day) (n=3) or 0.1% (180 mg/kg/day) (n=3). Blood samples were collected on several days at different times of the day. Plasma was prepared within 30 minutes of blood sampling by centrifugation at 1,500× g for 10 minutes at 4°C. The plasma was transferred into tubes and stored at −20°C within 1 hour of sample collection. Plasma concentration of ticagrelor was determined by protein precipitation and liquid chromatography mass spectrometry as described previously.18
Main study
At 20 weeks of age, the female apoE−/− mice exhibited advanced atherosclerotic lesions within the aortic sinus. Twenty-five, 20-week-old female mice were randomized to receive chow supplemented with ticagrelor (0.15%, 270 mg/kg/day, n=13) or standard chow (control, n=12) (chow prepared by Altromin, Lage, Germany). The dose of ticagrelor was based on the clinically relevant plasma exposure and was modulated on the basis of the data of our prior dose-finding study. All animal procedures were approved by the Federal Animal Care and Use Committee of the Regierungspraesidium Karlsruhe. All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the Guidelines of the Animal Welfare and approved by the local government (Regierungspraesidium Karlsruhe).
Animal sacrifice and preparation of tissues
Mice were sacrificed after 25 weeks of treatment (at 45 weeks of age) by deep sedation (Ketamine (CP Pharma, Burgdorf, Germany)/Xylazine (Alvetra, Neumuenster, Germany), intraperitoneal) and exsanguination while blood was collected from the inferior vena cava. The animals were perfused via left ventricle with 10 mL phosphate-buffered saline solution followed by resection of the aorta. Then, the mice were perfused with 4% buffered formalin for paraffin sections of the aortic root. The heart was dissected from each animal, and the aortic sinuses were embedded in paraffin followed by serial sectioning (5 μm). Every third section was stained with a modified Movat’s pentachrome stain.19
Determination of plasma lipid concentration
Total plasma cholesterol and low-density lipoprotein (LDL) levels were enzymatically determined; Siemens Healthcare Diagnostics GmbH, Eschborn, Germany) at the time of sacrifice.
Evaluation of lesion size and lesion composition
Cross-sectional area of Movat’s pentachrome stained sections was determined by using computer-assisted morphometry (Image J; Media Cybernetics, Inc., Rockville, MD, USA). Therefore, all the parts of the lesions of a section were measured and the median was calculated for each animal. Results of lesion sizes are reported as median and interquartile range (IQR) of cross-sectional lesions per group (data expressed in μm2). We further evaluated each section for characteristic features of plaque morphology/composition: thickness of the fibrous cap (data given in μm) and size of the necrotic core (ratio of mean size of necrotic core/mean lesion area) by morphometry.
Immunohistochemistry
Tissue sections of the aortic sinus adjacent to the sites of maximum lesion area were dewaxed and rehydrated. Endogenous peroxidase activity was inhibited by incubation with peroxoblock (Zytomed Systems GmbH, Berlin, Germany). Monocytes/macrophages were detected by using a monoclonal rat anti-mouse Mac-2-antibody (WAK-Chemie Medical GmbH, Steinbach, Germany). Anti-Mac-2 or isotype control was incubated for 1.5 hours at room temperature. The sections were then incubated with the biotinylated secondary antibodies for 30 minutes, rinsed three times with phosphate-buffered saline, and incubated for 10 minutes with streptavidin at room temperature. Acetate buffer, 3-amino-9-ethylcarbazole (AEC)-chromogen substrate (Zytomed Systems GmbH) was used for visualization. In order to detect apoptosis, “In Situ Cell Death Detection Kit TMR red” was used by the protocol of the manufacturer (Roche Diagnostics GmbH Mannheim, Germany). For quantification of staining we used computer-assisted morphometry. The results of Mac-2 staining is presented as ration extend stained area/total area of the lesion. Results of apoptosis are given as the ratio of Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive areas/total lesion area (Image J; Media Cybernetics, Inc.).
Cell culture
A murine macrophage cell line, RAW 264.7 cells (ATCC, Manassas, VA, USA), was grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco BRL, Karlsruhe, Germany) supplemented with 10% heat-inactivated fetal calf serum, glutamine (100 U/mL), penicillin (100 U/mL), and streptomycin (100 mg/mL). For incubation, cells were plated in a six-well plate.
Detection and quantification of apoptosis in RAW 264.7 cells
In order to induce apoptosis, cells were washed three times with phosphate-buffered solution and were grown in a serum-free medium (DMEM) for 8 hours. At the beginning of the experiment, ticagrelor (dissolved in Dimethylsulfoxid) was added at a final concentration of 0.1 μM. Following 8 hours of serum starvation, the cells were stained and fixed according to the manufacturers’ protocol (In Situ Cell Death Detection Kit; Roche Diagnostics). Positive staining was determined using computer-assisted morphometry. Data are presented as the ratio of TUNEL-positive cells per total amount of cells (Image J; Media Cybernetics, Inc.).
