Back to Journals » Biologics: Targets and Therapy » Volume 8
Therapeutic vaccines and cancer: focus on DPX-0907
Authors Karkada M, Berinstein N, Mansour M
Received 1 October 2013
Accepted for publication 27 November 2013
Published 10 February 2014 Volume 2014:8 Pages 27—38
DOI https://doi.org/10.2147/BTT.S55196
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Mohan Karkada,1,2 Neil L Berinstein,3 Marc Mansour1
1ImmunoVaccine Inc, 2Department of Microbiology/Immunology, Dalhousie University, Halifax, NS, Canada; 3Ontario Institute for Cancer Research, Toronto, ON, Canada
Abstract: In an attempt to significantly enhance immunogenicity of peptide cancer vaccines, we developed a novel non-emulsion depot-forming vaccine platform called DepoVax™ (DPX). Human leukocyte antigen (HLA)-A2 restricted peptides naturally presented by cancer cells were used as antigens to create a therapeutic cancer vaccine, DPX-0907. In a phase I clinical study, the safety and immune-activating potential of DPX-0907 in advanced-stage breast, ovarian, and prostate cancer patients were examined, following encouraging results in HLA-A2 transgenic mice. The DPX-0907 vaccine was shown to be safe and well tolerated, with injection-site reactions being the most commonly reported adverse event. Vaccinated cancer patients exhibited a 61% immune response rate, with higher response rates in the breast and ovarian cancer patient cohorts. In keeping with the higher immune efficacy of this vaccine platform, antigen-specific responses were detected in 73% of immune responders after just one vaccination. In 83% of responders, peptide-specific T-cells were detected at two or more time points post-vaccination, with 64% of these patients showing evidence of immune persistence. Immune monitoring also demonstrated the generation of antigen-specific T-cell memory, with the ability to secrete multiple type 1 cytokines. The novel DPX formulation promotes multifunctional effector/memory responses to peptide-based tumor-associated antigens. The data support the capacity of DPX-0907 to elicit type-1 biased immune responses, warranting further clinical development of the vaccine. In this review, we discuss the rationale for developing DPX-based therapeutic cancer vaccine(s), with a focus on DPX-0907, aimed at inducing efficient anti-tumor immunity that may eventually be shown to prolong patient survival.
Keywords: immunotherapy, DepoVax™, cancer vaccine, DPX-0907
Introduction
Cancer immunotherapy is gaining importance in disease management, given the demonstrated clinical benefit to patients in recent clinical trials. The renewed interest in the field is followed by US Food and Drug Administration approval of cell-based therapy for prostate cancer and cytotoxic T-lymphocyte associated antigen-4-blocking antibody for the treatment of patients with melanoma.1,2 However, considering the number of attempts in recent years, success in terms of clinical benefit to the patients has been limited. There are many critical issues that still need to be addressed, such as weak immunogenicity of tumor-associated self-antigens (TAAs), T helper (Th)-1 polarization of the immune response, inefficient trafficking of responder T-cells (cytotoxic T-lymphocytes [CTL]) into tumor beds, and inhibitory effects of tumor-induced suppressor cells. Immunogenic vaccines and effective combination treatment strategies of cancer vaccines with immune modulators, with appropriate patient selection and integration of immunotherapy with standard of care treatments, is essential for success. We sought to optimize a vaccination-delivery platform as a first step to develop an effective vaccination strategy for patients with cancer.
In order to be effective, cancer vaccines must induce specific, functional, and persistent cluster of differentiation (CD)-8 T-cells of central memory (TCM) and effector memory (TEM) phenotypes.3,4 Moreover, recently described T-memory stem cells (TSCM) with robust proliferative and self-renewal capacities might maintain vaccine-induced immunity/immune surveillance for longer periods of time, probably helping to improve progression-free survival of patients by acting as a continuous source for anti-tumor TCM and TEM CD8 T-cells.5 Given the mounting evidence for the role of CD4 T-cells in shaping the nature of CD8 CTL responses in cancer vaccine studies, measures to induce effector CD4 T-cells are also important to ensure overall success of a given clinical product.6,7
Peptides from TAAs can be selected on the basis of their ability to bind to specific major histocompatibility complex (MHC) molecules, and multi-peptide vaccines compatible for use in patients with different human leukocyte antigen (HLA) types can be rapidly designed and produced with minimum toxicity or regulatory concerns. Cancer-associated self-peptides have been used as targets for CTL response; however, these peptides are generally weak immunogens and are unable to break immune tolerance, probably because of their low MHC-binding affinity.8,9 Although many peptide-based vaccines have been able to induce some degree of immune response, many have failed to generate long-lasting memory CD8 T-cells. Despite the use of appropriate CTL peptide epitopes in cancer vaccines, success is not guaranteed when formulated in a conventional oil medium such as Montanide. To achieve clinical benefit from the cancer peptide vaccine, there is an unmet need for the development of novel vaccine platforms that can enhance immunogenicity.
DepoVax™ concept
To enhance the potency of a peptide vaccine, we developed a novel vaccine platform called DepoVax™ (Halifax, NS, Canada) (DPX), a liposome-in-oil platform containing stable components that does not require creation of an emulsion, simplifying the use of oil-based depot vaccines in the clinic.10,11 This platform is capable of inducing robust immune responses, applicable for use in both infectious disease and cancer indications. For therapeutic cancer vaccine design, DPX can be custom formulated with mixtures of CD8+ T-cell peptide epitopes, a Th epitope derived from tetanus toxoid,12 and an adjuvant of choice (such as a toll-like receptor agonist) to provide signals for improved antigen presentation. The liposomes carry incorporated hydrophilic antigens and adjuvant directly into an oil medium such as Montanide ISA51 VG, entrapping all vaccine ingredients in a form suitable for efficient uptake, processing and presentation by antigen-presenting cells (APCs).
