Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Thromboelastography with Platelet Mapping Detects Platelet Dysfunction in Patients with Aneurysmal Subarachnoid Hemorrhage with Rebleeding
Authors He Q, Zhou Y, Liu C, Zhang X, Huang N, Wang F, Liu G, Cheng Y, Xie Z
Received 31 August 2019
Accepted for publication 18 November 2019
Published 16 December 2019 Volume 2019:15 Pages 3443—3451
DOI https://doi.org/10.2147/NDT.S229284
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Qiuguang He,1 You Zhou,2 Chang Liu,3 Xiang Zhang,1 Ning Huang,1 Feng Wang,1 Guodong Liu,1 Yuan Cheng,1 Zongyi Xie1
1Department of Neurosurgery, The Second Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 2Department of Critical Care Medicine, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 3Department of Neurology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China
Correspondence: Zongyi Xie
Department of Neurosurgery, The Second Affiliated Hospital of Chongqing Medical University, Chongqing 400010, People’s Republic of China
Email [email protected]
Background: Aneurysmal subarachnoid hemorrhage (aSAH) has high rates of disability and mortality, and aneurysm rebleeding is associated with poor functional outcomes. Thrombelastography with platelet mapping (TEG-PM) measures platelet function; however, it has not yet been researched in aSAH. We aimed to use TEG-PM to detect changes in platelet function in patients with aSAH and the difference in patients with and without rebleeding.
Methods: We retrospectively included patients with aSAH who underwent a TEG-PM test on admission. Rebleeding was diagnosed according to clinical and imaging data. TEG-PM data of patients with unruptured intracranial aneurysms (UIA) were also obtained as controls. Univariate and multivariate logistic regression models were performed to investigate the relationship between the platelet function and rebleeding.
Results: A total of 245 aSAH patients and 32 UIA patients were included in our study. Compared with controls, patients with aSAH demonstrated higher arachidonic acid (AA) and adenosine diphosphate (ADP) inhibition of platelet function (P<0.05). Among them, 27 patients with Hunt-Hess grade IV or V were classified as the severe SAH group. There was a significant correlation between the severe SAH group and the degree of pathway inhibition (P<0.05). Furthermore, AA (Spearman’s r=0.264, P<0.001) and ADP (Spearman’s r=0.183, P=0.004) inhibition were elevated in Hunt–Hess grade-dependent manners. The AA (Spearman’s r=0.169, P=0.008) and ADP (Spearman’s r=0.233, P<0.001) inhibition were also significantly correlated with Fisher grade. Thirty-five patients (14.3%) suffered rebleeding. Rebleeding was significantly correlated with systolic blood pressure (P=0.011), diastolic blood pressure (P=0.008), Hunt–Hess grade (P=0.034), Fisher grade (P=0.015), AA inhibition (P<0.001), and ADP inhibition (P<0.001). Multivariate logistic regression analysis model revealed that both AA (P=0.037) and ADP inhibition (P=0.008) were independent determinants for rebleeding.
Conclusion: TEG-PM may assess platelet dysfunction in patients with aSAH, and the diminished platelet response to ADP and AA may be associated with rebleeding. These findings deserve further investigation.
Keywords: aneurysmal subarachnoid hemorrhage, thromboelastography, platelet function, rebleeding
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) comprises about 5% of all strokes, with high morbidity and mortality.1 If a patient survives the initial aSAH, the major early complication is aneurysmal rebleeding with an incidence of 8% to 23% during the first 72 hrs.2 Urgent aneurysm occlusion after initial SAH is essential to prevent rebleeding, with a reported mortality rate of up to 50%.3 Dissolution of the thrombosis at the original bleeding site is thought to be the major cause of rebleeding. Antifibrinolytic drugs, such as epsilon amino caproic acid, reduce the rate of rebleeding, but this effect is offset by increasing the rate of cerebral ischemia, with no improvement in functional outcome.4 Unlike many previous studies focusing on changes in coagulation and fibrinolysis, platelet function after aSAH has not been paid enough attention.5
Thromboelastography (TEG), a measure of the dynamics of clot formation and dissolution, provides comprehensive real-time analysis of the status of coagulation and fibrinolysis.6 In addition to the standard TEG parameters, platelet function is an available result of the TEG combined with platelet mapping (TEG-PM). TEG-PM has been available since 1948; however, it has not been reported in detecting platelet function in patients with aSAH.7,8
The purpose of the present study was to detect any difference in clotting characteristics and platelet function in patients with aSAH compared with unruptured intracranial aneurysm (UIA), and whether TEG-PM differed in patients with and without rebleeding. We hypothesized that abnormal TEG-PM might correlate with rebleeding. Thus, TEG-PM could be used as a predictor of rebleeding and provide possible direction concerning appropriate therapeutic interventions to prevent rebleeding.