OxLDL uptake in RAW 264.7 cells
Cells were pre-treated with ticagrelor at a final concentration of 0.1 μM (dissolved in Dimethylsulfoxid) for 4 hours. OxLDL (Hycultec, Beutelsbach, Germany) at a dose of 80 μg/mL was added for another 20 hours. Cells were washed with phosphate-buffered solution and fixed, followed by an oil-red-o staining. A representative picture out of the plate was taken, and positive staining was determined using computer-assisted morphometry. Results of the two groups are presented as the ratio of oil-red-o positive granula per cell (Image J; Media Cybernetics, Inc.).
Statistical analysis
Ticagrelor levels, lipid levels, and body weight are expressed as mean ± standard error of the mean. Differences between mean values are determined with the two-tailed unpaired Student’s t-test. For analyses of the lesion size Mann-Whitney U-test is performed and data are given as median and IQR. For the analysis of plaque morphometry and areas of positive staining, the groups were compared using the Mann–Whitney U-test as the data could not be assumed to be normally distributed. P-values <0.05 were considered as statistically significant. As this was an exploratory analysis, no adjustments for multiple testing were performed. Statistical analyses were done using GraphPad Prism version 6.00 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Dose finding
Ticagrelor levels
A dose-dependent increase in ticagrelor plasma levels was observed. Ticagrelor plasma exposure in mice receiving 0.05% (90 mg/kg/day) and 0.1% (180 mg/kg/day) ticagrelor was 0.28±0.2 μM and 0.99±0.7 μM (P=0.0005; Figure S1), respectively.
Animals
There was no relevant mortality during the treatment period (n=1, control group). For lesion evaluation, 24 mice were included.
Effect on plasma lipid level and body weight
At the time of necropsy, there were no significant differences in total plasma cholesterol, LDL, or body weight between the groups (Table 1).
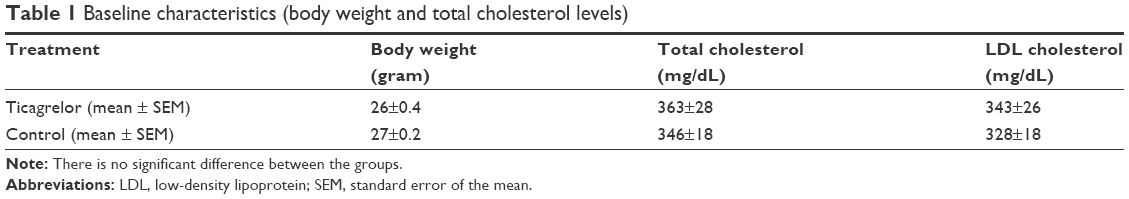 | Table 1 Baseline characteristics (body weight and total cholesterol levels) |
Effect on maximum lesion area within the aortic sinus
Ticagrelor treatment for 25 weeks (from 20 to 45 weeks of age) induced a numerical reduction in lesion size, which was not statistically significant (control, n=11, 565,881 [IQR 454,778–603,925 μm2] versus ticagrelor, n=13, 462,595 [IQR 379,740–546,037 μm2], P=0.1; Figures 1A and 2A).