During the early stages of development, the liquid water-in-oil liposome emulsion called VacciMax was tested in both infectious disease and cancer pre-clinical models. It was found to be very effective in inducing not only high antibody titers against antigens from infectious agents but also in generating effective cell-mediated immunity that can protect against cancer-induced death. Building on the success in pre-clinical models and the promise offered by such a novel vaccine platform to be applicable in human diseases, we further refined the platform as a water-free DPX vaccine. This improvement not only increased the stability of vaccine components for prolonged periods of time (>4 years at present) but also allowed simple preparation of vaccine for injection by mixing the dry vaccine cake in oil at the point of application, avoiding the need for preparing cumbersome emulsion in the clinic. Both water-in-oil VacciMax and water-free DPX-formulated vaccines can induce effective immune responses after a single-dose administration.10,13–16
DPX-0907: therapeutic cancer vaccine
The novel cancer vaccine DPX-0907 contains a polynucleotide-based adjuvant and a universal Th peptide, along with seven HLA-A2 restricted peptides derived from tumor-associated antigens. Proprietary peptide antigens, presented on tumor cells, were identified at Immunotope, Inc (Doylestown, PA, USA) by isolating MHC-associated peptides from human breast and ovarian cancer cell lines using an immune-affinity purification method. Proprietary techniques were used to release, separate, and purify the peptides from the MHC molecules. The purified peptide mixture was simplified by high-performance liquid chromatography (HPLC) fractionation followed by liquid chromatography–mass spectrometry (LC/MS) analysis to generate peptide sequence data (Table 1). The biological relevance of these peptides was validated by screening synthetic peptides to generate/activate CTL in vitro and the ability of CTL to recognize HLA-A2+ ovarian cancer cell lines. These peptides were among several peptides described as HLA-A2-bound peptides from HLA-A2+ ovarian cancer cell lines.17 The vaccine-incorporated peptides are differentially presented in MHC class I on the cell surface of breast, ovarian, and prostate cancer cells, but not on the cells derived from normal human tissues.17,18 Induction of antigen-specific, functional CD8 CTL in DPX-0907 vaccine recipients may result in anti-tumor immune responses and progression-free survival; however, this needs to be demonstrated in randomized clinical trials.
 | Table 1 TAA peptides included as antigens in DPX-0907 therapeutic cancer vaccine and their sequences and species homologies |
Of 12 original tumor-associated peptides identified in screening and considered important for tumor survival, seven peptides were selected as the DPX-0907 vaccine candidates based on a re-evaluation of the available literature as well as their chemical compatibility as a mixture in the DPX platform. These peptides, listed in Table 1, are involved in five out of six cancer survival and proliferation pathways.19 Three of the peptides share <90% homology to peptides in the corresponding mouse proteins, and three peptides have homologous counterparts in both species. These antigens are involved in multiple critical cancer pathways such as tissue invasion and metastasis (P5, integrin β8 subunit precursor; P14, Junction plakoglobin, and P15, EDDR1), evading apoptotic cell death (P3, BAP31), and providing the ability to resist anti-growth signals (P7, Abl binding protein C3).17–21 Because of multi-molecule targeting, specific immune responses that can be induced following DPX-0907 vaccination are expected to reduce the chances for progression of tumor escape variants.19,22
Vaccine peptides and a Th (A16L) peptide epitope derived from tetanus toxoid (830-844; AQYIKANSKFIGITEL) were formulated either as a conventional emulsion in Montanide ISA51 with granulocyte macrophage colony-stimulating factor (GM-CSF) or in the novel DPX formulation.10 Briefly, for the DPX-vaccine, liposomes were formulated by mixing phosphatidylcholine and cholesterol in a 10:1 ratio (w:w) with the seven cancer peptides, A16L, and a proprietary poly-nucleotide-based adjuvant in phosphate buffer (50 mM, pH 7.4). The liposomal solution was lyophilized and stored, and re-suspended in Montanide ISA51 VG (SEPPIC, Paris, France) just before injection. The control vaccine was prepared by emulsifying the seven breast/ovarian cancer-associated peptides, A16L, and 5 μg GM-CSF with Montanide ISA51. All vaccines contained 50 μg of each peptide and 25 μg of A16L peptide per 50 μL dose injected in pre-clinical studies. In the pre-clinical study, we directly compared the efficacy of immune responses against cancer peptides induced by conventional emulsion vaccine with GM-CSF (broadly used in peptide vaccine trials) and the novel DPX-based vaccine formulation.
Pre-clinical proof-of-concept studies
The peptides used in DPX-0907 are HLA-A2 restricted and thus the vaccine was tested in HLA-A*0201/H2Dd (AAD) transgenic mice. These AAD transgenic mice express a chimeric MHC class I molecule containing human α1 and α2 domains from human HLA-A*0201 and the transmembrane/cytoplasmic α3 domain from mouse H2Dd. Subcutaneous injection was provided either a single time or three times at weekly intervals. Draining lymph nodes (LNs) and spleens were harvested from mice 8 days following the final vaccination in each case. To fully capture the immune response to human peptides in humanized mice, we used two methods to measure immune response: 1) a modified enzyme-linked immunospot (ELISPOT) assay using bone marrow-derived mature dendritic cells as APC, a method that improves sensitivity of detection of immune response and provides better inter-assay control; 2) intracellular cytokine staining (ICS) and flow cytometry assay to determine both effector CD8 T-cells as well as suppressor cells such as regulatory T-cells (Treg) and Tr1 cells secreting suppressor cytokines such as interleukin (IL)-10 and transforming growth factor (TGF)-β.
