")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Therapeutic Potential of Janus Kinase Inhibitors for the Management of Interstitial Lung Disease
Authors Huo R, Guo Q, Hu J, Li N, Gao R, Mi L, Zhang Z, Liu H, Guo Z , Zhao H, Zhang L, Xu K
Received 24 December 2021
Accepted for publication 17 March 2022
Published 2 April 2022 Volume 2022:16 Pages 991—998
DOI https://doi.org/10.2147/DDDT.S353494
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Rongxiu Huo,1,* Qianyu Guo,1,* Junping Hu,2 Na Li,1 Rui Gao,1 Liangyu Mi,1 Zhaoliang Zhang,1 Hechao Liu,1 Zhiying Guo,1 Hanxi Zhao,3 Liyun Zhang,1 Ke Xu1
1Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences; Tongji Shanxi Hospital, Department of Rheumatology and Immunology, Taiyuan, 030032, People’s Republic of China; 2Department of Immunology, Shanxi Medical University, Taiyuan, 030000, People’s Republic of China; 3SILC Business School, Shanghai University, Shanghai, 200444, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ke Xu, Email [email protected]
Abstract: Interstitial lung disease (ILD) refers to a heterogeneous group of diseases characterized by lung fibroblast proliferation, interstitial inflammation, and fibrosis-induced lung damage. The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is known to be activated by pro-fibrotic/pro-inflammatory cytokines such as IL-6 and IL-13, whose levels are elevated in ILD. The overexpression of growth factors such as transforming growth factor β 1 in ILD activates the JAK/STAT pathway through classical or non-classical pathways, promotes macrophage activation, increases the release of pro-inflammatory and pro-fibrosis factors, and facilitates fibroblast differentiation into myofibroblasts. These findings implicate that the JAK/STAT pathway plays an important role in the course of ILD. Recent evidence also suggests that JAK inhibition alleviates excessive inflammation and pulmonary fibrosis. Accordingly, the JAK inhibitors may serve as promising drugs for the treatment of JAK/STAT-induced ILD.
Keywords: interstitial lung disease, Janus kinase, signal transducers and activators of transcription, JAK inhibitor, animal model
Introduction
Interstitial lung disease (ILD) is a group of diseases that originate from a variety of etiologies including infections, drugs, radiation-induced lung diseases, and autoimmune diseases such as systemic sclerosis (SSc) and rheumatoid arthritis (RA). These conditions cause damage to the alveolar epithelium and lung parenchyma, ultimately leading to inflammation and extensive fibrosis.1 ILD is characterized by varying degrees of inflammation or fibrosis in the pulmonary parenchyma. For instance, in inflammatory diseases, the histology of the pulmonary parenchyma is characterized by mechanical pneumonia or nonspecific interstitial pneumonia, while fibrotic diseases are characterized by common interstitial pneumonia.2
Pro-fibrotic/pro-inflammatory cytokines including interleukin-6 (IL6), IL13, and interferon-γ (IFNγ); and growth factors such as transforming growth factor β1 (TGFβ1) are overexpressed in different ILDs.3 Under these circumstances, the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is known to be activated, which further polarizes the macrophages into classic pro-inflammatory M1 macrophages. This phenomenon is characterized by the upregulation of key polarization markers (CD86), and elevated secretion of key pro-inflammatory cytokines and driving factors such as IL6, CXC motif chemokine 10 (CXCL10), and tumor necrosis factor (TNFα). These events, in turn, lead to inflammatory changes in the lungs or pro-fibrotic M2a macrophages, which exhibit enhanced expression of surface markers such as CD206.4 Furthermore, the amplified secretion of pro-fibrotic IL6 induces STAT3- mediated fibroblast-to-mesenchymal transition (FMT),5 thereby resulting in de-differentiation of fibroblasts into myofibroblasts with a mesenchymal phenotype, ultimately leading to ILD.6 In addition to JAK/STAT, JAK can activate other signaling pathways such as phosphoinositol 3-kinase/protein kinase B (PI3K/Akt) and Ras/Raf/MAPK/ERK, which are known to cause bone marrow proliferative diseases.7