Methods
Patient Population
Patients aged more than 18 years with aSAH or UIA admitted to our hospital and underwent TEG-PM between May 2016 and May 2019 were identified retrospectively. Inclusion criteria of aSAH patients include: patients with spontaneous SAH, which was identified by computerized tomography (CT) or xanthochromia of cerebrospinal fluid; aneurysmal cause of SAH; blood drawn for TEG-PM analysis before rebleeding. Exclusion criteria include: SAH due to trauma, known vascular malformation, hematological system disease; received any hemostatic agents before TEG-PM draw; patients on antiplatelet or anticoagulation therapy before symptom onset. The patients with UIA not receiving anticoagulants or nonsteroidal anti-inflammatory drugs were included in the UIA group as controls. This study was approved by the institutional review board of The Second Affiliated Hospital of Chongqing Medical University, in accordance with the Declaration of Helsinki. All patient data were obtained using a retrospective review of the medical records and were reported without patient identifiers. Therefore, the institutional review board waived the need for patient consent.
Examination and Treatment
We retrospectively reviewed clinical data on admission in our institution, including age, gender, systolic blood pressure (SBP), diastolic blood pressure (DBP), CT-Fisher grade, Hunt–Hess grade, aneurysm location and size, intracranial hematoma and clinical outcomes. We defined severe SAH as Hunt–Hess grade IV or V and nonsevere SAH as Hunt–Hess grade I-III. In our hospital, all patients with aSAH or UIA routinely underwent TEG-PM on admission to evaluate the platelet function for dose adjustment of antiplatelet drugs in the perioperative period of endovascular coiling. All patients were treated according to the standardized aSAH treatment protocol of our hospital, consisting of absolute bed rest until aneurysm treatment, strict blood pressure control, intravenous administration of hemostatic agents and nimodipine and regular assessment of clinical status.9 However, emergency clipping or coiling was not typically performed unless patients presented with space-occupying hemorrhages requiring immediate evacuation, because of the required time for examinations, surgery appointment and expense provision at our institution.
Diagnosis of Rebleeding
Patients who exhibited acute clinical deterioration, such as sudden headache and coma, accompanied with more blood in the subarachnoid cavity, brain parenchyma or ventricular system detected by repeated CT scanning, were diagnosed as rebleeding.10 All rebleeding events occurred before endovascular coiling or clipping were involved in this study.
Blood Sampling
Whole blood drawn from the median cubital vein was collected in citrated and heparinized tubes within 1 hr of the patient’s arrival at the neurosurgery department. The sample was stored at room temperature and taken to TEG and TEG-PM assays within 2 hrs. The platelet counts were detected using routine laboratory assays in the clinical laboratory of our hospital.
