")
Back to Journals » OncoTargets and Therapy » Volume 13
Therapeutic Potential of Apatinib Against Colorectal Cancer by Inhibiting VEGFR2-Mediated Angiogenesis and β-Catenin Signaling
Authors Cai X, Wei B, Li L, Chen X , Yang J, Li X, Jiang X, Lv M, Li M, Lin Y, Xu Q, Guo W , Gu Y
Received 25 June 2020
Accepted for publication 5 October 2020
Published 29 October 2020 Volume 2020:13 Pages 11031—11044
DOI https://doi.org/10.2147/OTT.S266549
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Leo Jen-Liang Su
Xiaomin Cai,1,* Bin Wei,1,2,* Lele Li,1,* Xiaofeng Chen,1 Jing Yang,1 Xiaofei Li,1 Xiaozheng Jiang,1 Mu Lv,1 Mingyang Li,1 Yumeng Lin,3 Qiang Xu,3 Wenjie Guo,3 Yanhong Gu1
1Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, People’s Republic of China; 2Department of Oncology, The Affiliated Huaian No.1 People’s Hospital of Nanjing Medical University, Huai’an 223300, People’s Republic of China; 3State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanhong Gu
Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, People’s Republic of China
Email [email protected]
Wenjie Guo
State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, 163 Xianlin Avenue, Nanjing 210023, People’s Republic of China
Email [email protected]
Purpose: Apatinib is an inhibitor of VEGFR2 (vascular endothelial growth factor receptor 2) that has attracted a great deal of attention due to its promotion of anticancer activity. In the present study, we investigated the therapeutic effects of apatinib against colorectal cancer (CRC) and examined the underlying mechanism.
Materials and Methods: Both in vivo and in vitro assays were conducted to study the effect of apatinib on CRC. To elucidate the associated mechanism, RNA-seq (transcriptome) analysis was conducted on apatinib-treated HCT116 cells.
Results: Apatinib showed antiproliferative and proapoptotic effects, induced G0/G1 arrest and blocked cell migration and invasion in CRC. An analysis of the mechanism associated with apatinib activity demonstrated that by interacting with VEGFR2, apatinib decreased p-Src, p-Akt, and p-GSK3β levels, which further increased β-catenin ubiquitination and reduced the nuclear translocation of β-catenin. Furthermore, apatinib strongly suppressed CT26 cell growth in mouse xenograft models by inhibiting β-catenin signaling and angiogenesis.
Conclusion: Overall, the results of the present study here indicated that by inhibiting the VEGFR2-β-catenin-mediated malignant phenotype, apatinib significantly suppresses the growth of CRC, suggesting that the use of apatinib is a promising therapeutic strategy for CRC.
Keywords: apatinib, colorectal cancer, VEGFR2, angiogenesis, β-catenin
Introduction
Colorectal cancer (CRC) is the third most common cancer and a major cause of death.1 In the United States, CRC incidence and mortality have decreased as a result of colonoscopy screening and therapeutic developments. However, an increasing number of young patients are diagnosed in the advanced stage of CRC.1,2 For patients with metastatic colorectal cancer (mCRC), the current emphasis is fluoropyrimidine-based chemotherapy combined with biological immunity or targeted therapy.3 After the failure of all standard therapy, regorafenib,4 TAS-102,5 and fruquintinib6 are available.4,7,8 Programmed cell death-1 (PD-1) blockade has shown high efficacy in mismatch-repair deficient tumors, which account for approximately 5% of mCRC cases.9,10 Thus, novel and safe treatment strategies for CRC are urgently needed.
Apatinib, a VEGFR2 inhibitor recognized in China for its effect against progressive gastric cancer,11 has been reported to have anticancer activity in various malignancies.12,13 In a previous study, we conducted a clinic trail assessing apatinib monotherapy treatment in mCRC, the results of which showed that the median PFS (Progression-free survival) of apatinib was 3.9 months.14 Although the therapeutic effect of apatinib against CRC has been reported, the associated mechanism has not been fully elucidated.
In our present study, the antitumor activity of apatinib against CRC was investigated. Our data revealed that apatinib arrests cell cycle progression, increases apoptosis and inhibits the proliferation, migration and invasion of CRC cells. Moreover, we observed that deactivation of VRGFR2-mediated angiogenesis and β-catenin signaling are principle mechanisms by which apatinib inhibits CRC. These findings highlighted the potential for using apatinib as a novel candidate for treating CRC in the future.
Materials and Methods
Antibodies and Materials
Apatinib was contributed by HengRui Medicine Co., Ltd (Lianyungang, Jiangsu, China). The storage and usage of apatinib are implemented as previously reported.15 (0.1% concentration of DMSO was used to dissolve apatinib at 100 mM for a stock solution and diluted into the work concentration with a culture medium.) Primary antibodies including anti-PARP, anti-Axin 2, anti-β-catenin, anti-cyclin D1, anti-vimentin, anti-α-SMA, anti-Survivin, anti-p-GSK-3β (ser9), anti-Ub, anti-p-VEGFR2, anti-VEGFR2, anti-p-Erk and anti-Lamin B1 were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-Actin, anti-Tubulin, anti-CD31 and anti-PCNA antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, California, USA). Alexa Fluor 488 goat anti-rabbit IgG was bought from Thermo Fisher Scientific (Waltham, MA, USA). Annexin V/PI (Propidium Iodide) staining kit was purchased from Beyotime Company (Nantong, Jiangsu, China). TUNEL assay kits were got from Vazyme Biotech Co., Ltd (Nanjing, Jiangsu, China). GTVisinTM anti-mouse/anti-rabbit immunohistochemical analysis KIT was bought from Gene Company (Shanghai, China).
Cell Lines
The HCT116, SW480, SW620, HT29 (human origin), CT26 (mouse origin, derived from BALB/c mice) colorectal cancer cell lines were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cell culture conditions are the same as reported.15
Animals
BALB/c mice, female, aged from 6 to 8 weeks, were purchased from Model Animal Research Center of Nanjing University (Nanjing, Jiangsu, China). The breeding environment is the same as reported.15 All animals suffered the minimum harm, as the experimental procedures were executed rigorously under the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, the United States) and the related ethical regulations of Nanjing Medical University.
Syngeneic Model
CT26 (2 × 106) cells were injected into the right flank of BALB/c mice by subcutaneous. After tumors had grown to about 100 mm3, mice were administered with apatinib by gavage daily (3, 10, 30 mg/kg) or with the vehicle solution (0.9% CMC.Na) or injected intraperitoneally with 5-FU (25 mg/kg) once every 3 days. The measurement and calculation of tumor volume are the same as previously described.15 After 15 days of administration, the animals were sacrificed.
Cell Viability Assay and Clone Formation Assay
The cells (5,000 cells/well in a 96-well plate) were incubated overnight in a medium with 10% FBS. Cells were incubated with various concentrations of apatinib at 37°C for 24 h, 48 h, 72 h, respectively. At the end of incubation, 10 μL MTT solutions (5 mg/mL in PBS) were added and incubation for another 4 h, and then the supernatants were discarded and 200 μL DMSO was added into each well. Then, OD570nm was measured by Microplate Reader (BioTek, Winooski, VT, USA). For clone formation assay, 500 cells were seeded into 6-well plates and incubated for 14 days (with medium replaced every 3 days). Then, the cells were fixed and stained with 1% crystal violet at room temperature for 20 min. Photos were taken and the number of clones was counted.
Cell Apoptosis Assay
Incubated cells were gathered and subjected to Annexin V-FITC/PI (propidium iodide) staining assay and then analyzed by using FACS Calibur flow cytometry (Becton-Dickinson, Franklin Lakes, NJ, USA).16 Annexin V +/PI− and Annexin V +/PI + cells were regarded as apoptotic cells.