TGFβ is considered to be a key pro-fibrotic cytokine.8 After binding to its receptor, it has been reported to phosphorylate SMAD2/3 in the cytoplasm through the classical pathway, which subsequently combines with SMAD4 to form a trimer complex. The trimeric complex then binds with DNA and regulates the expression of transcription factors that promote the expression of fibrotic genes, collagen production, and proliferation of lung fibroblasts.9 In addition to activating SMAD proteins, TGFβ enhances the binding of tumor necrosis factor receptor-associated factor 6 (TRAF6) to TGFβ receptor I and promotes its activation in a receptor kinase-independent manner. Furthermore, TRAF6 has been documented to ubiquitinate TGFβ-activated kinase 1 (TAK1), which is a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family, thereby leading to its activation.10,11 TAK1 subsequently phosphorylates and activates the MAP kinase kinases (MAPKKs), MAPKK3 and MAPKK6, which activate p38 MAP kinase (MAPK).12 P38 is an important mediator of TGFβ-induced epithelial-mesenchymal transition (EMT).12 EMT has been recognized as a source of pulmonary muscle fibroblasts during pulmonary fibrosis in various in vitro studies, animal models, and patients.13 These findings thus indicate that TGFβ promotes the occurrence of pulmonary fibrosis through both the classical and non-classical approaches. In addition, TGFβ receptors can activate non-SMAD signaling pathways, including the extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), PI3K/Akt, and Ras/Raf/MAPK/ERK pathways.12 In this manner, the pulmonary cells affected by the ILDs display a cross-talk between the non-classical TGFβ signaling and other pathways, such as the JAK pathway.
ILD is one of the most common and serious lung diseases. The study found that the overall incidence of ILD was 19.4/100,000 with the most common diagnoses being sarcoidosis (42.6%), ILD associated with connective tissue disease (16%), and idiopathic pulmonary fibrosis (IPF) (11.6%).14 While the common ILD symptoms include dry cough, shortness of breath, and fatigue, the management and treatment options for the same remain challenging for clinicians. Currently, the common treatment strategies include glucocorticoids and immunosuppressants such as cyclophosphamide.1 However, occasionally the disease is refractory, rapidly progressive, and resistant to hormonal and immunosuppressive therapies.15 As a result, it is necessary to develop alternative drug therapies based on the current understanding of ILD pathogenesis and the role of the JAK/STAT pathway in it. The existing knowledge and scientific rationale on the efficacy of JAK inhibitors (JAKi) suggest that they may provide clinicians with a more precise treatment strategy for reducing interstitial inflammation thereby preventing the progression of fibrosis and the continuous deterioration of lung function.
The Distribution of JAK and STAT in Lung Tissues
JAK is a group of intracellular tyrosine kinases including JAK1, JAK2, JAK3, and TYK2, in mammals that are critical for the signal transduction initiated by membrane receptors on the cell surface.16 JAKs are widely expressed in various tissues and cells, which is the basis for their ubiquitous participation in diverse signal transduction pathways.17 Studies on signal transduction of cytokines and growth factors suggest that multiple JAKs can be activated in a single cytokine signaling pathway. Moreover, the JAKs have been documented to participate in the signal transduction of various cytokines and growth factors. For instance, IL2, IL13, and IFNγ can activate JAK1 and JAK2, while IL6 can activate JAK1, JAK2, and TYK2.17,18 In contrast, STATs such as STAT1, STAT2, STAT3, STAT4, STAT5, and STAT6, act as transcription factors in various cell types.4 However, the STATs demonstrate a certain level of cytokine specificity as compared with that of JAKs.3 For instance, IL31 and vascular endothelial growth factor (VEGF) activate STAT1; IL6, IL11, TGFβ1, and epidermal growth factor activate STAT3; and IL4 and IL13 reportedly activate STAT6.3