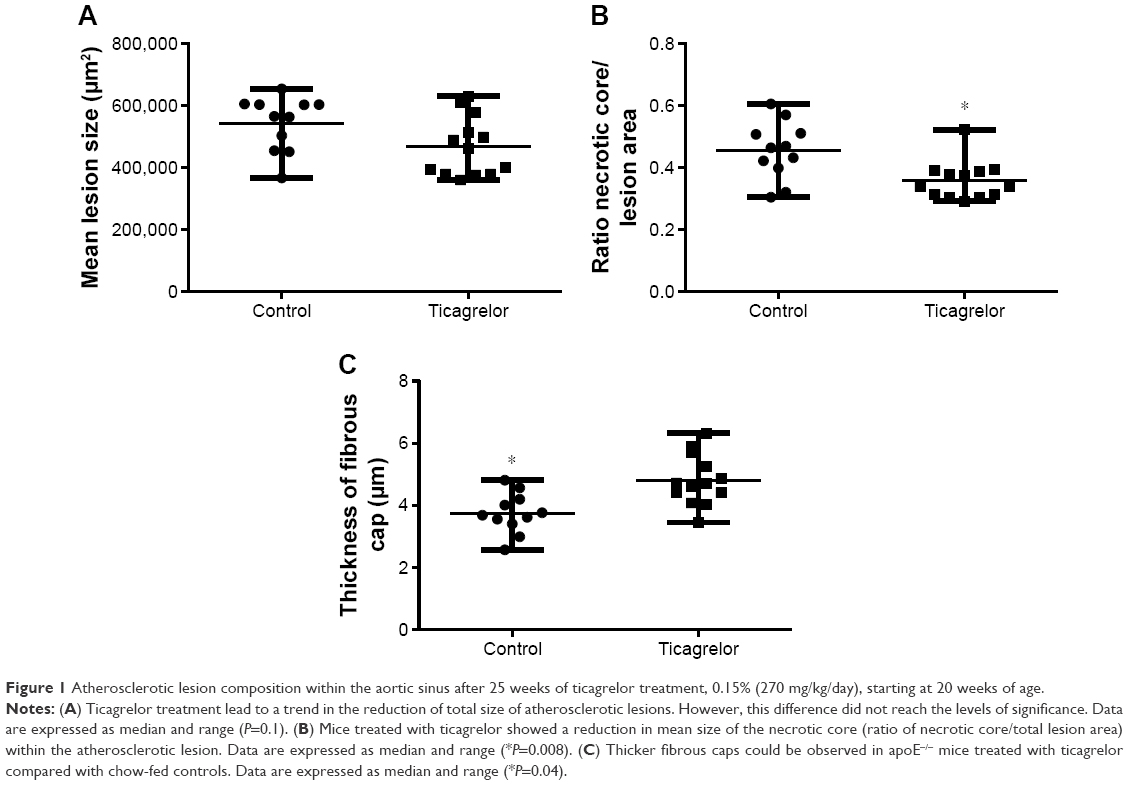 | Figure 1 Atherosclerotic lesion composition within the aortic sinus after 25 weeks of ticagrelor treatment, 0.15% (270 mg/kg/day), starting at 20 weeks of age. |
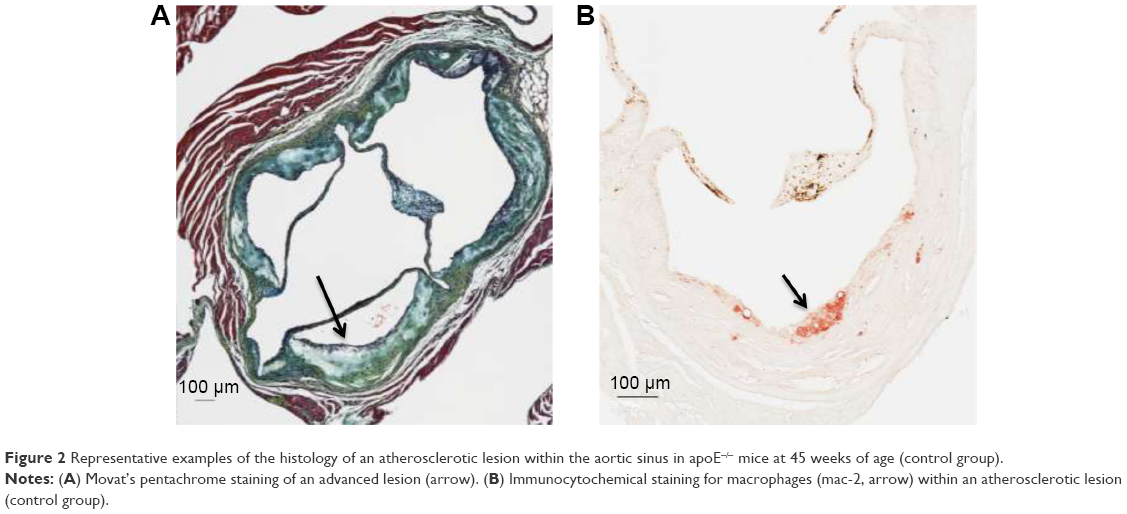 | Figure 2 Representative examples of the histology of an atherosclerotic lesion within the aortic sinus in apoE−/− mice at 45 weeks of age (control group). |
Features of lesion composition
Size of the necrotic core within the aortic sinus
Ticagrelor treatment resulted in a reduction of the average necrotic core area within the aortic sinus, presented as the ratio of mean necrotic core area/mean lesion area (control, n=11, 0.46 [IQR 0.4–0.51] versus ticagrelor, n=13, 0.34 [IQR 0.31–0.39], P=0.008; Figure 1B).
Thickness of fibrous caps
Ticagrelor treatment resulted in significantly thicker fibrous caps compared with controls (control, n=11, 3.7 [IQR 3.4–4.2 μm] versus ticagrelor, n=13, 4.7 [IQR 4.3–5.5 μm], P=0.04; Figure 1C).
