")
Back to Journals » OncoTargets and Therapy » Volume 12
Therapies targeting the signal pathways of pheochromocytoma and paraganglioma
Received 11 June 2019
Accepted for publication 14 August 2019
Published 4 September 2019 Volume 2019:12 Pages 7227—7241
DOI https://doi.org/10.2147/OTT.S219056
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr XuYu Yang
Yalin Liu1, Longfei Liu2, Feizhou Zhu1
1Department of Biochemistry and Molecular Biology, Xiangya School of Medicine, Central South University, Changsha, People’s Republic of China; 2Department of Urology, Xiangya Hospital, Central South University, Changsha, People’s Republic of China
Correspondence: Feizhou Zhu
Department of Biochemistry and Molecular Biology, Xiangya School of Medicine, Central South University (CSU), Tongzipo Road 172, Changsha, Hunan 410013, People’s Republic of China
Tel +86 01 370 749 6211
Fax +86 8 265 0230
Email [email protected]
Abstract: Pheochromocytoma and paraganglioma (PCC/PGL) are rare tumors that originate from adrenal or extra-adrenal chromaffin cells. A significant clinical manifestation of PCC/PGL is that the tumors release a large number of catecholamines continuously or intermittently, causing persistent or paroxysmal hypertension and multiple organ functions and metabolic disorders. Though majority of the tumors are non-metastatic, about 10% are metastatic tumors. Others even have estimated that the rate of metastasis may be as high as 26%. The disease is most common in individuals ranging from 20 to 50 years old and the age of onset strongly depends on the genetic background: patients with germline mutations in susceptible genes have an earlier presentation. Besides, there are no significant differences in the incidence between men and women. At present, traditional treatments, such as surgical treatment, radionuclide therapy, and chemotherapy are still prior choices. However, they all have several deficiencies so that the effects are not extremely significant. Contemporary studies have shown that hypoxia-associated signal pathway, associated with the cluster 1 genes of PCC/PGL, and increased kinase signal pathways, associated with the cluster 2 genes of PCC/PGL, are the two major pathways involving the molecular pathogenesis of PCC/PGL, indicating that PCC/PGL can be treated with targeted therapies in emerging trends. This article reviews the progress of molecular-targeted therapies for PCC/PGL.
Keywords: pheochromocytoma, paraganglioma, targeted therapies, signal pathways
Introduction
Pheochromocytoma and paraganglioma (PCC/PGL) are neuroendocrine tumors arising from the chromaffin cells which are derived from the embryonic neural crest, including adrenal medulla PCC and extra-adrenal sympathetic and parasympathetic paraganglia (PGL). PCC/PGL often show an increase in catecholamines (adrenalin, norepinephrine, and/or dopamine), which affects the cardiovascular system and metabolic processes, thus causing high blood pressure.1 Though hypertension is the most critical clinical symptom of PCC/PGL, this disease may also be associated with orthostatic hypotension. Besides, other common symptoms include recurrent headaches, excessive sweating, tachycardia as well as weight loss. It is reported that the features of headache, sweating, and palpitations appeared in 30–40% of the cases and could be seen as the best clue to suspect PCC/PGL.2 There are also some clinical atypical presentations of PCC/PGL including sustained hypertension and incidental mass without associated symptoms. If PCC/PGL are not diagnosed in time, delaying in treatment can cause serious heart, brain, kidney vascular damages, and even death. In terms of catecholamines in PCC/PGL, different patients may have different levels. Recent researchers have found that catecholamine excretion varied according to gene mutations.3 For example, mutations in NF1 and RET genes are almost always associated with PCC/PGL that produce catecholamine.4,5 In opposite, some tumors due to mutations in VHL and SDHx genes lack significant excretion of catecholamine.5 Some PCC/PGL express high levels of tyrosine hydroxylase (TH) which is the rate-limiting enzyme for catecholamine biosynthesis. For instance, the level of endogenous VHL tumor suppressor protein (pVHL) in PC12 cells expressing VHL antisense RNA reduced by 5–10 folds while the levels of TH protein and mRNA increased by 2–3 folds. Therefore, loss of pVHL function may be responsible for PCC-related hyper-catecholemia.6
The prevalence of PCC/PGL is estimated to be 1:6500–1:2500. Autopsy results show that the prevalence is as high as approximately 1:2000, indicating that many PCC/PGL were not diagnosed. The annual incidence rate is reported to be 2–10:1,000,000.7 Most PCC/PGL are discovered at 30–50 years old, and the incidence rate of males and females is basically equal.
It has been reported that PCC/PGL have the highest heritability in human tumors.8 Furthermore, it is a kind of human tumor model that has inherited mutations in a metabolic enzyme gene, succinate dehydrogenase subunit D (SDHD). In addition to classic mutations in the genes encoding for the subunits of SDH, in the past five years more germline or somatic mutations have been found in genes encoding for other enzymes catalyzing pivotal steps of the tricarboxylic cycle acid (TCA), such as fumarate hydratase (FH), malate dehydrogenase 2 (MDH2), glutamic-oxaloacetic transaminase 2 (GOT2), and dihydrolipoamide S-succinyltransferase (DLST), in PCC/PGL.9 In regard to the molecular pathogenetic mechanism of PCC/PGL, the hypoxia-related signal pathway (Figure 1) and the increased kinase signal pathways (Figure 2) are two main pathways involving the tumor.10 It has been proved that the mutations of subunits of succinate dehydrogenase (SDH) (including SDHA, SDHB, SDHC, SDHD, SDHAF2), FH, prolyl hydroxylase domain protein 2 (PHD2), von Hippel Lindau (VHL), and hypoxia-inducible factor 2A (HIF2A), which are the cluster 1 genes of PCC/PGL, influenced the hypoxia‑related signal pathway, while the mutations of rearranged during transfection proto-oncogene (RET), myc-associated factor X (MAX), transmembrane protein 127 (TMEM127), neurofibromin 1 (NF1), and kinesin family member1B β (KIF1Bβ), which are the cluster 2 genes of PCC/PGL, influenced the increased kinase signal pathways.10
If it were diagnosed and treated in a timely and early manner, PCC/PGL could be cured. Traditional therapies (surgical treatment, radionuclide therapy, and chemotherapy) are the most commonly used treatments for PCC/PGL nowadays, but they have shown suboptimal results in shrinking tumors and improving survival. Currently, effective molecular-targeted therapies aiming at permanent control of these highly complex neoplasms are the research hotspot and the aim of efforts and molecular characterization of PCC/PGL suggests the targeted therapies should be optional treatments (Table 1). The current progress in the treatments of PCC/PGL is summarized as follows.
