")
Back to Journals » The Application of Clinical Genetics » Volume 15
Three-Decade Successive Establishment of Care for Women/Girls from Families with Haemophilia
Authors Chuansumrit A , Sasanakul W, Sirachainan N , Santiwatana S, Kadegasem P, Wongwerawattanakoon P, Tungbubpha N, Chantaraamporn J
Received 21 July 2022
Accepted for publication 13 September 2022
Published 1 October 2022 Volume 2022:15 Pages 133—143
DOI https://doi.org/10.2147/TACG.S381683
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Ampaiwan Chuansumrit,1 Werasak Sasanakul,1 Nongnuch Sirachainan,1 Suttikarn Santiwatana,1 Praguywan Kadegasem,1 Pakawan Wongwerawattanakoon,2 Noppawan Tungbubpha,1 Juthamard Chantaraamporn1
1Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 2Department of Nursing, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Ampaiwan Chuansumrit, Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, 10400, Thailand, Tel +66 2 2011749, Fax +66 2 2011748, Email [email protected]
Objective: The study aimed to report a 3-decade successive establishment of care for women/girls from families with haemophilia.
Methods: A retrospective analysis was conducted on 462 women/girls from 243 families from 1987 to 2021.
Results: Combining phenotypic analysis of coagulation factor and genotypic analysis of either linkage analysis or mutation detection confirmed the status of all obligate haemophilia carriers (A118, B19). For potential carrier, 159 proven carriers (A130, B29) and 146 noncarrier status (A126, B20) were diagnosed except 20 potential carriers (A16, B4). Only 54 prenatal diagnoses were requested resulting in normal males (n = 21), males with haemophilia A (n = 12) and females with either normal or carrier status (n = 21). Additionally, 40 women/girls with haemophilia carrier received a diagnosis of severe haemophilia A with Turner’s syndrome (n = 2) and mild haemophilia (A31, B7). The skewed X-chromosome inactivation of the nonmutant factor VIII/IX carrying X-chromosome of 8% (2/25) was found in mild haemophilia. Factor concentrate and desmopressin are prescribed for these affected women/girls. The response of women/girls with either haemophilia carrier or haemophilia was amazement with their religious beliefs and cultural acceptance.
Conclusion: Appropriate care for women/girls from families with haemophilia concerning diagnosis and management of haemophilia and carrier has been successively established.
Keywords: women and girls, families with haemophilia, haemophilia carrier, severe haemophilia, mild haemophilia
Introduction
Haemophilia is considered one of the rare diseases inherited by the X-linked recessive pattern. Haemophilia is mainly found among males while females are carriers. Each patient with haemophilia may have several female relatives at risk of being haemophilia carriers. In 2021, the Scientific Standard Committee of the International Society of Thrombosis and Hemostasis (ISTH) defined haemophilia carrier in two groups depending on the levels of factor VIII or factor IX activity (FVIII:C, FIX:C): group 1 with level <0.4 IU/mL, categorised as women/girls with mild haemophilia (>0.05-<0.4 IU/mL), moderate haemophilia (0.01–0.05 IU/mL) and severe haemophilia (<0.01 IU/mL) and group 2 with level ≥0.4 IU/mL, categorised by bleeding phenotype as symptomatic haemophilia carrier and asymptomatic haemophilia carrier.1 Women/girls with haemophilia carrier status and haemophilia were at risk to exhibit excessive bleeding from heavy menstrual bleeding, postpartum haemorrhage, trauma and postoperative bleeding from dental and surgical procedures.2–4 Apart from counselling among the patients and family members especially immediate female relatives of mothers, sisters and daughters, the facility to accurately diagnose haemophilia carrier status and haemophilia using laboratory investigation of the phenotypic and genotypic analyses are essentially required. Women/girls with haemophilia are commonly found with possible genetic mechanisms involving abnormalities of the X-chromosome in structure and number,5,6 homozygosity for the factor VIII gene mutation when consanguinity is present,7,8 concomitant occurrence of two de novo factor VIII gene mutations9 and the most frequently found is among heterozygous females with nonrandom X-chromosome inactivation (XCI).10–15
This study aimed to present a three-decade successive establishment of care for women/girls from families with haemophilia. The set-up investigations to diagnose haemophilia carrier status and haemophilia, available medical services and prenatal diagnosis illustrated the recognition and awareness among health care personnel in handling these women/girls at risk. Also, the impact of religious beliefs and cultural acceptance of women with a diagnosis of either haemophilia carrier or haemophilia were presented.
Materials and Methods
A retrospective analysis was conducted of women/girls from families with haemophilia attending the Division of Hematology, Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand from 1987 to 2021. The severity of haemophilia is based on the FVIII:C and FIX:C levels: severe <0.01 IU/mL, moderate 0.01 to 0.05 IU/mL and mild >0.05 to <0.4 IU/mL.16 The study was approved by the Faculty Ethics Committee Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand (COA.MURA2020/1091) and conducted following the Declaration of Helsinki. Written informed consent was obtained from subjects and parents.
The haematologists provided counseling in a non-directional way to every patient with newly diagnosed haemophilia and their family members using face-to-face interactive communication concerning the natural history of haemophilia, inheritance, establishment of a family tree, laboratory investigation of coagulation factor assay and DNA analysis, treatment, available resources, prevention and possible alternatives. Then, they had their blood drawn for the phenotypic analysis of FVIII:C, vWF:Ag and FIX:C accordingly. EDTA blood were stored for the subsequent genotypic analysis supported by the Faculty Foundation, namely, Ramathibodi Foundation, from 1990 to 2017. Backing included DNA extraction, linkage analysis, mutation detection, heteroduplex screening before sequencing, intron 22 and intron 1 inversion on the factor VIII gene and all exons of FVIII/IX genes sequenced and X-chromosome inactivation (XCI). A verified translated-Thai self-assessment of bleeding scores has been used since 2018.