Previous studies have shown that STAT activation results in the transcription and expression of signaling proteins with diverse functions. For instance, STAT1 is involved in the expression of IL12-related proteins and promotes inflammation.16 STAT2 is involved in the expression of CD40- and CD80-related proteins and increases the expression of TNF receptor and CD80 ligand to promote apoptosis.16 STAT3 is involved in the expression of IL6-related proteins and encourages cell proliferation.16 STAT4 is involved in the expression of IFNγ-related proteins and stimulates immune regulation. STAT5 consists of two heterogeneous forms, STAT5a and STAT5b, whose protein structures are more than 95% identical. STAT5 participates in the expression of the suppressor of cytokine signaling 1 (SOCS1) and promotes inflammation.16 STAT6 is involved in the expression of B-cell lymphoma-2-related proteins and plays an anti-apoptotic role.16 In general, after cytokines and growth factors bind to their respective cell surface receptors, the receptor-associated JAK is activated and phosphorylated to form p-JAK, which binds to the docking site of STAT and phosphorylates it to form p-STAT.3
The JAK/STAT signaling pathway is reportedly responsible for the regulation of cell homeostasis; consequently, the excessive activation of this signaling axis can lead to autoimmune diseases.5 Moreover, the JAK2/STAT3 is considered to play a principal role in ILD development. In animal models, STAT3 has been shown to regulate IL6- and TGFβ 1- mediated differentiation of lung fibroblasts into myofibroblasts.6 In humans, the STAT3 has been reported to regulate the secretion of type I collagen in idiopathic pulmonary fibrosis (IPF) lung fibroblasts. Furthermore, the enhanced STAT3 expression in IPF fibroblasts is also attributed to their characteristic fibrotic phenotype.19,20 Likewise, the same mechanism has been reported in SSc patients, where TGFβ1 induces JAK2 phosphorylation that subsequently results in the phosphorylation of STAT3 to induce a fibrotic response. Interestingly, JAK2 may not only be a downstream target of TGFβ1 in fibroblasts but may also provide positive feedback, resulting in the amplification of the TGFβ1 signaling by stimulating TGFβ1 expression21 and further aggravating the ILD.
In terms of tissue distribution, JAK1 has been reported to be overexpressed in lung tissues and localized in inflammatory and epithelial cells in bleomycin (BLM)-induced fibrosis mouse models.22 In IPF patients, JAK2 is mainly distributed in the hyperplasia of alveolar epithelial type II cells, fibroblasts, and intima, as well as in the middle layer of small pulmonary artery,23 while the expression of p-JAK2 is augmented in the lung tissues and pulmonary arteries of IPF patients.
The expression and distribution of STAT1 have been mainly studied in animal models. Previous studies have shown that STAT1 is localized in the inflammatory and epithelial cells of BLM-induced fibrosis mouse models24 and in alveolar macrophages of rats.25 In addition, a recent study has reported that p-STAT1 is expressed in mouse lung tissues.26
STAT3 is highly distributed in alveolar macrophages, endothelial cells, and neutrophils in the BLM-induced fibrosis mouse models. Its phosphorylated form is also known to exist in myofibroblasts and alveolar macrophages.23 Moreover, STAT3 was detected in the inflammatory and epithelial cells of the same mouse model.27 Likewise, the p-STAT3 was found to be expressed in dense fibrotic areas, alveolar macrophages, myofibroblasts, and alveolar epithelial cell nuclei in IPF patients. Moreover, STAT3 is also present in the intima and media of small pulmonary arteries, hyperplastic alveolar cells, and fibroblasts in IPF patients.20,23
JAK/STAT Signaling Stimulates the Progression of ILD
Activation of the JAK/STAT pathway commences with the binding of ligands to their receptors. These ligands can be cytokines that promote fibrosis including IL2, IL4, IL6, IL11, and IL13; pro-inflammatory cytokines such as IL23 and IFNɣ; or growth factors such as TGFβ and platelet-derived factor (PDGF). These ligands bind to their receptors and thereby induce the dimerization of JAK. The receptor-associated JAK is then activated and phosphorylates the tyrosine residues in the tail of its receptor to form p-JAK (Figure 1). Subsequently, these phosphorylated sites act as docking sites for STATs and bind to them through their SH2 domain, resulting in tyrosine phosphorylation and activation of STAT to form p-STAT. The STATs thus form dimers and translocate from the cytoplasm to the nucleus, where they act as transcription factors.16 Most STATs form homodimers; however, heterodimers such as STAT1/2, STAT1/3, and STAT5a/b have also been reported.28 Thus, STATs are activated by JAKs and translocate from the cytoplasm to the nucleus to regulate gene expression.