Cell Cycle Assay
HCT116, CT26 cells (1×106 cells/well in 6-well plate) were stimulated with 0, 3, 10 and 30 μM apatinib for 24 h. For cell cycle analysis, after fixed in 70% ethanol overnight at 4°C, the cells were washed and stained with 5 μg/mL RNase and 20 μg/mL propidium iodide (PI) at 37°C for 20 min and analyzed by FACS Calibur flow cytometry (Becton Dickinson) as reported before.17
Real-Time PCR
Real-time PCR was carried out as reported.18 The primer sequence applied is as follows:
α-Sma: forward: 5’-GGCCAGATCCTGTCCAAGC-3’; reverse: 5’-GTGGGTTTCCACCATTAGCAC-3’;
Axin2 : forward: 5’‑CAACACCAGGCGGAACGAA‑3’; reverse: 5’‑GCCCAATAAGGAGTGTAAGGACT‑3’;
c-Myc: forward: 5’‑ATGGCCCATTACAAAGCCG‑3’; reverse: 5’‑TTTCTGGAGTAGCAGCTCCTAA‑3’;
CylinD1 : forward: 5’-GCTGCGAAGTGGAAACCATC-3’; reverse: 5’-CCTCCTTCTGCACACATTTGAA-3’;
Cxcr4: forward: 5’-ACTACACCGAGGAAATGGGCT-3’; reverse: 5’-CCCACAATGCCAGTTAAGAAGA-3’;
Survivin: forward: 5’-AGGACCACCGCATCTCTACAT-3’; reverse: 5’-AAGTCTGGCTCGTTCTCAGTG-3’;
Vimentin: forward: 5’-GACGCCATCAACACCGAGTT-3’; reverse: 5’-CTTTGTCGTTGGTTAGCTGGT-3’.
RNA Sequencing (RNA-Seq)
The RNA was extracted and sequenced. RNA sequencing of control and apatinib-treated (30μM) HCT116 (each with 3 biological replicates) was performed and analyzed by Beijing Novagene company. Feature Counts v1.5.0-p3 was applied to count the reads numbers mapped to each gene.19 Then, FPKM (Fragments Per Kilobase Million) of each gene was calculated based on the length of the gene and reads count mapped to this gene. Differential expression analysis of control and apatinib-treated HCT116 were performed using the DESeq2 R package (1.16.1).20 KEGG pathway enrichment analysis and Gene Ontology analysis of DEGs were conducted.21 Raw data have been uploaded to the Sequence Read Archive (SRA) database with accession code PRJNA602151.
Western Blot and Co-Immunoprecipitation
Western blot was conducted in accordance with the standard protocols as previously reported.18 Co-immunoprecipitation assay was done as reported.22
Isolation of Nuclear and Cytoplasmic Compartments
A Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Company, Nantong, Jiangsu, China) was taken to isolate the nuclear and cytoplasmic compartment proteins of cells. The experimental procedure was under the manufacturer’s instruction.
Immunohistochemistry and Immunofluorescence Analysis
Immunohistochemistry or immunofluorescence staining was done as reported.22 Regents used were listed as follows: streptavidin-HRP (Shanghai Gene Company, GK500705), DAB (Shanghai Gene Company, GK500705), Alexa Fluor 546 goat anti-rabbit IgG (1:500, Thermofisher). TUNEL-FITC (1:100) (Vazyme Biotech Co., Ltd Nanjing, Jiangsu, China).
Trans-Well Cell Invasion Assay
The trans-well chambers with Matrigel (BD Biosciences, San Jose, CA, USA) were used to evaluate the invasion of tumor cells. Cells (2.5×105 cells/mL) resuspend in 100 μL serum-free medium were added to the upper chamber, while the lower wells were filled with 600 μL medium containing 10% FBS and incubated in a humidified atmosphere with 5% CO2 at 37°C. After 24 h, the chambers were washed with PBS, fixed with 4% paraformaldehyde (in PBS, pH 7.4) for 30 min, and stained with 0.1% crystal violet dye for 20 min. Finally, five fields of view were randomly selected under an inverted phase-contrast microscope to observe cell migration and count the cells.
Wound Healing
Cells were cultured to 90% confluence in 6 well plates and incubated with 3, 10, 30 μM apatinib. After 24 h, a fine scratch was made using a sterile yellow 200 µL Eppendorf pipette tip. Then, the cells were cultured in a serum-free medium. The width of the scratch was recorded under phase-contrast light microscopy immediately (0 h) and after 6, 12, 24 h to determine the extent of wound closure.
Cellular Thermal Shift Assay (CETSA)
HCT116 cells were incubated with or without apatinib (30 μM) for 2 h, and then the cells were collected and submitted to CETSA assay.23 Incubated cells were divided into ten (50 μL each) parts and heated for 3 min under 43, 46, 49, 52, 55, 58, 61, 64, 67, 70°C, then put into −80°C overnight. Samples were taken out to room temperature for 2 h (repeated one more time), then centrifuged at 20000 g for 25 min. The level of VEGFR2 was detected by Western blot.
Statistical Analysis
Statistical analysis was performed as previously described.15
Results
Apatinib Inhibits the Proliferation and Colony Formation of Colorectal Cancer Cells in vitro
First, we performed MTT and colony formation assays on four CRC cell lines (HCT116, SW480, SW620 and CT26) to assess the effect of apatinib in vitro. Compared to the control group, the proliferation of CRC cells was significantly suppressed by the apatinib treatment (Figure 1A). As shown in Figure 1B, apatinib treatment significantly reduced clone formation in CRC cells, an effect that was most pronounced in the cells treated with 30 μM apatinib. In addition, the effect of apatinib towards the normal colon epithelial cell line NCM460 was examined, with the data showing that apatinib was not toxic to NCM460 at concentrations less than 100 μM (Supplementary Figure 1A and B).