Macrophage content within the aortic sinus
No difference was seen in the area of positive staining for macrophages within the atherosclerotic lesions, which was expressed as the ratio of positive staining/total lesion (control, n=5, 0.04 [IQR 0.007–0.05] versus ticagrelor, n=13, 0.02 [IQR 0.02–0.04]; P=0.9; Figure 2B).
Effect on apoptosis – in vivo
There was no difference in apoptotic cells within the atherosclerotic plaque assessed by TUNEL staining (control 0.03 [IQR 0.01–0.05] versus ticagrelor 0.03 [IQR 0.01–0.04], P=0.6).
Effect on apoptosis – in vitro
Apoptosis in RAW 264.7 macrophages was induced via serum starvation for 8 hours leading to a 1.5-fold increase in the ratio of TUNEL-positive cells/total amount of cells (0.03±0.05 versus 0.04±0.07, P=0.03; Figure S2). Presence of 0.1 μM ticagrelor during the 8-hour serum starvation resulted in the reduction of apoptotic cells compared with controls (0.07±0.2 versus 0.03±0.2; P=0.0002; Figures 3A and 4).
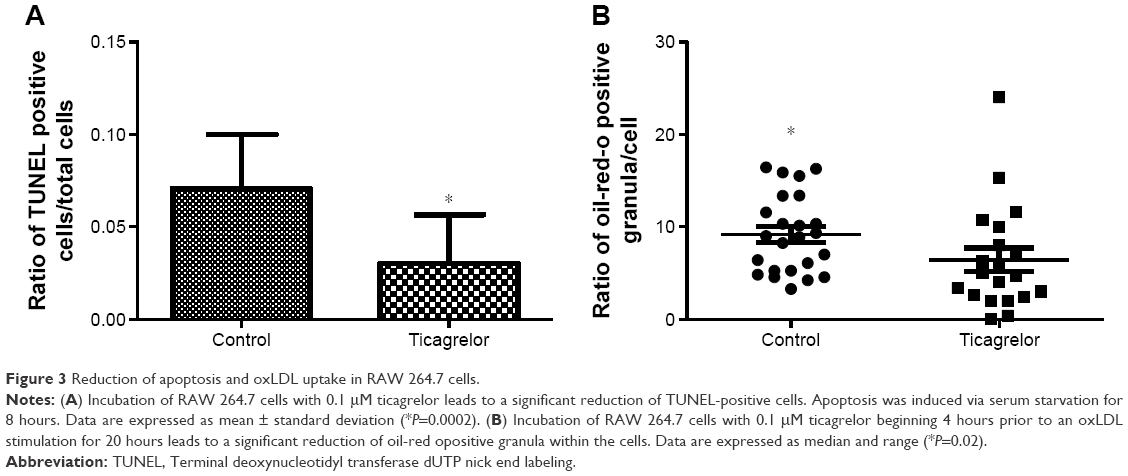 | Figure 3 Reduction of apoptosis and oxLDL uptake in RAW 264.7 cells. |
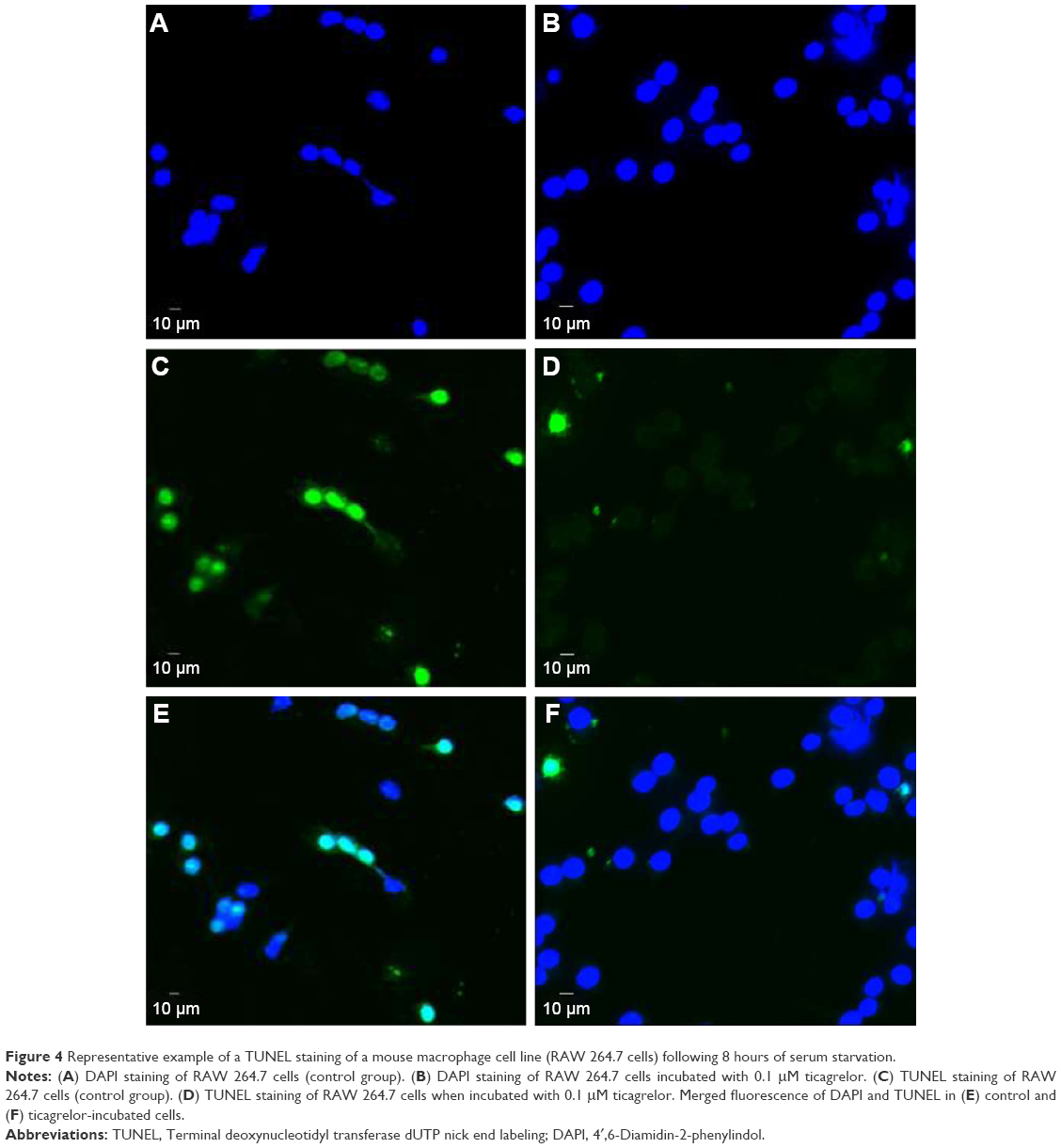 | Figure 4 Representative example of a TUNEL staining of a mouse macrophage cell line (RAW 264.7 cells) following 8 hours of serum starvation. |
Ticagrelor inhibits oxLDL-uptake in RAW 264.7 macrophages
Cells, pre-treated with ticagrelor and stimulated with oxLDL showed a significant reduction in oxLDL-uptake compared with controls, which is given as the ratio of stained granula/cell (control 9.2 [IQR 5.3–12.9] versus ticagrelor 6.4 [IQR 2.5–9.5]; P=0.02; Figures 3B and S3A and B).
Discussion
Increasing evidence supports the role of platelet activation which leads to vascular disease. Platelet interaction with the endothelium initiates atherogenesis, which leads to the recruitment of leukocyte and the release of pro-inflammatory mediators.1–3,20–22 These effects are mediated, in part, by the Adenosine diphosphate-activated G-protein-coupled receptor P2Y12.4 Inhibition of the P2Y12 receptor, by the antagonist ticagrelor, leads to several anti-inflammatory effects that are not necessarily mediated by the inhibition of platelet activation or aggregation. It sounds reasonable that there are also other mechanisms within the vascular wall since P2Y12 is also expressed on vascular smooth muscle cells and can be upregulated within the