We wanted to compare the immunogenicity of peptides formulated in a highly adjuvanted emulsion vaccine or in the DPX formulation. For safety evaluation of the two vaccine formulations, rats were immunized on a weekly basis with a conventional formulation or with a DPX vaccine formulation containing the lipid components, toll-like receptor agonist adjuvant, the Th epitope, and Montanide ISA51. Site reactions were recorded based on observations made in a detailed physical examination of the site of immunization. The vaccine was found to be well tolerated in this model, with no detectable adverse reactions at the site of vaccination after one immunization, or short-lived mild adverse reactions such as erythema after two or three vaccinations that lasted for 2–3 days. After three immunizations with DPX-0907, only one animal showed a moderate increase in tissue density at the previous immunization sites that was improved within 48 hours. However, the control emulsion vaccine containing GM-CSF exhibited mild immunization-site reactions after even one injection, and, after two injections, the site reactions progressed to well-defined erythema and marked increase in tissue density. After three immunizations with emulsion vaccine, most rats showed signs of well-defined and palpable tissue induration.10 There was no evidence of granuloma or necrosis in DPX-vaccinated animals. In contrast, sites of immunization with the emulsion vaccine showed signs of granuloma and fibrosis, as well as some necrosis. Thus, the DPX-platform was proved safe in pre-clinical models, and was even better tolerated than the GM-CSF-containing Montanide-emulsion vaccine most commonly used in cancer vaccine trials.
Although all the seven peptides induced in vitro T-cell responses from human peripheral blood mononuclear cells (PBMCs),18 their ability to induce in vivo immune responses in HLA-A2 transgenic mice was variable. Peptides P5, P14, and P15 produced the most consistent responses as measured by ELISPOT, with P5 typically producing the highest ELISPOT counts. When comparing the peptide sequence of P5 with its mouse counterpart, we observed not only one amino acid mismatch but most notably an amino acid deletion in the mouse. This reduced sequence homology may explain the higher immunogenicity of this particular sequence. Given the weak responsiveness of HLA-A2-transgenic mice against the remaining four peptides used in the vaccine, we focused our analyses on the P5, P14, and P15 peptides to determine immunogenicity of this vaccine.
Immune responses induced by the DPX-0907 vaccine in the pre-clinical model provided some important insights into how the vaccine is expected to behave in humans. DPX-0907-vaccinated transgenic mice, after just one injection, generated more than two-fold higher frequencies of peptide-specific CD8+interferon (IFN)-γ+ T-cells when compared with conventional emulsion-based vaccines (Figure 1A). Immunization with the DPX-platform alone, performed in initial experiments without cancer peptides, did not noticeably alter T-cell populations (data not shown). To assess the impact of repeated DPX-0907 immunization (prime-boost), we injected mice with DPX-based versus conventional oil-emulsion-based vaccines, either once or three times, on a weekly schedule. While both vaccine formulations promoted specific type-1 T-cell responses after a single injection (Figure 1B), the frequency of IFN-γ-secreting cells in the vaccine draining LN was significantly higher in booster DPX-0907-injected mice than in mice that received the multiple doses of conventional emulsion vaccine (Figure 1C). Most importantly, it was noted that mice immunized with DPX-0907 maintained strong IFN-γ responses after booster immunizations, whereas mice immunized with the conventional emulsion vaccine displayed significantly weaker responses after three immunizations. This suggests that conventional, but not DPX-0907-based, vaccines might be susceptible to treatment-associated induction of immune-regulatory cells, such as Treg cells. Indeed, the percentage of CD4+Foxp3+ Treg cells was significantly higher in mice vaccinated with conventional emulsion-based vaccine than in those that received DPX-based vaccines and in naïve mice.10 No such differences were observed between the DPX-0907 vaccinated and control naïve cohorts. Furthermore, we showed in this study that repeated vaccination with the conventional, but not DPX-0907-based, formulation led to a more pronounced level of splenic CD4+Foxp3+ Treg cells.10
 | Figure 1 Intracellular IFN-γ in CD8+ T-cells (A) and IFN-γ producing cells (B and C) from draining LNs of HLA-A2 transgenic mice immunized with single- (A and B) or three-dose (C) vaccination, either in DPX-0907 or conventional emulsion vaccine. Draining LN cells from naïve and vaccinated mice were incubated with either unloaded or peptide-loaded DC and stained by intracellular immunofluorescence or in a DC-ELISPOT assay for detecting induction of IFN-γ. |
Analysis of spleen of vaccinated mice showed that mice vaccinated with three doses of DPX-0907 did not induce significantly higher levels of CD4+IL-10+ Tr1 cells than did naïve mice, but levels were significantly lower than in conventional emulsion-vaccinated mice (P<0.03; Figure 2A). Repeated experiments have confirmed that vaccination with the emulsion vaccine results in a more than 3-fold increase in IL-10-producing suppressor cells compared with mice vaccinated with DPX-0907. Furthermore, Tr1-cell proliferation was specific for injected peptide antigens, since mice immunized with vehicle control alone (adjuvant and Th peptide) did not produce IL-10+ cells relative to naïve animals. Although not significantly different, DPX-0907-vaccinated mice showed higher frequencies of IFN-γ+ CD4 T-cells in the vaccine-draining LNs than those that received the emulsion vaccine (Figure 2B). The higher frequency of Th1 type CD4 T-cells in treated animals suggests a possible role for these cells in the efficient expansion of antigen-specific CD8 T-cells following DPX-0907 vaccination.
 | Figure 2 CD4+IL-10+ cells from spleen and CD4+IFN-γ+ cells from draining LNs of mice immunized with three doses of DPX-0907, oil emulsion vaccine or naïve mice. Splenocytes and LN cells from indicated groups of mice were isolated and activated for 48 hours in the presence of peptides and used for staining to detect suppressor Tr1 and effector CD4 T-cells. |
Use of mouse models for studying immune response to human antigen targets has provided useful insights into host response against cancer.23 In the present context, since the peptide selection was based on their association with class I MHC on tumor cells, we made use of the mouse model, which is transgenic for HLA-A2 so that the host APCs can present the peptides to a ‘humanized’ CD8+ T-cell repertoire in vivo.24 Of the seven peptides incorporated into the DPX-0907-based vaccine, we consistently observed type-1 immune responses against three peptides, among which, the anti-P5 response appeared dominant. In all cases, peptide vaccines based on conventional oil emulsions proved inferior to DPX-0907-based immunizations in promoting type-1 CD8+ T-cell responses. Given concerns for endogenous, as well as treatment-induced Treg/Tr1 activity as an impediment to successful immunotherapy in the cancer setting, this suggests a clear rationale for implementing vaccine platforms such as DPX-0907 that, unlike GM-CSF adjuvanted Montanide- emulsion vaccines, differentially induce type-1 over Treg/Tr1-type immunity. Lack of appropriate mouse tumor cell line(s) that express HLA-A2-restricted DPX-0907 peptides limited our ability to experimentally test this vaccine in a tumor challenge model. Nevertheless, based on the promise provided by the pre-clinical model in terms of safety and immunogenicity, DPX-0907 was taken to the clinic to further test the vaccine in cancer patients.