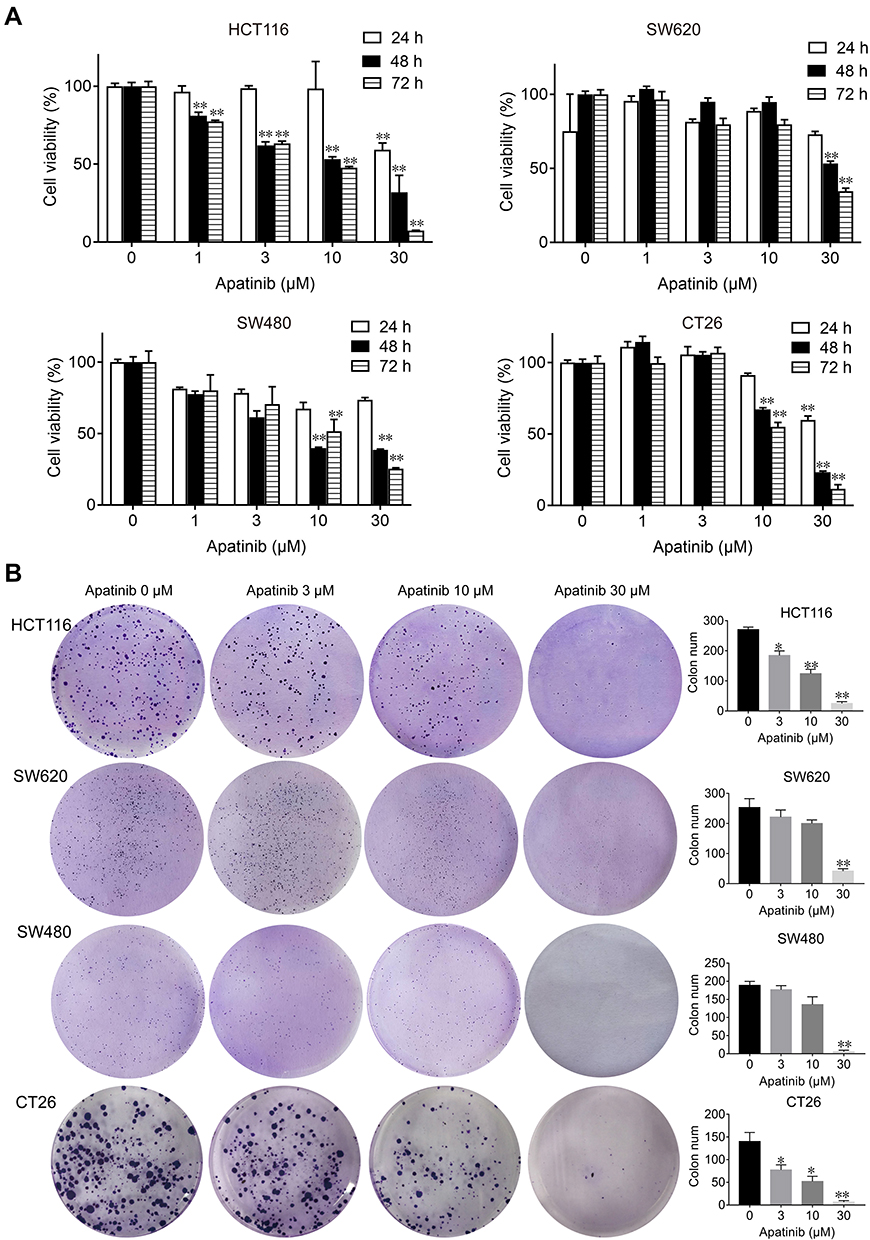 |
Figure 1 Apatinib inhibited proliferation and colony formation of colorectal cancer cells in vitro. (A) HCT116, SW620, SW480 and CT26 cells were treated with indicated concentrations of apatinib. Cell viability was tested by MTT assay after 24, 48 and 72 h. (B) Colorectal cancer cells were pretreated with indicated doses of apatinib for 24 h and then cells were seeded at a density of 500 cells per well into a 6-well plate. After cultured for 14 days, the clones were fixed and stained with crystal violet. The data were expressed as mean ± SEM obtained from triplicate experiments. *P < 0.05, **P < 0.01 vs control group. |
Apatinib Induces the Apoptosis of Colorectal Cancer Cells
Since the downregulated cell proliferation induced by apatinib may be attributed to apoptosis, we assessed the apoptotic rate of cells by using Annexin V/PI staining via flow cytometry. HCT116 and CT26 cells were co-cultured with apatinib for 24 h. Compared to the control group, the apatinib-treated cells exhibited a significantly higher level of apoptosis (Figure 2A and B). Then, the expression of apoptosis-related molecules in apatinib-treated cells was further examined by Western blot analysis. As shown in Figure 2C and D, PARP was abundantly activated after treatment with 30 μM apatinib for 24 h. Subsequently, Z-VAD-FMK, an inhibitor of apoptosis, was used to treat cells in combination with apatinib for 24 h. MTT assay results showed that the Z-VAD-FMK treatment reversed the inhibition of cell proliferation induced by apatinib (Figure 2E). These findings suggest that the observed downregulation of CRC cell proliferation by apatinib was partially due to the induction of cell apoptosis.
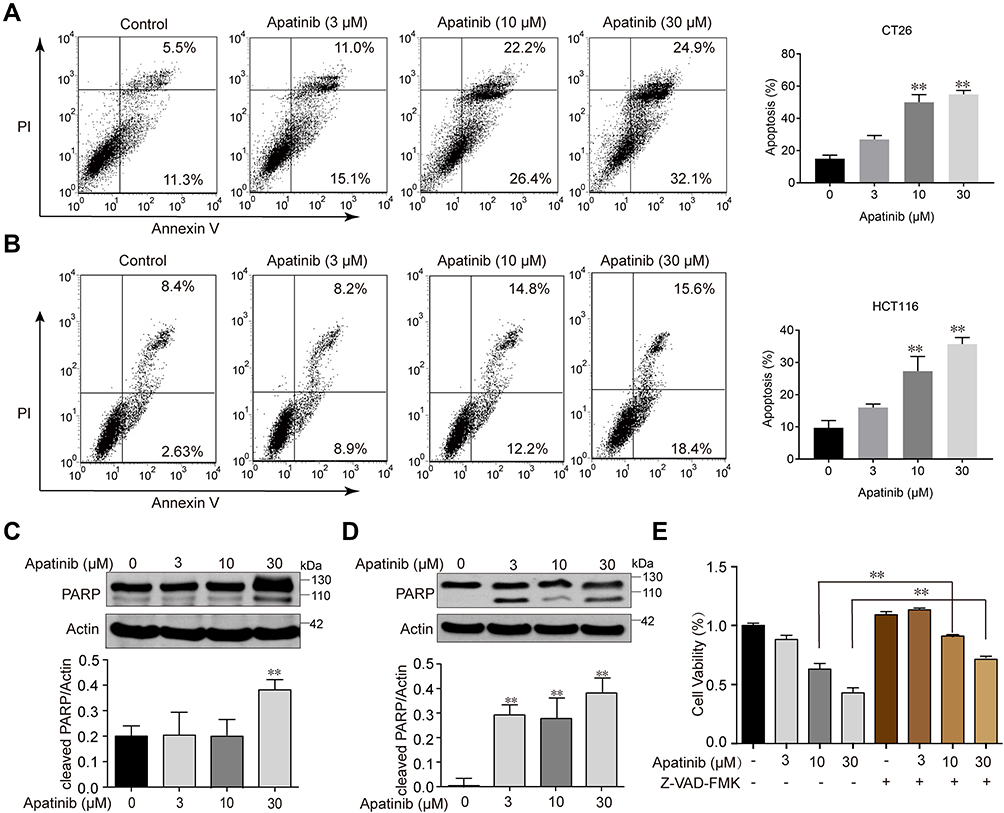 |
Figure 2 Apatinib induced apoptosis of colorectal cancer cells. CT26 cells (A) and HCT116 cells (B) were treated with indicated doses of apatinib or control (vehicle: 0.1% DMSO) for 24 h. Cells were then analyzed by Annexin V/PI staining. Data shown are representative of three experiments. CT26 cells (C) and HCT116 cells (D) were treated with indicated doses of apatinib for 24h. PARP expression was detected by immunoblotting, and β-Actin was used as a loading control. Results represent three repeated experiments. (E) HCT116 cells were treated with indicated doses of apatinib alone or in combination with Z-VAD-FMK (20 μM) for 24 h. Cell viability was tested by MTT assay. Data are expressed as means ± SEM of three experiments. **P < 0.01 vs control group. |
Apatinib Inhibits the Invasion, Migration, and Arrested Cell Cycle Progression of Colorectal Cancer Cells
Based on the observed antiproliferative and proapoptotic effects of apatinib, we next assessed whether apatinib affects the migration and invasion or cell cycle progression of CRC cells. To this end, we investigated the role of apatinib in cell cycle progression via PI staining. The mean numbers of cells at different phases are shown in Figure 3A. The number of CT26 cells in G0/G1 phase increased from 48.8% (control) to 68.2% after treatment with 30 μM apatinib. Furthermore, HCT116 cells treated with apatinib also showed an increase in the number of G0/G1 stage cells.
