")
Back to Journals » International Journal of General Medicine » Volume 16
Thrombotic Pathogenesis and Laboratory Diagnosis in Cancer Patients, An Update
Received 10 August 2022
Accepted for publication 4 January 2023
Published 22 January 2023 Volume 2023:16 Pages 259—272
DOI https://doi.org/10.2147/IJGM.S385772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
David Bolaji Akinbo,1,2 Olutayo Ifedayo Ajayi3
1Department of Medical Laboratory Science, College of Medicine and Health Sciences, Afe Babalola University, Ado – Ekiti, Ekiti State, Nigeria; 2Department of Food, Nutrition, Dietetics and Health, College of Health and Human Sciences, Kansas State University, Manhattan, KS, USA; 3Department of Physiology, School of Basic Medical Sciences, College of Medical Sciences, University of Benin, Benin City, Edo State, Nigeria
Correspondence: David Bolaji Akinbo, Email [email protected]
Abstract: Cancer-associated thrombosis (CAT) is a leading cause of mortality in cancer patients and its incidence varies in different parts of the world. Venous thromboembolism (VTE) is a prominent manifestation of CAT, and significantly impacts morbidity and survival compared to arterial thrombosis in cancer patients. Several risk factors for developing VTE such as chemotherapy and immobilization have also been found co-existing with cancer patients and contributing to the increased risk of VTE in cancer patients than in non-cancer patients. This review highlights recent mechanisms in the pathogenesis of hypercoagulable syndromes associated with cancer, multiple mechanisms implicated in promoting cancer-associated thrombosis and their diagnostic approaches. Cancer cells interact with every part of the hemostatic system; generating their own procoagulant factors, through stimulation of the prothrombotic properties of other blood cell components or the initiation of clotting by cancer therapies which can all directly activate the coagulation cascade and contribute to the VTE experienced in CAT. It is our hope that the multiple interconnections between the hemostatic system and cancer biology and the improved biomarkers reported in this study can be relevant in establishing a predictive model for VTE, optimize early detection of asymptomatic microthrombosis for more personalized prophylactic strategies and incorporate effective therapeutic options and patient management to reduce mortality and morbidity, and improve the quality of life of affected cancer patients.
Keywords: thrombosis, hemostasis, venous thromboembolism, Trousseau’s syndrome, laboratory diagnosis, cancer
Introduction
Cancer is the second most common cause of mortality in the United States and recently in some other countries too. Thrombosis has been established as having a close association with cancer and thus represents a common complication of malignancy and the second leading cause of death in cancer patients.1 The association of cancer with thrombosis has been recognized over the years, with numerous postmortem studies identifying an increased incidence of thromboembolic deaths in cancer including mucinous carcinoma of the pancreas, lung, and gastrointestinal tract.2 It is commonly called cancer-associated thrombosis (CAT), and was first described by Armand Trousseau due to increased incidence of phlegmasia alba dolens particularly affecting patients with gastrointestinal cancers.3 The abnormalities of coagulation tests have been increasingly observed in cancer patients, indicating the presence of an ongoing subclinical hypercoagulable condition in this group. Several laboratory tests results have also demonstrated that a fibrin formation and removal process parallels the development of malignancy and this becomes paramount considering that fibrin and other clotting products are essential components of thrombogenesis and tumor progression.4
The incidence of thrombotic complications in cancer can vary from arterial or venous thromboembolism to disseminated intravascular coagulation. Cancer patients have an increased occurrence of both venous thromboembolism (4–20%) and arterial thrombosis (2–5%) when compared to the general population.5,6 However, cancer patients have recently had an increased risk of deep venous thrombosis (DVT); significantly impacting morbidity and survival than with arterial thrombosis.7 The presence of clinically detectable venous thromboembolism (VTE) has also been reported in 15% of all cancer patients, with a likelihood of the number being more when subclinical thromboembolism (TE) is added. Venous thromboembolism (VTE) is a collective term for broadly describing acute superficial vein thrombosis, acute deep venous thrombosis (DVT), acute pulmonary embolism (PE), and splanchnic vein thrombosis (SPVT).8 It is the third most prominent cardiovascular disorder with an estimated annual incidence of 0.1%, and affecting 2% to 5% of the entire population during their lifetimes. Venous thromboembolism is the second highest cause of mortality among cancer patients.1,9 Cancer patients represent 20% of all patients with deep venous thrombosis (DVT) and pulmonary embolism (PE) diagnosis.2
There is a ninefold increase in the risk for VTE across all cancer patients, with cancer being majorly responsible for 18% of all incident VTE. There have been cases of the VTE risks being elevated to as much as 28-fold in certain types of malignancies.10,11 Consistent with other previous studies, certain cancers were also discovered to be more predisposed to prothrombotic tendencies; and associated with disease staging, bed rest, as well as the therapeutic interventions employed. Evidence of TE has also been described in 5–10% of patients with breast cancer despite undergoing adjuvant chemotherapy with about 15% in those having metastatic disease. The recurrence of TE is also twice as probable in cancer patients despite being established on oral anticoagulant therapy when compared with the general population.2 These patients were also observed to have had longer hospitalization, poor response to oral anticoagulant therapy alongside a poorer prognosis following their first episode of VTE. Regardless of the lack of thrombotic presentations, some cancer patients such as those with solid tumors and leukemia however, still commonly present with aberrant laboratory coagulation tests which are characterized by varying degrees of clotting activation; suggesting the presence of a subclinical hypercoagulable condition. Such complications like the recurrent VTE, postthrombotic syndrome, and chronic thromboembolic pulmonary hypertension are costly and common occurrences even among the patients who survive an episode of VTE, and thereby having a profound impact on the patient’s quality of life.12
Despite numerous reports establishing a correlation between cancer and thrombotic complications for over two decades, it is still largely unclear if there is a similarity between the mechanisms that trigger cancer-associated DVT and those found in nonmalignant thrombotic incidences in the ordinary population.7 The mechanisms underlying cancer-associated thrombosis much like cancer itself have been described as being multifactorial and incompletely understood.13 Generally, cancer patients are in a hypercoagulable state characterized by abnormalities in each component of the Virchow’s triad, thereby enhancing thrombogenesis. The triad comprises three broad categories which includes stasis of blood flow, endothelial injury, and hypercoagulability, with the latter including abnormalities in the coagulation and fibrinolytic pathway and platelet activation.14 The specific mechanisms resulting in Virchow’s triad in cancer patients, particularly the effect on the host hemostatic system to promote the prothrombotic state, still remain poorly understood as the cancer types and stages, tumor-derived factors and genetics all pose varying risks rate for cancer-associated thrombosis. This review is aimed at giving an overview of the current factors implicated in the pathogenesis and capable of contributing to the hypercoagulable syndromes associated with cancer (arterial and venous thrombosis), the risk factors for developing cancer-associated thrombosis, patterns of thrombosis associated with cancer, the multiple mechanisms (direct and indirect) implicated in promoting cancer-associated thrombosis and their diagnostic approaches.