DPX-0907: safety and immunogenicity in a human clinical trial
The goals around the clinical development of DPX-0907 are to bring together 1) a vaccine platform that has been shown in preclinical models to generate CD8+/Th1 responses preferentially over Treg responses, and 2) to target multiple antigens that may be playing a role in the neoplastic phenotype to reduce the likelihood of antigen escape variants from emerging. With regard to DPX-0907 vaccine-targeted antigens (Table 1), topoisomerase IIa is associated with rapidly progressing cancers and poor survival and is a target for anthracyclines such as doxorubicin.25 Epidermal discoidin receptor (EDDR)-1 is a protein receptor kinase found to be overexpressed in ovarian cancers and associated with poor prognosis and overall survival.26,27 Tumor necrosis factor-alpha converting enzyme (TACE/ADAM-17) generates soluble epidermal growth factor receptor ligands such as EGF and heparin-binding-EGF that are important in tumorigenesis/tumor progression. The epidermal growth factor receptor pathway is targeted by humanized antibodies (cetuximab) and tyrosine kinase inhibitors (gefitinib, erlotinib).19,28 In the following sections, we discuss the safety and immunogenicity of a phase I trial of DPX-0907 in patients with advanced breast, ovarian, and prostate cancer.
Although DPX-0907 included antigens targeting multiple TAAs, the vaccine directed at peptides that bind to MHC HLA-A02 and hence can be used only in HLA-A2 cancer subjects. However, since HLA-A2 is the most common allele in the Caucasian population (40%–50%), this not only allows for availability of subjects for clinical testing but also a significant proportion of patient population will be eligible for treatment with DPX-0907. The primary objective of phase I study was to assess safety of DPX-0907 in late-stage metastatic cancer patients, with a secondary objective to analyze the immune-response profile against the vaccine antigens. Subjects were recruited into the study among several cancer centers in the USA based on competitive enrollment, rather than disease subtype preference. The majority of subjects enrolled had prostate cancer (n=13), followed by ovarian (n=6) and breast (3) cancer.
Specific eligibility criteria for DPX-0907 study for each type of cancer has been described in an earlier report.11 Briefly, ovarian cancer patients with stage III or IV disease with evidence of a complete or partial response by radiological imaging after front-line de-bulking surgery and platin-based cytotoxic therapy, or patients with metastatic disease, or clinically or radiologically stable disease for more than 3 months after completion of first-line therapy were included. Patients were considered eligible regardless of their CA-125 results. Breast cancer patients with stage IV disease who had received at least one course of hormonal or cytotoxic therapy for metastatic cancer and had completed their course of cytotoxic therapy and had stable disease for ≥3 months were considered eligible for the study. Continuation of hormonal therapy was allowed for recruited breast cancer subjects. Prostate cancer patients with rising prostate-specific antigen levels or increases in measurable disease after at least one course of an accepted hormonal therapy (castrate testosterone levels <50 ng/dL) or who had received previous courses of cytotoxic chemotherapy were included. Where applicable, patients with prostate cancer remained on anti-androgen therapy during the trial. All patients recruited for the study were positive for HLA-A2 expression and met other standard inclusion and exclusion criteria, with a life expectancy of at least 6 months. A summary of recruited patient demographics is published elsewhere.11
Following initial screening for inclusion/exclusion criteria, subjects received three subcutaneous injections of the DPX-0907 vaccine 3 weeks apart in the same upper thigh region, either at 0.25 mL (dose A) or 1.0 mL (dose B), according to the schedule shown in Figure 3. A total of 23 cancer patients qualified to participate in the study, which included cohorts of 13 prostate cancer, seven ovarian cancer, and three breast cancer patients. One of the ovarian cancer patients in the dose B group discontinued due to an unrelated serious adverse effect following a single vaccination, leaving a total of 22 patients with 11 patients in each of the dosage groups.
 | Figure 3 Outline of clinical protocol followed in DPX-0907 trial with details on the time-line for screening, treatment and follow-up of late-stage breast, ovarian, and prostate cancer patients. Blood collected at the indicated time points are used for immune monitoring, and the samples up to study day 73 are analyzed. |
Safety outcome
The primary endpoint of safety was achieved in this first-in-man study. Repeated administration of DPX-0907 did not result in any dose-limiting toxicities or serious adverse events related to vaccine treatment. Similar to our observations in the pre-clinical model, the most frequent side effects were grade 1–2 injection-site erythema and induration, consistent with other vaccines that incorporate Montanide ISA51 VG as the carrier medium.29–31 There was no relationship between immune responses and the grade or frequency of injection-site reactions. When 0.25 mL (dose A) is compared with other vaccines that incorporate antigens in Montanide ISA51 VG emulsion with a similar oil content, DPX-0907 treatment also results in lower reports of pain and no reports of grade 3 induration and swelling.29 However, we did note a greater frequency of pain and edema, and the presence of grade 3 induration, erythema, and transient ulceration in patients receiving multiple injections of 1.0 mL DPX-0907. However, most injection-site reactions in these patients had resolved to grade 1 during follow-up. Nevertheless, it is not unreasonable to assign these reactions as due to the volume of Montanide ISA51 injected. Although the adverse events seen at the 1.0 mL vaccine dose were largely transient in nature, further studies with DPX-0907 might use a dose intermediate between the two (0.5 mL) to maintain immunogenicity and reduce the injection-site side effects.