atherosclerotic plaque leading to an induction of different inflammatory and pro-atherogenic mechanisms.23 Furthermore, a reduction of serum C-reactive protein or CD 40 ligand has been shown in atherothrombotic patients when treated with P2Y12 anatgonists.24 Recently, West et al16 demonstrated a role for vessel wall that expressed P2Y12 in the initiation of atherosclerosis. However, no inhibition was seen with platelet-derived P2Y12 inhibition, leading to the conclusion that in early disease pro-atherogenic effects are mediated only by vessel P2Y12 receptors. Furthermore, Li et al (2012)17 showed an atherosclerotic lesion reduction in apoE−/−/P2Y12−/− mice at 25 weeks of age after 12 weeks on a high-fat diet.17
Considering clinical conditions, nearly all the cardiovascular patients have established atherosclerosis. Therefore, it is important to focus on the effects of P2Y12 inhibition in advanced vascular disease. The experimental design employed in this study models advanced stages of atherosclerosis, in line with clinical presentation. A trend toward the reduction in total atherosclerotic lesion size was seen after 25 weeks of ticagrelor treatment. However, this result was not statistically significant, which might also be due to the use of standard mouse chow instead of a high-fat “western type” diet. Standard chow was specifically used to avoid artefacts by cholesterol-induced massive inflammatory triggering.17 In contrast, some investigators prefer high-fat diet–induced lesions for the evaluation of very advanced and complex plaques. We did not add a high-fat diet group in this study, which might be considered as a limitation in our experimental setup. Besides total vessel narrowing, the stability of an atherosclerotic lesion determines the clinical outcome. Therefore, we further focused on lesion composition after ticagrelor treatment. Previously, an increase in fibrous content was observed by Li et al17 in apoE−/−/P2Y12−/− double knockout mice. In line with this, we were able to demonstrate thicker fibrous caps as well as a reduction in necrotic core size in ticagrelor-treated mice. To our knowledge, this is the first data to demonstrate plaque-stabilizing effects with ticagrelor or any P2Y12 antagonist. Furthermore, it is important to emphasize that this observation is made in an animal model of very advanced atherosclerosis.