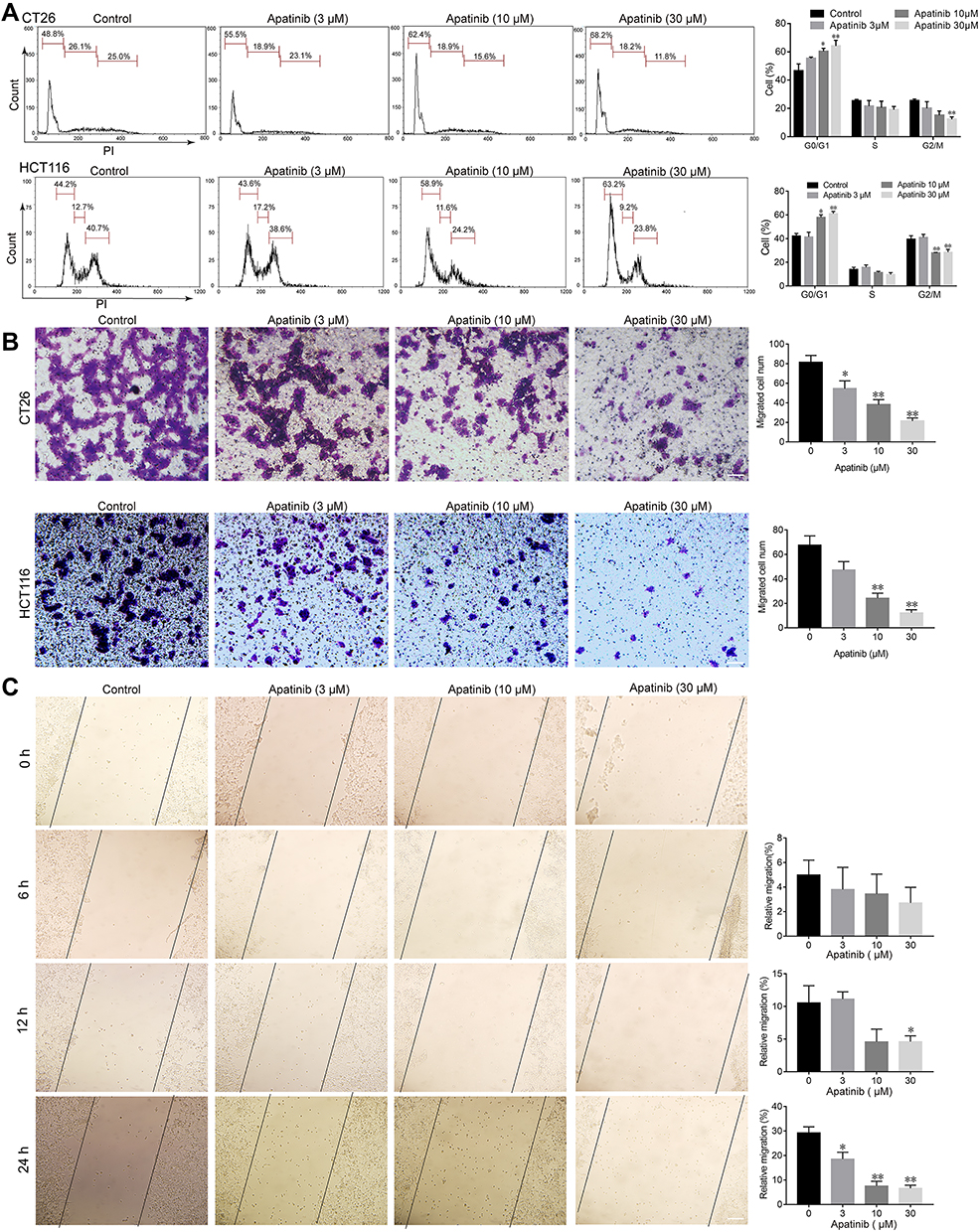 |
Figure 3 Apatinib arrested cell cycle progression and inhibited the invasion and migration of colorectal cancer cells. (A) CT26 cells and HCT116 cells were treated with indicated doses of apatinib for 24 h, after which the cells were washed, fixed, stained with PI, and analyzed for DNA content by flow cytometry. (B) HCT116 cells and CT26 cells were pretreated with apatinib at the different concentrations for 24 h. After 24 h, trans-well assay invasion was performed as described in the methods. (C) HCT116 cells were pretreated with apatinib at the different concentrations for 24 h. After 24 h, the wound healing assay was performed as described in the methods. Data are expressed as means ± SEM of three experiments. *P < 0.05, **P < 0.01 vs control group. |
Subsequently, we performed trans-well invasion and wound healing assays to evaluate the effect of apatinib on the invasion and migration of CRC cells and observed that apatinib treatment suppressed CRC cell invasion and migration (Figure 3B and C).
Apatinib Inhibits the VEGFR2-β-Catenin Pathway
To further investigate how apatinib inhibits CRC cell proliferation, we used RNA-seq technology to assess RNA expression changes. RNA from apatinib-treated HCT116 cells (30 μM, 24 h) was extracted and sequenced. The Pearson correlation coefficient (R) between the three replicates (C1, C2, and C3) in the control group was greater than 0.95, which is nearly as high as the 0.99 observed in the apatinib treatment group (A1, A2, and A3) (Supplementary Figure 2A). This result indicates the outstanding quality of the samples and that they could be used for subsequent analysis. Subsequent cluster analysis of differentially expressed genes also showed that the reproducibility of the three samples was good (Supplementary Figure 2B). KEGG pathway enrichment analysis and Gene Ontology analysis of DEGs further confirmed that apatinib arrested the cell cycle of HCT116 cells (Supplementary Figure 2C and D). We analyzed the sequencing results and observed that among the 155 genes regulated by the-β-catenin signaling pathway (https://web.stanford.edu/group/nusselab/cgi-bin/wnt/target_genes), 46 were downregulated after treatment with apatinib. Furthermore, 23 of these genes (such as Cxcr4, Wnt6, Myc, Axin2, Acta1, Bmp4 and Cacna1g) were downregulated by more than 2-fold (Figure 4A), which was confirmed by RT-PCR (Figure 4B). In addition, the downstream components of the β-catenin pathway were further examined at protein level in HCT116 cells and CT26 cells. The expression of the examined genes (Cyclin D1, Axin2, α-Sma, Vimentin, Survivin) decreased in response to apatinib treatment (Figure 4C and D). Taken together, the above results showed that apatinib suppresses β-catenin pathway signaling.
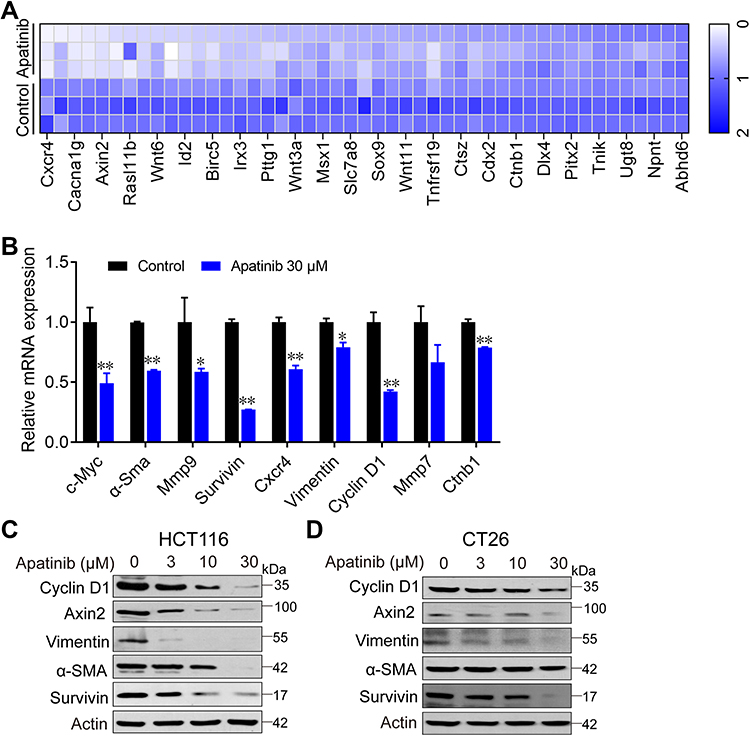 |
Figure 4 Apatinib inhibited the β-catenin pathway. Apatinib decreased mRNA and protein levels of endogenous β-catenin target genes in HCT116 and CT26 cells. HCT116 cells were treated with 30 μM apatinib for 24 h, and different expression genes were analyzed by RNA-seq. mRNA levels of β-catenin target genes were analyzed (A) and further confirmed by RT-PCR (B). Data shown represent the mean of three independent RT-PCR reactions, graphed as relative expression level compared to that of DMSO-treated control. (C and D) The effects of apatinib on β-catenin target gene products in HCT116 (C) and CT26 cells (D) were examined through Western blot analyses. The level of β-actin was used as a loading control. Data are expressed as means ± SEM of three experiments. *P < 0.05, **P < 0.01 vs control group. |
Apatinib Increases the Ubiquitination of β-Catenin and Reduces Its Nuclear Localization
Subsequently, we further examined the expression of β-catenin after apatinib treatment. To this end, HCT116 and CT26 cells were treated with apatinib for 24 h and then analyzed for β-catenin expression, which was significantly decreased by treatment with 30 μM apatinib. We further investigated this response and observed decreased expression levels of p-GSK3β after apatinib treatment (Figure 5A). In addition, Western blot analysis of immunoprecipitated β-catenin revealed that apatinib significantly increased β-catenin ubiquitination (Figure 5B). Then, we analyzed the cytosolic and nuclear fraction of apatinib-treated cells, and a significant decrease in nuclear β-catenin was observed (Figure 5C). Moreover, immunofluorescence results also confirmed the decreased localization of β-catenin in the nucleus in HCT116 cells (Figure 5D). Taken together, these results revealed that the β-catenin pathway is inhibited by apatinib.