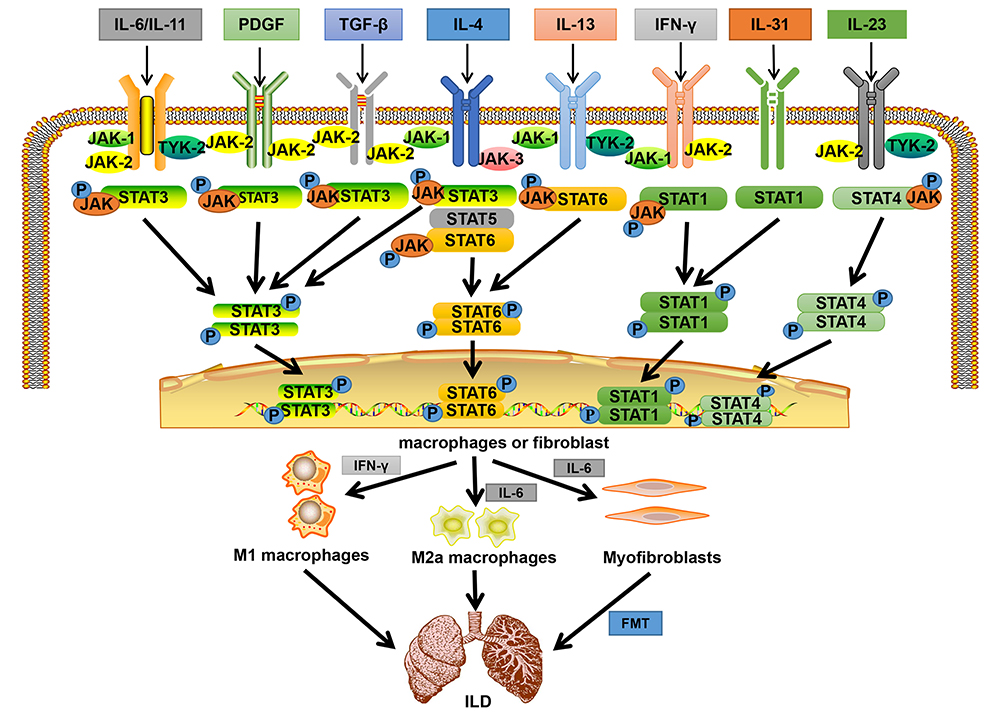 |
Figure 1 Binding of different cytokines and growth factors to corresponding receptors phosphorylates STATs and JAKs, activating STATs and transferring them to the nucleus to regulate gene expression. The secretion of pro-inflammatory and pro-fibrosis factors increased, eventually leading to the formation of ILD. Abbreviations: IL, Interleukin; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor β; IFN-γ, interferon-γ; P, phosphate; JAK, janus tyrosine kinase; STAT, signal transducers and activators of transcription; ILD, interstitial lung disease; FMT, fibroblast to mesenchymal transition. Note: Adapted from Montero P, Milara J, Roger I, et al. Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int J Mol Sci. 2021;22(12):6211. Creative Commons license and disclaimer available from: http://creativecommons.org/licenses/by/4.0/legalcode.3 |
Various studies have reported that activation of the JAK/STAT pathway is associated with the induction of ILD. Present evidence indicates that the JAK/STAT path is found to be active in alveolar epithelial cells, fibroblasts, macrophages, and other cells.3 Furthermore, this pathway is reported to upregulate the expression of macrophage polarization markers, such as CD86 and CD206. This results in the differentiation of macrophages into pro-inflammatory M1 macrophages that release inflammatory factors such as IFNɣ, and into pro-fibrotic M2a macrophages that release pro-fibrotic cytokines such as IL6. The released factors are further amplified via a positive feedback mechanism, ultimately promoting lung inflammation and fibrosis.4 Furthermore, the lung fibroblasts have been reported to instigate their own proliferation through this pathway or transform into mesenchymal cells. This transformation results in the abnormal expression of α-smooth muscle actin and extracellular matrix components, which induce fibroblasts to de-differentiate and acquire the mesenchymal phenotype of myofibroblasts, ultimately leading to the development of ILD.3 In summary, the existing findings suggest that JAK/STAT pathway activation plays an integral role in the development and progression of ILD (Figure 1).