Methods
Coagulation Factors
The subjects’ whole blood was mixed with 3.2% sodium citrate at the ratio of 9:1, immediately placed on ice and centrifuged at 3000 rpm for 15 minutes to obtain platelet poor plasma. The FVIII:C, FIX:C and vWF:Ag were assayed within 4 hours or in the plasma stored at −70°C for less than six months before testing. Initially, the FVIII:C and FIX:C levels were determined using a manual tilt tube technique based on one-stage assay17 using in-house factor VIII and factor IX deficient plasma and switched to using an automated coagulometer with commercial factor VIII and factor IX deficient plasma in 1996. Similarly, vWF antigen was measured using a modified electroimmunoassay18 and switched to the ELISA technique in 1996.19
The carrier status of haemophilia A and B was diagnosed using the levels of FVIIIC or FIX:C ≤0.5 IU/mL, respectively. For haemophilia A carrier, the additional criteria of the average ratio of two successive results of FVIII:C and vWF:Ag less than 0.8 was used before 199620 and the cutoff ratio was revised to 0.7 in 1996.21
DNA Extraction
In 1990, DNA was initially extracted from 10 mL of EDTA blood using proteinase-K digestion followed by phenol/chloroform extraction.22 Starting in 1994, an in-house salting-out DNA extraction23 was modified from a commercial kit. Also, EDTA blood or buffy coat for DNA extraction could be transported to the comprehensive centre by mail without ice.24,25
Linkage Analysis
The linkage analysis associated with intron 18 of the factor VIII gene and 5’ flanking region of the factor IX gene using restriction enzymes of Bcl I and Mse I were performed among patients with haemophilia A and B as well as women/girls at risk in their families, respectively using the previous published primers26 starting in 1991.
Mutation Detection
Starting in 1995, mutation detection was initially performed among some patients with haemophilia A and B. In 2008, intron 22 and intron 1 inversion was the initial mutation detection for patients with severe and moderate haemophilia A using the inverse-shifting PCR technique.27 For patients with haemophilia A without intron 22 and intron 1 inversion and patients with haemophilia B, all exons were sequenced by direct Sanger sequencing.28
Heteroduplex Screening by Conformation Sensitive Gel Electrophoresis (CSGE)
Starting in 1998, the heteroduplex screening by CSGE before sequencing was performed using the method of Ganguly et al.29 The coding regions, intron/exon boundaries and 5’ and 3’ regions of the factor VIII and IX genes of the patient and a normal control were amplified in 33 and 9 separate reactions using the previously published primers,30,31 respectively. The individual PCR product of the patient was mixed with the control to allow heteroduplex/homoduplex formation. Finally, the heteroduplex/homoduplex was run on the denaturing acrylamide gel and the bands were visualized using silver staining. Abnormal CSGE of the specific site displaying more than one band was subsequently sequenced to identify the mutation.
Fetal Specimen Samples in Prenatal Diagnosis (PND)
A multidisciplinary team of haematologists, obstetricians and laboratory scientists planned for each PND starting in 1996. The PND was performed among pregnant women with obligate carrier, proven potential carrier and untested potential carrier status who were at risk of bearing a haemophilia son with moderate and severe degrees. The complete genotypic analysis was performed in the index patient and woman at risk. All women had their levels of FVIII:C or FIX:C measured before PND procedures. Factor VIII or IX concentrate was administered to those with levels of FVIII:C or FIX:C less than 0.6 IU/mL.
Before PND procedures, each woman underwent an ultrasonography examination to confirm the gestational age and to perform a complete fetal anatomic survey. The selection of fetal sampling methods depended on the gestational age under ultrasonography guidance.32 Chorionic villus samples were obtained by transcervical route for women with 11 to 14 weeks of gestation. Amniocentesis was performed for those with 16 to 20 weeks of gestation and transabdominal cordocentesis was performed for those with 20 to 24 weeks of gestation. The purity of fetal blood was checked using the acid elution test.33 The amplification for amelogenin was initially performed to determine the fetal sex,34 then amplification of the informative RFLP sites, intron 22 inversion and other specific mutations were also performed, accordingly.
X-Chromosome Inactivation (XCI)
In 2017, the degree of XCI was determined using the modified methylation-specific PCR assay.35 Briefly, DNA was double-digested using methylation-sensitive restriction enzymes, HpaII and HhaI. Then amplification of the polymorphic CAG repeats in the androgen receptor gene was performed. The products were separated using capillary electrophoresis and the peak intensity was analyzed. The ratio of the intensity of the shorted allele to the summation of the intensities of both alleles was calculated and converted to percentage of the allele with stronger intensity (% of XCI). A degree ≥80% was defined as having skewed XCI. By matching the CAG repeat size to those of the male relatives with haemophilia, the mutation carrying X chromosome could be distinguished from the nonmutant one.
Statistical Analysis
Student’s paired t-test was used for paired samples. The Chi-Square or Fisher’s exact test was used for discrete data while the Mann–Whitney U or Wilcoxon Signed rank test was used for continuous data. The level of significance was set at p ≤0.05.
Results
A total of 462 women/girls from 243 haemophilia families (A199, B44) defined as known (A87, B13) and without family history as sporadic (A112, B31) were retrospectively studied. One to eight subjects from each family enrolled in the study are shown in Table 1. Altogether, 390 and 72 women/girls were from families with haemophilia A and B, respectively, defined as 137 obligate (A118, B19) and 325 potential (A272, B53) carriers. Most were immediate female relatives of mothers and sisters of patients with haemophilia as shown in Table 2.