TEG and TEG-PM
TEG-PM analyzer (Haemoscope, Model 5000) was used to detect platelet function. The analyzer measured the percent inhibition of the platelet activated by adenosine diphosphate (ADP) and arachidonic acid (AA). This parameter was calculated by comparing the maximum amplitude (MA), representing the viscoelastic strength of the thrombus, created through three TEG channels as follows: 1) The MA reflects thrombin-activated platelets (MAThrombin) measured by a kaolin/Ca2+ activated citrated blood sample; 2) a reptilase and activator F activated heparinized blood sample, representing fibrin-only contribution to the clot strength (MAFibrin); and 3) the heparinized blood mixed with 2-mM ADP (MAADP) or 1-mM AA (MAAA) combined with activator F as platelet agonists reflects platelet responsiveness to ADP and AA.11 The equation 100-((MAADP or MAAA)-MAFibrin)/(MAThrombin-MAFibrin))×100 was used to calculate the percentage of platelet inhibition in response to ADP and AA (Figure 1B).12
 |
Figure 1 Explanation of TEG and TEG-PM. (A) TEG parameters: K, Angle, MA, EPL and LY30. (B), TEG-PM parameters: MAThrombin, MAADP/AA, and MAFibrin. R, reaction time to clot formation; K, clotting time until 20-mm amplitude is achieved; Angle, time to reach a maximum speed of initial clot formation. Abbreviations: MA, maximum amplitude; EPL, percent of clot lysis 30 mins after MA; LY30, percent of amplitude decay 30 mins after MA; AA, arachidonic acid; ADP, adenosine diphosphate. |
Specific TEG parameters were as follows: reaction time to clot formation (R); clotting time until 20-mm amplitude is achieved (K); time to reach maximum speed of initial clot formation (angle); maximum clot strength (MA); percent of clot lysis 30 mins after MA (EPL); percent of amplitude decay 30 mins after MA (LY30) (Figure 1A).13
Statistical Analysis
Continuous variables with normal distributions were summarized using mean±SDs and compared using a two-tailed, unpaired student t-test. The median and the interquartile range for each continuous variable with skewed distributions were calculated, and the Mann–Whitney U-test was used for comparisons. Categorical data were expressed as proportions and compared using χ2-test or Fisher exact test as appropriate. The relationship between severe SAH, rebleeding, and these parameters was investigated. The platelet count and TEG-PM parameters were compared in the following groups: 1) all aSAH versus UIA patients; 2) severe SAH versus UIA patients and 3) severe versus nonsevere SAH patients using the Mann–Whitney U-test. Spearman’s rank correlation coefficient was performed to analyze the correlations between Hunt–Hess grade and the degree of pathway inhibition. The correlations between Fisher grade and the degree of pathway inhibition were also analyzed using Spearman’s rank correlation coefficient. The associations of baseline characteristics and parameters with rebleeding were analyzed using univariable logistic regression models. All variables with P<0.10 were included in the multivariable logistic regression models to evaluate the association of the platelet function and rebleeding. Statistical analyses were performed with SPSS 19.0 software, and a P<0.05 was considered statistically significant.
Results
Baseline Characteristics
A total of 245 patients with aSAH were included in the analysis (Table 1). There were 79 men and 166 women, and the mean patient age was 55.2±10.7 years. The SBP on admission was 145.9±25.4 mmHg, and DBP on admission was 84.3±14.2 mmHg. There were 232 (94.7%) patients who carried anterior circulation aneurysm, and 13 (5.3%) patients carried posterior circulation aneurysm. Hunt–Hess grade was as follows: grade I, 3 cases; grade II, 176 cases; grade III, 39 cases; grade IV, 22 cases; grade V, 5 cases. Fisher grade was as follows: grade I, 2 cases; grade II, 39 cases; grade III, 128 cases; grade IV, 76 cases. Thirty-two patients (13.1%) had intracranial hematoma. A total of 186 patients (75.9%) with aSAH received endovascular coiling or clipping, and the mean timing was 4.4±1.6 days after symptom onset.
 |
Table 1 Patient Characteristics |
Parameters in Controls and aSAH Patients
We also observed 32 patients with UIA. With the estimation of the initial assessment of Hunt–Hess grade, 27 patients (11.0%) were classified as the severe SAH group (Hunt–Hess grade IV or V). The TEG and TEG-PM parameters of individuals with UIA, severe SAH and nonsevere SAH were compared and listed in Table 2. Patients with aSAH demonstrated higher AA (P=0.013) and ADP (P<0.001) inhibition of platelet function than controls. There was no significant difference in platelet count and TEG values of R, K, Angle, MA, EPL and LY30 when comparing aSAH patients with controls (P>0.05). Patients with severe SAH had significantly higher levels of AA and ADP inhibition when compared with controls and patients with nonsevere SAH (P<0.05).
 |
Table 2 Platelet Count and TEG-PM Parameters in Controls and Subarachnoid Hemorrhage Patients Stratified by Neurological Severity |
Hunt–Hess grade and Fisher grade were related to the degree of pathway inhibition, respectively. AA (Spearman’s r=0.264, P<0.001) and ADP (Spearman’s r=0.183, P=0.004) inhibition were elevated in Hunt–Hess grade-dependent manners. The AA (Spearman’s r=0.169, P=0.008) and ADP (Spearman’s r=0.233, P<0.001) inhibition was also significantly correlated with Fisher grade (Figure 2).