Clinical outcome
Since long-term monitoring of patients was not part of this phase I clinical plan, the maximum time to progression period recorded in the study was 8–9 months starting from the day of patients’ first vaccination. Hence, patients who reached the 6-month follow-up visit and remained disease free at that time were assigned a time to progression of >8–9 months. During the study period, 14 patients progressed; most of them were prostate cancer patients (10 of 14 patients). In addition, one prostate cancer patient died of progressive disease 3.5 months into this study. Information on disease progression is not available for the remaining two prostate cancer patients, since they opted out of the study. In contrast, disease progression was reported for only two of seven ovarian cancer patients. Based on general health, the breast cancer and ovarian cancer patient populations are less likely to be compromised immunologically and thus were evaluated together. Among patients who completed follow-up, one of three breast cancer patients and four of six ovarian cancer patients had a time to progression of >8–9 months. Among these five breast/ovarian cancer patients, two were treated in the dose A cohort and three were treated with the dose B vaccine. Given the small sample size and absence of a control arm, it was not possible to conclude on the effect of vaccine dose on time to progression. Furthermore, these patients were still within the median progression-free survival period for their previous treatment, which made it difficult to ascertain clinical benefit provided by DPX-0907 vaccination. Similar to our study, Morse et al18 reported the results of emulsion vaccine using 12 HLA-A2 peptides (that included the seven peptides in DPX-0907), where four of eight ovarian and one of seven breast cancer subjects progressed during the study, although median survival had not been reached at the time of reporting. To assign clear clinical benefits to patients provided by DPX-0907, longer follow-up of a larger number of subjects and comparison with a prospective control population (in a randomized trial) is required in each disease cohort.
Immune outcome
An important objective of this trial was to determine the magnitude and diversity of immune responses induced by vaccination against the seven TAA peptides. This would also define the relative immunogenicity of individual vaccine antigens included in DPX-0907 and can potentially provide a means of correlating immune response against a given antigen and patient survival in future studies. Previous studies have suggested that T-cell responses to peptide vaccination may consist of different T-cell subsets associated with different effector functions.6,32–35 We included multiple methods in immune monitoring of the subjects in order to identify the different CD8+ T-cell populations induced by peptide vaccination. This was measured primarily by 1) MHC-multimer staining for antigen-specific CD8+ T-cells, both ex vivo and in vitro, and 2) multi-parametric flow cytometry using the ICS assay for multi-functional T-cells. An exploratory ELISPOT assay was also performed on select samples to confirm findings in the other immune assays. Although a total of 22 patients received the full cycle of vaccinations, because of the poor quality of PBMC from four prostate cancer patient samples, only 18 patients were deemed ‘immune-evaluable’.
In this dose-ranging trial with patients receiving only three vaccinations, vaccine recipients were considered responders to the vaccine antigens if they responded to one or more antigens at one or more time points post-vaccination in one or more immune-monitoring assays. Among the 18 patients evaluable for immune endpoints in this study, 89% of the breast and ovarian cancer patients responded (Table 2). However, among prostate cancer patients, only three of nine (33%) showed a positive immune response to vaccination. Taken together, 11 of 18 patients (61%) showed measurable immune responses against one or more of the vaccine antigens. While the responses of two patients were measurable only by MHC-multimer testing and three patients were positive only using the ICS assay, the majority of patient responses were detectable by both the methods used (Table 3). Immune responses were detectable as early as study day (SD) 21 in 8 of 11 (73%) responders, suggesting that just one immunization was sufficient to elicit specific T-cell responses in most of these patients. In 7 of 11 (64%) responders, specific response to cancer antigens was observed at SD73, supporting the persistence of immune memory in peripheral blood. Vaccination with DPX-0907 induced a specific response against six of seven tumor-associated peptides. The most frequent responses were seen against peptide P5, followed by P7 and P15. Five of 11 responding patients were positive for responses to two or more peptides contained in DPX-0907.
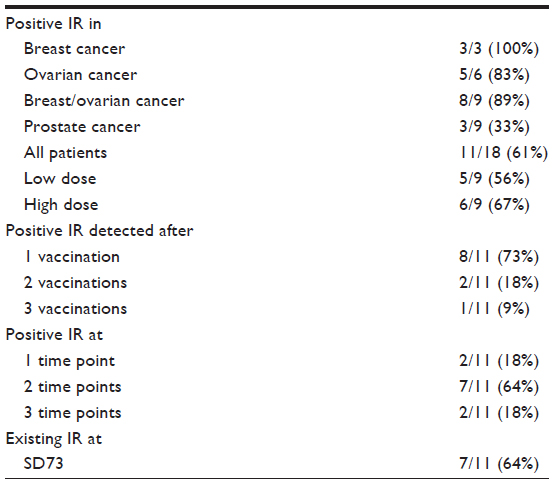 | Table 2 Summary of the immune responses in DPX-0907-treated patients |
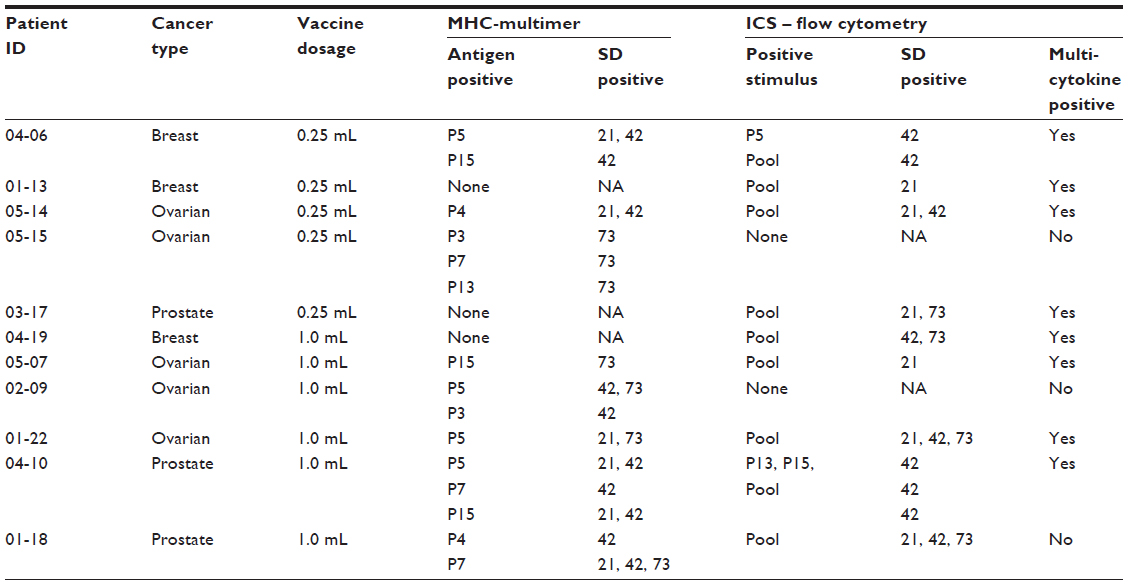 | Table 3 Responders among DPX-0907 vaccine recipients and their immune response to vaccine antigens as determined by MHC-multimer assay and multi-parametric flow cytometry |