Application of JAK Inhibitors (JAKi) in ILD Treatment
Most of the evidence on the use of JAKi to treat ILD has been documented in autoimmune disease-related ILD, particularly refractory and progressive ILD. For instance, Hornig et al29 had reported that the lung involvement in a 32-year-old male dermatomyositis patient with ILD continued to progress under several immunosuppressive therapy regimens, including high-dose steroids, cyclophosphamide, rituximab, immunoglobulin, plasmapheresis, cyclosporine, and mitomycin ester (MMF). However, JAKi (tofacitinib) treatment, post discontinuation of MMF and cyclophosphamide, significantly improved his clinical symptoms. The improved clinical prognosis was consistent with augmented carbon monoxide diffusing capacity (DLCO) and forced vital capacity (FVC); in addition to significant improvements in the fiber density and range, in a second high-resolution chest CT (HR-TCT) performed two months later. In addition, in another accompanied by rapid progress of sex without myopathy patients with dermatomyositis-associated ILD, high-dose glucocorticoids (GC), cyclophosphamide, and phosphatase inhibitor of calmodulin nerve triple therapy to treat the effect not beautiful, in Canada with JAKi (for the occasional method) after treatment with rapid improvement in,30 ILD show Overall, these findings indicate that JAKi therapy has a promising curative effect against ILD. Consequently, the JAKi have been considered as a viable option for the treatment of ILD caused by activation of the JAK/STAT pathway, which mainly includes the drugs listed in Table 1; nevertheless, their clinical application requires comprehensive studies to rule out the possibility of adverse reactions, if any.
 |
Table 1 Overview of JAKis for the Treatment of ILD |
Therapeutic Effect of JAKi on ILD in Animal Models
In autoimmune pulmonary fibrosis (eg, SSc-ILD), the M1 and M2 macrophages play key pathogenic roles. Lescoat et al4 had shown that treatment with JAKi (ruxolitinib 20 mg/kg, twice daily) prevented the upregulation of pro-inflammatory markers in M1 macrophages (TNF α and CXCL10) and pro-fibrotic markers in M2 macrophages (Arg1), and improved the lung inflammation and fibrotic lesions in SSc-ILD mouse model.21 In addition, another study had demonstrated that intraperitoneal injection of tofacitinib (20 mg/kg) thrice per week for two months inhibited the progression of RA-ILD in mice by promoting the expansion of myeloid suppressor cells in the lungs.36 In addition, intraperitoneal injection of tofacitinib (30 mg/kg, twice daily) was found to ameliorate the IL6-induced skin fibroblasts in mice with BLM-induced pulmonary fibrosis thereby playing an anti-fibrotic role. However, the study also reported no significant effect on the regression of pulmonary fibrosis.37 Overall, these findings suggest that JAKi could serve as potential ILD drugs by inhibiting the JAK/STAT pathway. Regrettably, the adverse effects caused by JAKi were not evaluated in these studies as the mice were sacrificed in these studies.
Efficacy of JAKi Treatment in Human Patients with ILD
JAKi have been documented to not only improve the respiratory symptoms of ILD in animal models but also reduce the levels of serological markers associated with ILD. As a result, we performed a literature search to understand the ILD treatment efficacy of JAKi in humans. Accordingly, we came across a single-arm open-label trial that had evaluated the efficacy of tofacitinib in patients with early anti-melanoma differentiation-associated gene 5 (MDA5) antibody-positive amyopathic dermatomyositis (ADM) associated with ILD (ADM-ILD).38 This study prospectively treated 18 patients with ADM-ILD, who were anti-MDA5 antibody-positive, with glucocorticoid and tofacitinib (10 mg/day). The trial compared the survival rates of patients who received tofacitinib with those of patients who received conventional treatment of glucocorticoids as a historical control. The results revealed that the six-month survival rate of the tofacitinib-treated group (100%) was significantly higher than that of the historical control group (78%). Moreover, the DLCO and HR-TCT results in the tofacitinib-treated group improved significantly over time as compared with that of the historical control group. In addition, the adverse events in patients administered tofacitinib were mostly low-grade, such as mild elevation in the levels of liver enzymes.38 This study thus suggests that tofacitinib can be considered as an effective and safe drug for the treatment of ILD.