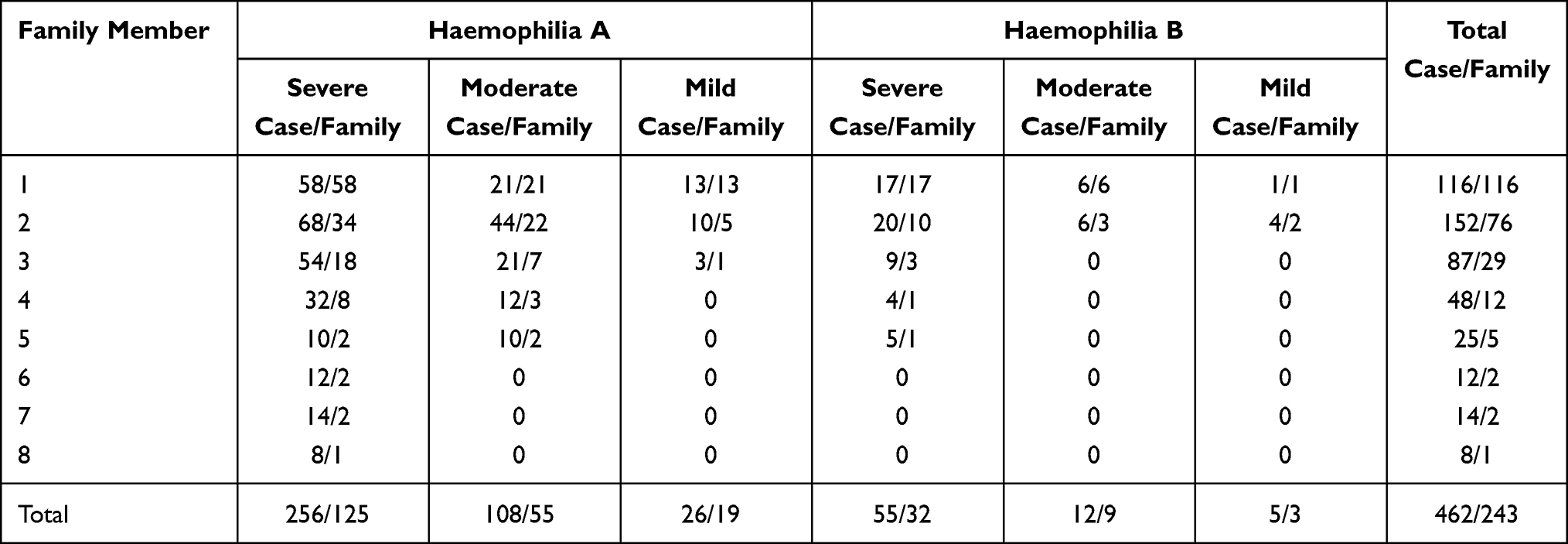 |
Table 1 Number of Women/Girls and Families Enrolled in the Study |
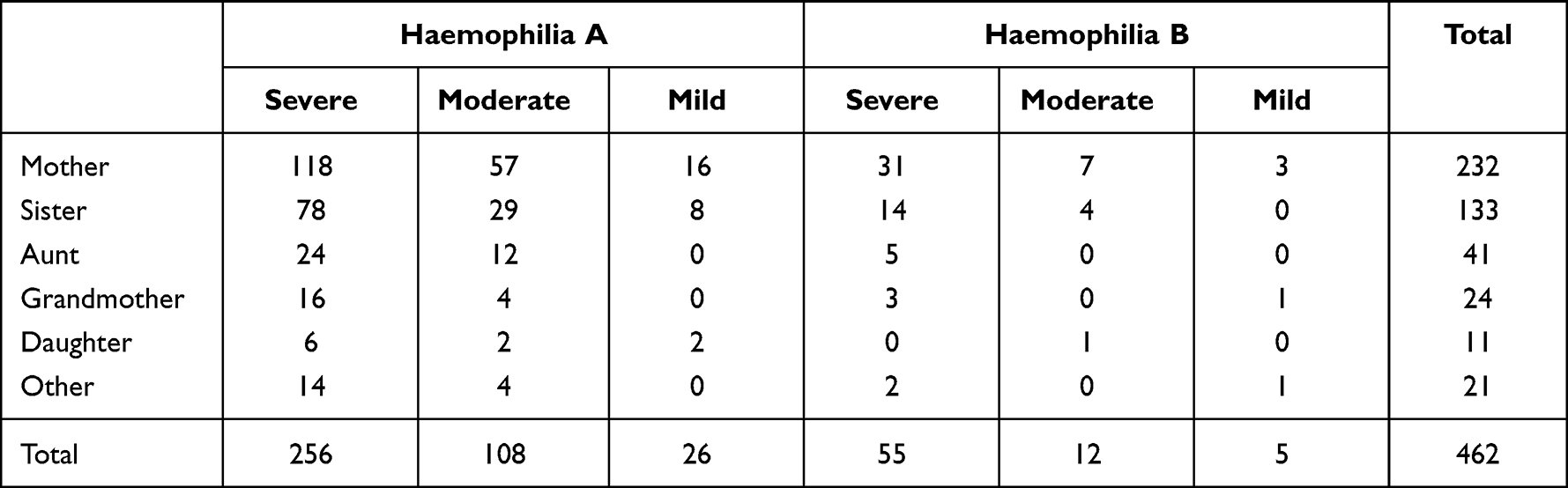 |
Table 2 Relationship between Women/Girls at Risk and Patients |
Diagnosis of Haemophilia Carrier Status Among Women/Girls
A combination of phenotypic analysis of coagulation factor and genotypic analysis of either linkage analysis or mutation detection was used to determine haemophilia carrier status. The results confirmed the status of all obligate carriers (A118, B19). However, one obligate haemophilia A carrier had no mutation of c.3637 del A, p.Ile 1213 Phe fs*5 detected. She might have had somatic or gonadal mosaicism. For potential carrier, 159 (A130, B29) proven carrier status and 146 (A126, B20) noncarrier status were diagnosed but 20 potential carriers (A16, B4) with only phenotypic analysis could not have their carrier status determined. The factor VIII gene mutations revealed prominent intron 22 inversion (53.9%) followed by frameshift (13.9%), missense (13.9%), nonsense (10.0%), large structural change >50 base pairs (3.8%), intron 1 inversion (1.5%), splice site change (1.5%) and unidentified mutation (1.5%) while the factor IX gene mutations revealed mainly missense (61.0%) followed by nonsense (19.5%), frameshift (7.3%), promoter site change (5.0%), splice site change (2.4%), small structural change <50 bp (2.4%) and unidentified mutation (2.4%).