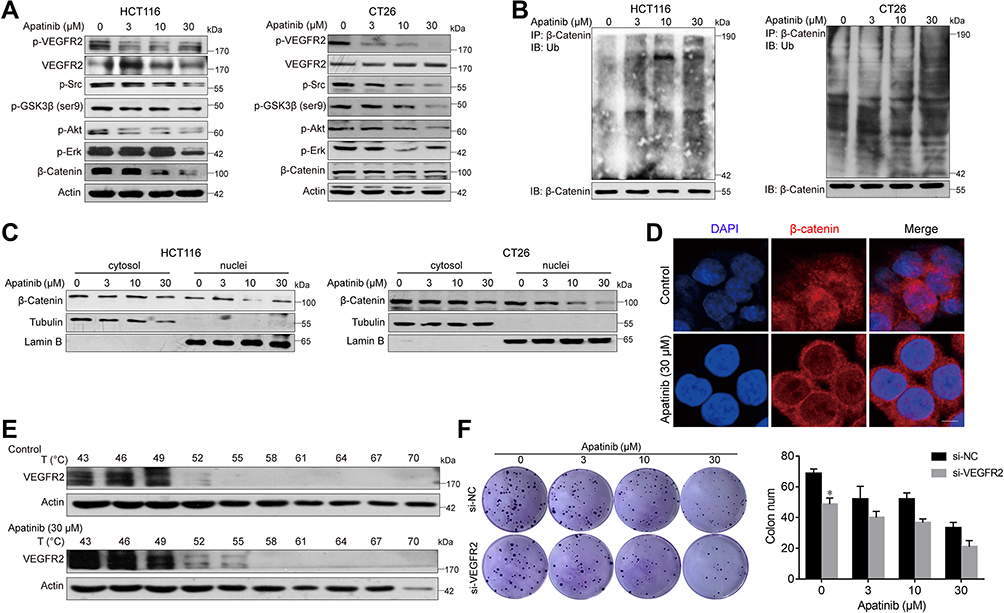 |
Figure 5 Apatinib increased the ubiquitination of β-catenin and inhibited the nuclear translocation of β-catenin. (A) HCT116 and CT26 cells were treated with apatinib for 24h; the expression of related proteins was detected by immunoblotting. The level of β-Actin was used as a loading control. (B) Apatinib increased the ubiquitination of β-catenin. β-catenin was immunoprecipitated using the anti-β-catenin antibody in HCT116 and CT26 cells, immunoprecipitated fractions were analyzed by Western blot analysis to examine the protein level of Ub (ubiquitin). (C) HCT116 and CT26 cells were treated with 3, 10, 30 µM Apatinib for 24 h. The nucleus/cytoplasmic fractionation was separated and the localization of beta-catenin was corroborated by Western blot. (D) Apatinib inhibited nuclear translocation of β-catenin. HCT116 cells were treated with 30 µM Apatinib for 24 h. The image shows β-catenin (red)-stained Fluor-conjugated secondary antibody and the nucleus (blue) stained with DAPI, and the merged image of apatinib-treated cells shows the cytoplasm location of the β-catenin protein. Scale bar 10 μm. (E) HCT116 cells were incubated with or without apatinib (30 μM) for 2 h, and then the cells were collected and subjected to CETSA assay. (F) HCT116 cells with or without VEGFR2 knockdown were treated with apatinib for 24 h, and then cells were seeded at a density of 500 cells per well into a 6-well plate. After cultured for 14 days, the clones were fixed and stained with crystal violet. The data were expressed as mean ± SEM obtained from triplicate experiments. *P < 0.05 vs control group. |
As apatinib has been reported to act on VEGER2, we subsequently verified this activity. As shown in Figure 5A, p-VEGFR2 levels were significantly decreased, as were those of the downstream signaling proteins Erk, PI3K-Akt and Src. Upon binding with small drug molecules, proteins become resistant to denaturation at higher temperatures. Therefore, CETSA was conducted on HCT116 cell to further confirm the direct interaction between apatinib and VEGFR2. VEGFR2 began to degrade at 49°C and disappear at 55°C in vehicle‐treated cells, both of which occurred at higher temperatures in apatinib-treated cells (Figure 5E). VEGFR2 knockdown decreased the HCT116 cell growth, which acted in synergy with the inhibitory functions of apatinib (Figure 5F). Collectively, these data confirmed the on-target effect of apatinib.
Apatinib Inhibits the Growth of Colorectal Cancer in vivo
Subsequently, we tested the antitumor effects of apatinib in vivo using xenograft mouse models that were established as described in the methods. We observed that in contrast to that observed in the control group, the mice in the apatinib-treated group had slower tumor growth and lower tumor volume. Interestingly, an inhibitory effect on tumor growth was analogous to that of 5-FU (Figure 6A and B). The results showed that the mice in the 5-FU group had the lowest tumor weight, while that observed in the apatinib-treated group was lower than that measured in the control group (Figure 6C). In addition, no significant decrease in body weight was observed in each group during treatment (Figure 6D).