A retrospective study had evaluated the efficacy of tofacitinib (10 mg/day) in patients with anti-MDA5 antibody-positive DM-ILD and resistant to triple therapy, which consisted of high-dose GC, cyclosporine A, and cyclophosphamide.39 The results showed that 60% (3 out of 5) of the patients treated with tofacitinib responded well and survived. In contrast, none of the six historical controls treated without tofacitinib survived, despite both the groups presenting similar adverse prognostic factors including serum ferritin levels greater than 1000 ng/mL and lung infiltration, prior to triple therapy. Furthermore, the lung infiltration was determined to be worse during triple therapy as compared to those treated with tofacitinib,39 suggesting that tofacitinib is a potential treatment for refractory DM-ILD candidates with anti-MDA5 antibodies. Remarkably, the main adverse event associated with tofacitinib reported in this study was viral infection; 100% of patients treated with tofacitinib experienced cytomegalovirus reactivation and 60% experienced herpes zoster infection as compared to that in patients belonging to the historical control group.39 Interestingly, other studies have also reported the superiority and safety of tofacitinib in the treatment of ILD.29,30
There are no large clinical trials of ruxolitinib, except for a few published case reports. For instance, ruxolitinib was found to be effective in three ILD patients with acquired functional mutations in STAT. The three patients were treated with two daily doses of 5 mg, 10 mg, and 20 mg ruxolitinib, respectively. The findings showed that the ruxolitinib intervention improved the hypoxia symptoms of the patients, along with evident cystic changes and ground-glass opacity improvements in stable chest CT scans. Moreover, the lung function was stabilized or improved however, the patients did not report any improvement with the bead sheet resistance monotherapy that they had received previously.40 In this case report, the patients did not report significant adverse events, which suggests that ruxolitinib might be considered to be safe and effective in the treatment of ILD. Another case report describes the treatment of patients with refractory ILD associated with systemic juvenile idiopathic arthritis using ruxolitinib doses of 1 mg/kg/day. After 15 months, the patient’s fever symptoms were relieved, and the chest CT scans indicated alleviated ILD, and the hormone use was reduced. The case report suggests that ruxolitinib was well tolerated by the patients, implying that it may be an effective and safe drug for the treatment of ILD.41 However, large clinical trials are needed to confirm the efficacy and safety of ruxolitinib through statistically supported conclusions.
Baricitinib has been evaluated for the treatment of ILD in two studies. One is a case report of ILD associated with STING vascular lesions. The patient did not respond to ruxolitinib (2.5 mg/day) and discontinued use owing to elevated liver enzyme levels and severe rotavirus enteritis. However, when the patient received 2 mg/day of baricitinib, his clinical presentation improved without adverse reactions.42 The second study was a retrospective analysis of four patients with RA-ILD, who were administered 4 mg/day of baricitinib for six months. The study reported that as compared with the previous treatment, baricitinib significantly increased the DLCO levels and significantly decreased the levels of pro-inflammatory cytokines such as IL6, and serum marker salivation glycochain antigen, thus reflecting the stability of these patients after lung interstitial involvement. Although alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were slightly elevated in these patients, the treatment with baricitinib was considered to be safe.43
The case reports previously discussed showed positive effects of treatments with JAKi. However, large-scale prospective randomized controlled trials are still needed to confirm the effectiveness and safety of these inhibitors, as well as probable adverse events (Table 1).
Outlook
The JAK/STAT pathway plays a significant role in the pathogenesis of ILD. Moreover, the JAK2/STAT3 is a major pathway found to be activated in ILD patients and animal models. In ILD, this signaling pathway is activated by a variety of upregulated cytokines and growth factors. While most of the relevant studies have focused on the autoimmune disease-related ILDs, research on how this pathway is associated with other types of ILDs is still lacking. Recently, preliminary findings have revealed the therapeutic potential of JAKi such as tofacitinib, ruxolitinib, and baricitinib; however, larger prospective studies are essential to validate these findings and provide novel insights for the development of innovative and effective treatment strategies for ILD patients.