Upon informing of the carrier status, the response of most women was amazement because this had been their feared expectation. The decision of the carrier was initially evaluated among 54 married women with at least one son with haemophilia and seven single women at risk.36 Nineteen of the 54 married women with an additional normal son and daughter decided to undergo sterilisation. The remaining women had different attitudes including adopting a daughter (n = 1), taking oral contraceptive pills (n = 13), and no action concerning birth control (n = 21). Eleven of these 21 women challenged the pregnancy. They had six daughters, three normal sons and two sons with haemophilia. Nine women with an additional normal son and daughter underwent sterilisation while two women with the second son with haemophilia remained on oral contraceptive pills. Ultimately, only eight of 54 married women (14.8%) on oral contraceptive pills requested to have PND in their next pregnancy. Divorce was found in five of 54 married women (9.2%). For single women, four proven potential carriers and one noncarrier decided to remain single. They stated that they had already received the stigma of being labeled a haemophilia carrier after confirming haemophilia in their elder or younger brother. The remaining two noncarriers have not decided yet.
Phenotypic and Genotypic Analyses
Using the ratio of FVIII:C and vWF:Ag at 0.8 and 0.7 in the laboratory tests determined before and after 1996 as cutoff criteria for haemophilia A carrier, obligate carriers with positive result (63/83=75.9%) were significantly more numerous than those potential carriers (79/166=47.6%) with a p-value of 0.0001. Then phenotypic analysis of FVIII ≤0.5 IU/mL and indicated cutoff ratio of FVIII:C/vWF:Ag for diagnosing haemophilia A carrier status was compared with the mutation identification among 73 potential carriers. The results revealed a sensitivity of 89.6% (26/29) and specificity of 84.1% (37/44). Importantly, the false negative rate of the coagulation factor assay was 9.6% (7/73) found among two mothers and three elder sisters with intron 22 inversion, one mother with c.5751_5752 insTA, p.Cys1918 fs*29 and one aunt with c.4794G>T, p.Glu1598Asp. Additionally, the false positive rate of the coagulation factor assay was 4.1% (3/73) found in one aunt and two elder sisters without mutations of intron 22 inversion, c.6380 del A, p.Asp2127Val fs*16 and c.4794G>T, p.Glu1598Asp found in the index patients, respectively. However, these specific mutations should be screened in further pregnancies among these women.
Prenatal Diagnosis
A total of 54 pregnancies among 49 women at risk of offspring with moderate (A9, B1) and severe haemophilia (A33, B6) requested the PND from 1996 to 2021. Forty-two PND were reported in 2016.32 They were obligate (A10, B3), proven potential (A28, B4) and untested potential carriers (A4). The mean ±SD age of pregnant women was 29.8±5.0 years. Twenty-one women were primigravida and 33 were multigravida ranging from the second to the fourth pregnancies (2nd=25, 3rd=6, 4th=2). Forty-nine women received one episode of PND while one and three women received three and two episodes of PND, respectively. They comprised relatives of the patients including mothers (n = 25), sisters (n = 18), daughters (n = 3), aunts (n = 2) and cousins (n = 1).
Ultrasonography was initially performed among 51 pregnant women and the noninvasive prenatal test for fetal sex was performed among three pregnant women. Sixteen women with female offspring and two women with male offspring refused to have further PND procedures. The postnatal testing revealed males with haemophilia (n = 2), normal female (n = 4) and females with either normal or potential carrier status (A9, B3). The remaining 36 pregnancies continued PND by chorionic villi sampling (n = 16), amniocentesis (n=2), and cordocentesis (n = 18) for fetal FVIII:C or FIX:C. Linkage analysis employed restriction enzymes of Bcl I, Mse I and Taq I (n = 13), and intron 22 inversion (n = 4) and other mutation (n = 1) were searched. The results revealed normal males (n = 21), males with haemophilia A (n = 10), normal females (n = 3), and females with haemophilia A carrier status (n = 2). Therapeutic abortion was performed among nine women with a haemophilia son except one woman who continued her pregnancies and had birth control after delivering a haemophilia son safely at hometown hospital. However, one woman with a normal male offspring, affected by Down’s syndrome, decided to terminate her pregnancy.
Diagnosis of Women/Girls with Haemophilia
Forty of 296 women/girls (13.5%) with 137 obligate (A118, B19) and 159 proven potential haemophilia carrier status (A130, B29) received a diagnosis of severe haemophilia A (n = 2) by FVIII:C less than 0.01 IU/mL and mild haemophilia (A31, B7) by FVIII:C and FIX:C levels >0.05 to <0.4 IU/mL. No consanguineous marriage among their parents was known. Twenty-seven females were from severe haemophilia (A23, B4), ten from moderate haemophilia (A8, B2) and three from mild haemophilia (A2, B1) families. Fifteen females were obligate carriers (A13, B2) while 25 females were proven potential carriers (A20, B5).