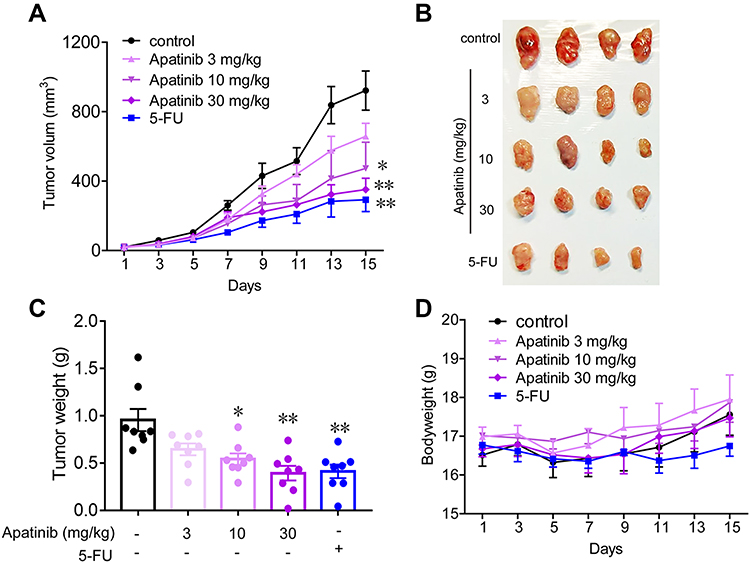 |
Figure 6 Apatinib inhibited the growth of colorectal cancer cells in vivo. 2 × 106 CT26 cells were transplanted subcutaneously into the armpit of the BALB/c mice. Three days after transplanted, mice were randomly allocated to either control or treatment groups, with 6 mice per group. Drugs were intraperitoneal injected (see in Materials and methods). Bodyweight and tumor volumes were measured every two days (A and D). After mice were sacrificed, solid tumors were separated and weighted (B and C). Data are expressed as means ± SEM, n=8. *P < 0.05, **P < 0.01 vs control group. |
Apatinib Promotes Tumor Cell Apoptosis and Inhibits the Proliferation of Tumor Cells in vivo
We further assessed the effect of apatinib on the proliferation and apoptosis of tumor cells in vivo. Resected specimens from the apatinib-treated group displayed typical pathological characteristics of malignancy via H&E staining, such as condensation of the cytoplasm and pyknosis of the nuclei (Figure 7A). The PCNA expression was decreased in the tumor tissues from the apatinib and the 5-FU groups compared to that observed in the control group (Figure 7B and D). To assess whether apatinib promotes tumor reduction by increasing tumor cell apoptosis, we performed TUNEL staining, and a significant increase in TUNEL-positive apoptotic cells in the apatinib-treated group was observed (Figure 7C and E).
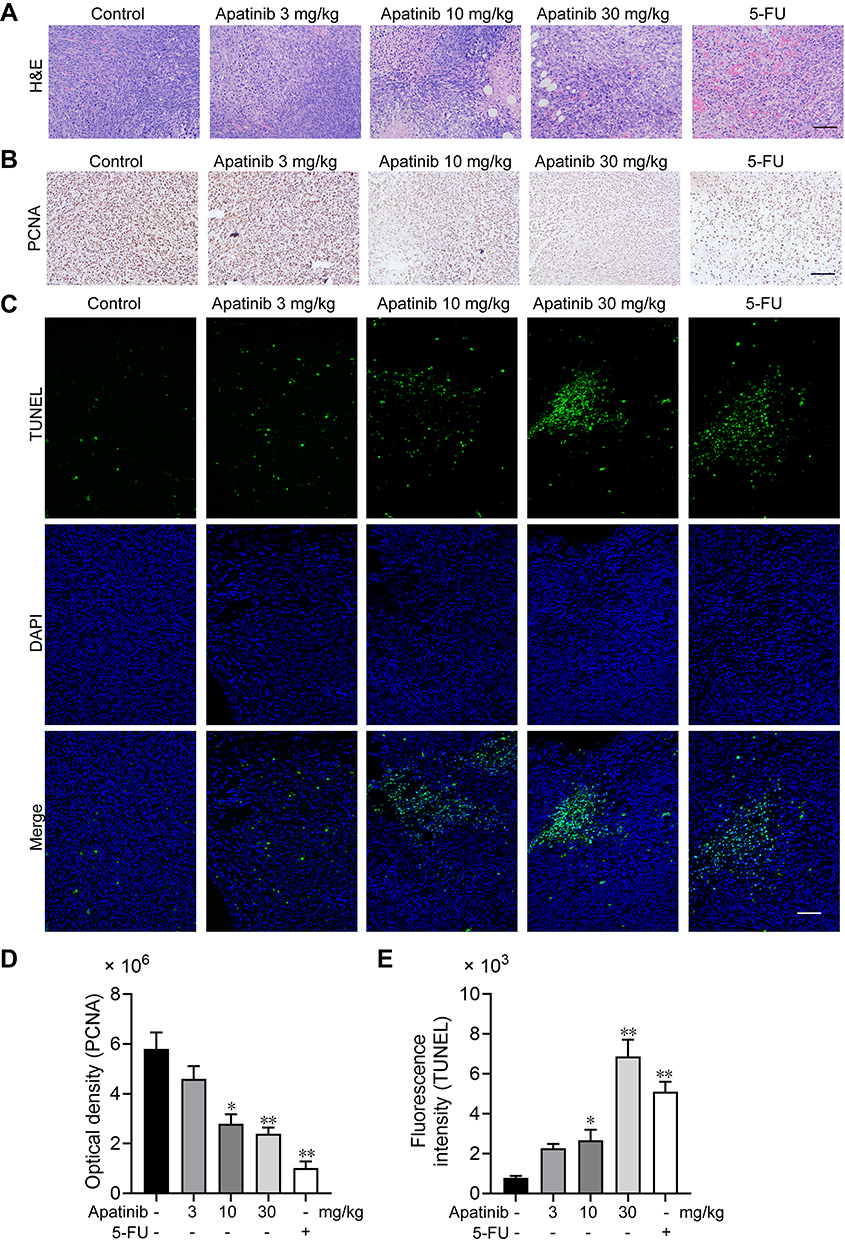 |
Figure 7 Apatinib promoted tumor cell apoptosis and inhibited the proliferation of tumor cells in vivo. (A) The tumor tissues of the mice were fixed and sliced. The morphology and density of the tumor cells of each group were observed by H&E staining. (B and D) Immunohistochemistry staining and quantitative analysis of PCNA in tumor tissues. (C and E) TUNEL staining and quantitative analysis of apoptotic tumor cells in each group. Scale bar 100 μm, *p<0.05, **p<0.01 vs control group. |
Apatinib Inhibits the Activation of VEGFR2 and β-Catenin in vivo
Subsequently, we assessed whether apatinib affects the activation of VEGFR2 and β-catenin in vivo. Immunoblotting results showed that the expression of p-VEGFR2 and β-catenin levels were markedly decreased in response to apatinib treatment in a dose-dependent manner (Figure 8A), which was further confirmed by immunohistochemistry staining results (Figure 8B). Moreover, the levels of proteins downstream of VEGFR2 and β-catenin, including cyclin D1, p-Erk and CD31, were also confirmed to be suppressed by apatinib treatment (Figure 8C). These results demonstrated that in addition to the in vitro activity observed using CRC cell lines, apatinib can inhibit the activity of VEGFR2 and β-catenin in CRC in vivo.