Since macrophages play a pivotal role in initiation and early development of atherosclerosis, we also evaluated the macrophage content within atherosclerotic lesions after ticagrelor treatment. In contrast to the study of Li et al17 where a reduction in monocyte/macrophage infiltration in the apoE−/−/P2Y12−/− mice at 25 weeks of age was detected, we could not measure any difference between the groups. This is explained by our chosen model of very advanced atherosclerosis in mice at 45 weeks of age. The more advanced and necrotic the lesions of apoE−/− mice get, the cellular survival decreases and cells are therefore undetectable within the plaque.25 Using the aortic root for atherosclerotic lesion evaluation is a standard method, next to lesion evaluation “en-face” or within the innominate artery. As a limitation, we cannot exclude that site-specific effects can bias our data. To explore the potential pathophysiological mechanisms behind our observations, we performed in vitro experiments with the macrophage cell line RAW 264.7 for the detection and quantification of apoptosis. Macrophages are an important cell type in plaque physiology. Evidence suggests the involvement of P2Y12 receptors in the regulation of cell death.26,27 Webster et al28 observed less neuronal injury in an ischemia model due to inhibition of P2Y12. Using serum starvation as an appropriate approach to induce apoptosis in vitro, we were able to demonstrate a ticagrelor-induced inhibition of apoptosis in RAW 264.7 cells. However, we could not detect any effect within the lesions of the aortic roots, which might also be explained by the advanced, cell-depleted fibrous plaques of older mice. One cannot exclude that apoptosis might be affected in early or intermediate atherosclerotic lesions. In addition, we set up an experiment to model oxLDL uptake by macrophages. We were able to demonstrate a ticagrelor-dependent reduction in oxLDL uptake, evaluated by oil-red-o staining. Knowing that these are only in vitro data, oxLDL-dependent transformation to foam cells is an important step in early and intermediate fatty streak/plaque development. It triggers lesion size and composition and therefore also plaque stability.
Considering the data of West et al,16 where they could not show any effect of ticagrelor and clopidogrel on mice aged 12–14 weeks, there might also be age- or better disease-stage-dependent effects of P2Y12 inhibition.
Clinical data from the Platelet Inhibition and Patient Outcomes Trial study clearly demonstrated a reduction in overall mortality with ticagrelor as compared with clopidogrel in patients with acute coronary syndroms.10 Furthermore, recent data from the Prevention of Cardiovascular Events in Patients With Prior Heart Attack Using Ticagrelor Compared to Placebo on a Background of Aspirin-Thrombolysis in Myocardial Infarction trial showed a reduction in the risk for cardiovascular events in patients with a history of heart attack when treated with ticagrelor. More than half of the enrolled study subjects had a multivessel disease, which means a stage of advanced coronary atherosclerosis.12 While considering these data, it is necessary to focus on the effects of P2Y12 inhibition in models of advanced stages of atherosclerosis. Following new clinical outcome data, our study demonstrates for the first time a potential plaque-stabilizing effect of ticagrelor in a model of advanced vascular disease, and this is possibly due to the inhibition of apoptosis or oxLDL-uptake.
Conclusion
P2Y12 inhibition is a guideline-recommended treatment in patients with a prior myocardial infarction. Furthermore, clinical data demonstrated the beneficial effects by long-term administration of the P2Y12 inhibitor ticagrelor. However, effects on already existing atherosclerotic lesions have not been evaluated yet.
Our study demonstrates for the first time a potential plaque-stabilizing effect of ticagrelor in a model of advanced vascular disease which is consistent with its clinical effect in patients. Besides the effects on platelet aggregation, further studies will be necessary to focus on platelet-independent P2Y12 effects.