Thrombotic Complications in Cancer
Venous Thromboembolism
Venous thromboembolism (VTE) primarily comprises deep vein thrombosis (DVT) and pulmonary embolism (PE) and their development is commonly initiated in the valve sinus. A number of features surrounding these valves such as abnormal and reduced blood flow, reduced shear stress, and hypoxia leading to an intact but dysfunctional endothelium; all contribute to the prothrombotic nature of the sites.15 Platelets and leukocytes are also capable of becoming trapped in valve pockets, tumors are capable of compressing veins in cancer patients thereby resulting in venous stasis and subsequently encouraging thrombosis. A VTE diagnosis is a serious complication of cancer, adversely affecting the quality of life of the patient by decreasing their overall survival rates and subsequently contributes significantly to the morbidity and mortality of this patient group.16 Fatal PE has been reported to be thrice more common in cancer patients, and a five- to sevenfold increased risk of VTE development in cancer patients than in noncancer patients.17 Cancer patients who develop VTE at diagnosis of their cancers or within the diagnosis year usually have a worse prognosis compared to those without VTE. Approximately 4–20% of cancer patients experience VTE at some stage, and the highest rate occurring in the initial period following diagnosis. Furthermore, 0.5% of cancer patients will experience thrombosis annually compared to a 0.1% incidence rate in the general population.14
Arterial Thrombosis
Arterial thrombosis in cancer are less common compared to the VTE as multiple case reports indicative of acute arterial thrombosis in the setting of a new malignancy exists. The incidence of arterial thrombosis at six months was recently assessed in a large retrospective matched-cohort study, and it was reported at 4.7% in cancer patients compared with 2.2% in the matched controls.18 Arterial thrombosis differs substantially in their pathogenesis from venous thrombosis as it typically occurs with endothelial damage. Atherosclerotic plaques having lipid-rich core with thin fibrous caps are prone to thrombosis, and a ruptured plaque or an intact plaque with superficial endothelial erosion can result in the formation of a thrombus. Persistent platelet activation at the rupture site promotes thrombosis by exposing the procoagulant molecules within the plaque core.19 The high shear rates in stenosed arteries contribute to a thrombus predominantly composed of platelets unlike in the low venous shear rates in VTE development. Platelets are the only blood elements that are capable of adhering at high shear stress and the high shear rates has a positive feed-forward activity for platelet activation which further enhance thrombogenesis.14 However, arterial thrombosis in cancer can also occur in the absence of an atherosclerotic plaque as seen in cardiovascular patients, where several secreted factors from cancer cells which includes thrombin and vascular endothelial growth factor (VEGF) can induce systemic hypercoagulation which results in the promotion of platelet activation and coagulation.20
There have also been multiple case reports establishing an association between chemotherapy and arterial thrombosis with numerous chemotherapy agents known to be prothrombotic such as the platinum-based agents (cisplatin), vascular endothelial growth factor (VEGF) inhibitors (bevacizumab), and VEGF tyrosine kinase receptor inhibitors (sorafenib/sunitinib/pazopanib) have been associated with increased rates of arterial thrombosis.20 Vascular damage resulting from hypertension, atherosclerosis, or vascular anomalies are other major risk factors for arterial thrombosis by inducing turbulence and blood flow perturbation, thereby enhancing platelet adhesion which is a key aspect of thrombotic pathogenesis.19
Chronic Disseminated Intravascular Coagulation
Cancer-associated thrombotic complications are not restricted to VTE or arterial thrombosis, as other more severe procoagulant manifestations such as the disseminated intravascular coagulation (DIC) and thrombotic microangiopathy (TMA) are also types of CAT complications.21 Simply put, DIC refers to a relatively rare but severe complication of cancer manifesting as a consumptive coagulopathy that leads to microvascular thrombosis with tendency for severe bleeding, thrombocytopenia, and subsequent organ failure.22 The bleeding is, however, believed to result from hyperfibrinolysis which dominates microvascular thrombosis.23 There have been varying reports of incidence of DIC with 7% recorded for solid tumors and other reports indicating a higher incidence of about 85% in acute promyelocytic leukemia also. Defects in the three constituents of normal host defense against thrombosis tend to be commonly implicated in DIC and thrombogenesis in cancer patients and these include: (1) blood flow leading to stasis, (2) balance of the procoagulant and anticoagulant proteins within the blood, resulting in the activation of circulating procoagulant proteins, and (3) vessel wall activation, resulting in an increase in its procoagulant contribution.14 DIC in cancer patients often has less severe clinical presentations with a more delayed onset, sequel to which DIC progresses in a gradual yet chronic manner leading to systemic activation of coagulation and subsequently resulting in the quick exhaustion of coagulation factors and platelets. Bleeding would usually result at the first clinical symptom to indicate DIC.21
Thrombotic microangiopathy (TMA) manifests as thrombotic purpura and hemolytic uremic syndrome and often difficult to distinguish from DIC as it shares similar clinical presentations as DIC which subsequently result in microvascular thrombosis with increased predisposition for bleeding and organ failure. A distinctive diagnosis of TMA is highly essential in order for accurate treatments of the condition since TMA and DIC both have different therapeutic approaches despite their similarities and close associations.22 Unlike DIC which results from the marked activation and consumption of the coagulation system and subsequent secondary fibrinolytic activation, the initiation of TMA occurs by the marked activation and consumption of platelets in response to numerous factors which results in the activation and subsequent damage to the vascular endothelium.14
Thrombotic Thrombocytopenic Purpura
The complication of thrombotic thrombocytopenic purpura can occur in patients with cancer, and various cancer chemotherapeutic agents have been associated with both TTP and hemolytic uremic syndrome. TTP is commonly characterized by pulmonary manifestations, especially pulmonary edema and neurologic changes have also been frequently observed. The condition has been observed to have resulted directly from many different types of cancer and found only in a minute fraction of these cancer patients, most commonly gastric adenocarcinoma patients followed by carcinoma of the breast, colon, and small cell lung carcinoma patients.24 The deficiency of a von Willebrand factor (vWF)-cleaving protease (ADAMTS13) resulting from an inhibitor(s) present in the patient or congenital deficiency and the presence of unusually large (uL)vWF multimers in the TTP patients have been associated with the pathophysiology of this condition.25 The protease have also been discovered to be responsible for cleaving uLvWF multimers to smaller ones, which is capable of promoting the activation and aggregation of platelets if not cleaved.26 uLvWF multimers can also be released into the circulation in the case of a disease or injury that involves the endothelial cell, thereby promoting the activation and aggregation of platelets in the arteriolar capillaries which results in thrombocytopenia and microangiopathic hemolytic anemia if the vWF-cleaving protease is not available.