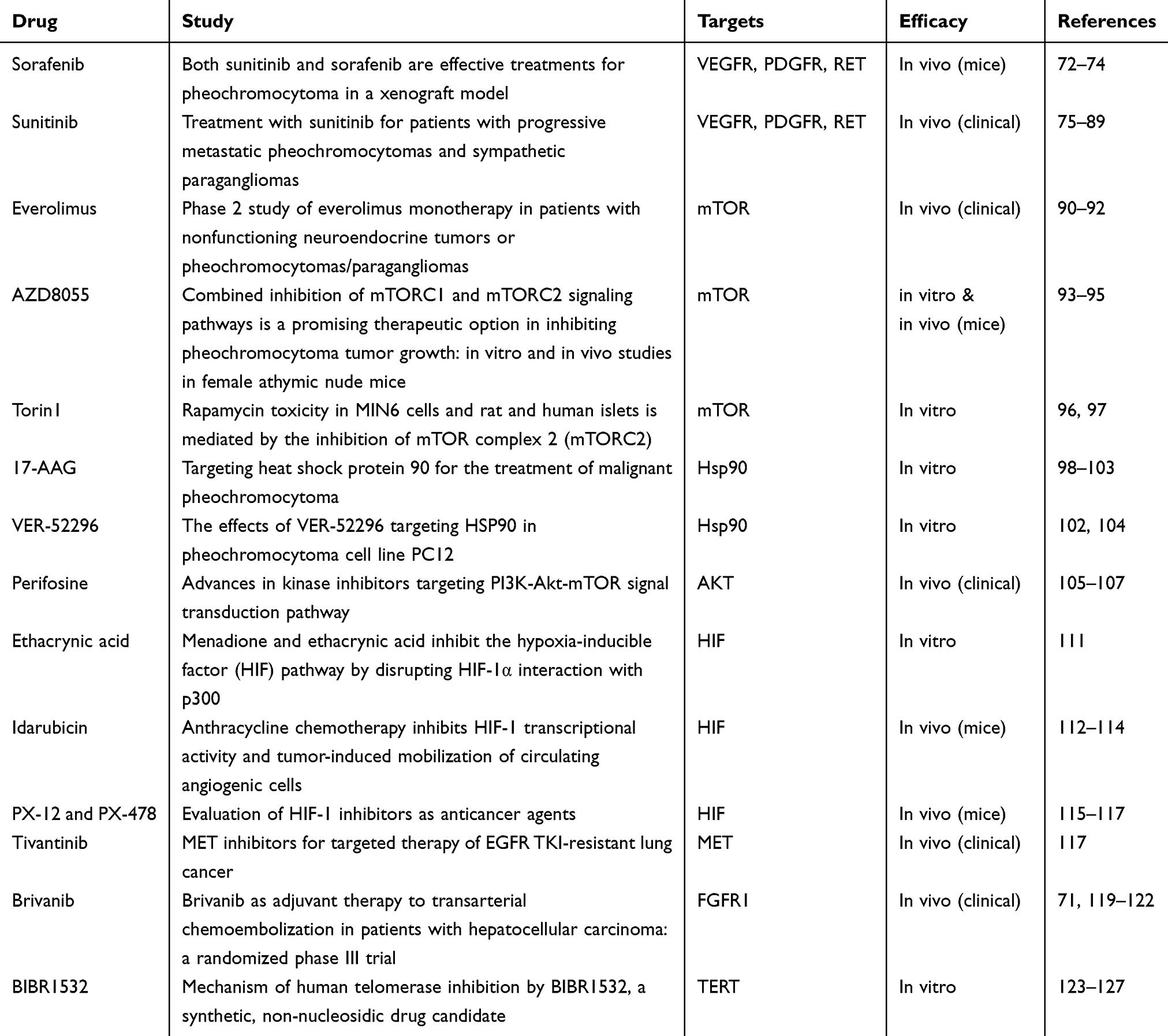 |
Table 1 Targeted drugs in PCC/PGL |
Traditional therapies
Surgical treatments
In terms of surgical methods, laparoscopic adrenalectomy (LA) has become the choice of more and more surgeons. Open adrenalectomy (OA) has a great advantage in large tumor resection and the invention of robot-assisted adrenalectomy (RA) undoubtedly provides surgeons with more options.11
LA treatment of PCC is still controversial. On the one hand, laparoscopic surgery is a great advantage for adrenal tumors with deep, small, and difficult exposures. And LA is regarded as the gold standard for the treatment of non-metastatic adrenal tumors. On the other hand, adrenal PCC is characterized by abundant blood supply and tumor volume is usually larger than other adrenal tumors.12 Complications including uncontrolled high blood pressure, hemodynamic instability, invasion of surrounding tissues, and local recurrence are likely to occur during surgeries. It may lead to difficulties in LA, which has to be transited to OA then, not to mention that the establishment of LA itself may also stimulate the secretion of catecholamines, thereby increasing the risk of surgery.12
OA as a traditional surgical method has unfavorable factors such as high intraoperative blood loss, slow recovery, and long hospital stay, but it is still unable to be replaced in the resection of large tumors (>6 cm in diameter) and uncertain non-metastatic or metastatic tumors before the operation.13 Blood supply of giant PCC is abnormal, and there is plenty of collateral circulation. Because the tumor supply vessels are ligated and blocked, the pressure inside the tumor will increase when the blood flow of the tumor blocked. Therefore, the blood will ooze more during the operation and the blood loss is greater when the tumor is isolated. At that time, timely blood transfusion is the key of guaranteed successful surgeries. The use of autologous blood recovery has the advantages of rapid, timely avoiding the loss of allogeneic blood as well as washed red blood cells are fresh thus can immediately exert oxygen-carrying function and the adverse reactions are small.14
Robot-assisted adrenalectomy is as safe as LA, with less bleeding, faster recovery, and shorter hospital stay. First, surgeons can adjust the angle of the endoscope to get a good view of the operation according to their own needs, allowing deep adrenal tumor surgery to achieve a better surgical field and provides protection for surgical resection. Second, the operator can use the operating tool to operate the surgical instruments freely and flexibly, making the surgery more sensitive and the operation more precise and rapid. The robotic system also allows the surgery to be performed in a relatively relaxed and comfortable environment so that the operator is less prone to be fatigue; thus, the quality of the operation is guaranteed. But it is generally believed that RA has a longer operation time and a higher cost than LA. Moreover, the operator lacks an intuitive touch to the organ during the operation, increasing the possibility of potentially damaging adjacent organs.15–17
In principle, the main purpose of surgical treatment is to eradicate the primary tumor and to remove local and distant metastases. However, due to the tendency of distant metastasis and recurrence of metastatic tumors, surgeries often fail to achieve the desired results. It is recommended that plasma or urinary adrenaline should be tested annually to screen for local or metastatic recurrence or new tumors and all patients undergoing PPGL surgery should be followed for at least 10 years. High-risk patients even need to receive annual follow-up during lifetime.18
Radionuclide therapy
I131-metaiodoenzylguanidine (I131-MIBG) is a kind of radiopharmaceutical which acts as a norepinephrine analog taken up by cells in the sympathomedullary system19 and high doses of I131-MIBG can prolong survival and relieve symptoms.20 Since PCC expresses somatostatin receptors, radiopharmaceuticals based on the somatostatin analogs octreotide and lanreotide have also begun to be used.21 However, these treatments are only suitable for cancer patients who have high intake of radionuclides and there is still insufficient evidence to determine radionuclide therapy doses and normal tissue tolerated doses. The dose segmentation of radionuclide therapy is also controversial and still requires a larger sample size as well as further follow-up confirmation.22
Chemotherapy
Since the late 1960s, a range of chemotherapies have been described as potential treatments for PCC/PGL. These include temozolomide, dacarbazine, streptozotocin, doxorubicin, vincristine, methotrexate, 5-fluorouracil, ifosfamide, cyclophosphamide, and platinum compounds. They inhibit different stages of the cell cycle, resulting in cancer cell death so they are effective in the treatment of many cancers.23 O-6-methylguanine-DNA methyltransferase (MGMT) promoter displaying hypermethylation in SDHB-related tumors leads to the silencing of MGMT expression.24 MGMT is at high risk of progression with DNA hypermethylation and is a prognostic factor for the response to temozolomide. When tested temozolomide in PCC/PGL patients, 67% of the patients showed clinical benefits and 80% of them showed tumors of low-level MGMT.24 Moreover, the CVD regimen (a combination of cyclophosphamide, vincristine, and dacarbazine) recommended for chemotherapy had an effective rate of about 50%, but most of them relapsed within 2 years.21 A meta-analysis showed that about 37% of the patients had partial response on reducing tumor volume after using chemotherapy drugs.20 A study from Japan showed that in 17 patients with metastatic PCC, the partial response rate of chemotherapy was 47.1% and no patients were fully cured.25 Huang et al26 showed that in 18 metastatic PCC patients who received chemotherapy, 11% achieved complete remission and 44% had partial remission. After 22 years of follow-up, their 5-year survival was less than 50%. The study by Nomura et al27 also showed that the survival of the chemotherapy group was not superior to the control group. Therefore, the advantage of chemotherapy is to improve symptoms, but may not extend the long-term survival time.
Signal pathways and genes associated with hereditary PCC/PGL
Before studying the targeted therapy, we try to make clear the signal pathways involving PCC/PGL in which proteins may be the selected targets for therapies. There are two kinds of signal pathways leading to PCC/PGL.28–33 To be specific, the hypoxia-associated signal pathway, associated with the cluster 1 genes of PCC/PGL, and increased kinase signal pathways, associated with the cluster 2 genes of PCC/PGL, are the two major pathways involving the molecular pathogenesis of PCC/PGL, proven by the Cancer Genome Atlas (TCGA) sequencing study.34
Hypoxia-related signal pathway and cluster 1 genes
In the process of rapid proliferation, tumor cells need to consume a large amount of oxygen, leading to the gradual formation of chronic hypoxic microenvironment in tumor tissues due to insufficient blood oxygen supply. In response to the hypoxic microenvironment, cancer cells will regulate themselves through a series of energy metabolism signal pathways, angiogenesis, proliferation, survival, invasion, and tumor metastasis. Hypoxia-inducible factor (HIF) is a kind of transcription factor that controls energy, erythropoiesis, iron metabolism, and development29 and when dysregulated, it will result in tumorigenesis.35 In the hypoxia-related signal pathway, the HIF is a kind of transcription factor found in mammalian cells and consists of two subunits heterodimeric, which are oxygen-sensitive HIF-α and stably expressed HIF-β. HIF-α is one of the key proteins that regulate these signal pathways in tumor cells in a hypoxic environment.36 HIF-α has two transactivation domains at the carboxyl terminus, N-terminus and C-terminus. Under hypoxic conditions, the transactivation region at the C-terminus interacts with the transcriptional coactivator p300 and is involved in the regulation of transcriptional activation. Specifically, HIF-α has normal physiological functions after being modified by heat shock protein Hsp90 and then forms a dimer with HIF-β. After entering the nucleus, the dimer binds to the co-transcription factor p300 and cyclic AMP response element-binding protein (p300/CBP) to form HIF-p300/CBP complex which binds to a hypoxia-responsive element (HRE) of DNA and regulates transcription of downstream genes.37 It is worth mentioning that in 2013, David et al first associated the somatic HIF2A gain-of-function mutation with PCC/PGL.38
What influences the hypoxia-related signal pathway is the mutations of Cluster 1 genes. In the presence of oxygen, the PHD2 catalyzes the proline hydroxylation of HIF, which is recognized and targeted to degrade by VHL that constitutes a part of the E3 ubiquitin ligase complex.39,40 Since the discovery of VHL disease tumor suppressor gene VHL in 1993 as the genetic basis of VHL disease, VHL has been proven to have broad medical and scientific value. pVHL plays an important role in cellular oxygen sensing through targeting HIF for ubiquitination and proteasomal degradation.41 Besides, in PCC/PGL, VHL mutations reported were often missense mutations.42 These mutations result in the activation of HIF signal pathways at normal oxygen levels, elevated erythropoietin levels, and overproduction of red blood cells.43 Somatic VHL mutations in PGLs were first reported by Merlo,44 which was tested in 53 PGL tissues through gene sequencing and other methods. The results indicated that VHL mutations could predict the clinical diagnosis of PCCs and play a crucial role in the pathogenesis of sporadic head and neck paragangliomas.