Funding
This work was supported by grants from the National Natural Science Foundation (81871292) and the Key Research and Development (R&D) Projects of Shanxi Province (201803D31136).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Shao T, Shi X, Yang S, et al. Interstitial lung disease in connective tissue disease: a common lesion with heterogeneous mechanisms and treatment considerations. Front Immunol. 2021;12:684699. doi:10.3389/fimmu.2021.684699
2. Mikolasch TA, Garthwaite HS, Porter JC. Update in diagnosis and management of interstitial lung disease. Clin Med. 2017;17(2):146–153. doi:10.7861/clinmedicine.17-2-146
3. Montero P, Milara J, Roger I, et al. Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int J Mol Sci. 2021;22(12):6211. doi:10.3390/ijms22126211
4. Lescoat A, Lelong M, Jeljeli M, et al. Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: perspectives for scleroderma-associated interstitial lung disease. Biochem Pharmacol. 2020;178:114103. doi:10.1016/j.bcp.2020.114103
5. Kagan P, Sultan M, Tachlytski I, et al. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS One. 2017;12(5):e0176173. doi:10.1371/journal.pone.0176173
6. Pedroza M, Le TT, Lewis K, et al. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016;30(1):129–140. doi:10.1096/fj.15-273953
7. Levine RL, Pardanani A, Tefferi A, et al. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7(9):673–683. doi:10.1038/nrc2210
8. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. doi:10.1038/nrd3810
9. Ma BN, Li XJ. Resveratrol extracted from Chinese herbal medicines: a novel therapeutic strategy for lung diseases. Chin Herb Med. 2020;12(4):349–358. doi:10.1016/j.chmed.2020.07.003
10. Sorrentino A, Thakur N, Grimsby S, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10(10):1199–1207. doi:10.1038/ncb1780
11. Kim SI, Kwak JH, Na HJ, et al. Transforming growth factor-beta (TGF-beta1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-beta receptor kinase activity in mesangial cells. J Biol Chem. 2009;284(33):22285–22296. doi:10.1074/jbc.M109.007146
12. Heldin CH, Moustakas A. Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol. 2016;8(8):a022053. doi:10.1101/cshperspect.a022053
13. Li M, Luan F, Zhao Y, et al. Epithelial-mesenchymal transition: an emerging target in tissue fibrosis. Exp Biol Med. 2016;241(1):1–13. doi:10.1177/1535370215597194
14. Duchemann B, Annesi-Maesano I, Jacobe de Naurois C, et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur Respir J. 2017;50(2):1602419. doi:10.1183/13993003.02419-2016
15. Selva-O’Callaghan A, Romero-Bueno F, Trallero-Araguás E, et al. Pharmacologic treatment of anti-MDA5 rapidly progressive interstitial lung disease. Curr Treatm Opt Rheumatol. 2021:1–15. doi: 10.1007/s40674-021-00186-x
16. Bousoik E, Montazeri Aliabadi H. “Do we know Jack” about JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. 2018;8:287. doi:10.3389/fonc.2018.00287
17. Bolen JB, Brugge JS. Leukocyte protein tyrosine kinases: potential targets for drug discovery. Annu Rev Immunol. 1997;15:371–404. doi:10.1146/annurev.immunol.15.1.371
18. Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers. 2019;11(12):2002. doi:10.3390/cancers11122002
19. Banerjee S, Biehl A, Gadina M, et al. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77(5):521–546. doi:10.1007/s40265-017-0701-9
20. Pechkovsky DV, Prêle CM, Wong J, et al. STAT3-mediated signaling dysregulates lung fibroblast-myofibroblast activation and differentiation in UIP/IPF. Am J Pathol. 2012;180(4):1398–1412. doi:10.1016/j.ajpath.2011.12.022
21. Beyer C, Distler JH. Tyrosine kinase signaling in fibrotic disorders: translation of basic research to human disease. Biochim Biophys Acta. 2013;1832(7):897–904. doi:10.1016/j.bbadis.2012.06.008
22. Ma X, Chen R, Liu X, et al. Effects of matrine on JAK-STAT signaling transduction pathways in bleomycin-induced pulmonary fibrosis. Afr J Tradit Complement Altern Med. 2013;10(3):442–448. doi:10.4314/ajtcam.v10i3.10
23. Milara J, Hernandez G, Ballester B, et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir Res. 2018;19(1):24. doi:10.1186/s12931-018-0728-9
24. Shi K, Jiang J, Ma T, et al. Pathogenesis pathways of idiopathic pulmonary fibrosis in bleomycin-induced lung injury model in mice. Respir Physiol Neurobiol. 2014;190:113–117. doi:10.1016/j.resp.2013.09.011
25. Fan XM, Wang ZL, Li ZH. STAT1 activation and STAT1-dependent immune-response gene ICAM-1 expression in alveolar macrophages of rats suffered from interstitial pulmonary fibrosis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2003;19(1):3–6.