Acknowledgments
We would like to thank Annette Buttler and Claudia Felbinger for expert technical assistance. This study was supported by research grants from AstraZeneca GmbH, Wedel, Germany, and University of Heidelberg, Faculty of Medicine (to Ingo Staudacher). We further acknowledge financial support by Deutsche Forschungsgemeinschaft and Ruprecht-Karls-Universität Heidelberg within the funding programme Open Access Publishing.
Disclosure
The authors report no conflicts of interest in this work.
References
Bombeli T, Schwartz BR, Harlan, JM. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J Exp Med. 1998;187(3):329–339. | ||
Gawaz M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res. 2004;61(3):498–511. | ||
Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9(1):61–67. | ||
Projahn D, Koenen RR. Platelets: key players in vascular inflammation. J Leukoc Biol. 2012;92(6):1167–1175. | ||
Massberg S, Brand K, Grüner S, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196(7):887–896. | ||
Paul A, Calleja L, Camps J, et al. The continuous administration of aspirin attenuates atherosclerosis in apolipoprotein E-deficient mice. Life Sci. 2000;68(4):457–465. | ||
Karshovska E, Zhao Z, Blanchet X, et al. Hyperreactivity of junctional adhesion molecule A-deficient platelets accelerates atherosclerosis in hyperlipidemic mice. Circ Res. 2015;116(4):587–599. | ||
CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet. 1996;348(9038):1329–1339. | ||
Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST segment elevation. N Engl J Med. 2001;345(7):494–502. | ||
Wallentin L, Becker R, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–1057. | ||
Wiviott SD, Braunwald E, McCabe CH, et al.; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–2015. | ||
Bonaca MP, Bhatt DL, Cohen M, et al; PEGASUS-TIMI 54 Steering Committee and Investigators. Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med. 2015;372(19):1791–800. | ||
Liu O, Jia L, Liu X, et al. Clopidogrel, a platelet P2Y12 receptor inhibitor, reduces vascular inflammation and angiotensin II induced-abdominal aortic aneurysm progression. PLoS One. 2012;7(12):51707. | ||
Guns PJ, Hendrickx J, Van Assche T, Fransen P, Bult H. P2Y receptors and atherosclerosis in apolipoprotein E deficient mice. Br J Pharmacol. 2010;159(2):326–336. | ||
Thomas MR, Storey RF. Effect of P2Y12 inhibitors on inflammation and immunity. Thromb Haemost. 2015;114(3):490–497. | ||