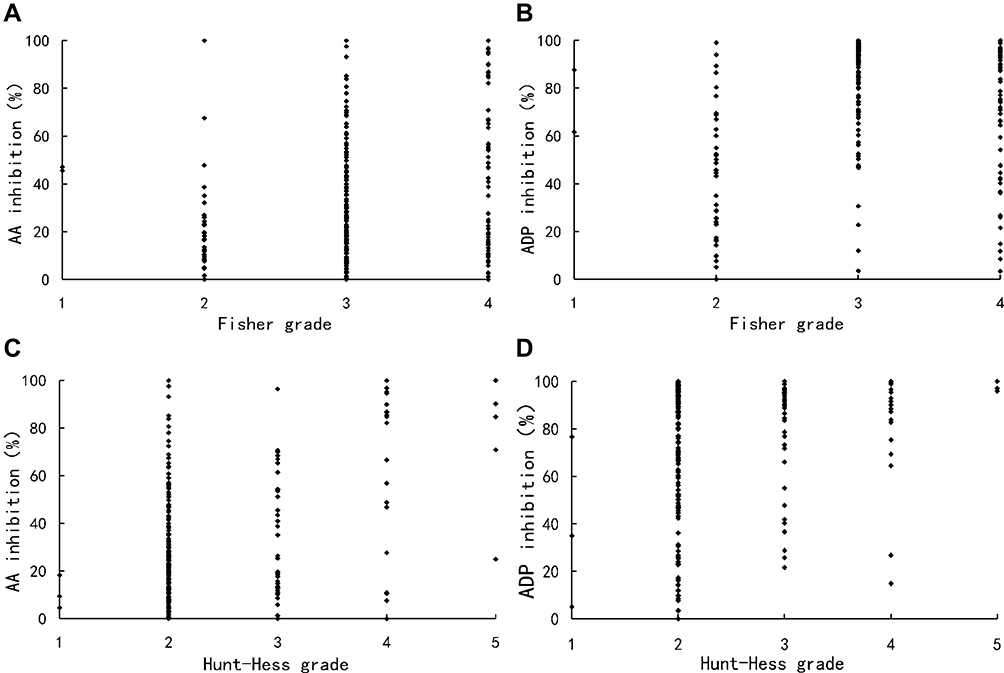 |
Figure 2 Scatter plots showing the correlations between the degree of pathway inhibition and clinical grades. (A) AA inhibition was significantly correlated with Fisher grade (Spearman’s r=0.169, P=0.008); (B) ADP inhibition was significantly correlated with Fisher grade (Spearman’s r=0.233, P<0.001); (C) AA inhibition was significantly correlated with Hunt–Hess grade (Spearman’s r=0.264, P<0.001); (D) ADP inhibition was significantly correlated with Hunt–Hess grade (Spearman’s r=0.183, P=0.004). Abbreviation: AA, arachidonic acid; ADP, adenosine diphosphate. |
Predictors of Rebleeding
Thirty-five patients (14.3%) with aSAH developed rebleeding after admission. The clinical characteristics and parameters of patients with and without rebleeding were compared and are listed in Table 3. Patients with rebleeding were more likely to have higher SBP (P=0.011), DBP (P=0.008), Hunt–Hess grade (P=0.034), Fisher grade (P=0.015), AA (P<0.001) and ADP inhibition (P<0.001). Multivariable logistic regression model analysis showed SBP (P=0.050), AA (P=0.037) and ADP inhibition (P=0.008) were independent risk factors for rebleeding in aSAH patients (Table 4).