Metastasis of the bone marrow may also be associated with increased and abnormal angiogenesis for the growth of cancer as observed in cancer metastasis to other organs. It is therefore possible for the aggressive growth of these tumors and secondary myelofibrosis to injure the endothelial cells of the vessels in the marrow by direct encroachment thus causing the release of uLvWF multimers in advanced cancer, thereby contributing to the aggregation of platelets. The production of mucin is however believed to contribute to the TTP as it is capable of exerting a direct detrimental effect on the pathologic endothelial cell to change the endothelial function.25,26
The Mechanisms of Cancer-associated Thrombosis
Cancer patients have an increased predisposition to thrombosis, and this can be viewed from multiple perspectives. There is nonetheless a poor understanding of the molecular mechanisms underlying the risk of thromboembolic complications in cancer patients. In addition, it still remains unclear whether the mechanisms triggering cancer-associated DVT and mechanisms behind nonmalignant thrombosis have any similarity. Various studies have attempted unraveling the relationship between these two processes since the initial discovery by Bouillard and further insights by Trousseau over two decades ago.27 Numerous other mechanisms capable of promoting a hypercoagulable state in cancer patients do exist; mostly traceable to cancer cells being able to alter the various components of Virchow’s triad and resulting in an increased susceptibility to thromboembolic events. The proper identification of the pathophysiology of cancer-associated DVT could proffer crucial therapeutic outcomes requiring certain pharmacological approaches distinct from others used for prophylaxis or treatment of nonmalignant thrombosis. This study highlights the mechanisms of VTE in cancer, emphasizing VTE as the most common clinical presentation of CAT and broadly classifying them into direct and indirect mechanisms as summarized in Figures 1 and 2, respectively.
 |
Figure 1 Schematic representation of the direct mechanisms for cancer-associated thrombosis. |
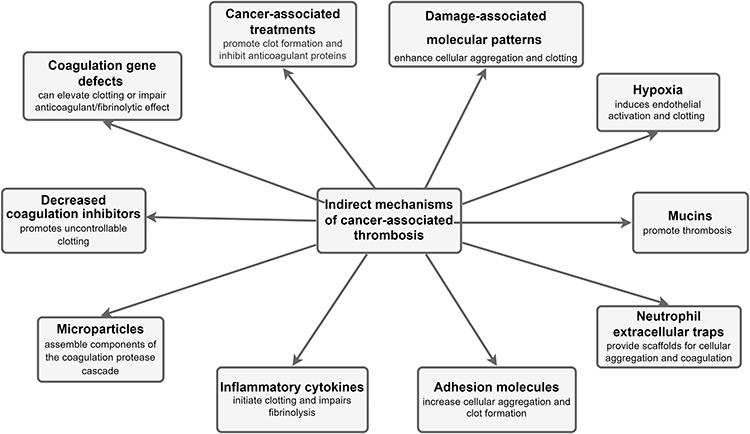 |
Figure 2 Schematic representation of the indirect mechanisms for cancer-associated thrombosis. |
Direct Mechanisms of Cancer-associated Thrombosis
Tissue Factor
Tissue factor (TF) is considered the best characterized tumor-derived procoagulant protein abundantly expressed on subendothelial cells such as fibroblasts, pericytes, and vascular smooth muscle cells triggering hemostatic response upon vascular injury. It is usually not expressed on normal quiescent endothelium, but becomes constitutively expressed by malignant tissue involving endothelial and tumor cells. Tumor TF expression has only been associated with VTE risk in pancreatic and ovarian cancers despite the other numerous reports of TF expression in several types of cancers.1 An elevated TF expression has also been positively correlated with advanced stages of pancreatic cancer and poorer prognosis. In addition, TF has also been observed on the surface of microvesicles released by pancreatic tumors; and their presence associated with increased VTE in pancreatic cancer patients.28
Microparticles (MP)
MPs are small membrane vesicles having 0.1–1 µm diameter and are released from apoptotic or activated normal cells or resting malignant cells. They are capable of directly and indirectly enhancing prothrombotic mechanisms in cancer when released from cancer cells. The components of the coagulation protease cascade are assembled on their membrane surface. Breast and hepatocarcinoma cell lines were reported to show procoagulant activity both in vivo and in vitro associated with tumor-derived vesicles, alongside increased in vivo thrombus formation by circulating MP in several other cancer types more recently.14 One mechanism of MP-induced VTE is the surface expression of active TF and the presence of phosphatidylserine which supports the assembly of coagulation complexes on its negatively charged surface.29 The externalization of phosphatidylethanolamine from pancreatic cancer MP has recently been reported to play a crucial role in cancer-associated DVT too.3 This association of TF-positive MP with VTE complications has however, only been reported in patients with pancreatic cancer.30
Podoplanin (PDP)
Podoplanin (PDP) is a platelet activating and aggregating protein, which occurs through tumor-cell-induced platelet activation using the C-type lectin receptor 2 (CLEC-2).31 It is produced by cancer-associated fibroblasts, and its expression observed in pancreatic cancer cell lines.32 CLEC-2 depletion in platelets was observed to have resulted in a decreased venous thrombosis using an inferior vena cava (IVC) stenosis model of DVT, which was however restored following the transfusion of wild-type platelets. This increased level of circulating podoplanin in the IVC wall after stenosis correlated with the degree of thrombosis.33 Tumor-associated podoplanin has only been associated with VTE in brain cancer patients, while no association has been established yet in other cancer patients.34 A few studies have postulated that podoplanin is expressed into the bloodstream by cancer cells in order to regulate thrombosis at distant sites correlating with other studies where tumor-derived MPs bearing podoplanin were detected in the peripheral circulation of pancreatic and colorectal cancer patients.35,36
Plasminogen Activator Inhibitor-1 (PAI-1)
PAI-1 inhibits fibrinolysis, and is reported to be highly expressed in cancer cells such as the pancreatic cancer cells. Increase in PAI-1 plasma level reduces fibrinolytic activity, and subsequently predisposes the affected persons to thrombosis.1,37 Excessive PAI-1 levels correlated with the degree of thromboembolism in pancreatic cancer patients, inhibiting plasminogen activator and impairing fibrinolysis which resulted in disseminated intravascular coagulation, circulatory hypoperfusion, and organ dysfunction in affected cancer patients.38 Studies have shown a significantly elevated thrombi level when treated with an anti-VEGF drug, bevacizumab, thereby resulting in an increased circulating levels of PAI-1 in the tumor and plasma. PAI-1 inhibitor significantly lowered the thrombotic effect of bevacizumab, suggesting a possible use for PAI-1 inhibitor in enhancing the clearance of VTE in cancer-associated thrombosis.39 Further studies are however essential in validating its role and elucidating the mechanism.