Besides, SDHx genes encode succinate dehydrogenase (SDH), a mitochondrial enzyme complex, which is part of the citric acid cycle and electron transport chain.45 SDH oxidizes succinate to fumarate in the citric acid cycle and delivers electrons to coenzyme Q in the electron transport chain.45 Harmful mutations in SDHx lead to energy metabolism disorders and succinate accumulation, thereby inhibiting the activity of PHD2. These, in turn, lead to increased activation of HIF-α.46 The SDHx consists of four subunits: SDHA, SDHB, SDHC, and SDHD, and germline mutations in the SDHx gene result in hereditary PCC/PGL syndrome. The discovery of SDH complex assembly in 2009 involved two factors, SDHAF1 and SDHAF2, which may play a part in the development of cancers related to this pathway.47,48 SDHA gene acts as tumor suppressor and is considered as a new PCC/PGL susceptibility gene.49 SDHB mutations and SDHC mutations were also proven to have relationship with PCC/PGL.50,51 In 2000, SDHD mutations were found in sporadic and hereditary PGL/PCC.50,52,53
Last but not least, FH is a fumarate hydratase in the citric acid cycle, catalyzing the hydration of fumarate to malate. The mutations of FH result in the accumulation of fumarate, and thus activate the oncogenic HIF pathway through inhibiting PHD2.54 FH mutations can be regarded as a rare source of susceptibility to PCC/PGL32 and FH-deficient PCC/PGL have similar genetic developmental pathways to SDHB-mutated metastatic PCC/PGL. In conclusion, mutations of FH result in metastatic PCC/PGL with a great possibility.31
Increased kinase signal pathways and cluster 2 genes
PI3K/AKT/mTOR signal pathway is an important intracellular kinase signal pathway that regulates the cell cycle and is directly related to cell dormancy, proliferation, carcinogenesis, and longevity. PI3K is a heterodimer of the regulatory subunit P85 and the catalytic subunit P110. Various growth factors and cytokines, such as insulin-like growth factor (IGF), nerve growth factors (NGF), and platelet-derived growth factors (PDGF), activate PI3K via tyrosine kinase and G protein-coupled receptors. Subsequently, activated PI3K catalyzes the phosphorylation of phosphatidylinositol biphosphate 2 (PIP2) to phosphatidylinositol biphosphate 3 (PIP3). On the other hand, phosphatase and tensin homolog deleted on chromosome ten (PTEN) that is a tumor suppressor gene can induce the dephosphorylation of PIP3 into PIP2, thereby antagonizing PI3K.55 PIP3 acts as a second messenger, allowing the phosphorylation and activation of the AKT with the help of 3-phosphoinositol-dependent protein kinases (PDK1 and PDK2). Then, the activated AKT further activates the downstream mTOR which is one of the most important substrates of AKT and is closely related to the occurrence and development of tumors.56
Another increased kinase signal pathway of PCC/PGL is the Ras/Raf/MEK/ERK that is a typical mitogen-activated protein kinase (MAPK) pathway through which extracellular signals can be transmitted into cells to participate in physiological activities such as cell proliferation and survival. It is mainly composed of Ras (a G protein) and three bispecific protein kinases like Raf, MEK, and ERK. ERK is involved in the regulation of life activities such as cell proliferation, survival, and migration by phosphorylating downstream substrates in the cytoplasm and nucleus.
Actually, the Cluster 2 genes have fundamental impacts on PI3K/AKT/mTOR and Ras/Raf/MEK/ERK signal pathways. To begin with, RET is an oncogene in PCC/PGL susceptibility gene. In 1993, Mulligan et al found that RET was a kind of receptor tyrosine kinase gene expressed in PCC and acted as a candidate gene for multiple endocrine neoplasia type 2A (MEN 2A), a syndrome in which two or more endocrine gland neoplasms including PCC occur simultaneously or successively in the same patient.57 The RET gene is principally expressed in the genitourinary system and neural crest precursor cells.58 The oncogenic activation of RET has been verified to activate both PI3K/AKT/mTOR- and Raf/Raf/MEK/ERK-dependent pathways.59
When it comes to MAX, its mutations were first reported in susceptibility to hereditary PCC/PGL in 2011. MAX plays a crucial role in the MYC/MAX pathway, the deregulation of which contributes to activate PI3K. The isolation of MAX mutations, immunohistochemical analysis of MAX protein deletions in tumors, and loss of wild-type MAX alleles in tumors implied that MAX is a tumor suppressor gene.60 Burnichon sequenced MAX using extracted DNA from blood leukocytes of 1694 patients with PCC/PGL, of which the MAX mutation was first discovered in PGL.61
As for TMEM127, it was identified as a new PCC susceptibility gene in 2010 by Yuejuan Qin et al,62 its tumor suppressor properties cause changes in the function of endolysosomal mTOR. Therefore, mutations in TMEM127 result in an increase in the mTOR signal, which may contribute to the development of PCC/PGL.63,64 These results suggest that abnormal activation of mTOR signal pathway may promote the occurrence of PCC, and the degree of its activation may be related to tumor invasion and metastasis.65
On account of KIF1Bβ, it influences Ras/Raf/MEK/ERK signal pathway by acting on the Ras. It was reported that a family with KIF1Bβ gene mutations developed neuroblastoma, ganglioneuroma, PCC, and lung cancer.66 Similarly, in 2002, Kimura et al found that NF1 is associated with the tumorigenesis of composite PCC.67 NF1 gene acts as a tumor suppressor gene and its main function is to inhibit the oncogenic Ras/Raf/MAPK signal pathway by converting Ras protein into an inactive form to inhibit cell proliferation. About one-fifth of sporadic PCCs may have somatic NF1 mutations and the loss of heterozygosity in NF1 suggests that loss of function may be the primary mechanism of NF1 alteration in PCC/PGL.68,69
There are many other recently discovered mutations that converge in this pathway, such as HRas proto-oncogene (HRAS), MET proto-oncogene (MET), and fibroblast growth factor receptor 1 (FGFR1). The first HRAS mutation found in a patient with PCC was reported by Yoshimoto in 1992.70 Belonging to the Ras oncogene family, it is associated with Ras/Raf activation thus results in PCC/PGL. In addition, MET may also have a relationship with PCC/PGL predisposition.71 MET is a proto-oncogene whose transmembrane receptor protein MET has tyrosine kinase activity. The binding of the MET receptor to hepatocyte growth factor induces the activation of MET dimer, which in turn phosphorylates its substrate to activate downstream signaling pathways: PI3K/AKT/mTOR and Ras/Raf/MEK. Furthermore, FGFR1 encodes FGFR1 protein, which is a kind of transmembrane protein belonging to receptor tyrosine kinase. When fibroblast growth factor binds to the extracellular segment of FGFR1, the intracellular segment of the receptor affects the downstream kinase signaling pathway. Therefore, the dysregulation of FGFR1 signaling leads to the development, proliferation, survival, and metastasis of tumors.71
Targeted therapies
Multi-target inhibitors: sorafenib and sunitinib
Sorafenib
Angiogenesis, the formation of capillaries, is critical for tumor growth and metastasis, as tumors require independent blood supply with oxygen, glucose, and other nutrients to cancer cells.72 By this token, it has already been proven that drugs blocking angiogenesis can be effective in anti-cancer therapies. In the past 10 years, a variety of drugs have been developed, including tyrosine kinase inhibitors which have anti-angiogenic activity. Vascular endothelial growth factor receptor (VEGFR) tyrosine kinases involve tumor growth, progression, metastasis as well as angiogenesis; thus, it is a suitable target for drugs. Sorafenib is a new multi-target oral anti-cancer medicine, which can inhibit serine/threonine kinase activity and tyrosine kinase activity of VEGFR-2, VEGFR-3, PDGF-β, stem cell factor receptor (KIT), and FMS-like tyrosine kinase (FLT)−3 receptors, RET, Raf.73 Therefore, sorafenib has dual anti-tumor effects. For one thing, it can directly inhibit the proliferation of tumor cells by blocking the signal transduction pathway mediated by Raf/MEK/ERK. For another, it can also inhibit the formation of neovascularization and cut off the nutritional supply of tumor cells by acting on VEGFR, so as to achieve the purpose of restraining the growth of tumors.74
Sorafenib has been used to treat kidney cancer and liver cancer in the clinic. At present, sorafenib treating metastatic PCC are limited to several case reports, so more clinical studies and long-term follow-up are needed to confirm its efficacy.