MHC-multimer reagents incorporating individual DPX-0907 peptides were used to detect specific CD8+ T-cells. While most patients required in vitro stimulation of PBMCs to detect multimer-positive cells because of the low frequency of such cells in circulation, it was possible to detect such specific CD8 T-cells ex vivo in two patients, suggesting the ability of DPX-0907 to induce a robust T-cell response that can be detected in circulation. Among 11 responders in this study, eight were positive in MHC-multimer testing, and in some instances, a >50-fold increase in specific CD8 T-cells was seen at post-vaccination time point compared with baseline. Moreover, evidence of pre-existing immunity was also seen in some subjects and such immunity was further enhanced following DPX-0907 treatment. Sequential changes in the frequency of antigen-specific T-cells at pre- and post-vaccination time points are shown in Figure 4. Although the peak response occurred on different post-vaccination days for different responders, in almost all patients the frequency of such cells remained higher at SD73 than at SD0.
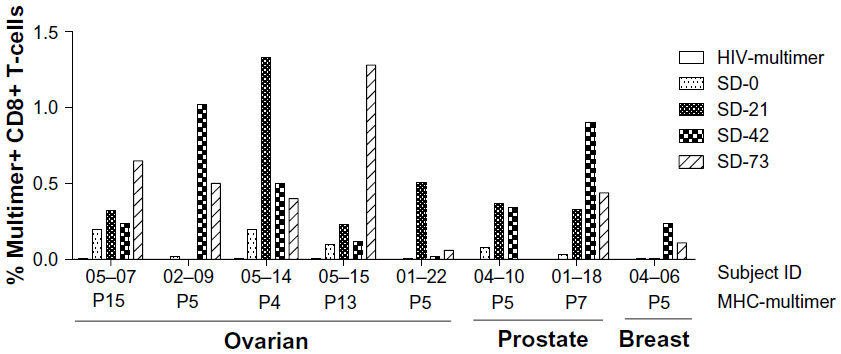 | Figure 4 DPX-0907-induced increase in the frequency of antigen-specific CD8+ T-cells in vaccinated ovarian, prostate, and breast cancer patients. Patient PBMC were stimulated with indicated peptides for 10 days and were stained with corresponding MHC-multimer reagents to detect CD8+ T-cells with peptide-specific TCR repertoire. HIV-multimer served as a negative control and CMV-specific multimer was used on a known CMV-positive donor PBMC as internal positive control for the assay (data not shown). |
The ICS assay allows a broader analysis of functionally relevant antigen-specific T-cells following vaccination. ‘Multi-functional’ T-cells, secreting a diverse array of cytokines in response to antigen(s), have been increasingly associated with host protection.11 Multi-parametric flow cytometry was performed to identify the phenotype of responding T-cells, along with their ability to secrete multiple Th1 type cytokines such as IFN-γ, tumor necrosis factor-α, and IL-2. Nine of 11 subjects were found to be responders in the ICS assay. TCM CD8+ T-cells (CD45RA−CD27+) were detected in these patients, suggesting that the vaccine-induced antigen-specific memory responses. Cytokine-positive cells were more frequent in the TCM compartment than among total CD8+ T-cells, in keeping with previous activation of such TCM cells by vaccine antigens. Importantly, most of the responder total CD8 and/or CD8+ TCM T-cells secreted more than one cytokine. The triple cytokine-positive cells are induced only after vaccination, suggesting de novo induction of multi-functional, anti-tumor effector cells by DPX-0907. At least four patients showed expression of CD107a in combination with IFN-γ, tumor necrosis factor-α, or both, another functional attribute of vaccine-induced T-cells. Our finding that the majority of patients responding to DPX-0907 generate such multi-functional T-cells is encouraging and suggests that the vaccine may be generating clinically relevant immune responses and that the immunogenic antigens have been combined with a suitably immunogenic delivery mechanism.
DPX-0907 also includes a Th epitope that has previously been used to augment peptide vaccine immunogenicity.12,36 As part of the ICS assay, we also examined the response of CD4 T-cells to the Th epitope. In 13 patients, we were able to detect CD4 T-cell response, while four patients were considered non-responders. Interestingly, only two patients had pre- and post-vaccination responses to A16L, while 11 patients had a CD4 response to A16L only post-vaccination. Furthermore, all nine patients who had a positive cytokine response in their CD8 T-cell also had CD4-A16L response, while only four patients showed CD4 T-cell response to A16L without mounting a CD8 T-cell response. Although the focus of this study was to study antigen-specific CD8 T-cells, CD4 responses detected more frequently correlated with CD8+ T-cell responses. While it could not be definitively concluded that inclusion of a Th peptide facilitated the generation of the specific CD8 T-cells in these patients, the results confirm the observations in the pre-clinical model that the DPX vaccine-induced increases in IFN-γ+CD4+ T-cells.10
DPX-0907 generates rapid and sustained immune responses that are detectable at two or more time points post-vaccination in more than half of the immune-evaluable subjects. Such immune responses could be detected by more than one assay in the majority of subjects (Table 3). Furthermore, positive responses detected in MHC-multimer and ICS assay were confirmed in the functional IFN-γ ELSPOT assay and the responses were antigen specific.11 There were no major differences in patient response rate in either of the dose cohorts, suggesting the lack of a vaccine dose/immune response relationship similar to some reports.32,37 However, other studies have demonstrated dose-dependent immune responses for peptide antigens,38,39 which may be related to the antigen(s), formulation, peptide length, adjuvant used, vaccination regimen, and patient population.