Cancer Procoagulant (CP)
Cancer procoagulant (CP) is a cysteine protease capable of activating the coagulation cascade by its direct activation of factor X independently of FVII. It was initially isolated from rabbit malignant tissue and later isolated from human carcinomas carrying procoagulant activity too. CP cleaves the FX heavy chain at a site different from the other known FX activators.12 A study, however, reported a lack of correlation between CP and procoagulant markers in breast cancer patients which led to the purity of the CP preparations utilized in earlier studies being doubted and potential contamination of TF/factor VIIa complex being considered.40
Tumor-derived Platelet Agonists
Cancer cells cause the secretion of adenosine diphosphate (ADP) responsible for the activation and aggregation of platelets via the P2Y1 and P2Y12 receptors. Thrombin synthesis has also been reported in pancreatic tumors, with an increased plasma level in pancreatic cancer patients.41 ADP and thrombin are platelet aggregation agonists which mediate primary hemostasis. This may, therefore, explain the enhancement of platelet activation and coagulation in cancer patients by these tumor-derived products, correlating with other studies on the effect of tumor-derived agonists on platelets.14
Indirect Mechanisms of Cancer-associated Thrombosis
The cancer-derived factors and some of its associated mechanisms capable of enhancing interactions with host cells can result in thromboembolic complications in cancer patients as shown in Figure 2.
Microparticles
Microparticles (MP) expressing TF in cancer patients are reportedly derived from the cancer cells while activated endothelial cells and monocytes have also been observed to release TF-positive MPs in response to the condition. Cancer cells produce and secrete numerous pro-inflammatory cytokines which can cause the release of TF-positive MP from activated endothelial cells and monocytes. The tumor and host cells’ relative contribution to the total amount of TF-positive MP observed in cancer patients, however, still remain unconfirmed.14
Inflammatory Cytokines
Majority of the inflammatory cytokines synthesized and secreted by the tumor cells are phenotypically procoagulant, capable of enhancing thrombogenesis and disrupting host endothelial cells. A reactive response is also induced in the host’s inflammatory tissues by the tumor presence causing an excessive cytokine release.13 The tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β) are considered the most well-defined pro-inflammatory cytokines, they have been reported to exert prothrombotic effects and possible induction of TF and von Willebrand factor expression on vascular endothelial cells.2 However, TNF-α and IL-1β also downregulate and attenuate thrombomodulin; a receptor on endothelial cells that binds with thrombin and is also responsible for protein C activation thus serving as an antithrombotic regulator. These inflammatory cytokines also act on endothelial cells to inhibit the release of nitric oxide and prostacyclin while also promoting the prothrombotic activity of the endothelial cells by stimulating the fibrinolysis inhibitor; PAI-1 production.
The release of pro-angiogenic and growth factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor, and granulocyte colony stimulating factor (G-CSF) induce procoagulant phenotype in host cells. Excessive TF is produced by macrophages, which is induced and activated upon exposure to VEGF produced by various tumor cells. Hemostatic activation is mediated by endothelial activation markers (thrombomodulin and von Willebrand factor) and coagulation markers and their level has been reported to increase when in the presence of G-CSF, indicating hypercoagulation. TF expression on endothelial cells has also been reported to be aggravated by basic fibroblast growth factor.42
Adhesion Molecules
Cancer cells have been reported to promote blood vessel walls attachments, interactions with blood cells and activation of the procoagulant properties of the hosts’ endothelial cells, leukocytes, and platelets. The attachment of endothelial cells to tumor cells can trigger a localized clotting near the blood vessel wall and subsequent thrombus formation. Numerous adhesion molecules account for various types of tumor cells adhesion to endothelial cells. They include the E-selectin which enabled the HT-29M colon carcinoma cells attachment and rolling on activated endothelial cells under shear stress, the A375M cells 29M used the vascular cell adhesion molecule-1 for its adhesion; with all these molecules capable of increasing aggregate formation and promoting thrombosis by obstructing vascular patency. Further, P-selectin synthesized by the endothelial cells and activated platelets facilitates binding with cancer cells. However, the P-selectin ligand on cancer cells still remains unconfirmed. These interactions between cancer cells, platelets and endothelial cells results in the formation of cell–cell aggregates, which then disrupts blood flow and promote clotting and vessel occlusion.4
Neutrophil Extracellular Traps
Neutrophil extracellular traps (NETs) are DNA-associated mesh of histones and neutrophil-derived proteases first recognized for their antimicrobial functions and have been observed to be produced in vitro under the influence of pancreatic-cancer-derived factors. Recently, NETs are also shown to enhance venous and arterial thrombosis in mice and serve as a platform for direct platelet adhesion and aggregation.43,44 In addition, cancer-associated NETs could further enable the activation of host cells to promote thromboembolic events such as the activation of endothelial cells by NET-associated histones, subsequently increasing the release of von Willebrand factor.45,46 Elevated citrullinated histone H3 (a biomarker for NET formation) increased VTE in cancer patients. Other NET biomarkers including cell-free DNA and nucleosomes also positively correlated with increased VTE predisposition within the first three-to-six months.47
Mucins
Numerous cancer types exhibit aberrant expression and altered glycosylation of many mucins.48 Mucins possess heavy O-linked glycosylation sites which act as ligands for selectins. They utilize adhesion-dependent signaling to promote thrombosis in cells. Mucins are incapable of self-activating platelets directly in vitro, but caused platelet activation when incubated with whole blood, suggesting a possible link with the L-selectin. Purified mucin also caused widespread platelet-rich intravascular microthrombi in mice, correlating with previous reports on further mechanistic insights into mucin-induced microthrombi.49 The L-selectin and P-selectin glycoprotein ligand-1 interaction on neutrophils and P-selectin on platelets to produce cathepsin G from neutrophils dictates their prothrombotic potential in cancer patients.