Sunitinib
Sunitinib is another multi-target tyrosine kinase inhibitor with anti-angiogenic and anti-tumor effects.75 As it has been mentioned earlier, HIF is a transcription factor responsible for regulating cellular responses to hypoxia, and its expression and regulation are influenced by intracellular oxygen tension. Both VHL mutations and SDHx mutations can cause HIF-α accumulation, leading to elevated VEGF and PDGF.76 Targets of sunitinib are mainly VEGFR, PDGFR, and RET, and it is usually used to alleviate/treat metastatic PCC.77,78 Experiments have shown that knocking out VEGFR-2 attenuated the effects of sunitinib.79 In addition, for another mechanism of PCC, the PI3K/AKT/mTOR pathway, sunitinib inhibits phosphorylation of AKT and mTOR, thereby inhibiting cell proliferation, angiogenesis, and apoptosis escaping. An important trait of metastatic PCC showed most VHL and SDHX-associated tumors are usually of high vascularization, suggesting that anti-angiogenic drugs can represent an effective treatment.80–83 A clinical study and case report showed that oral sunitinib was effective in treating PCC/PGL.84–86 In particular, Ayala-Ramirez et al reported some of the benefits of sunitinib in tumor shrinkage and disease stabilization in some patients with progressive metastatic PCC.84 Several studies have also shown that can be used for the treatment of metastatic PCC, renal cell carcinoma, as well as pancreatic neuroendocrine tumors and achieved relatively good results.75,87–89 Importantly, it has been approved in Europe and America.
mTOR inhibitor: everolimus, AZD8055, and Torin1
Everolimus
Everolimus is an mTOR inhibitor. The PI3K/AKT/mTOR pathway regulates cell growth and survival, resulting in high expression of mTOR, leading to cell proliferation, angiogenesis, and apoptosis evasion.90 Druce et al91 reported that 4 patients who underwent everolimus had progression and considered that everolimus was not effective in treating metastatic PCC/PGL. Oh et al92 used everolimus to treat 5 patients with metastatic PCC and 2 patients with paraganglioma. After treatment, the patients’ conditions remained stable. Though 2 patients’ tumors progressed, 4 patients had significantly smaller tumor volume. It can be seen that the efficacy of everolimus in the treatment of metastatic PCC/PGL is still uncertain.
AZD8055
AZD8055 is a novel ATP-competitive mTOR inhibitor that can be administered orally and has good selectivity.93 Furthermore, in inhibitory effects of AZD8055, mTOR is approximately 1000 times more than all class I PI3K isoenzymes and other PI3K-like kinase family members.
AZD8055 can inhibit S6K1 and 4e-bp-1 which are the downstream factors of mTOR complex 1. Besides, it can also inhibit the phosphorylation of mTOR complex 2 substrate AKT and downstream proteins. In vitro, it inhibited hyperplasia and induced autophagy in H838 and A549 cells. In a mouse xenograft model, AZD8055 inhibited the phosphorylation of S6K1 and AKT in a concentration-dependent manner thus effectively inhibited tumor growth.94
Alessio Giubellino95 found that AZD8055 could significantly reduce volume of tumor in a female athymic mouse of metastatic PCC/PGL.
Torin1
Torin1 is one of mTOR kinase inhibitors and others including Torin2, PP242, PP30, KU0063794, WAY-600, WYE-687, WYE-354, OSI-027, AZD-8055, KU-BMCL-200908069–1, Wyeth-BMCL- 200908069–2, XL-388, INK-128he, and AZD-2014 and some have entered different clinical trials.96 In addition, Torin1 can also inhibit glycolysis of tumor cells in hypoxic and energy-poor environments, thereby making tumor cells starved and further enhancing their anti-tumor effects.97
H90 inhibitors: 17-AAG and VER-52296
HSP90 has turned out to be involved in the maturation and stable folding of a variety of proteins, which are critical for tumor cell growth and metastasis. Therefore, HSP90 may be a potential for tumor therapies.98,99 Several Hsp90 inhibitors have been developed in the past few years and several clinical trials are in progress. Geldanamycin (17-AAG) is one of the most widely studied HSP90 inhibitors in cancer treatment in recent years. One study has demonstrated that 17-AAG reduced the expression of Hsp90-dependent phospho-AKT.100,101 Moreover, experiments have proven that 17-AAG can affect client proteins of Hsp90 in MTT (a kind of metastatic mouse PCC cell-derived cell line) cells and inhibit metastasis of PCC/PGL.102 They have a significant anti-tumor activity against both subcutaneous and metastatic tumor growth. However, its clinical application is restricted by adverse liver toxicity and adverse effects such as resistance.103
Another Hsp90 inhibitor VER-52296 can be used as a better small molecule inhibitor that is toxic to tumor cells while avoiding adverse effects of 17-AAG medicines.104 VER52296 mainly inhibits the activity of HSP90 protein by binding to the N-terminal domain of HSP90 protein and promotes the anti-tumor effect of its client protein activity. In addition, Xu et al102 found that VER-52296 can effectively inhibit the growth of tumors and the effects of inducing apoptosis on metastatic PCCs are shown both in vitro. Therefore, VER-52296 will be a very reasonable and effective option for the treatment of metastatic PCCs, either alone or in combination with other medicines. Cell experiments showed that VER-52296 significantly inhibited the proliferation and migration of PC12 cells in a time- and dose-dependent manner, and apoptosis and cell cycle arrest were observed in cells exposed to VER-52296. Therefore, it was speculated that VER-52296 might play a regulatory role by phosphorylation of HSP90-specifically related client proteins. Noteworthily, exposure of HSP70 to VER-52296 for 24 hrs showed a relationship with upregulation of dose-dependent expression, which is a typical marker for HSP90 inhibition. In sharp contrast, phosphorylated AKT, MEK, and ERK were significantly downregulated after exposure to VER-52296 while the expression of total protein of these three signal proteins did not change significantly. The study indicated that the inhibition of PI3K/AKT and MEK/ERK signal pathways produced by VER-52296 could provide a reasonable explanation for its obvious promotion in apoptosis and significant promotion in cell cycle quiescence.102
AKT inhibitor: perifosine
Perifosine is a class of phosphatidylinositol analogs developed by Aeterna Zentaris and it is the first AKT inhibitor. It prevents the recruitment and activation of AKT on cell membrane, thereby inhibiting the activation of AKT. Studies have shown that perifosine inhibits AKT-mediated cellular signal pathways. In preclinical studies, perifosine inhibits the proliferation of immortalized keratinocytes (HaCaT cells) and squamous cell carcinoma of the head and neck. Also, it significantly reduces AKT phosphorylation and cell cycle arrest in G1 and G2. Besides, it causes dose-dependent growth inhibition of mouse glial progenitor cells.105 In addition, perifosine exhibits good anti-tumor activity against mouse neuroblastoma.
Perifosine has shown excellent efficacy and tolerability in clinical phase II studies of recurrent breast, pancreatic, prostate, head and neck, and lung cancer.106 Unlike the majority of kinase inhibitors that target the adenosine triphosphate-binding region, perifosine targets the homeodomain of AKT, preventing its transport to the plasma membrane. Additionally, the efficacy of perifosine has been observed in patients with sarcoma. However, the response rate of common solid tumors to perifosine as a single medicine has been disappointing, lowering expectations and prompting further research into its mechanism of action. Although many preclinical studies have documents on AKT inhibition by perifosine, clinical validation of these findings is still lacking.107
PI3K inhibitor: wortmannin
For PI3K inhibitors, the present clinical evidence is more limited due to their early stages of development. In most phase I studies, the quantity of responses was too small to determine any clear association with the candidate PI3K/AKT/mTOR pathway biomarkers currently.108,109
Wortmannin is an inhibitor of the PI3K’s catalytic subunit. Although wortmannin has a good anti-tumor effect, it is poorly water-soluble and highly toxic, which limits its further development into clinical use. Although PI3K inhibitors can avoid activation of AKT, they do not show significant clinical advantages which may be related to the early stage of PI3K inhibitor development.110
HIF inhibitors: ethacrynic acid, idarubicin, PX-12, and PX-478
Ethacrynic acid
Na et al111 found that ethacrynic acid has a blocking effect on HIF-α/p300 protein–protein interaction. Ethacrynic acid is a diuretic that increases urination mainly by inhibiting the active reabsorption of NaCl in thick sections of the renal tubules. It does not affect the expression of HIF-1α but can reduce the transcriptional expression of downstream VEGF. As a commonly used diuretic, the safety of ethacrynic acid has been clinically proven. Whereas, the structure–activity relationship and target selectivity need to be further studied to increase protein–protein interaction blocking activity and decrease diuretic activity.