Two girls from known severe haemophilia A families received a diagnosis of severe haemophilia A and a cytogenetic study revealed 45XO of Turner’s syndrome at the ages of 1 year and 7 months and 1 year and 6 months, respectively. One girl showed clinical features on physical examination of webbing of the neck, low hairline, cubitus valgus and broad-spaced nipples while another girl did not. However, both exhibited frequent bruises since 1 month of age and haemarthrosis when they started to crawl, similar to their elder brothers with haemophilia A. They were born to obligate carrier mothers and the linkage analysis of RFLPs at the intron 18 using restriction enzyme Bcl I and variable dinucleotide tandem repeats at intron 22 of the factor VIII gene revealed that patients inherited only the maternal X-chromosome without the paternal X-chromosome. The factor VIII mutations revealed intron 22 inversion and deletion exon 22, respectively.
For women with mild haemophilia, their mean±SD levels of FVIII:C and FIX:C were 0.31±0.07 and 0.25±0.11 IU/mL, respectively. No significant difference was found among the levels of FVIII:C and FIX:C, either of obligate and potential carriers or in severe, moderate and mild haemophilia families. All women/girls with mild haemophilia comprised heterozygous factor VIII and factor IX gene mutations, accordingly except unidentified mutation (n = 2). Further, no mutation was detected with suspected somatic or gonadal mosaicism (n = 1) as shown in Table 3. The XCI determination among 26 (A22, B4) of 38 women with mild haemophilia was performed. Skewed XCI (≥80%) was found among 6 (A3, B3) of 26 studied women (23.1%), similar to a related study.37 Unexpectedly, when the mutant factor VIII/IX carrying X-chromosome among the studied women was specifically identified by matching their X-chromosomes with related male patients with haemophilia, skewed inactivation of the nonmutant factor VIII/IX carrying X-chromosome was found among only two of 25 women (8%) excluding one woman with unidentified factor IX mutation (patient No.40 in Table 3).
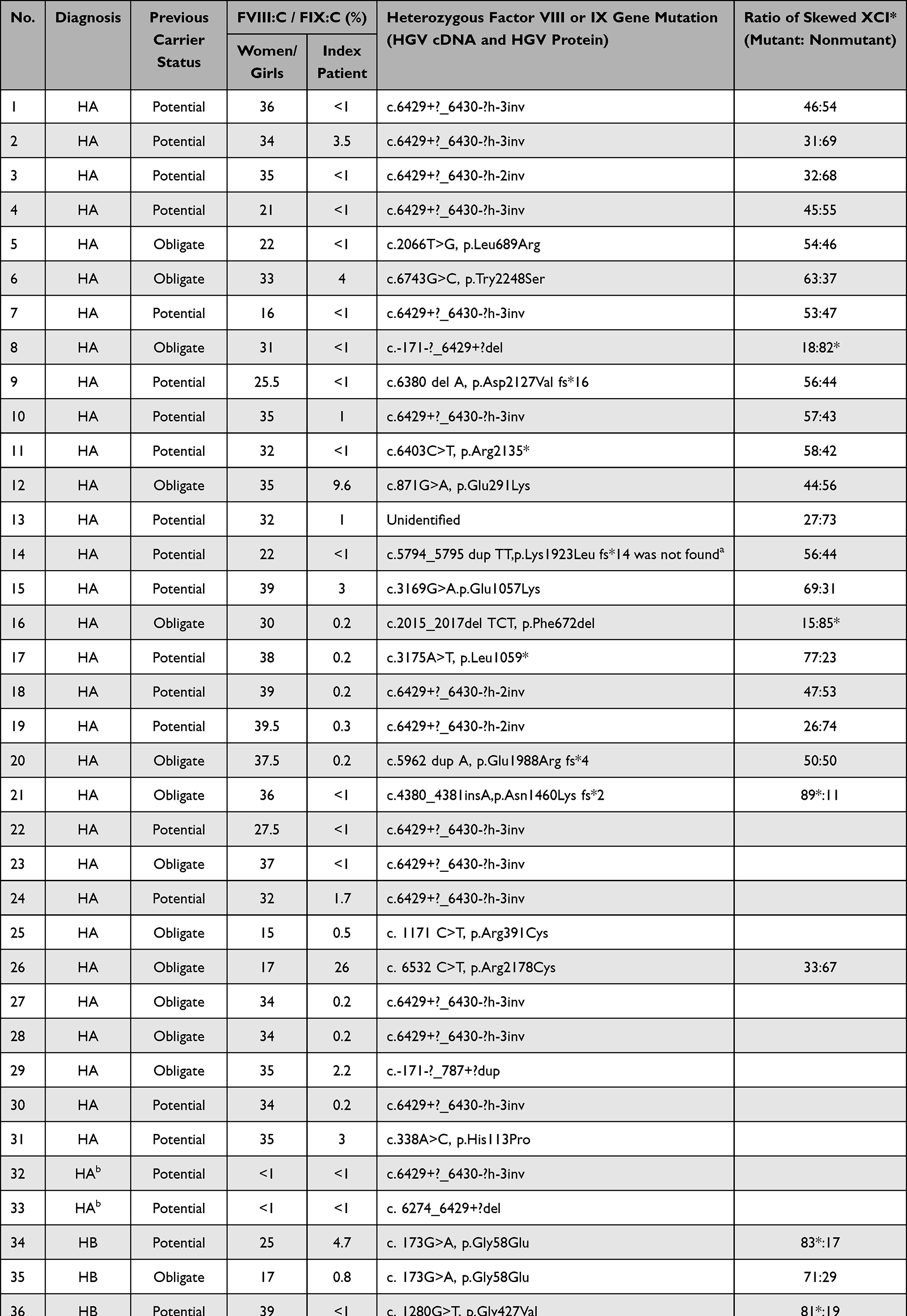 |
Table 3 Women/Girls with Two Severe Haemophilia A and 38 Mild Haemophilia (A31, B7) |
Upon informing the haemophilia status to the previously diagnosed haemophilia carriers, the response was well accepted and gratitude was expressed for registering them in the high cost treatment of the National Health Security Office to receive similar treatment as males with haemophilia.