Hypoxia
Tumors result in a very hypoxic microenvironment which promotes endothelial perturbation. Endothelial cells release elevated levels of phospholipase A2 during hypoxia, causing the excessive production of prostaglandins and platelet-activating factor (PAF) synthesis. Besides activating platelets, PAF also activates neutrophils; thereby promoting their endothelial adhesion under hypoxic conditions. The adhesion of neutrophils to hypoxic endothelial cells was reported to have declined upon obstructing the release of PAF.50 Hypoxia causes exocytosis of Weibel–Palade bodies from the endothelial cells, the release of von Willebrand factor and P-selectin overexpression, and escalates the indirect procoagulant response. The increased synthesis of ADP during hypoxia in tumor patients could also lead to increased platelet activation.51
Damage-associated Molecular Patterns (DAMPs)
DAMPs are molecules comprising histones, high mobility group box 1 (HMGB1), S100 proteins, and heat shock proteins localized within the cell and produced by senescent tumor cells upon cell death or during cell stress. They coordinate protective responses via innate immune pattern recognition receptors upon release, and propagate a host response. However, DAMPs are also capable of inducing chronic inflammation and immune cell activation in the host resulting in thrombotic events and promoting tumor growth and survival. Cancer patients reportedly had an increased plasma circulating nucleosomes when compared with a healthy control group.52 Histones and HMGB1 are the DAMPs that have so far been recognized as having potent procoagulant activity and their presence in peripheral circulation of cancer patients indicates an enhanced platelet activation and aggregation and neutrophil activation, inducing neutrophil extracellular traps (NETs) release and enhancing thrombosis.15 Increased extracellular DNA levels have also been observed in patients with PE, suggesting an association of extracellular DNA with thromboembolic complications.53 However, their role in cancer patients and noncancer patients is yet to be fully delineated.
Cancer-associated Treatments
Various cancer treatments exist; however, chemotherapy is currently the commonest and most readily available form. Cisplatin-based chemotherapy has been reported to promote thromboembolic events in cancer patients; the mechanism underlying cisplatin-associated thrombosis, however, still remains unclear. The treatment of two human endothelial cell lines with cisplatin led to endothelial cell apoptosis while provoking procoagulant endothelial microparticles release and resulted in thrombin formation independent of TF.54 A series of distinct mechanisms seem to be involved in this prothrombotic activity, including the direct drug-induced damage to the endothelium and the elevated expression of TF procoagulant activity of monocytes and macrophages propagating a procoagulant response from the host cells. The direct hepatotoxicity of chemotherapy may play another major role in promoting hypercoagulation by the inhibition of natural anticoagulant proteins, such as the protein S, protein C, and antithrombin. Furthermore, chemotherapy induces apoptosis of both tumor and host endothelial cells resulting in cytokine release and increasing TF expression and activity.55
Coagulation Gene Defects
Oncogenic mutations and some coagulant effectors have been reported to deregulate hemostatic genes in cancer cells.56 Regardless of the tumor types; oncogenic mutations in cancer cells have been associated with the incidence of CAT in some cancer patients. Certain mutations such as the STK11, KRAS, CTNNB1, KEAP1, CDKN2B, and MET, were reported to predict striking increase in the occurrence of CAT in the patients for about a year prior to primary diagnosis.57 In addition, elevated factor V Leiden presence and prothrombin gene G20210A mutation were also reported to upregulate the risk of thromboembolic complications in gastrointestinal cancers.58 Single nucleotide polymorphisms of factors V, X and the endothelial protein C receptor (EPCR) had a novel association with susceptibility to hypercoagulability in breast cancer patients.59 The homozygosity of the fibrinogen gamma gene (FGG) rs2066865 has been associated with increased risk of VTE in active cancer, especially increasing pulmonary embolism risks in cancer patients.60 It is noteworthy that other coagulation‐related genes polymorphisms including FII G20210A, FIII –603A/G, FIII +5466A>G, FXIII Val34Leu, and methylenetetrahydrofolate reductase C667T have been reported not to have any effect on the incidence of VTE in patients with and without cancer.61,62
Decreased Coagulation Inhibitors
Coagulation inhibitors including the antithrombin III, heparin cofactor II, protein C, free protein S, and thrombomodulin were significantly reduced as pancreatic cancer progresses after diagnosis in a patient.14
Current Biomarkers for the Diagnosis of Venous Thromboembolism in Cancer
In establishing the laboratory diagnosis of cancer-associated thrombosis, a series of coagulation diagnostic assays is recommended than a single laboratory investigation in order to arrive at a more precise diagnosis and effective treatment strategies.7 These laboratory investigations are classified into clinical biomarkers currently in use for VTE risk assessments in cancer patients and novel clinical biomarkers still under investigation for VTE risk assessments in cancer patients as summarized in Tables 1 and 2.
 |
Table 1 Biomarkers Currently in Use for Venous Thromboembolism Risk Assessments in Cancer Patients |
 |
Table 2 Novel Biomarkers Still under Investigation for Venous Thromboembolism Risk Assessments in Cancer Patients |
Tissue Factor
Tissue factor (TF) is an integral membrane protein and a major trigger of blood clot formation when exposed on the endothelium upon injury. It complexes with FVIIa to generate a FIXa and FXa burst activating the extrinsic coagulation pathway which results in the formation of a stable fibrin clot.63 Numerous cancer types have been found expressing TF, with TF-positive extracellular vesicles reported as being released by cancer cells expressing TF.64 Elevated TF level enhances the predisposition of cancer patients to increased activation of the extrinsic coagulation pathway.
D-dimer
D-dimers are products of fibrin degradation present in the peripheral circulation upon active coagulation like the presence of a clot. It may be measured in a qualitative or quantitative manner. Elevated D-dimer levels have been largely associated with specificity for VTE.64 Various commercially available assays can now be used for the assessment of D-dimer levels, with the sensitivity and specificity varying for the different assays.