Idarubicin
Idarubicin is one of the anthracyclines, which is a well-known class of chemotherapy medicines mainly including daunorubicin, doxorubicin, epirubicin, and idarubicin. When administered to mice at low doses, it had been shown to inhibit tumor growth and angiogenesis by blocking the HIF signal pathway.112,113 Anthracyclines’ effects on the progression of metastatic PCC have been explored in vitro and in vivo.114 It has been demonstrated that anthracyclines, particularly idarubicin, could suppress hypoxia signaling by preventing the binding of HIF to the hypoxia response element sites on DNA. This contributed to reduced transcriptional activation of HIF target genes, including vascular endothelial growth factor A (VEGFA), erythropoietin (EPO), kinase 1 (PGK1), lactate dehydrogenase A (LDHA), endothelin 1 (EDN1), and phosphoglycerate glucose transporter 1 (GLUT1), and consequently it inhibited the growth of metastatic PCC. What’s more, idarubicin interferes with the transcriptional activation of HIF-α to downregulate hypoxia signaling. Moreover, the animal model demonstrates the dose-dependent inhibition of idarubicin on metastatic PCC growth in vivo. Thus, anthracyclines are promising candidates for inclusion in metastatic PCC therapies, especially for patients who have gene mutations in the hypoxia signal pathway.114
PX-12 and PX-478
HIF is a key regulator of tumor environment, so some agents like PX-12and PX-478 play a role in inhibiting tumor activity by inhibiting HIF. Although there is no reported data on metastatic PCC, these medicines have shown significant antineoplastic activity in mouse xenografts of various human tumors and appear to be promising for metastatic PCC, but there is no conclusive data.115–117
Inhibitors of MET, FGFR1, and TERT
MET inhibitors have three categories: the small molecule MET receptor inhibitors such as tivantinib, crizotinib, tepotinib, savolitinib, cabozantinib, and foretinib; MET receptor monoclonal antibodies such as onartuzumab; antibodies against its ligand. They have shown survival benefit in the treatment of PCC/PGL.118
As for FGFR1, in the past few years, there has been considerable progress in the development of selective FGFR1 inhibitors for use.71 At present, more than 10 FGFR1 inhibitors have been in clinical trials. Under the experimental conditions, several agents have achieved encouraging results.119 The use of clinical grade tyrosine kinase inhibitors targeting FGFR1, as well as agents for chromatin modifications may extend the number of experimental treatment regimens to malignant or inoperable PCC/PGL.120,121 Brivanib is a kind of FGFR1 inhibitor developed as therapeutics and has already been in clinical phrase III.122
Last but not least, telomerase reverse transcriptase (TERT) activation is also a potential target, particularly for metastatic tumors,123 because immortalization in primary PCC/PGL could be an important risk factor for metastasis. Immortalization is achieved by a recombination-based alternative lengthening of telomeres (ALT) pathway124 which can be assessed by evaluation of TERT expression.125 BIBR1532 is one of the most promising TERT-specific active site inhibitors to date. It is a synthetic compound of non-nucleotide small molecules that inhibits telomere extension by noncompetitive binding to the active site of TERT.126,127
Combined medicine therapies
The growth-inhibiting effects of AZD8055 and Torin-1 that are the inhibitors of mTOR were evaluated in human primary cells derived from PCC/PGL patients’ donated tumor tissue. Combined use of AZD8055 with Torin-1 reduced the number of tyrosine hydroxylase positive cells to 50% compared with the control cells, confirming the cytotoxic effect on human PCC/PGL cells. In addition, they can inhibit the glycolysis of tumor cells and further enhance its anti-tumor effect. Some have entered different clinical trials.96
Additionally, activation of ERK contributes to the acquired resistance and poor prognosis in everolimus-treated renal cell carcinoma (RCC) patients. The combination of SCH772984, an ERK inhibitor, with everolimus, an mTOR inhibitor, inhibited the proliferation of RCC cells synergistically by blocking the G1 phase cell cycle. Meanwhile, the combined therapy significantly downregulated the transcription of ribonucleotide reductase M1 (RRM1) and ribonucleotide reductase M2 (RRM2) by weakening the expression of E2F. Overexpression of E2F1 or supplementation of dNTP saved the anti-proliferative activity of Ivermos-SCH772984 combination.128
What’s more, PI3K/AKT/mTOR signal pathway has cross-talking with Ras/Raf/MEK/ERK signal pathway. To be specific, the PI3K/AKT/mTOR and Ras/Raf/ERK signal pathways are not independent of each other in transducing survival signals and there are many interactions between them.129 As a result, inhibiting both these signal pathways at the same time may be a promising strategy for treating PCC/PGL. For instance, the combination of BKM120 (a selective inhibitor of PI3K) and GSK1120212 (a MEK inhibitor) has entered the clinical trial phase I.130
Last but not least, combination of olaparib and temozolomide can reduce the tumor burden and metastatic lesions of SDHB mutant PCC/PGL.131,132 Olaparib is a kind of poly ADP-ribose polymerase (PARP) inhibitor and temozolomide is a kind of chemotherapeutic agent which triggers tumor cell apoptosis through damaging genomic DNA.133,134 The PCC/PGL with SDHB mutants exhibited a reprogrammed mitochondrial complex I. Elevation of NAD+ is an important cofactor that supports the PARP DNA repair pathway, leading to resistance to SDHB-related PCC/PGL. The use of olaparib and temozolomide to target the NAD+/PARP DNA repair pathway not only makes cluster I PCC/PGL cells sensitive to genotoxic drugs but also inhibits metastatic xenograft damage and improves overall survival.135
Expectations and conclusion
In this review, we introduced the genes associated with PCC/PGL and summarized two types of signal pathways about their composition and roles. Besides, how these two types of signal pathways lead to the onset of PCC/PGL was also detailedly described. For the relevant targets in these two signal pathways, we introduced the targeted medicines and their mechanism as comprehensively as possible. Therefore, our review would provide a reference for clinical targeted therapy on PCC/PGL.
Targeted therapy has many advantages, such as less toxic and side effects, longer survival time, and better quality of life. The molecular pathogenetic mechanism of PCC/PGL shows that the mutations of SDHx, FH, PHD2, VHL, and HIF influenced the hypoxia-related signaling, while the mutations of RET, MAX, TMEM127, NF1, KIF1Bβ, HRAS, MET, and FGFR1 influenced the increased kinase signaling. The review provides new ideas for targeted therapies of PCC/PGL by acting on the signal pathway of PCC. In a word, valuable progress has been achieved.
However, despite many new treatments for PCC/PGL have appeared, these data are still limited or experimental. Though molecular-targeted therapy is a promising strategy, but due to the complexity of the pathogenesis of tumors, further research on PCC/PGL tumor biology is needed to discover new targeting genes and targeted medicines to develop more effective treatments, optimize clinical trial designs as well as improve the prognosis and survival of patients with PCC/PGL.