Immune responses were detected against six of the seven antigens (P3, P4, P5, P7, P13, and P15) contained in DPX-0907. Pre-existing immune responses to a few of these antigens suggest that patients’ T-cells have been primed prior to patient inclusion on this study. This provides further validation that the chosen vaccine epitopes are biologically relevant targets. The multi-peptide approach is expected to minimize the impact of immune escape mechanisms such as immune editing of antigens by cancer cells under immune pressure. It is unclear whether enhanced and more frequent immune response to DPX-0907 in ovarian and breast cancer than prostate cancer patients is due to immune responsiveness or whether this reflects other confounding clinical factors in each cancer type. The more advanced nature of the disease and immune suppression associated with advanced disease in the prostate cancer patients could have contributed to the lower immune response rate. Indeed, we have observed significantly higher levels of myeloid-derived suppressor cells in prostate cancer subjects than in breast/ovarian cancer subjects in a related study (unpublished observations).
Summary and future directions
In summary, the novel cancer vaccine DPX-0907, employing an innovative liposomal formulation, is safe and able to break self-tolerance and induce robust immune response to cancer over-expressed antigens. Among reported studies, comparable levels of CD8+ T-cell responses were seen in patients with epithelial ovarian cancer using NY-ESO1 peptides or a mix of TAA peptides, but required multiple vaccinations.36,40,41 DPX-0907 induced immune response in a majority of subjects with one vaccination, with higher response rates after three vaccinations in similar patient populations. It is not clear in the vaccine trials mentioned above whether these vaccines could generate immune responses after just one vaccination and, unlike in the DPX-0907 study, multi-functional T-cell responses were not documented to assess the quality of immune response.
This first-in-man study suggests it is worthwhile to test this in additional studies to evaluate clinical benefits, especially in breast and ovarian cancer subjects. It should be noted that most of the breast and ovarian cancer patients had stable disease or responses to their previous treatment, whereas the prostate cancer patients enrolled had progressive disease. As proposed, perhaps patients with a history of favorable responses to previous therapies who are in a state of minimal residual disease are likely to benefit from DPX-0907 immunotherapy.42 Future strategies will explore other peptide antigen vaccines utilizing the DPX formulation, by optimizing the patient populations and by incorporating an immune modulator. This approach of combination treatment is expected to further optimize antigen-specific immune responses and generate functionally relevant clinical responses aimed at reducing or eliminating metastatic disease.
Our recent pre-clinical and clinical studies have provided strong evidence that combining a DPX-based cancer vaccine with low-dose cyclophosphamide can induce very strong antigen-specific immunity (unpublished data). A low oral dose of cyclophosphamide in combination with a DPX-based vaccine enhances immune response and protects mice in pre-clinical models. We have recently taken this approach to the clinic to test the safety and immunogenicity of DPX-based, TAA-survivin-targeted vaccine in late-stage ovarian cancer patients. The results from the phase I trial of DPX-0907 described here suggest that DPX is a very immunogenic vaccine formulation with adjuvant properties. Further studies with this vaccine and other vaccines containing tumor antigens such as survivin are underway.
Disclosure
MK and MM are full time employees of ImmunoVaccine Inc. NLB has received consultancy fee from Immuno Vaccine Inc. as medical consultant but declares no conflicts of interest. The funding for the study was provided by Immuno Vaccine Inc. from its research and development program.
References
Kantoff PW, Higano CS, Shore ND, et al; IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. | |
Perret R, Ronchese F. Memory T cells in cancer immunotherapy: which CD8 T-cell population provides the best protection against tumours? Tissue Antigens. 2008;72(3):187–194. | |
Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12(10):671–684. | |
Dobrzanski MJ. Expanding roles for CD4 T cells and their subpopulations in tumor immunity and therapy. Front Oncol. 2013;3:63. | |
Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol. 1998;10(5):588–594. | |
van der Burg SH, Bijker MS, Welters MJ, Offringa R, Melief CJ. Improved peptide vaccine strategies, creating synthetic artificial infections to maximize immune efficacy. Adv Drug Deliv Rev. 2006;58(8):916–930. | |
van der Burg SH, Visseren MJ, Brandt RM, Kast WM, Melief CJ. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide complex stability. J Immunol. 1996;156(9):3308–3314. | |
Karkada M, Weir GM, Quinton T, et al. A novel breast/ovarian cancer peptide vaccine platform that promotes specific type-1 but not Treg/Tr1-type responses. J Immunother. 2010;33(3):250–261. | |
Berinstein NL, Karkada M, Morse MA, et al. First-in-man application of a novel therapeutic cancer vaccine formulation with the capacity to induce multi-functional T cell responses in ovarian, breast and prostate cancer patients. J Transl Med. 2012;10:156–168. | |