Soluble P-selectin
P-selectin (CD62p) is a glycoprotein largely expressed by activated platelets and endothelial cells. It is rapidly mobilized from α-granules to the platelet surface, causing interaction between platelets and vascular endothelium, cross-linking platelets and leukocytes, resulting in inflammation, thrombosis, and atherogenesis upon the release of pro-inflammatory cytokines.65 Elevated levels of soluble P-selectin have been reported to be associated with an increased rate of VTE in cancer patients, suggestive of a role for P-selectin in cancer prophylaxis and management.66
Microparticles
Microparticles (MPs) are microvesicles or fragments derived from platelets, leucocytes and endothelial cells. The different MPs types participate in fibrin clot formation, enhancement of platelet leukocyte interactions and influencing other plasma proteins such as von Willebrand’s factor. Increased circulating MPs have been implicated in numerous inflammatory, thrombotic and vascular diseases, with elevated levels of MPs reported in an experimental VTE model.64 Elevated MPs levels have also been reported in cancer patients with VTE, and their increased levels used for predicting VTE in patients with known malignancy.67
Prothrombin Fragment 1+2 (F1+2)
Prothrombin fragment 1+2 (F1+2) is a by-product of prothrombin conversion to thrombin and can be utilized in assessing coagulation status by measuring its plasma levels. It has a molecular weight of about 31,000 D and is usually excreted in urine (uF1+2) due to its size.68
Thrombin Generation Test (TGT)
The thrombin generation test (TGT) is aimed at evaluating the overall balance between procoagulant and anticoagulant forces which has provided new insights into the coagulation cascade, including diagnosing hypocoagulability and hypercoagulability conditions. It is also useful in identifying novel genetic risk factors for venous thromboembolism. A high level of TGT therefore implies increased thrombin generation resulting from pathological activation of in vivo coagulation such as venous thrombosis, and can be measured using several commercially available methods.69
Thrombin-anti-thrombin Complexes (TATC)
The combination of the prothrombin complex with calcium and phospholipid surface results in the release of thrombin leading to the formation of a local clot. Fibrinogen conversion to fibrin through the action of thrombin ends the prothrombotic process. The excess production of thrombin, however, is a major cause of VTE. Antithrombin is therefore activated as a biological defense reaction to inhibit thrombin in the face of thrombophillia leading to the formation of the TAT generated as a result of these reactions. The TAT complex is simply a molecular complex made up of thrombin and AT; a primary thrombin inhibitor and an increased TAT indicate excess thrombin production thereby serving as a biomarker of prothrombotic status.70
CD40 Ligand
CD40L is expressed on vascular cells and activated platelets which interact with soluble CD40L (sCD40L) released into the circulation by activated T-lymphocytes and membrane-bound CD40L and sCD40L to promote inflammatory and prothrombotic responses in the host. The CD40-CD40L interaction has also been observed to produce many angiogenesis-associated factors like the vascular endothelial growth factor (VEGF) which further contributes to the promotion of prothrombotic responses.71
Platelet Factor-4 (PF4)
Platelet factor 4 (PF4) is released into the circulation upon platelet activation; it is constitutively and abundantly expressed by platelets. A major consequence of PF4 expression is inflammation and wound healing. The PF4 enhances procoagulant activities by disrupting the stable heparin-antithrombin III-thrombin ternary complex.72
Thrombospondin-1
Thrombospondin-1 (TSP1) is a major constituent of α-granules responsible for the modulation of a wide range of biological functions including modulation of endothelial cell adhesion, platelet aggregation enhancement, angiogenesis inhibition, and latent transforming growth factor-β (TGF-β) activation. When released, it induces chemo-attraction impacting various inflammatory cells which when extensive, results in tissue damage and inflammatory conditions.73
Molecular Diagnosis
Molecular techniques can be used in identifying pathogenic mutations in genes encoding corresponding clotting factors. Mutations such as STK11, KRAS, CTNNB1, KEAP1, CDKN2B, and MET and prothrombin gene G20210A mutation can all be detected using molecular techniques. The SNPs of FV, FX and EPCR resulting in hypercoagulability can also be useful biomarkers in in breast cancer patients.59 A combined deficiency of FV and FVIII has been described to be due to mutations in genes that encode proteins responsible for intracellular transport of the two coagulation factors which are the multiple coagulation factor deficiency and the lectin mannose binding 1, respectively.74 Direct oncogenes activation and repressor gene-mediated neoplastic transformations which activate blood clotting and/or suppress fibrinolysis, produce thrombosis and/or DIC have also been detected in experimental models via molecular diagnosis.75 Molecular genotyping can also be conducted to determine homozygosity of the FGG rs2066865 gene in cancer patients.60
Conclusion
Cancer patients are increasingly predisposed to thrombosis, with venous thromboembolism as one of the most common causes of mortality and morbidity among them. The interactions of various mechanisms resulting in the activation of an array of hemostatic components contribute to the hypercoagulable state in cancer. Malignant cells interact with every part of the hemostatic system, generating their own procoagulant factors, or through stimulation of the prothrombotic properties of other blood cell components which can directly activate the coagulation cascade. The cytotoxic chemotherapy and other cancer therapies are additional mechanisms by which clotting is also initiated. Early diagnosis as well as effective prophylaxis and treatment of VTE can help reduce mortality and morbidity, and improve the quality of life of affected cancer patients.
Acknowledgments
The authors thank the authorities of Afe Babalola University Ado-Ekiti; Ekiti State; Nigeria and Kansas State University for providing some of the facilities and resources used in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Khorana AA, Mackman N, Falanga A, et al. Cancer-associated venous thromboembolism. Nat Rev Dis Primers. 2022;8(1):1–18. doi:10.1038/s41572-022-00336-y