West LE, Steiner T, Judge HM, Francis SE, Storey RF. Vessel wall, not platelet, P2Y12 potentiates early atherogenesis. Cardiovasc Res. 2014;102(3):429–435. | ||
Li D, Wang Y, Zhang L, et al. Roles of purinergic receptor P2Y, G protein-coupled 12 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32(8):81–89. | ||
Sillen H, Cook M, Davis P. Determination of ticagrelor and two metabolites in plasma samples by liquid chromatography and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(25):2299–2306. | ||
Movat HZ. Demonstration of all connective tissue elements in a single section; pentachrome stains. AMA Arch Pathol. 1955;60(3):289–295. | ||
Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123(18):2759–2767. | ||
Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–274. | ||
Amison R, Page C, Pitchford S. Pharmacological modulation of the inflammatory actions of platelets. Handb Exp Pharmacol. 2012;210:447–468. | ||
Rauch BH, Rosenkranz AC, Ermler S, et al. Regulation of functionally active P2Y12 ADP receptors by thrombin in human smooth muscle cells and the presence of P2Y12 in carotid artery lesions. Arterioscler Thromb Vasc Biol. 2010;30:2434–2442. | ||
Steinhubl SR, Badimon JJ, Bhatt DL, et al. Clinical evidence for anti-inflammatory effects of antiplatelet therapy in patients with atherothrombotic disease. Vasc Med. 2007;12:113–122. | ||
Rosenfeld ME, Averill MM, Bennett BJ, Schwartz SM. Progression and disruption of advanced atherosclerotic plaques in murine models. Curr Drug Targets. 2008;9(3):210–216. | ||
Mamedova LK, Gao ZG, Jacobson KA. Regulation of death and survival in astrocytes by ADP activating P2Y1 and P2Y12 receptors. Biochem Pharmacol. 2006;72(8):1031–1041. | ||
Zhang S, Ye J, Zhang Y, et al. P2Y12 protects platelets from apoptosis via PI3k-dependent Bak/Bax inactivation. J Thromb Haemost. 2013;11(1):149–160. | ||
Webster CM, Hokari M, McManus A, et al. Microglial P2Y12 deficiency/inhibition protects against brain ischemia. PLoS One. 2013;8(8):70927. |
Supplementary materials
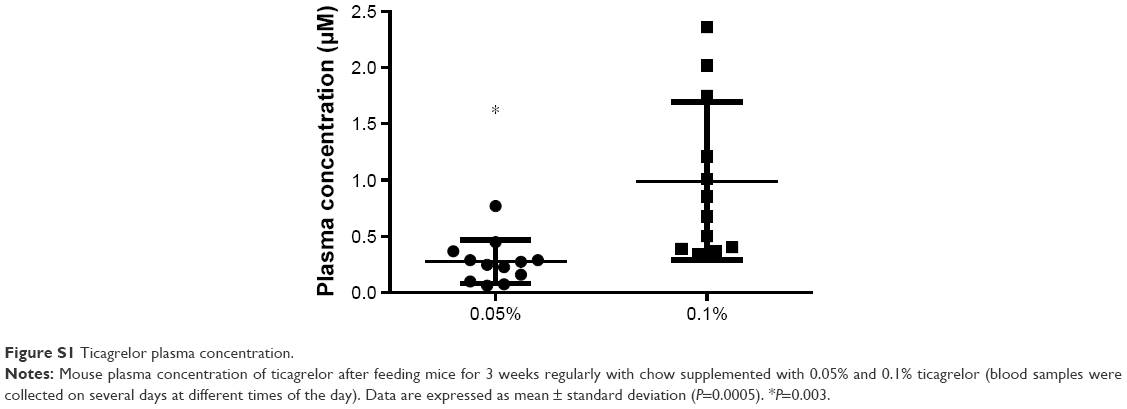 | Figure S1 Ticagrelor plasma concentration. |
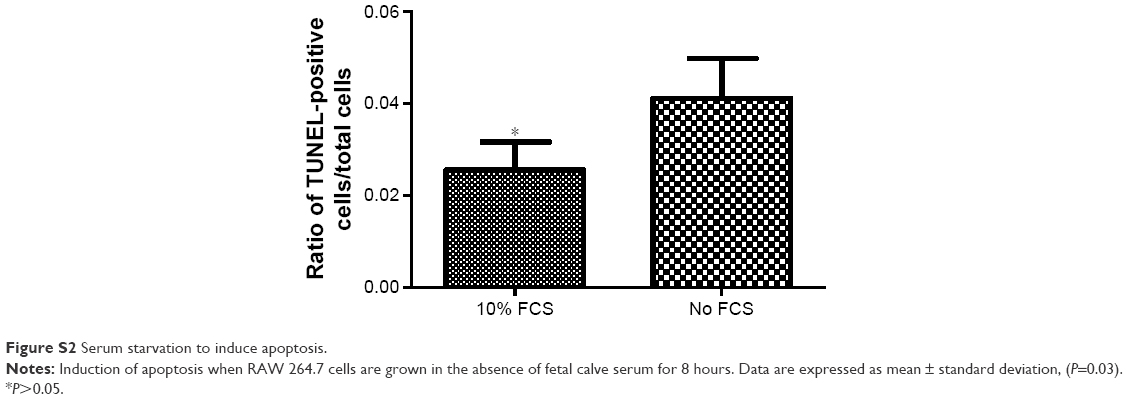 | Figure S2 Serum starvation to induce apoptosis. |
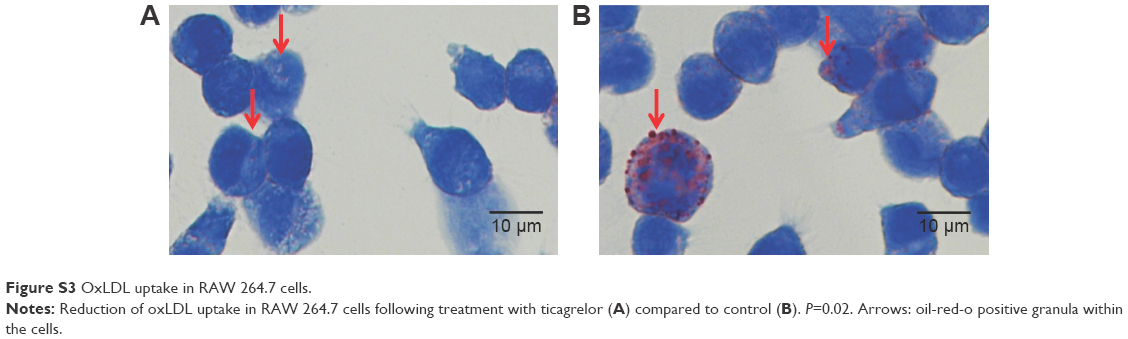 | Figure S3 OxLDL uptake in RAW 264.7 cells. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.