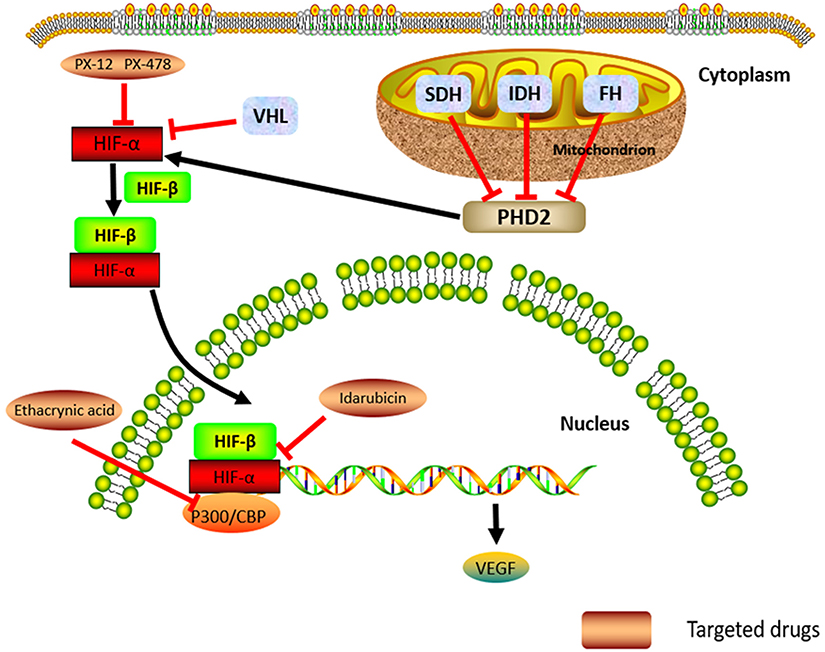 |
Figure 1 The hypoxia-related signal pathway, Cluster 1 genes, and their potential molecular-targeted medicines. Abbreviations: HIF, hypoxia-inducible factor; VEGF, vascular endothelial growth factor; PHD2, prolyl hydroxylase domain protein 2; SDH, succinate dehydrogenase; IDH, isocitrate dehydrogenase; FH, fumarate hydratase; VHL, von Hippel Lindau. |
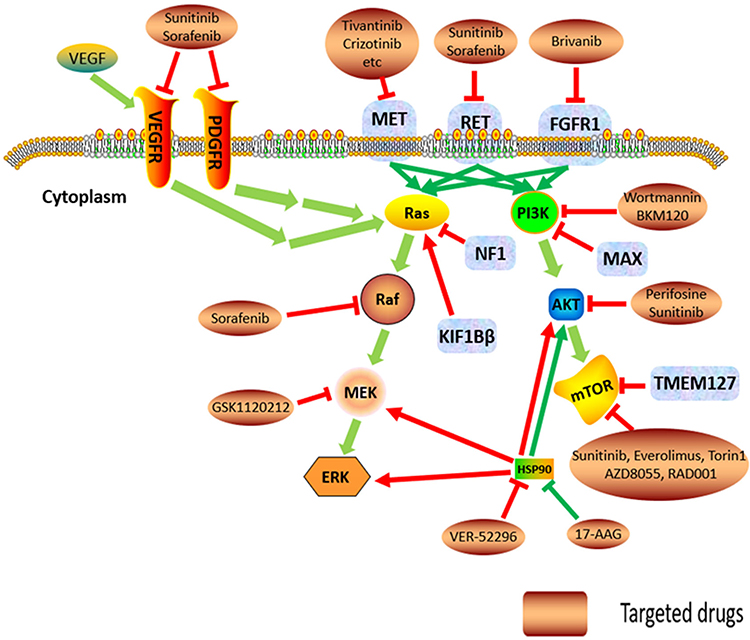 |
Figure 2 The increased kinase signal pathways, Cluster 2 genes, and their potential molecular-targeted medicines. Abbreviations: VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; PDGF, platelet-derived growth factors receptor; MAX, myc-associated factor X; TMEM127, transmembrane protein 127; NF1, neurofibromin 1; KIF1Bβ, kinesin family member1B β; RET, rearranged during transfection proto-oncogene; FGFR1, fibroblast growth factor receptor 1; MET, MET proto-oncogene; PI3K, phosphatidylinositol 3-kinase; mTOR, mammalian target of rapamycin; MEK, MAPK/ERK kinase; ERK, extracellular regulated protein kinases; HSP, heat shock protein. |
Acknowledgment
This study was supported by the Science and Technology Plan Project of Hunan Province, China (no. 2017SK2092).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Gunawardane PTK, Grossman A. Phaeochromocytoma and paraganglioma. Adv Exp Med Biol. 2017;956:239–259. doi:10.1007/5584_2016_76
2. Cotesta D, Petramala L, Serra V, et al. Clinical experience with pheochromocytoma in a single centre over 16 years. High Blood Press Cardiovasc Prev. 2009;16(4):183–193. doi:10.2165/11530430-000000000-00000
3. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. doi:10.1210/jc.2014-1498
4. Eisenhofer G, Huynh TT, Hiroi M, Pacak K. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev Endocr Metab Disord. 2001;2(3):297–311.
5. Graeme E, Lenders JWM, Henri T, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57(3):411–420. doi:10.1373/clinchem.2010.153320
6. Bauer AL, Paulding WR, Striet JB, Schnell PO, Czyzyk-Krzeska MF. Endogenous von Hippel-Lindau tumor suppressor protein regulates catecholaminergic phenotype in PC12 cells. Cancer Res. 2002;62(6):1682–1687.
7. Jenny W, Peter SD, Oliver G. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18(6):253–276. doi:10.1530/ERC-11-0170
8. Dahia PLM. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nature Rev Cancer. 2014;14(2):108–119. doi:10.1038/nrc3648
9. Cascon A, Remacha L, Calsina B, Robledo M. Pheochromocytomas and paragangliomas: bypassing cellular respiration. Cancers (Basel). 2019;11(5):1–22. doi:10.3390/cancers11050683
10. Pillai S, Gopalan V, Smith RA, Lam AK. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit Rev Oncol Hematol. 2016;100:190–208. doi:10.1016/j.critrevonc.2016.01.022
11. Zijin Shen ZS. Progress in surgical treatment of adrenal pheochromocytoma. Shanghai Med J. 2009;32(2):154–157.
12. Shen ZJ, Chen SW, Wang S, et al. Predictive factors for open conversion of laparoscopic adrenalectomy: a 13-year review of 456 cases. J Endourol. 2007;21(11):1333–1337. doi:10.1089/end.2006.450
13. Shen WT, Sturgeon C, Clark OH, Duh QY, Kebebew E. Should pheochromocytoma size influence surgical approach? A comparison of 90 malignant and 60 benign pheochromocytomas. Surgery. 2004;136(6):1129–1137. doi:10.1016/j.surg.2004.05.058
14. Fernandez MC, Gottlieb M, Menitove JE. Blood transfusion and postoperative infection in orthopedic patients. Transfusion. 2010;32(4):318–322. doi:10.1046/j.1537-2995.1992.32492263444.x
15. Brunaud L, Bresler L, Ayav A, et al. Robotic-assisted adrenalectomy: what advantages compared to lateral transperitoneal laparoscopic adrenalectomy? Am J Surg. 2008;195(4):433–438. doi:10.1016/j.amjsurg.2007.04.016
16. Winter JM, Talamini MA, Stanfield CL, et al. Thirty robotic adrenalectomies: a single institution’s experience. Surg Endosc. 2006;20(1):119–124. doi:10.1007/s00464-005-0082-0
17. Bentas W, Wolfram M, Brautigam R, Binder J. Laparoscopic transperitoneal adrenalectomy using a remote-controlled robotic surgical system. J Endourol. 2002;16(6):373–376. doi:10.1089/089277902760261419
18. Plouin PF, Amar L, Dekkers OM, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. 2016;174(5):G1–G10. doi:10.1530/eje-16-0033
19. Jimenez P, Tatsui C, Jessop A, Thosani S, Jimenez C. Treatment for malignant pheochromocytomas and paragangliomas: 5 years of progress. Curr Oncol Rep. 2017;19(12):83. doi:10.1007/s11912-017-0643-0
20. Yanqun N, Zhangqun Y, Sun Y, Sun G. 2014 CUA Guidelines.
21. Kaltsas GA, Stefanidou Z, Papadogias D, Grossman AB. Treatment of advanced neuroendocrine tumours with the radiolabelled somatostatin analogue octreotide. Hormones (Athens). 2002;1(3):149–156.
22. Imhof A, Brunner P, Marincek N, et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol. 2011;29(17):2416–2423. doi:10.1200/jco.2010.33.7873
23. Niemeijer ND, Alblas G, Hulsteijn LT, Van Dekkers OM, Corssmit EPM. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf). 2014;81(5):642–651. doi:10.1111/cen.12542
24. Hadoux J, Favier J, Scoazec JY, et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer. 2014;135(11):2711–2720. doi:10.1002/ijc.28913
25. Tanabe A, Naruse M, Nomura K, Tsuiki M, Tsumagari A, Ichihara A. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Horm Cancer. 2013;4(2):103–110. doi:10.1007/s12672-013-0133-2
26. Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer. 2008;113(8):2020–2028. doi:10.1002/cncr.23812
27. Nomura K, Kimura H, Shimizu S, et al. Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab. 2009;94(8):2850–2856. doi:10.1210/jc.2008-2697
28. Joakim C, Alberto DV, Rajani M, et al. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab. 2013;98(7):E1266–E1271. doi:10.1210/jc.2012-4257
29. Zhengping Z, Chunzhang Y, Felipe L, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2015;367(10):922–930.
30. Yang C, Zhuang Z, Fliedner SM, et al. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J Mol Med. 2015;93(1):93–104. doi:10.1007/s00109-014-1205-7
31. Castro-Vega LJ, Buffet A, De Cubas AA, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23(9):2440–2446. doi:10.1093/hmg/ddt639
32. Letouze E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–752. doi:10.1016/j.ccr.2013.04.018
33. Toledo RA, Qin Y, Srikantan S, et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2013;20(3):349–359. doi:10.1530/ERC-13-0101
34. Akbani R, Ally A, Amar L, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31(2):181–193. doi:10.1016/j.ccell.2017.01.001
35. Chunzhang Y, Sun MG, Joey M, et al. Novel HIF2A mutations disrupt oxygen sensing, leading to polycythemia, paragangliomas, and somatostatinomas. Blood. 2013;121(13):2563–2566. doi:10.1182/blood-2012-10-460972
36. Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47. doi:10.1038/nrc704
37. Xiaofei M, Xiaoli XU, Wang Y, You Q. Advances in the HIF-1α/p300 protein-protein interaction inhibitors. J China Pharm Univ. 2017;48(5):515–522.