Management for Women/Girls with Haemophilia Carrier Status and Haemophilia
Women/girls with haemophilia carrier status and haemophilia held an identification card and a referral letter to receive medical service at their hometown hospital and maintained contact with the comprehensive team at the IHTC-Bangkok. The 24-hour hotline telephone consultation of the IHTC-Bangkok for both patients and medical personnel has been actively operated. Follow-up calls to discover the situation progression have been routinely performed. Further, desmopressin and limited factor concentrate have been provided to these affected women/girls.
To assess bleeding manifestations, the verified translated-Thai self-assessment bleeding scores38 were initially sent to 52 women with FVIII:C or FIX:C levels ≤0.5 IU/mL. The response rate was 53.8% (28/52) including 17 of 30 women with mild haemophilia and 11 of 22 women with haemophilia carrier status. The median of bleeding assessment score was 0 (range −3 to 10) which was lower than that reported in the ISTH-BAT study of 4 (range 0 to 35).39 However, the number of women with bleeding assessment score ≥3 reflecting their bleeding tendency among women with mild haemophilia was significantly higher than those with haemophilia carrier status (6/17 vs 2/11) with a p-value of <0.0001.
Discussion
Haemophilia is one of the rare hereditary bleeding conditions involving serious bleeding manifestations. The X-linked recessive inheritance affects males while females are carriers. Women with haemophilia carrier status have experienced a wide range of emotional and psychosocial impacts including feelings of guilt, sorrow, self-blame and being stigmatized.40 Also, women/girls in families with haemophilia become victims in this tragedy. The religious beliefs and cultural acceptance involving fate in human life produces stigma and labeling concerning these women/girls. Married women with carrier status underwent sterilization when they had one normal son or daughter. Some women challenged their pregnancies because they had a 75% chance of not having a haemophilia son from each pregnancy. When they failed, they sought help from medical services. Upon receiving PND, they faced the heart breaking guilty feeling to terminate the pregnancy regarding bearing a male with haemophilia. Finally, some women decided to follow their fate and bore a son with haemophilia. PND was not a proper solution for every pregnant woman that could explain quite a low number of PND requested during the previous 25 years of establishment. Most primigravida or multigravida pregnant women decided not to have a further pregnancy after one episode of PND, no matter that they did not have any offspring or normal son. They accepted their fate. For single women, some with proven haemophilia carrier or noncarrier status decided to remain single. Their lives would be more relaxed and peaceful, not having to face all the related challenges. Probably, this was also their own fate to accept being a single woman, and not marry. Therefore, psychosocial support for women/girls with haemophilia carrier status is essentially required.
Thai patients and family members have a tight knit among family members with an extended family pattern. The grandmother constitutes one of the key individuals in the family handling the tasks and conflicts within their family. Importantly, the counselor had to clarify at the beginning that her daughter-in-law, whose son was diagnosed with haemophilia, was not a scapegoat. The grandmother would be advised to learn how to raise a haemophilia grandson properly. The National Health Security Office in Thailand has provided limited factor concentrate to treat early bleeding episodes and low dose prophylaxis. It has shown an effectiveness in decreasing bleeding episodes and preventing deformity.41,42
The combination of phenotypic analysis of coagulation factor and genotypic analysis of either linkage analysis and mutation detection were required to accurately diagnose carrier or noncarrier status. The mutation detection was an important informative finding. Simple methods of DNA extraction and transport of blood samples for DNA analysis by mail without ice were helpful. The PCR technique is preferable compared with those using the Southern blot requiring a radioactive substance. In addition, the allelic frequencies of polymorphisms associated with factor VIII and IX genes at 30% to 40% of the population should be selected for the linkage analysis. In the current study, Bcl I RFLP at the intron 18 of the factor VIII gene and Mse I RFLP at the 5’ flanking region of the factor IX gene with the allelic frequencies of 34% and 44%, respectively among the Thai population were used.43 Finally, the size of undigested and digested bands should be easily distinguished. The variable tandem repeat of CA at introns 13 and 22 with the minimal difference of 2 base pairs was frequently not used. Importantly, the discovery of the intron 22 inversion found in 40% of patients with severe haemophilia A and their mothers constituted an essential step in mutation detection. Modifying the intron 22 inversion determination from the Southern blot to the PCR technique has been one of the main tools. Furthermore, mutation detection by sequencing would constitute an essential informative finding. At the beginning, the sequencing technique was complicated and costly. The addition of CSGE as a screening tool for the specific site of the mutation was helpful at that time. Further simplifying the sequencing technique and lowering costs have been appreciated. Finally, the widely available sequencing of all exons in factor VIII and IX genes opens a new era of genotypic analysis services. To our knowledge, the current study constitutes the first report of extremely low skewed inactivation of the nonmutant factor VIII/IX carrying X-chromosome at 8% (2/25). Other genetic mechanisms causing low levels of FVIII:C and FIX:C among women/girls with mild haemophilia require further intensive investigation.
Managing women/girls with haemophilia is similar to that of males with haemophilia. For patients with mild degree, desmopressin is suggested to treat mild bleeding episodes.44 Factor concentrate will be used to treat more serious bleeding episodes. Tranexamic acid is additionally prescribed as an adjuvant therapy. Hormonal therapy and tranexamic acid should be used to control heavy menstrual bleeding among women who do not wish or wish to conceive, respectively. The communication among experts at comprehensive haemophilia treatment centre and general practitioners, surgeons and dentists providing care to these women/girls with haemophilia carrier and haemophilia in their hometown is compulsively needed. Education concerning bleeding disorders among women/girls with haemophilia carrier status and haemophilia should be regularly provided to health care personnel to maintain their awareness and recognition.