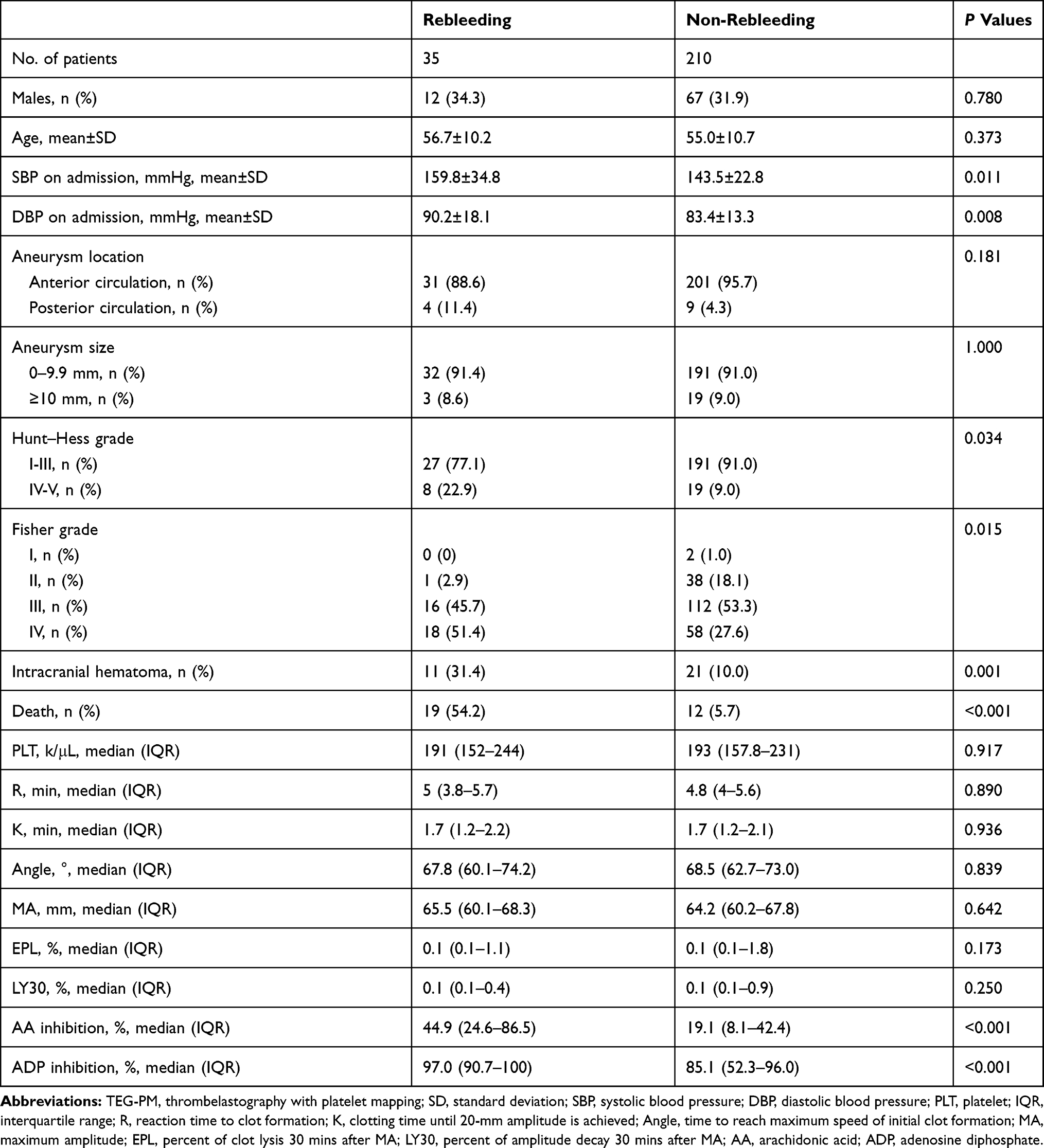 |
Table 3 Comparison of Baseline Demographic, Clinical Characteristics and TEG-PM Parameters Between Patients with and Without Rebleeding |
 |
Table 4 Multivariate Analysis for Risk Factors of Rebleeding |
Discussion
To our knowledge, this is the first study to evaluate coagulation, fibrinolysis status and platelet function as reflected by TEG-PM in aSAH patients. The primary finding of this study is that platelet dysfunction existed in individuals with aSAH and was significantly correlated with Fisher grade and Hunt–Hess grade on admission. Moreover, AA and ADP inhibition was independently associated with rebleeding in patients with aSAH. These results indicate that platelet dysfunction may play a significant role in rebleeding with important clinical value and potential therapeutic applications. Although the risk of platelet dysfunction itself may not be changed, a sufficient awareness of the risk enables us to pay more attention to aSAH patients with high AA and ADP inhibition.