Slingluff CL Jr, Yamshchikov G, Neese P, et al. Phase I trial of a melanoma vaccine with gp100(280-288) peptide and tetanus helper peptide in adjuvant: immunologic and clinical outcomes. Clin Cancer Res. 2001;7(10):3012–3024. | |
Daftarian P, Mansour M, Benoit AC, et al. Eradication of established HPV 16-expressing tumors by a single administration of a vaccine composed of a liposome-encapsulated CTL-T helper fusion peptide in a water-in-oil emulsion. Vaccine. 2006;24(24):5235–5244. | |
Karkada M, Quinton T, Blackman R, Mansour M. Tumor inhibition by DepoVax-based cancer vaccine is accompanied by reduced regulatory/suppressor cell proliferation and tumor infiltration. ISRN Oncol. 2013;2013:753427. | |
Mansour M, Pohajdak B, Kast WM, et al. Therapy of established B16-F10 melanoma tumors by a single vaccination of CTL/T helper peptides in VacciMax. J Transl Med. 2007;5:20–26. | |
MacDonald LD, Fuentes-Ortega A, Sammatur L, Mansour, M. Efficacy of a single dose hepatitis B depot vaccine. Vaccine. 2010;28(44):7143–7145. | |
Ramakrishna V, Ross MM, Petersson M, et al. Naturally occurring peptides associated with HLA-A2 in ovarian cancer cell lines identified by mass spectrometry are targets of HLA-A2-restricted cytotoxic T cells. Int Immunol. 2003;15(6):751–763. | |
Morse MA, Secord AA, Blackwell K, et al. MHC class I-presented tumor antigens identified in ovarian cancer by immunoproteomic analysis are targets for T-cell responses against breast and ovarian cancer. Clin Cancer Res. 2011;17(10):3408–3419. | |
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. | |
Sinnathamby G, Zerfass J, Hafner J, et al. ADAM metallopeptidase domain 17 (ADAM17) is naturally processed through major histocompatibility complex (MHC) class I molecules and is a potential immunotherapeutic target in breast, ovarian and prostate cancers. Clin Exp Immunol. 2011;163(3):324–332. | |
Sinnathamby G, Zerfass J, Hafner J, et al. EDDR1 is a potential immunotherapeutic antigen in ovarian, breast, and prostate cancer. J Clin Cell Immunol. 2011;2(1):1–8. | |
Pilla L, Rivoltini L, Patuzzo R, Marrari A, Valdagni R, Parmiani G. Multipeptide vaccination in cancer patients. Expert Opin Biol Ther. 2009;9(8):1043–1055. | |
Lévy F, Colombetti S. Promises and limitations of murine models in the development of anticancer T-cell vaccines. Int Rev Immunol. 2006;25(5–6):269–295. | |
Newberg MH, Smith DH, Haertel SB, Vining DR, Lacy E, Engelhard VH. Importance of MHC class 1 alpha2 and alpha3 domains in the recognition of self and non-self MHC molecules. J Immunol. 1996;156(7):2473–2480. | |
Binaschi M, Bigioni M, Cipollone A, et al. Anthracyclines: selected new developments. Curr Med Chem Anticancer Agents. 2001;1(2):113–130. | |
Heinzelmann-Schwarz VA, Gardiner-Garden M, Henshall SM, et al. Overexpression of the cell adhesion molecules DDR1, Claudin 3, and Ep-CAM in metaplastic ovarian epithelium and ovarian cancer. Clin Cancer Res. 2004;10(13):4427–4436. | |
Shimada K, Nakamura M, Ishida E, et al. Prostate cancer antigen-1 contributes to cell survival and invasion though discoidin receptor 1 in human prostate cancer. Cancer Sci. 2008;99(1):39–45. | |
Appay V, Douek DC, Price DA. CD8+ T cell efficacy in vaccination and disease. Nat Med. 2008;14(6):623–628. | |
Jäger E, Jäger D, Knuth A. Antigen-specific immunotherapy and cancer vaccines. Int J Cancer. 2003;106(6):817–820. | |
Chiang CL, Kandalaft LE, Coukos G. Adjuvants for enhancing the immunogenicity of whole tumor cell vaccines. Int Rev Immunol. 2011; 30(2–3):150–182. | |
Takahashi N, Ohkuri T, Homma S, et al. First clinical trial of cancer vaccine therapy with artificially synthesized helper/killer-hybrid epitope long peptide of MAGE-A4 cancer antigen. Cancer Sci. 2012;103(1):150–153. | |
Disis ML, Schiffman K, Guthrie K, et al. Effect of dose on immune response in patients vaccinated with an her-2/neu intracellular domain protein--based vaccine. J Clin Oncol. 2004;22(10):1916–1925. | |
Kanodia S, Kast WM. Peptide-based vaccines for cancer: realizing their potential. Expert Rev Vaccines. 2008;7(10):1533–1545. | |
Pejawar-Gaddy S, Finn OJ. Cancer vaccines: accomplishments and challenges. Crit Rev Oncol Hematol. 2008;67(2):93–102. | |
van der Burg SH, Piersma SJ, de Jong A, et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc Natl Acad Sci U S A. 2007;104(29):12087–12092. | |
Chianese-Bullock KA, Irvin WP Jr, Petroni GR, et al. A multipeptide vaccine is safe and elicits T-cell responses in participants with advanced stage ovarian cancer. J Immunother. 2008;31(4):420–430. | |
Perales MA, Yuan J, Powel S, et al. Phase I/II study of GM-CSF DNA as an adjuvant for a multipeptide cancer vaccine in patients with advanced melanoma. Mol Ther. 2008;16(12):2022–2029. | |
Bernhardt SL, Gjertsen MK, Trachsel S, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer. 2006;95(11):1474–1482. | |
Holmes JP, Benavides LC, Gates JD, et al. Results of the first phase I clinical trial of the novel II-key hybrid preventive HER-2/neu peptide (AE37) vaccine. J Clin Oncol. 2008;26(20):3426–3433. | |
Diefenbach CS, Gnjatic S, Sabbatini P, et al. Safety and immunogenicity study of NY-ESO-1b peptide and montanide ISA-51 vaccination of patients with epithelial ovarian cancer in high-risk first remission. Clin Cancer Res. 2008;14(9):2740–2748. | |
Odunsi K, Qian F, Matsuzaki J, et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci U S A. 2007; 104(31):12837–12842. | |
Spisek R. Immunoprevention of cancer: time to reconsider timing of vaccination against cancer. Expert Rev Anticancer Ther. 2006;6(12):1689–1691. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.