38. David TE, Chunzhang Y, Blandine D, et al. First report of bilateral pheochromocytoma in the clinical spectrum of HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab. 2013;98(5):E908–E913. doi:10.1210/jc.2013-1217
39. Maynard MA, Ohh M. The role of hypoxia-inducible factors in cancer. Cell Mol Life Sci. 2007;64(16):2170–2180. doi:10.1007/s00018-007-7082-2
40. Kaelin WG
41. Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015;15(1):55–64. doi:10.1038/nrc3844
42. Walther MM, Reiter R, Keiser HR, et al. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol. 1999;162(3 Pt 1):659–664. doi:10.1097/00005392-199909010-00004
43. Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114(10):2015–2019. doi:10.1182/blood-2009-05-189985
44. Merlo A, de Quiros SB, de Santa-Maria IS, et al. Identification of somatic VHL gene mutations in sporadic head and neck paragangliomas in association with activation of the HIF-1alpha/miR-210 signaling pathway. J Clin Endocrinol Metab. 2013;98(10):E1661–1666. doi:10.1210/jc.2013-1636
45. Rustin P, Rotig A. Inborn errors of complex II–unusual human mitochondrial diseases. Biochim Biophys Acta. 2002;1553(1–2):117–122. doi:10.1016/s0005-2728(01)00228-6
46. Guzy RD, Bhumika S, Eric B, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cel Biol. 2008;28(2):718–731. doi:10.1128/MCB.01338-07
47. Ghezzi D, Goffrini P, Uziel G, et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009;41(6):654–656. doi:10.1038/ng.378
48. Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325(5944):1139–1142. doi:10.1126/science.1175689
49. Nelly B, Jean-Jacques B, Rossella L, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19(15):3011–3020. doi:10.1093/hmg/ddq206
50. Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287(5454):848–851. doi:10.1126/science.287.5454.848
51. Niemann S, Müller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26(3):268–270. doi:10.1038/81551
52. Gimm O, Armanios M, Dziema H, Neumann HP, Eng C. Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res. 2000;60(24):6822–6825.
53. Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69(1):49–54. doi:10.1086/321282
54. Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi:10.1016/j.ccr.2004.11.022
55. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–13378. doi:10.1074/jbc.273.22.13375
56. SUN KF. Role of PI3K/AKT/mTOR signaling pathway in development and progress of pheochromocytoma and its combined targeted therapies. J Shanghai Jiao Tong Univ Med Sci. 2015;35(1):137–141.
57. Mulligan LM, Kwok JB, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363(6428):458–460. doi:10.1038/363458a0
58. Ichihara M, Murakumo Y, Takahashi M. RET and neuroendocrine tumors. Cancer Lett. 2004;204(2):197–211. doi:10.1016/s0304-3835(03)00456-7
59. Besset V, Scott RP, Ibanez CF. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J Biol Chem. 2000;275(50):39159–39166. doi:10.1074/jbc.M006908200
60. Peczkowska M, Kowalska A, Sygut J, et al. Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clin Endocrinol (Oxf). 2013;79(6):817–823. doi:10.1111/cen.12218
61. Burnichon N, Cascon A, Schiavi F, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. 2012;18(10):2828–2837. doi:10.1158/1078-0432.ccr-12-0160
62. Mulligan LM, Kwok JBJ, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363(6428):458–460. doi:10.1038/363458a0
63. Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42(3):229–233. doi:10.1038/ng.533
64. Neumann HP, Sullivan M, Winter A, et al. Germline mutations of the TMEM127 gene in patients with paraganglioma of head and neck and extraadrenal abdominal sites. J Clin Endocrinol Metab. 2011;96(8):E1279–E1282. doi:10.1210/jc.2011-0114
65. Chaux A, Schultz L, Albadine R, et al. Immunoexpression status and prognostic value of mammalian target of rapamycin and hypoxia-induced pathway members in papillary cell renal cell carcinomas. Hum Pathol. 2012;43(12):2129–2137. doi:10.1016/j.humpath.2012.01.009
66. Yeh IT, Lenci RE, Qin Y, et al. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet. 2008;124(3):279–285. doi:10.1007/s00439-008-0553-1
67. Kimura N, Fukase M, Wakita A, Kimura I. Loss of the neurofibromin-NF1 gene product and composite pheochromocytoma. Ann N Y Acad Sci. 2010;971(1):536–538. doi:10.1111/j.1749-6632.2002.tb04521.x
68. Burnichon N, Buffet A, Parfait B, et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet. 2012;21(26):5397–5405. doi:10.1093/hmg/dds374
69. Welander J, Larsson C, Backdahl M, et al. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet. 2012;21(26):5406–5416. doi:10.1093/hmg/dds402
70. Yoshimoto K, Iwahana H, Fukuda A, et al. ras mutations in endocrine tumors: mutation detection by polymerase chain reaction-single strand conformation polymorphism. Jpn J Cancer Res. 1992;83(10):1057–1062. doi:10.1111/j.1349-7006.1992.tb02722.x
71. Toledo RA, Qin Y, Cheng ZM, et al. Recurrent mutations of chromatin-remodeling genes and kinase receptors in pheochromocytomas and paragangliomas. Clin Cancer Res. 2016;22(9):2301–2310. doi:10.1158/1078-0432.ccr-15-1841
72. Folkman J. Tumor Angiogenesis. Adv Cancer Res. 1985;43(1):175–203.
73. Shi Zhang EA. Advances in pharmacokinetic interactions targeting antitumor drugs. China Pharm. 2016;27(20):2871–2874. doi:10.6039/j.issn.1001-0408.2016.20.44
74. Tod M, Mir O, Bancelin N, et al. Functional and clinical evidence of the influence of sorafenib binding to albumin on sorafenib disposition in adult cancer patients. Pharm Res. 2011;28(12):3199–3207. doi:10.1007/s11095-011-0499-1
75. Jimenez C, Cabanillas ME, Santarpia L, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab. 2009;94(2):386–391. doi:10.1210/jc.2008-1972
76. Shah U, Giubellino A, Pacak K. Pheochromocytoma: implications in tumorigenesis and the actual management. Minerva Endocrinol. 2012;37(2):141–156.
77. Gupta R, Maitland ML. Sunitinib, hypertension, and heart failure: a model for kinase inhibitor-mediated cardiotoxicity. Curr Hypertens Rep. 2011;13(6):430–435. doi:10.1007/s11906-011-0229-4
78. Aita Y, Ishii KA, Saito Y, et al. Sunitinib inhibits catecholamine synthesis and secretion in pheochromocytoma tumor cells by blocking VEGF receptor 2 via PLC-gamma-related pathways. Am J Physiol Endocrinol Metab. 2012;303(8):E1006–E1014. doi:10.1152/ajpendo.00156.2012
79. Saito Y, Tanaka Y, Aita Y, et al. Sunitinib induces apoptosis in pheochromocytoma tumor cells by inhibiting VEGFR2/Akt/mTOR/S6K1 pathways through modulation of Bcl-2 and BAD. Am J Physiol Endocrinol Metab. 2012;302(6):E615–E625. doi:10.1152/ajpendo.00035.2011
80. Favier J, Igaz P, Burnichon N, et al. Rationale for anti-angiogenic therapy in pheochromocytoma and paraganglioma. Endocr Pathol. 2012;23(1):34–42. doi:10.1007/s12022-011-9189-0
81. Oberg K, Casanovas O, Castano JP, et al. Molecular pathogenesis of neuroendocrine tumors: implications for current and future therapeutic approaches. Clin Cancer Res. 2013;19(11):2842–2849. doi:10.1158/1078-0432.ccr-12-3458
82. Parenti G, Zampetti B, Rapizzi E, Ercolino T, Giache V, Mannelli M. Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. J Oncol. 2012;2012:872713. doi:10.1155/2012/872713
83. Svenja NL, Alessio G, Yasin T, et al. Combination of 13-Cis retinoic acid and lovastatin: marked antitumor potential in vivo in a pheochromocytoma allograft model in female athymic nude mice. Endocrinology. 2014;155(7):2377–2390. doi:10.1210/en.2014-1027
84. Ayala-Ramirez M, Chougnet CN, Habra MA, et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J Clin Endocrinol Metab. 2012;97(11):4040–4050. doi:10.1210/jc.2012-2356
85. Bourcier ME, Vinik AI. Sunitinib for the treatment of metastatic paraganglioma and vasoactive intestinal polypeptide-producing tumor (VIPoma). Pancreas. 2013;42(2):348–352. doi:10.1097/MPA.0b013e31825c53fa
86. Prochilo T, Savelli G, Bertocchi P, et al. Targeting VEGF-VEGFR pathway by sunitinib in peripheral primitive neuroectodermal tumor, paraganglioma and epithelioid hemangioendothelioma: three case reports. Case Rep Oncol. 2013;6(1):90–97. doi:10.1159/000348429
87. Hahn NM, Reckova M, Cheng L, Baldridge LA, Cummings OW, Sweeney CJ. Patient with malignant paraganglioma responding to the multikinase inhibitor sunitinib malate. J Clin Oncol. 2009;27(3):460–463. doi:10.1200/jco.2008.19.9380
88. Joshua AM, Ezzat S, Asa SL, et al. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J Clin Endocrinol Metab. 2009;94(1):5–9. doi:10.1210/jc.2008-1836