While the phenomenon of AA and ADP receptor dysfunctional in aSAH has not been discovered previously, platelet dysfunction has been observed in traumatic brain injury (TBI) patients.12,14,15 The significant increase percentage of AA and ADP receptor inhibition has been noted in individuals with severe TBI,14 which is consistent with our findings. In addition, we have also demonstrated significant correlations between pathway inhibition and the amount of intracranial blood produced by initial bleeding, measured by the Fisher grade, and the severity of clinical symptoms, measured by the Hunt–Hess grade. This might be a specific response to bleeding.
Although we were unable to clarify the mechanisms underlying the association between aSAH and platelet dysfunction in this present study, several previous studies have explored the potential mechanism of platelet dysfunction in TBI patients.12,14–17 One possible mechanism is that the release of tissue factor, distributed abundantly throughout the brain, vessels, and into circulation secondary to disruption of the blood–brain barrier (BBB) after TBI may result in platelet activation.15 The prolonged refractory state, in which a fraction of activated platelets remain dysfunctional in circulation, might be a consequence of immediate platelet activation.12,14–16 When it comes to aSAH, one conceivable process is that the initial bleeding might be a result of mechanical injury to the cerebral vessel endothelium and brain, regardless of intracranial hematoma formation.18,19 Furthermore, the BBB disruption may also induce the release of tissue factor,20,21 leading to platelet activation.22–24 This hypothesis is supported by the observation that patients with high Fisher grade suffered increasing AA and ADP pathway inhibition. However, we did not measure those activation markers; further research is needed to explore the mechanism of AA and ADP receptor dysfunctional in aSAH.
Additionally, the significant AA receptor inhibition coupled to the level of ADP receptor dysfunction presents a clinical scenario similar to that of a patient taking a combination of aspirin and clopidogrel.25 Therefore, we explored the association of platelet dysfunction with rebleeding. This study revealed AA and ADP inhibition as independent determinants for rebleeding. Conversely, there was no difference in platelet count, activation of coagulation or fibrinolysis between patients with and without rebleeding.
The initial step of the coagulation cascade after aneurysm rupture is the release of tissue factor from the damaged brain tissue and the injured cerebral vessel endothelium. In addition, the sudden increase of intracranial pressure balances the internal and external pressures of blood vessels.26 Thrombin is activated by the coagulation cascade, which leads to platelet and fibrin aggregation in the damaged vessel, working as an immobile hemostatic plug and thereby preventing further hemorrhage.3 Following plug formation, the accumulation of activated platelets continues for quite a few days to avoid rebleeding.27 The ADP released by damaged red blood cell and thromboxane A2 plays an important part in the activation of platelets.28 Meanwhile, the deposition of fibrin and tissue ischemia ends with the activation of the fibrinolytic system. Hence, a lower strength of hemostatic plug in patients with high ADP or AA inhibition may be an explanation for the more rate of rebleeding especially with dysphoria and high BP.
Coagulation and fibrinolysis status after aSAH have been measured by the prothrombin time, activated partial thromboplastin time, D-dimer and thrombin-antithrombin in previous studies.29 However, these tests were unable to measure the dynamics of clot formation and dissolution. Furthermore, there was no difference in platelet count, activation of coagulation or fibrinolysis between patients with and without rebleeding in the current study. Juvela and Kaste30 found platelet aggregability was weakened during the early stage in patients with rebleeding after aSAH, but the sample detection was not widely used in various facilities in the clinical setting because of the complicated manual manipulation. On the contrary, TEG and TEG-PM can automatically detect the status of coagulation, fibrinolysis and platelet function rapidly, so it is more convenient in clinical practice.
With the remarkable progress that has been achieved in the diagnosis and treatment of aSAH, urgent repair of the ruptured aneurysm by neurosurgical clipping or endovascular coiling is feasible to reduce rebleeding. However, nonmodifiable factors such as transfer time, availability of neurovascular centers, serious underlying diseases, expense provision and surgery appointments may contribute to treatment delay.31 Moreover, poor-grade aSAH patients are often considered for delayed surgery. Once, for whatever reason, surgery is delayed, rebleeding is an unpredictably significant threat. TEG-MP as a commonly used test would be sufficiently precise to identify the patients at highest risk for developing rebleeding; these patients should receive intensive treatments such as strict control of blood pressure, hemostatic drugs and ultra-early surgical treatment. In addition, it is still unknown whether the application of antifibrinolytic therapy is effective.4 Our study implicates that early platelet transfusion may be a potential intervention to prevent rebleeding.