In conclusion, this study constitutes the successive establishment of care for women/girls from families with haemophilia including their religious beliefs and cultural acceptance concerning the diagnosis of either haemophilia carrier status or haemophilia. A comprehensive approach starting from history taking, self-assessing bleeding score, physical examination, and phenotypic and genotypic analyses would be helpful to confirm the definitive diagnosis of haemophilia carrier status and haemophilia among women/girls at risk. PND should be set up for some indicated pregnant women by sharing the facility with other common inherited diseases. Safe and appropriate management should be planned to control spontaneous and excessive bleeding after trauma, dental and surgical procedures.
Significance Statement
- Comprehensive investigation is essential to determine haemophilia carrier or haemophilia.
- Women accepted their fate of either haemophilia carrier or haemophilia due to religious and cultural beliefs.
- Skewed X-chromosome inactivation of the nonmutant factor VIII/IX carrying X-chromosome was extremely low at 8% (2/25) among mild haemophilia.
Data Sharing Statement
The data from the findings of the study are available from the corresponding author upon reasonable request.
Acknowledgments
This work has been previously presented at the World Federation of Hemophilia Congress 2022 as a virtual poster presentation entitled “Three-decade ongoing establishment of care for women/girls with haemophilia carriers and haemophilia” Montreal, Canada, May 8-11 2022.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
Ampaiwan Chuansumrit acts advisory board of Novo Nordisk and has received honoraria from Novo Nordisk, Grifols, Takeda and Roche while the other authors state that they have no interests which might be perceived as posing a conflict or bias for this study.
References
1. van Galen KP, d’Oiron R, James P, et al. A new hemophilia carrier nomenclature to define hemophilia in women and girls: communication from the SSC of the ISTH. J Thromb Haemost. 2021;19(8):1883–1887. doi:10.1111/jth.15397
2. Bellis EK, Li AD, Jayasinghe YL, et al. Exploring the unmet needs of parents of adolescent girls with heavy menstrual bleeding and dysmenorrhea: a qualitative study. J Pediatr Adolesc Gynecol. 2020;33:271–277. doi:10.1016/j.jpag.2019.12.007
3. Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood. 2006;108(1):52–56. doi:10.1182/blood-2005-09-3879
4. Paroskie A, Gailani D, DeBaun MR, Sidonio RF Jr. A cross-sectional study of bleeding phenotype in haemophilia A carriers. Br J Haematol. 2015;170(2):223–228. doi:10.1111/bjh.13423
5. Gilchrist GS, Hammond D, Melnyk J. Hemophilia A in a phenotypically normal female with XX-XO mosaicism. N Engl J Med. 1965;273(26):1402–1406. doi:10.1056/NEJM196512232732602
6. Chuansumrit A, Sasanakul W, Goodeve A, et al. Inversion of intron 22 of the factor VIII gene in a girl with severe hemophilia A and Turner’s syndrome. Thromb Haemost. 1999;82(4):1379. doi:10.1055/s-0037-1614406
7. Morita H, Kagami M, Ebata Y, Yoshimura H. The occurrence of homozygous hemophilia in a female. Acta Haematol. 1971;45(2):112–119. doi:10.1159/000208614
8. Piétu G, Thomas-Maison N, Sié P, Larrieu MJ, Meyer D. Haemophilia A in a female: study of a family using intragenic and extragenic restriction site polymorphisms. Thromb Haemost. 1988;60(1):102–106. doi:10.1055/s-0038-1647644
9. Windsor S, Lyng A, Taylor SA, Ewenstein BM, Neufeld EJ, Lillicrap D. Severe haemophilia A in a female resulting from two de novo factor VIII mutations. Br J Haematol. 1995;90(4):906–909. doi:10.1111/j.1365-2141.1995.tb05213.x
10. Mannucci PM, Coppola R, Lombardi R, Papa M, De Biasi R. Direct proof of extreme lyonization as a cause of low factor VIII levels in females. Thromb Haemost. 1978;39(2):544–545. doi:10.1055/s-0038-1646723
11. Favier R, Lavergne JM, Costa JM, et al. Unbalanced X-chromosome inactivation with a novel FVIII gene mutation resulting in severe hemophilia A in a female. Blood. 2000;96(13):4373–4375. doi:10.1182/blood.V96.13.4373
12. Bicocchi MP, Migeon BR, Pasino M, et al. Familial nonrandom inactivation linked to the X inactivation centre in heterozygotes manifesting haemophilia A. Eur J Hum Genet. 2005;13(5):635–640. doi:10.1038/sj.ejhg.5201386
13. Renault NK, Dyack S, Dobson MJ, Costa T, Lam WL, Greer WL. Heritable skewed X-chromosome inactivation leads to haemophilia A expression in heterozygous females. Eur J Hum Genet. 2007;15(6):628–637. doi:10.1038/sj.ejhg.5201799
14. Knobe KE, Sjörin E, Soller MJ, Liljebjörn H, Ljung RC. Female haemophilia A caused by skewed X inactivation. Haemophilia. 2008;14(4):846–848. doi:10.1111/j.1365-2516.2008.01754.x
15. Minks J, Robinson WP, Brown CJ. A skewed view of X chromosome inactivation. J Clin Invest. 2008;118(1):20–23. doi:10.1172/JCI34470
16. Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A. Subcommittee on Factor VIII, Factor IX and rare coagulation disorders of the scientific and standardization committee of the international society on thrombosis and hemostasis. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939. doi:10.1111/jth.12672
17. Hardisty RM, Macpherson JC. A one-stage factor VIII (antihaemophilic globulin) assay and its use on venous and capillary plasma. Thromb Diath Haemorrh. 1962;7:215–228.