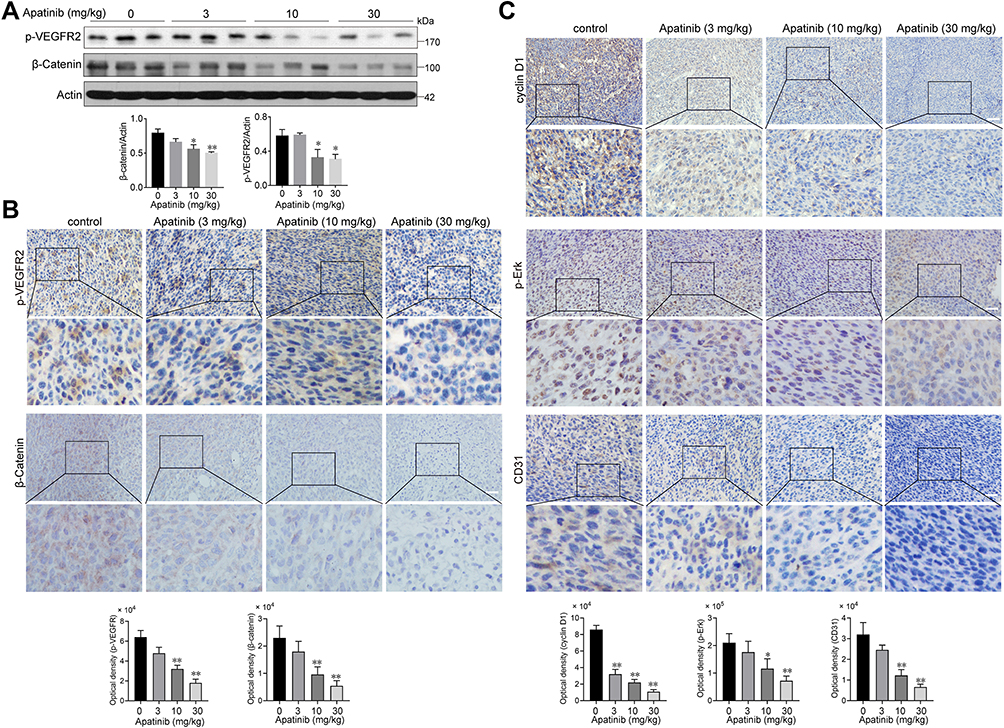 |
Figure 8 Apatinib inhibited the VEGFR2-β-catenin pathway in vivo. (A) The expression of β-catenin and p-VEGFR2 in tumor tissues was detected by immunoblotting. (B) The Immunohistochemistry staining and quantitative analysis of p-VEGFR2, β-catenin. (C) The Immunohistochemistry staining and quantitative analysis of cyclin D1, p-Erk and CD31 in tumor tissues of each group. *P < 0.05, **P < 0.01 versus control group. |
Discussion
Both the incidence and mortality of CRC are increasing every year in China.24 As more patients with mCRC can receive third-line therapy,8,25 new treatment strategies are urgently needed. Apatinib is a small-molecule kinase inhibitor that targets VEGFR2.26 In the present study, we investigated the antitumoral effects of apatinib on CRC in vitro and in vivo. The results indicate the involvement of a newly elucidated mechanism by which apatinib inhibits the β-catenin pathway.
We demonstrated that apatinib treatment blocks the cell cycle progression and inhibits the proliferation, migration and invasion of CRC cells in vitro, which is consistent with the results of previous research.27–29 Additionally, the xenograft assay results indicated that apatinib impeded the tumor growth of CRC, inhibited angiogenesis and promoted apoptosis in vivo. In our present study, the tumor lethality induced by apatinib in CRC was well demonstrated.
The canonical Wnt-β-catenin pathway is mutated in almost 90% of CRC, which is related to metastasis and chemotherapy resistance in CRC.30–35 The previous study has provided a rationale for targeting β-catenin signaling in the treatment of CRC.36 In the present study, we observed that apatinib can inhibit the β-catenin pathway, as evidenced by observations of increased ubiquitination of β-catenin and reduced nuclear translocation of β-catenin. As previously reported, the cytoplasmic levels of β-catenin are maintained through a multiprotein complex called the β-catenin-destroying complex, which consists of Axin1/2, adenomatous polyposis coli (APC), casein kinase I-alpha (CKIα) and GSK-3β.37–39 Ser-9 is one of the major phosphorylation sites of GSK3b, and phosphorylation of ser-9 inhibits GSK-3β activity. Our results demonstrated that apatinib treatment decreased the phosphorylation of ser-9 in GSK3β, thereby decreasing the nuclear localization of β-catenin.40,41
Studies have shown that apatinib can regulate multiple VEGFR2-mediated pathways to have activity against cancers.42–44 The VEGF/VEGFR axis has also been shown to promote β-catenin activation through modulation of SRC-PI3K-AKT-mediated GSK3β phosphorylation.45–48 Therefore, it is reasonable to suggest that by acting on the VEGFR2 expressed in cancer cells, which was confirmed by the CETSA assay results, apatinib inhibits β-catenin-dependent cancer growth. This conclusion was further strengthened by observations that VEGFR2 knockdown could decrease the cancer cell growth, which acted synergistically with the inhibitory functions of apatinib. In addition, we also observed that apatinib could inhibit the VEGFR2-Erk pathway, which is consistent with the results of a previous study on cholangiocarcinoma cells.42
To sum up, it could conclude that apatinib has a therapeutic effect on CRC in vivo and in vitro, and apatinib exerts the antitumor effect by inhibiting angiogenesis in endothelial cells as well as the VEGFR2-β-catenin pathway in cancer cells (Figure 9). These results reveal a mechanism of apatinib activity in CRC and provide a new candidate for the treatment of CRC in clinical practice.
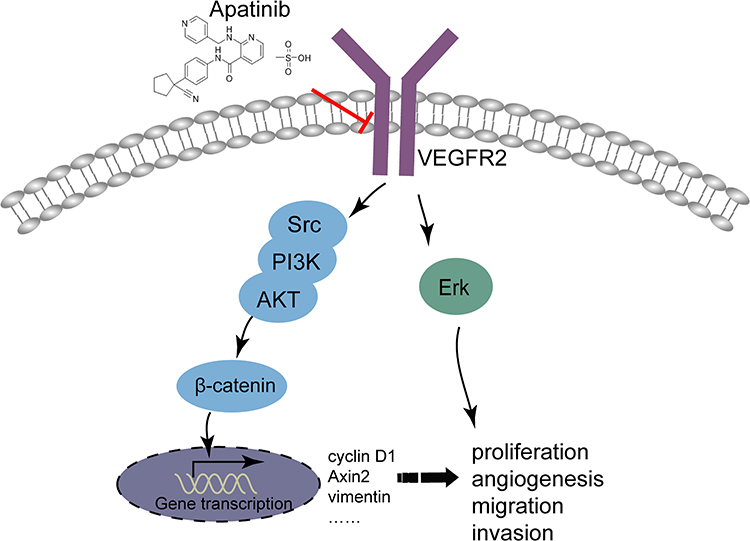 |
Figure 9 Illustration for the mechanism underlying apatinib induced colorectal cancer growth inhibition. By inhibiting VEGFR2-β-catenin mediated singling for colorectal cancer proliferation, invasion, migration and VEGFR2-mediated angiogenesis, apatinib successfully suppressed colorectal cancer growth, suggesting the utility of apatinib as an effective therapeutic strategy for colorectal cancer in the clinic. |
Conclusion
Apatinib has a therapeutic effect on CRC in vivo and in vitro, and apatinib exerts the antitumor effect by inhibiting angiogenesis in endothelial cells, as well as the VEGFR2-β-catenin pathway in cancer cells (Figure 9). These results reveal a mechanism of apatinib activity in CRC and provide a new candidate for the treatment of CRC in clinical practice.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81871944, 81572389), Jiangsu province key medical talents (ZDRCA2016026) and Priority Academic Program Development of Jiangsu Higher Education Institutions. Project was supported by the National Training Program for Innovation and Entrepreneurship for undergraduate students (G201810284152), Jiangsu Students’ Platform for innovation and entrepreneurship training program (201710312008Z), Fundamental Research Funds for the Central Universities (020814380114).
Disclosure
The authors declared no conflict of interest, and this study received support materials from HengRui Medicine Co., Ltd.