Several limitations in this present research should be considered. First, we made a single test on admission. The dynamic of ADP and AA inhibition was lacking, which might explain the phenomenon that rebleeding frequently occurs within the first 6 hrs after the initial bleed.31 Second, some causes, such as transfer to our hospital, may delay detection. The inconsistent test time might also affect the values. Last, this was a retrospective study with a small sample size, so further studies are required to support our findings.
In conclusion, TEG-PM may detect platelet dysfunction in patients with aSAH. Moreover, the degree of pathway inhibition was significantly correlated with Hunt–Hess grade and Fisher grade on admission, respectively. Additionally, the diminished platelet response to ADP and AA may be associated with rebleeding. These findings potentially provide a chance for identification and treatment of aSAH patients at high risk of rebleeding in clinical practice.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Ingall T, Asplund K, Mahonen M, Bonita R. A multinational comparison of subarachnoid hemorrhage epidemiology in the WHO MONICA stroke study. Stroke. 2000;31:1054–1061. doi:10.1161/01.str.31.5.1054
2. van Donkelaar CE, Bakker NA, Veeger NJ, et al. Predictive factors for rebleeding after aneurysmal subarachnoid hemorrhage: rebleeding aneurysmal subarachnoid hemorrhage study. Stroke. 2015;46:2100–2106. doi:10.1161/STROKEAHA.115.010037
3. Larsen CC, Astrup J. Rebleeding after aneurysmal subarachnoid hemorrhage: a literature review. World Neurosurg. 2013;79:307–312. doi:10.1016/j.wneu.2012.06.023
4. Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. 2013;8:CD001245. doi:10.1002/14651858.CD001245.pub2
5. Ray B, Pandav VM, Mathews EA, et al. Coated-platelet trends predict short-term clinical outcome after subarachnoid hemorrhage. Transl Stroke Res. 2018;9:459–470. doi:10.1007/s12975-017-0594-7
6. Wikkelso A, Wetterslev J, Moller AM, Afshari A. Thromboelastography (TEG) or rotational thromboelastometry (ROTEM) to monitor haemostatic treatment in bleeding patients: a systematic review with meta-analysis and trial sequential analysis. Anaesthesia. 2017;72:519–531. doi:10.1111/anae.13765
7. Wohlauer MV, Moore EE, Thomas S, et al. Early platelet dysfunction: an unrecognized role in the acute coagulopathy of trauma. J Am Coll Surg. 2012;214:739–746. doi:10.1016/j.jamcollsurg.2012.01.050
8. Ramchand P, Nyirjesy S, Frangos S, et al. Thromboelastography parameter predicts outcome after subarachnoid hemorrhage: an exploratory analysis. World Neurosurg. 2016;96:215–221. doi:10.1016/j.wneu.2016.04.002
9. Connolly ES
10. Yin C, Huang GF, Ruan J, He ZZ, Sun XC. The APOE promoter polymorphism is associated with rebleeding after spontaneous SAH in a Chinese population. Gene. 2015;563:52–55. doi:10.1016/j.gene.2015.03.009
11. Bochsen L, Wiinberg B, Kjelgaard-Hansen M, Steinbruchel DA, Johansson PI. Evaluation of the TEG platelet mapping assay in blood donors. Thromb J. 2007;5:3. doi:10.1186/1477-9560-5-3
12. Castellino FJ, Chapman MP, Donahue DL, et al. Traumatic brain injury causes platelet adenosine diphosphate and arachidonic acid receptor inhibition independent of hemorrhagic shock in humans and rats. J Trauma Acute Care Surg. 2014;76:1169–1176. doi:10.1097/TA.0000000000000216
13. Kawano-Castillo J, Ward E, Elliott A, et al. Thrombelastography detects possible coagulation disturbance in patients with intracerebral hemorrhage with hematoma enlargement. Stroke. 2014;45:683–688. doi:10.1161/STROKEAHA.113.003826
14. Davis PK, Musunuru H, Walsh M, et al. Platelet dysfunction is an early marker for traumatic brain injury-induced coagulopathy. Neurocrit Care. 2013;18:201–208. doi:10.1007/s12028-012-9745-6