2. Nasser NJ, Fox J, Agbarya A. Potential mechanisms of cancer-related hypercoagulability. Cancers. 2020;12(3):566. doi:10.3390/cancers12030566
3. Stark K, Schubert I, Joshi U, et al. Distinct pathogenesis of pancreatic cancer microvesicle–associated venous thrombosis identifies new antithrombotic targets in vivo. Arterioscler Thromb Vasc Biol. 2018;38:772–786. doi:10.1161/ATVBAHA.117.310262
4. Falanga A, Russo L, Milesi V, Vignoli A. Mechanisms and risk factors of thrombosis in cancer. Crit Rev Oncol Hematol. 2017;118:79–83. doi:10.1016/j.critrevonc.2017.08.003
5. Levi M. Cancer-related coagulopathies. Thromb Res. 2014;133:S70–S75. doi:10.1016/S0049-3848(14)50012-6
6. Eichinger S. Cancer associated thrombosis: risk factors. Thromb Res. 2016;140:S12–S17. doi:10.1016/S0049-3848(16)30092-5
7. Sheth RA, Niekamp A, Quencer KB, et al. Thrombosis in cancer patients: etiology, incidence, and management. Cardiovasc Diagn Ther. 2017;7(3):S178–S185. doi:10.21037/cdt.2017.11.02
8. Streiff MB, Holmstrom B, Angelini D, et al. Cancer-associated venous thromboembolic disease, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2021;19(10):1181–1201. doi:10.6004/jnccn.2021.0047
9. Wells P, Anderson D. The diagnosis and treatment of venous thromboembolism. Hematology Am Soc Hematol Educ Program. 2013;2013:457–463. doi:10.1182/asheducation-2013.1.457
10. Mulder FI, Horváth-Puhó E, van Es N, et al. Venous thromboembolism in cancer patients: a population-based cohort study. Blood. 2021;137(14):1959–1969. doi:10.1182/blood.2020007338
11. Hayashida K, Kawabata Y, Saito K, et al. Prevalence and risk factors of preoperative venous thromboembolism in patients with malignant musculoskeletal tumors: an analysis based on D-dimer screening and imaging. Thromb J. 2022;20(1):22. doi:10.1186/s12959-022-00382-2
12. Mandoj C, Tomao L, Conti L. Coagulation in brain tumors: biological basis and clinical implications. Front Neurol. 2019;10:181. doi:10.3389/fneur.2019.00181
13. Falanga A, Marchetti M, Russo L. The mechanisms of cancer-associated thrombosis. Thromb Res. 2015;135(1):S8–S11. doi:10.1016/S0049-3848(15)50432-5
14. Abdol Razak NB, Jones G, Bhandari M, Berndt MC, Metharom P. Cancer-associated thrombosis: an overview of mechanisms, risk factors, and treatment. Cancers. 2018;10(10):380. doi:10.3390/cancers10100380
15. Sun HJ, Wu ZY, Nie XW, Bian JS. Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Front Pharmacol. 2020;10:1568. doi:10.3389/fphar.2019.01568
16. Fuentes HE, Tafur AJ, Caprini JA. Cancer-associated thrombosis. Dis Mon. 2016;62(5):121–158. doi:10.1016/j.disamonth.2016.03.003
17. Connolly GC, Francis CW. Cancer-associated thrombosis. Hematology Am Soc Hematol Educ Program. 2013;2013:684–691. doi:10.1182/asheducation-2013.1.684
18. Navi BB, Reiner AS, Kamel H, et al. Risk of arterial thromboembolism in patients with cancer. J Am Coll Cardiol. 2017;70(8):926–938. doi:10.1016/j.jacc.2017.06.047
19. Luo X, Lv Y, Bai X, et al. Plaque erosion: a distinctive pathological mechanism of acute coronary syndrome. Front Cardiovasc Med. 2021;8:711453. doi:10.3389/fcvm.2021.711453
20. Tuzovic M, Herrmann J, Iliescu C, Marmagkiolis K, Ziaeian B, Yang EH. Arterial thrombosis in patients with cancer. Curr Treat Options Cardiovasc Med. 2018;20(5):40. doi:10.1007/s11936-018-0635-x
21. Levi M. Management of cancer-associated disseminated intravascular coagulation. Thromb Res. 2016;140(1):S66–70. doi:10.1016/S0049-3848(16)30101-3
22. Wada H, Matsumoto T, Suzuki K, et al. Differences and similarities between disseminated intravascular coagulation and thrombotic microangiopathy. Thromb J. 2018;16:14. doi:10.1186/s12959-018-0168-2
23. Thachil J, Falanga A, Levi M, Liebman H, Di Nisio M. Scientific and standardization committee of the international society on thrombosis and hemostasis. Management of cancer-associated disseminated intravascular coagulation: guidance from the SSC of the ISTH. J Thromb Haemost. 2015;13(4):671–675. doi:10.1111/jth.12838
24. Dirweesh A, Siddiqui W, Khan M, Iyer P, Seelagy M. Adenocarcinoma of the lung presenting as thrombotic thrombocytopenic purpura. Respir Med Case Rep. 2017;21:82–83. doi:10.1016/j.rmcr.2017.04.004
25. Zheng XL. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med. 2015;66:211–225. doi:10.1146/annurev-med-061813-013241
26. Nolasco L, Nolasco J, Feng S, Afshar-Kharghan V, Moake J. Human complement factor H is a reductase for large soluble von Willebrand factor multimers--brief report. Arterioscler Thromb Vasc Biol. 2013;33(11):2524–2528. doi:10.1161/ATVBAHA.113.302280
27. Piazza G. Venous thromboembolism and cancer. Circulation. 2013;128:2614–2618. doi:10.1161/CIRCULATIONAHA.113.002702
28. Hisada Y, Mackman N. Cancer cell-derived tissue factor-positive extracellular vesicles: biomarkers of thrombosis and survival. Curr Opin Hematol. 2019;26(5):349–356. doi:10.1097/MOH.0000000000000521
29. Gardiner C, Harrison P, Belting M, et al. Extracellular vesicles, tissue factor, cancer and thrombosis - discussion themes of the ISEV 2014 Educational Day. J Extracell Vesicles. 2015;4:26901. doi:10.3402/jev.v4.26901
30. Geddings JE, Mackman N. Tumor-derived tissue factor–positive microparticles and venous thrombosis in cancer patients. Blood. 2013;122:1873–1880. doi:10.1182/blood-2013-04-460139
31. Shindo K, Aishima S, Ohuchida K, et al. Podoplanin expression in cancer-associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol Cancer. 2013;12(1):168. doi:10.1186/1476-4598-12-168
32. Gagliano N, Celesti G, Tacchini L, et al. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma: characterization in a 3D-cell culture model. World J Gastroenterol. 2016;22(18):4466–4483. doi:10.3748/wjg.v22.i18.4466
33. Payne H, Ponomaryov T, Watson SP, Brill A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood. 2017;129(14):2013–2020. doi:10.1182/blood-2016-09-742999
34. Riedl J, Preusser M, Nazari PM, et al. Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood. 2017;129(13):1831–1839. doi:10.1182/blood-2016-06-720714
35. Mege D, Panicot-Dubois L, Ouaissi M, et al. The origin and concentration of circulating microparticles differ according to cancer type and evolution: a prospective single-center study. Int J Cancer. 2016;138(4):939–948. doi:10.1002/ijc.29837
36. Zwicker JI. Risking thromboembolism: podoplanin and glioma. Blood. 2017;129(13):1742–1743. doi:10.1182/blood-2017-02-763524
37. Urano T, Suzuki Y, Iwaki T, Sano H, Honkura N, Castellino FJ. Recognition of Plasminogen Activator Inhibitor Type 1 as the Primary Regulator of Fibrinolysis. Curr Drug Targets. 2019;20(16):1695–1701. doi:10.2174/1389450120666190715102510