89. Sun F. Preliminary study on the target treatment of malignant adrenal pheochromocytoma.
90. Yuan R, Kay A, Berg WJ, Lebwohl D. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. J Hematol Oncol. 2009;2:45. doi:10.1186/1756-8722-2-45
91. Druce MR, Kaltsas GA, Fraenkel M, Gross DJ, Grossman AB. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: experience with the mTOR inhibitor everolimus (RAD001). Horm Metab Res. 2009;41(9):697–702. doi:10.1055/s-0029-1220687
92. Oh DY, Kim TW, Park YS, et al. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer. 2012;118(24):6162–6170. doi:10.1002/cncr.27675
93. Lee M, Minaskan N, Wiedemann T, et al. Targeting PI3K/mTOR signaling exerts potent antitumor activity in pheochromocytoma in vivo. Endocr Relat Cancer. 2017;24(1):1–15. doi:10.1530/erc-16-0324
94. Zhang X, Wang X, Xu T, Zhong S, Shen Z. Targeting of mTORC2 may have advantages over selective targeting of mTORC1 in the treatment of malignant pheochromocytoma. Tumour Biol. 2015;36(7):5273–5281. doi:10.1007/s13277-015-3187-7
95. Giubellino A, Bullova P, Nolting S, et al. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: in vitro and in vivo studies in female athymic nude mice. Endocrinology. 2013;154(2):646–655. doi:10.1210/en.2012-1854
96. Zaytseva YY, Valentino JD, Gulhati P, Evers BM. mTOR inhibitors in cancer therapy. Cancer Lett. 2012;319(1):1–7. doi:10.1016/j.canlet.2012.01.005
97. Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121(4):1231–1241. doi:10.1172/jci44145
98. Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res. 2009;15(1):9–14. doi:10.1158/1078-0432.CCR-08-0132
99. Jarosz D. Hsp90: a global regulator of the genotype-to-phenotype map in cancers. Adv Cancer Res. 2016;129:225–247. doi:10.1016/bs.acr.2015.11.001
100. Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem. 1995;270(41):24585–24588. doi:10.1074/jbc.270.41.24585
101. Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277(42):39858–39866. doi:10.1074/jbc.M206322200
102. Giubellino A, Sourbier C, Lee MJ, et al. Targeting heat shock protein 90 for the treatment of malignant pheochromocytoma. PLoS One. 2013;8(2):e56083. doi:10.1371/journal.pone.0056083
103. Piper PW, Millson SH. Mechanisms of resistance to Hsp90 inhibitor drugs: a complex mosaic emerges. Pharmaceuticals (Basel). 2011;4(11):1400–1422. doi:10.3390/ph4111400
104. Gaspar N, Sharp SY, Pacey S, et al. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 2009;69(5):1966–1975. doi:10.1158/0008-5472.can-08-3131
105. Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65(16):7429–7435. doi:10.1158/0008-5472.can-05-1042
106. Chao Zhang NY. Advances in kinase inhibitors targeting PI3K-Akt-mTOR signal transduction pathway. China Oncol. 2006;16(12):1064–1070.
107. Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep. 2009;11(2):102–110.
108. Mayer IA, Balko JM, Kuba MG, et al. PD09-05: SU2C phase Ib study of pan-PI3K inhibitor BKM120 plus aromatase inhibitor letrozole in ER+/HER2− Metastatic Breast Cancer (MBC). Cancer Res. 2011;71(24 Supplement):
109. Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–290. doi:10.1200/jco.2011.36.1360
110. Gonzalez-Angulo AM, Blumenschein GR
111. Na YR, Han KC, Park H, Yang EG. Menadione and ethacrynic acid inhibit the hypoxia-inducible factor (HIF) pathway by disrupting HIF-1alpha interaction with p300. Biochem Biophys Res Commun. 2013;434(4):879–884. doi:10.1016/j.bbrc.2013.04.044
112. Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci U S A. 2009;106(7):2353–2358. doi:10.1073/pnas.0812801106
113. Yamazaki Y, Hasebe Y, Egawa K, Nose K, Kunimoto S, Ikeda D. Anthracyclines, small-molecule inhibitors of hypoxia-inducible factor-1 alpha activation. Biol Pharm Bull. 2006;29(10):1999–2003. doi:10.1248/bpb.29.1999
114. Pang Y, Yang C, Schovanek J, et al. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget. 2017;8(14):22313–22324. doi:10.18632/oncotarget.16224
115. Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12(19–20):853–859. doi:10.1016/j.drudis.2007.08.006
116. Welsh S, Williams R, Kirkpatrick L, Paine-Murrieta G, Powis G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2004;3(3):233–244.
117. Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther. 2003;2(3):235–243.
118. Wang Q, Yang S, Wang K, Sun SY. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J Hematol Oncol. 2019;12:63.
119. Yu T, Yang Y, Liu Y, et al. A FGFR1 inhibitor patent review: progress since 2010. Expert Opin Ther Pat. 2017;27(4):439–454. doi:10.1080/13543776.2017.1272574
120. Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Eur J Cancer Suppl. 2007;6(9):153–158.
121. Kristian H, Dashyant D. Chromatin proteins and modifications as drug targets. Nature. 2013;502(7472):480–488. doi:10.1038/nature12751
122. Masatoshi K, Guohong H, Finn RS, et al. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: a randomized phase III trial. Hepatology. 2014;60(5):1697–1707. doi:10.1002/hep.27163
123. Job S, Draskovic I, Burnichon N, et al. Telomerase activation and ATRX mutations are independent risk factors for metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. 2019;25(2):760–770. doi:10.1158/1078-0432.ccr-18-0139
124. Dilley RL, Greenberg RA. ALTernative telomere maintenance and cancer. Trends Cancer. 2015;1(2):145–156. doi:10.1016/j.trecan.2015.07.007
125. Sumit B, Linghe X, Zaug AJ, et al. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science. 2015;347(6225):1006–1010. doi:10.1126/science.1260200
126. Xuehua Guo NL. The new biological functions of telomerase reverse transcriptase. Chin J Biochem Mol Biol. 2018;34(9):927–934.
127. Emanuelle P, Christian W, Joachim L, et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J Biol Chem. 2002;277(18):15566–15572. doi:10.1074/jbc.M201266200
128. Zou Y, Li W, Zhou J, Zhang J, Huang Y, Wang Z. ERK inhibitor enhances everolimus efficacy through the attenuation of dNTP pools in renal cell carcinoma. Mol Ther Nucleic Acids. 2019;14:550–561. doi:10.1016/j.omtn.2019.01.001
129. De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16 Suppl 2:S17–S27. doi:10.1517/14728222.2011.639361
130. Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):121–130. doi:10.1517/14728222.2011.644788
131. Benhur E, Utsumi H, Elkind MM. Inhibitors of poly (ADP-ribose) synthesis enhance radiation response by differentially affecting repair of potentially lethal versus sublethal damage. Br J Cancer Suppl. 1984;6(1):39–42.
132. Schlicker A, Peschke P, Burkle A, Hahn EW, Kim JH. 4-Amino-1,8-naphthalimide: a novel inhibitor of poly(ADP-ribose) polymerase and radiation sensitizer. Int J Radiat Biol. 1999;75(1):91–100.
133. Chaudhuri AR, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nature Rev Mol Cell Biol. 2017;18(10):610–621. doi:10.1038/nrm.2017.53
134. Murcia JM, De Niedergang C, Trucco C, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA. 1997;94(14):7303–7307. doi:10.1073/pnas.94.14.7303
135. Pang Y, Lu Y, Caisova V, et al. Targeting NAD+/PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I pheochromocytoma and paraganglioma. Clin Cancer Res. 2018;24(14):3423–3432. doi:10.1158/1078-0432.CCR-17-3406
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.