26. Wang YJ, Li Y, Wang XL, et al. Effect of total flavonoids of oxytropis falcata bunge on the expression of p-JAK1-and p-STAT1-related proteins in idiopathic pulmonary fibrosis. Evid Based Complement Alternat Med. 2020;2020:2407239. doi:10.1155/2020/2407239
27. Shieh JM, Tseng HY, Jung F, et al. Elevation of IL-6 and IL-33 levels in serum associated with lung fibrosis and skeletal muscle wasting in a bleomycin-induced lung injury mouse model. Mediators Inflamm. 2019;2019:7947596. doi:10.1155/2019/7947596
28. Subramaniam PS, Torres BA, Johnson HM. So many ligands, so few transcription factors: a new paradigm for signaling through the STAT transcription factors. Cytokine. 2001;15(4):175–187. doi:10.1006/cyto.2001.0905
29. Hornig J, Weinhage T, Schmidt LH, et al. Ansprechen einer Dermatomyositis mit Lungenbeteiligung auf eine Janus kinase-Inhibitor-Therapie [Response of dermatomyositis with lung involvement to Janus kinase inhibitor treatment]. Z Rheumatol. 2018;77(10):952–957. German. doi:10.1007/s00393-018-0565-8
30. Nandy A, Gaïni S, Sore P. Rapidly progressive interstitial lung disease in a patient with anti-MDA5-positive amyopathic dermatomyositis. Scand J Rheumatol. 2018;47(4):334–335. doi:10.1080/03009742.2017.1369155
31. O’Shea JJ, Schwartz DM, Villarino AV, et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–328. doi:10.1146/annurev-med-051113-024537
32. Fragoulis GE, McInnes IB, Siebert S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology. 2019;58(Suppl1):i43–i54. doi:10.1093/rheumatology/key276
33. Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57(12):5023–5038. doi:10.1021/jm401490p
34. Markham A. Baricitinib: first global approval. Drugs. 2017;77(6):697–704. doi:10.1007/s40265-017-0723-3
35. You H, Xu D, Zhao J, et al. JAK inhibitors: prospects in connective tissue diseases. Clin Rev Allergy Immunol. 2020;59(3):334–351. doi:10.1007/s12016-020-08786-6
36. Sendo S, Saegusa J, Yamada H, et al. Tofacitinib facilitates the expansion of myeloid-derived suppressor cells and ameliorates interstitial lung disease in SKG mice. Arthritis Res Ther. 2019;21(1):184. doi:10.1186/s13075-019-1963-2
37. Zhang Y, Liang R, Chen CW, et al. JAK1-dependent transphosphorylation of JAK2 limits the antifibrotic effects of selective JAK2 inhibitors on long-term treatment. Ann Rheum Dis. 2017;76(8):1467–1475. doi:10.1136/annrheumdis-2016-210911
38. Chen Z, Wang X, Ye S. Tofacitinib in amyopathic dermatomyositis-associated interstitial lung disease. N Engl J Med. 2019;381(3):291–293. doi:10.1056/NEJMc1900045
39. Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57(12):2114–2119. doi:10.1093/rheumatology/key188
40. Silva-Carmona M, Vogel TP, Marchal S, et al. Successful treatment of interstitial lung disease in STAT3 gain-of-function using JAK inhibitors. Am J Respir Crit Care Med. 2020;202(6):893–897. doi:10.1164/rccm.201906-1204LE
41. Bader-Meunier B, Hadchouel A, Berteloot L, et al. Effectiveness and safety of ruxolitinib for the treatment of refractory systemic idiopathic juvenile arthritis like associated with interstitial lung disease: a case report. Ann Rheum Dis. 2020. doi:10.1136/annrheumdis-2020-216983
42. Balci S, Ekinci RMK, de Jesus AA, et al. Baricitinib experience on STING-associated vasculopathy with onset in infancy: a representative case from Turkey. Clin Immunol. 2020;212:108273. doi:10.1016/j.clim.2019.108273
43. d’Alessandro M, Perillo F, Metella Refini R, et al. Efficacy of baricitinib in treating rheumatoid arthritis: modulatory effects on fibrotic and inflammatory biomarkers in a real-life setting. Int Immunopharmacol. 2020;86:106748. doi:10.1016/j.intimp.2020.106748
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.