38. Tipoe TL, Wu WKK, Chung L, et al. Plasminogen activator inhibitor 1 for predicting sepsis severity and mortality outcomes: a systematic review and meta-analysis. Front Immunol. 2018;9:1218. doi:10.3389/fimmu.2018.01218
39. Chen N, Ren M, Li R, et al. Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol Cancer. 2015;14:140. doi:10.1186/s12943-015-0418-x
40. Lal I, Dittus K, Holmes CE. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. 2013;15(4):207. doi:10.1186/bcr3425
41. Woei-A-Jin FJ, Tesselaar ME, Garcia Rodriguez P, Romijn FP, Bertina RM, Osanto S. Tissue factor-bearing microparticles and CA19.9: two players in pancreatic cancer-associated thrombosis? Br J Cancer. 2016;115(3):332–338. doi:10.1038/bjc.2016.170
42. Wojtukiewicz MZ, Sierko E, Hempel D, Tucker SC, Honn KV. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017;36(2):249–262. doi:10.1007/s10555-017-9673-1
43. Leal AC, Mizurini DM, Gomes T, et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci Rep. 2017;7(1):6438. doi:10.1038/s41598-017-06893-7
44. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129(10):1357–1367. doi:10.1182/blood-2016-09-741298
45. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768–2776. doi:10.1182/blood-2013-10-463646
46. Lam FW, Cruz MA, Parikh K, Rumbaut RE. Histones stimulate von Willebrand factor release in vitro and in vivo. Haematologica. 2016;101(7):e277–279. doi:10.3324/haematol.2015.140632
47. Mauracher LM, Posch F, Martinod K, et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost. 2018;16(3):508–518. doi:10.1111/jth.13951
48. Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol. 2013;10(10):607–620. doi:10.1038/nrgastro.2013.120
49. Galgano L, Guidetti GF, Torti M, Canobbio I. The controversial role of LPS in platelet activation in vitro. Int J Mol Sci. 2022;23(18):10900. doi:10.3390/ijms231810900
50. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83–92. doi:10.2147/HP.S93413
51. Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36(3):293–303. doi:10.1038/onc.2016.206
52. Rojas A, Delgado-López F, Perez-Castro R, et al. HMGB1 enhances the protumoral activities of M2 macrophages by a RAGE-dependent mechanism. Tumour Biol. 2016;37(3):3321–3329. doi:10.1007/s13277-015-3940-y
53. Tadie JM, Bae HB, Jiang S, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L342–L349. doi:10.1152/ajplung.00151.2012
54. Abdel-Razeq H, Mansour A, Abdulelah H, et al. Thromboembolic events in cancer patients on active treatment with cisplatin-based chemotherapy: another look! Thromb J. 2018;16:2. doi:10.1186/s12959-018-0161-9
55. Rangaswamy C, Mailer RK, Englert H, Konrath S, Renné T. The contact system in liver injury. Semin Immunopathol. 2021;43(4):507–517. doi:10.1007/s00281-021-00876-7
56. Tawil N, Bassawon R, Rak J. Oncogenes and clotting factors: the emerging role of tumor cell genome and epigenome in cancer-associated thrombosis. Semin Thromb Hemost. 2019;45(4):373–384. doi:10.1055/s-0039-1687891
57. Rak J. Cancer genes and blood clots. Blood. 2021;137(15):1996–1997. doi:10.1182/blood.2020009967
58. Heraudeau A, Delluc A, Le Henaff M, et al. Risk of venous thromboembolism in association with factor V Leiden in cancer patients - The EDITH case-control study. PLoS One. 2018;13(5):e0194973. doi:10.1371/journal.pone.0194973
59. Tinholt M, Viken MK, Dahm AE, et al. Increased coagulation activity and genetic polymorphisms in the F5, F10 and EPCR genes are associated with breast cancer: a case-control study. BMC Cancer. 2014;14:845. doi:10.1186/1471-2407-14-845
60. Paulsen B, Skille H, Smith EN, et al. Fibrinogen gamma gene rs2066865 and risk of cancer-related venous thromboembolism. Haematologica. 2020;105(7):1963–1968. doi:10.3324/haematol.2019.224279
61. Ekim M, Ekim H, Yılmaz YK. The prevalence of Factor V Leiden, prothrombin G20210A, MTHFR C677T and MTHFR A1298C mutations in healthy Turkish population. Hippokratia. 2015;19(4):309–313.
62. Ünlü B, Versteeg HH. Cancer-associated thrombosis: the search for the holy grail continues. Res Pract Thromb Haemost. 2018;2(4):622–629. doi:10.1002/rth2.12143
63. Grover SP, Tissue Factor: MN. An essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. 2018;38(4):709–725. doi:10.1161/ATVBAHA.117.309846
64. Patel H, Sun H, Hussain AN, Vakde T. Advances in the diagnosis of venous thromboembolism: a literature review. Diagnostics. 2020;10(6):365. doi:10.3390/diagnostics10060365
65. Jenne CN, Urrutia R, Kubes P. Platelets: bridging hemostasis, inflammation, and immunity. Int J Lab Hematol. 2013;35(3):254–261. doi:10.1111/ijlh.12084
66. Hisada Y, Mackman N. Cancer-associated pathways and biomarkers of venous thrombosis. Blood. 2017;130(13):1499–1506. doi:10.1182/blood-2017-03-743211
67. Riondino S, Ferroni P, Zanzotto FM, Roselli M, Guadagni F. Predicting VTE in cancer patients: candidate biomarkers and risk assessment models. Cancers. 2019;11(1):95. doi:10.3390/cancers11010095
68. Borgen PO, Reikeras O. Prothrombin fragment F1+2 in plasma and urine during total Hip arthroplasty. J Orthop. 2017;14(4):475–479. doi:10.1016/j.jor.2017.08.001
69. Duarte RCF, Ferreira CN, Rios DRA, Reis HJD, Carvalho MDG. Thrombin generation assays for global evaluation of the hemostatic system: perspectives and limitations. Rev Bras Hematol Hemoter. 2017;39(3):259–265. doi:10.1016/j.bjhh.2017.03.009
70. Rimpo K, Tanaka A, Ukai M, Ishikawa Y, Hirabayashi M, Shoyama T. Thrombin-antithrombin complex measurement using a point-of-care testing device for diagnosis of disseminated intravascular coagulation in dogs. PLoS One. 2018;13(10):e0205511. doi:10.1371/journal.pone.0205511
71. Chung HW, Lim JB. Clinical significance of elevated serum soluble CD40 ligand levels as a diagnostic and prognostic tumor marker for pancreatic ductal adenocarcinoma. J Transl Med. 2014;12:102. doi:10.1186/1479-5876-12-102
72. Lord MS, Cheng B, Farrugia BL, McCarthy S, Whitelock JM. Platelet factor 4 binds to vascular proteoglycans and controls both growth factor activities and platelet activation. J Biol Chem. 2017;292(10):4054–4063. doi:10.1074/jbc.M116.760660
73. Gutierrez LS, Gutierrez J. Thrombospondin 1 in metabolic diseases. Front Endocrinol. 2021;12:638536. doi:10.3389/fendo.2021.638536
74. Castaman G, Linari S. Diagnosis and treatment of von Willebrand disease and rare bleeding disorders. J Clin Med. 2017;6(4):45. doi:10.3390/jcm6040045
75. Mussbacher M, Salzmann M, Brostjan C, et al. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front Immunol. 2019;10:85. doi:10.3389/fimmu.2019.00085
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.