15. Sillesen M, Johansson PI, Rasmussen LS, et al. Platelet activation and dysfunction in a large-animal model of traumatic brain injury and hemorrhage. J Trauma Acute Care Surg. 2013;74:1252–1259. doi:10.1097/TA.0b013e31828c7a6b
16. Kutcher ME, Redick BJ, McCreery RC, et al. Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg. 2012;73:13–19. doi:10.1097/TA.0b013e318256deab
17. Herbert JP, Guillotte AR, Hammer RD, Litofsky NS. Coagulopathy in the setting of mild traumatic brain Injury: truths and consequences. Brain Sci. 2017;7. doi:10.3390/brainsci7070092
18. Larsen CC, Hansen-Schwartz J, Nielsen JD, Astrup J. Blood coagulation and fibrinolysis after experimental subarachnoid hemorrhage. Acta Neurochir (Wien). 2010;152:1577–1581. doi:10.1007/s00701-010-0699-1
19. Friedrich V, Flores R, Muller A, Sehba FA. Escape of intraluminal platelets into brain parenchyma after subarachnoid hemorrhage. Neuroscience. 2010;165:968–975. doi:10.1016/j.neuroscience.2009.10.038
20. Fujimoto M, Shiba M, Kawakita F, et al. Deficiency of tenascin-C and attenuation of blood-brain barrier disruption following experimental subarachnoid hemorrhage in mice. J Neurosurg. 2016;124:1693–1702. doi:10.3171/2015.4.JNS15484
21. Doczi T, Joo F, Adam G, Bozoky B, Szerdahelyi P. Blood-brain barrier damage during the acute stage of subarachnoid hemorrhage, as exemplified by a new animal model. Neurosurgery. 1986;18:733–739. doi:10.1227/00006123-198606000-00010
22. Frontera JA, Provencio JJ, Sehba FA, et al. The role of platelet activation and inflammation in early brain injury following subarachnoid hemorrhage. Neurocrit Care. 2017;26:48–57. doi:10.1007/s12028-016-0292-4
23. Frontera JA, Aledort L, Gordon E, et al. Early platelet activation, inflammation and acute brain injury after a subarachnoid hemorrhage: a pilot study. J Thromb Haemost. 2012;10:711–713. doi:10.1111/j.1538-7836.2012.04651.x
24. Perez P, Lukaszewicz AC, Lenck S, Nizard R, Drouet L, Payen D. Platelet activation and aggregation after aneurysmal subarachnoid hemorrhage. BMC Neurol. 2018;18:57. doi:10.1186/s12883-018-1062-z
25. Collyer TC, Gray DJ, Sandhu R, Berridge J, Lyons G. Assessment of platelet inhibition secondary to clopidogrel and aspirin therapy in preoperative acute surgical patients measured by thrombelastography platelet mapping. Br J Anaesth. 2009;102:492–498. doi:10.1093/bja/aep039
26. Conzen C, Becker K, Albanna W, et al. The acute phase of experimental subarachnoid hemorrhage: intracranial pressure dynamics and their effect on cerebral blood flow and autoregulation. Transl Stroke Res. 2018. doi:10.1007/s12975-018-0674-3
27. Haining JL, Clower BR, Honma Y, Smith RR. Accumulation of intimal platelets in cerebral arteries following experimental subarachnoid hemorrhage in cats. Stroke. 1988;19:898–902. doi:10.1161/01.str.19.7.898
28. Siddiqui TI, Kumar KSA, Dikshit DK. Platelets and atherothrombosis: causes, targets and treatments for thrombosis. Curr Med Chem. 2013;20:2779–2797. doi:10.2174/0929867311320220004
29. Boluijt J, Meijers JC, Rinkel GJ, Vergouwen MD. Hemostasis and fibrinolysis in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: a systematic review. J Cereb Blood Flow Metab. 2015;35:724–733. doi:10.1038/jcbfm.2015.13
30. Juvela S, Kaste M. Reduced platelet aggregability and thromboxane release after rebleeding in patients with subarachnoid hemorrhage. J Neurosurg. 1991;74:21–26. doi:10.3171/jns.1991.74.1.0021
31. Boogaarts HD, van Lieshout JH, van Amerongen MJ, et al. Aneurysm diameter as a risk factor for pretreatment rebleeding: a meta-analysis. J Neurosurg. 2015;122:921–928. doi:10.3171/2014.12.JNS14931
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.