18. Laurell CB. Electroimmunoassay. Scand J Clin Lab Invest. 1972;29(Suppl124):21–27.
19. Fishman DJ, Jones PK, Menitove JE, et al. Detection of the carrier state for classic hemophilia using an enzyme-linked immunosorbent assay (ELISA). Blood. 1982;59:1163–1168. doi:10.1182/blood.V59.6.1163.1163
20. Chuansumrit A, Pintadit P, Isarangkura P, Hathirat P, Phongcharoen S. Detection of hemophilia A carrier. Rama Med J. 1993;13(1):1–6.
21. Pintadit P, Chuansumrit A, Chotsupakarn S, et al. Carrier detection in hemophilia A by using the ratio of factor VIII clotting activity and von Willebrand factor antigen. Thai J Hematol Transfus Med. 1996;6(1):24–31.
22. Kunkel LM, Smith KD, Boyer SH, et al. Analysis of human Y-chromosome-specific reiterated DNA in chromosome variants. Proc Natl Acad Sci USA. 1977;74:1245–1249. doi:10.1073/pnas.74.3.1245
23. Sasanakul W, Chuansumrit A, Kadegasem P, Hathirat P. A comparison of DNA extraction between conventional phenol-chloroform and in-house modified method. Rama Med J. 1997;20:119–124.
24. Sasanakul W, Chuansumrit A, Rurgkhum S, Udomsubpayakul U, Hathirat P. DNA extraction and amplification of 10-day, room-temperature blood samples. J Med Assoc Thai. 1999;82(Suppl 1):S186–189.
25. Kadegasem P, Rurgkhum S, Sasanakul W, Chuansumrit A. DNA extraction from buffy coat sent by mail without ice. Thai J Hematol Transfus Med. 2001;11:167–171.
26. Peake I, Lilicrap DP, Boulyjenkov V, et al. Report of a joint WHO/WFH meeting on the control of haemophilia: carrier detection and prenatal diagnosis. Blood Coagul Fibrinolysis. 1993;4:313–344. doi:10.1097/00001721-199304000-00013
27. Rossetti LC, Radic CP, Larripa IB, De Brasi CD. Developing a new generation of tests for genotyping hemophilia-causative rearrangements involving int22h and int1h hotspots in the factor VIII gene. J Thromb Haemost. 2008;6:830–836. doi:10.1111/j.1538-7836.2008.02926.x
28. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74(12):5463–5467. doi:10.1073/pnas.74.12.5463
29. Ganguly A, Rock MJ, Prockop DI. Conformation-sensitive gel electrophoresis for rapid detection of single-base differences in double-stranded PCR products and DNA fragment: evidence for solvent-induced bends in DNA heteroduplexes. Proc Natl Acad Sci USA. 1993;90:10325–10329. doi:10.1073/pnas.90.21.10325
30. Williams IJH, Abuzenadah A, Winship PR, et al. Precise carrier diagnosis in families with hemophilia A: use of conformation sensitive gel electrophoresis for mutation screening and polymorphism analysis. Thromb Haemost. 1998;79:723–726. doi:10.1055/s-0037-1615052
31. Hink JL, Winship PR, Makris M, et al. A rapid method for haemophilia B mutation detection using conformation sensitive gel electrophoresis. Br J Haematol. 1999;104:915–918. doi:10.1046/j.1365-2141.1999.01274.x
32. Chuansumrit A, Sasanakul W, Promsonthi P, et al. Prenatal diagnosis for haemophilia: the Thai experience. Haemophilia. 2016;22:880–885. doi:10.1111/hae.13002
33. Betke K, Sanguansermsri T. Cytological differentiation of blood pigments, possibilities, results and practical application. Munch Med Wochenschr. 1972;114:1099–1104.
34. Baily DMD, Affara NA, Ferguson-Smith MA. The X-Y homologous gene amelogenin maps to the short arms of both the X and Y chromosome and is highly conserved in primates. Genomics. 1992;14:14203–14205.
35. Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51(6):1229.
36. Chuansumrit A, Panthangkura W, Isarangkura P, Hathirat P, Mahaphan W. Genetic counseling for hemophilia carriers. Thai J Hematol Transfus Med. 1993;3(1):37–50.
37. Orstavik KH, Scheibel E, Ingerslev J, Schwartz M. Absence of correlation between X chromosome inactivation pattern and plasma concentration of factor VIII and factor IX in carriers of haemophilia A and B. Thromb Haemost. 2000;83(3):433–443. doi:10.1055/s-0037-1613833
38. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063–2065. doi:10.1111/j.1538-7836.2010.03975.x
39. James PD, Mahlangu J, Bidlingmaier C, et al. Evaluation of the utility of the ISTH-BAT in haemophilia carriers: a multinational study. Haemophilia. 2016;22(6):912–918. doi:10.1111/hae.13089
40. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia. Haemophilia. 2020;26(Suppl6):1–158. doi:10.1111/hae.14046
41. Sooksriwong C, Boonkerd L, Chanjaruporn F, Chuansumrit A. Incremental cost effectiveness analysis for haemophilia home-based care programme in Thailand. Haemophilia. 2012;18(4):548. doi:10.1111/j.1365-2516.2012.02871.x
42. Pattanaprateep O, Chuansumrit A, Kongsakon R. Cost-utility analysis of home-based care for treatment of Thai hemophilia A and B. Value Health Reg Issues. 2014;3:73–78. doi:10.1016/j.vhri.2014.02.008
43. Goodeve AC, Chuansumrit A, Sasanakul W, et al. A comparison of the allelic frequencies of ten DNA polymorphisms associated with factor VIII and factor IX genes in Thai and Western European populations. Blood Coagul Fibrinolysis. 1994;5(1):29–35. doi:10.1097/00001721-199402000-00005
44. Loomans JI, Kruip MJ, Carcao M, et al. Desmopressin in moderate hemophilia A patients: a treatment with considering. Haematologica. 2018;103(3):550–557. doi:10.3324/haematol.2017.180059
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.