References
1. Siegel RL, Miller KD, Goding Sauer A, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145–164.
2. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019;16(12):713–732. doi:10.1038/s41575-019-0189-8
3. Kelly H, Goldberg RM. Systemic therapy for metastatic colorectal cancer: current options, current evidence. J Clin Oncol. 2005;23(20):4553–4560. doi:10.1200/JCO.2005.17.749
4. Li J, Qin S, Xu R, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet Oncol. 2015;16(6):619–629. doi:10.1016/S1470-2045(15)70156-7
5. Marcus L, Lemery S, Khasar S, et al. FDA approval summary: TAS-102. Clin Cancer Res. 2017;23(12):2924–2927. doi:10.1158/1078-0432.CCR-16-2157
6. Shirley M. Fruquintinib: first global approval. Drugs. 2018;78(16):1757–1761. doi:10.1007/s40265-018-0998-z
7. Sanchez-Gundin J, Fernandez-Carballido AM, Martinez-Valdivieso L, Barreda-Hernandez D, Torres-Suarez AI. New trends in the therapeutic approach to metastatic colorectal cancer. Int J Med Sci. 2018;15(7):659–665. doi:10.7150/ijms.24453
8. Bekaii-Saab T, Kim R, Kim TW, et al. Third- or later-line therapy for metastatic colorectal cancer: reviewing best practice. Clin Colorectal Cancer. 2019;18(1):e117–e129. doi:10.1016/j.clcc.2018.11.002
9. Asaoka Y, Ijichi H, Koike K. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;373(20):1979.
10. Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–779. doi:10.1200/JCO.2017.76.9901
11. Geng R, Li J. Apatinib for the treatment of gastric cancer. Expert Opin Pharmacother. 2015;16(1):117–122. doi:10.1517/14656566.2015.981526
12. Scott AJ, Messersmith WA, Jimeno A. Apatinib: a promising oral antiangiogenic agent in the treatment of multiple solid tumors. Drugs of Today. 2015;51(4):223–229. doi:10.1358/dot.2015.51.4.2320599
13. Zhang H. Apatinib for molecular targeted therapy in tumor. Drug Des Devel Ther. 2015;9:6075–6081. doi:10.2147/DDDT.S97235
14. Chen X, Qiu T, Zhu Y, et al. A single-arm, phase II study of apatinib in refractory metastatic colorectal cancer. Oncologist. 2019;24.
15. Cai X, Wei B, Li L, et al. Apatinib enhanced anti-PD-1 therapy for colon cancer in mice via promoting PD-L1 expression. Int Immunopharmacol. 2020;88:106858. doi:10.1016/j.intimp.2020.106858
16. Chen Y-N, LaMarche MJ, Chan HM, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535(7610):148–152. doi:10.1038/nature18621
17. Guo WJ, Zhang YM, Zhang L, et al. Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy. 2013;9(7):996–1008. doi:10.4161/auto.24407
18. Guo W, Sun Y, Liu W, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10(6):972–985. doi:10.4161/auto.28374
19. Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi:10.1093/bioinformatics/btt656
20. Qian Y, Zhang L, Cai M, et al. The prostate cancer risk variant rs55958994 regulates multiple gene expression through extreme long-range chromatin interaction to control tumor progression. Sci Adv. 2019;5(7):eaaw6710. doi:10.1126/sciadv.aaw6710
21. Ji Q, Xu X, Kang L, et al. Hematopoietic PBX-interacting protein mediates cartilage degeneration during the pathogenesis of osteoarthritis. Nat Commun. 2019;10(1):313. doi:10.1038/s41467-018-08277-5
22. Guo W, Liu W, Chen Z, et al. Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat Commun. 2017;8(1):2168. doi:10.1038/s41467-017-02351-0
23. Geng J, Liu W, Gao J, et al. Andrographolide alleviates Parkinsonism in MPTP-PD mice via targeting mitochondrial fission mediated by dynamin-related protein 1. Br J Pharmacol. 2019;176(23):4574–4591. doi:10.1111/bph.14823
24. Feng R-M, Zong Y-N, Cao S-M, Xu R-H. Current cancer situation in China: good or bad news from the 2018 global cancer statistics? Cancer Commun. 2019;39(1):22. doi:10.1186/s40880-019-0368-6
25. Tampellini M, Di Maio M, Baratelli C, et al. Treatment of patients with metastatic colorectal cancer in a real-world scenario: probability of receiving second and further lines of therapy and description of clinical benefit. Clin Colorectal Cancer. 2017;16(4):372–376. doi:10.1016/j.clcc.2017.03.019
26. Li J, Qin S, Xu J, et al. Apatinib for chemotherapy-refractory advanced metastatic gastric cancer: results from a randomized, placebo-controlled, parallel-arm, phase II trial. J Clin Oncol. 2013;31(26):3219–3225. doi:10.1200/JCO.2013.48.8585
27. Cheng X, Feng H, Wu H, et al. Targeting autophagy enhances apatinib-induced apoptosis via endoplasmic reticulum stress for human colorectal cancer. Cancer Lett. 2018;431:105–114. doi:10.1016/j.canlet.2018.05.046
28. Yin L, Wang J, Huang F-C, et al. [Inhibitory effect of apatinib on HCT-116 cells and its mechanism]. Nan Fang Yi Ke Da Xue Xue Bao. 2017;37(3):367–372.
29. Lu W, Ke H, Qianshan D, Zhen W, Guoan X, Honggang Y. Apatinib has anti-tumor effects and induces autophagy in colon cancer cells. Iran J Basic Med Sci. 2017;20(9):990–995.
30. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi:10.1038/nature11252
31. Raji R, Sasikumar R, Jacob E. Multiple roles of adenomatous polyposis coli gene in Wnt signalling - a computational model. BioSystems. 2018;172:26–36. doi:10.1016/j.biosystems.2018.08.001
32. Hankey W, McIlhatton M, Ebede K, et al. Mutational mechanisms that activate Wnt signaling and predict outcomes in colorectal cancer patients. Cancer Res. 2018;78:617–630.
33. Vermeulen L, Melo FDSE, Heijden MVD, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2015;12(5):468–476. doi:10.1038/ncb2048
34. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. doi:10.1038/nature03319
35. Morin PJ. β-catenin signaling and cancer. BioEssays. 1999;21(12):1021–1030. doi:10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P
36. Cheng X, Xu X, Chen D, Zhao F, Wang W. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed Pharmacother. 2019;110:473–481. doi:10.1016/j.biopha.2018.11.082
37. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. doi:10.1016/j.cell.2017.05.016
38. Huang J, Guo X, Li W, Zhang H. Activation of Wnt/β-catenin signalling via GSK3 inhibitors direct differentiation of human adipose stem cells into functional hepatocytes. Sci Rep. 2017;7:40716. doi:10.1038/srep40716
39. Jope RS. Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003;24(9):441–443. doi:10.1016/S0165-6147(03)00206-2
40. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–131. doi:10.1016/j.pharmthera.2014.11.016
41. Tejeda-Muñoz N, Robles-Flores M. Glycogen synthase kinase 3 in Wnt signaling pathway and cancer. IUBMB Life. 2015;67(12):914–922.
42. Huang M, Huang B, Li G, Zeng S. Apatinib affect VEGF-mediated cell proliferation, migration, invasion via blocking VEGFR2/RAF/MEK/ERK and PI3K/AKT pathways in cholangiocarcinoma cell. BMC Gastroenterol. 2018;18(1):169. doi:10.1186/s12876-018-0870-3
43. Yu X, Fan H, Jiang X, et al. Apatinib induces apoptosis and autophagy via the PI3K/AKT/mTOR and MAPK/ERK signaling pathways in neuroblastoma. Oncol Lett. 2020;20(4):52.
44. Liu K, Ren T, Huang Y, et al. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis. 2017;8(8):e3015. doi:10.1038/cddis.2017.422
45. Luo M, Hou L, Li J, et al. VEGF/NRP-1 axis promotes progression of breast cancer via enhancement of epithelial-mesenchymal transition and activation of NF-κB and β-catenin. Cancer Lett. 2016;373(1):1–11. doi:10.1016/j.canlet.2016.01.010
46. Maes C, Goossens S, Bartunkova S, et al. Increased skeletal VEGF enhances β-catenin activity and results in excessively ossified bones. EMBO J. 2010;29(2):424–441. doi:10.1038/emboj.2009.361
47. Lee M, Choy WC, Abid MR. Direct sensing of endothelial oxidants by vascular endothelial growth factor receptor-2 and c-Src. PLoS One. 2011;6(12):e28454. doi:10.1371/journal.pone.0028454
48. Yang J, Caldwell RB, Behzadian MA. Blockade of VEGF-induced GSK/β-catenin signaling, uPAR expression and increased permeability by dominant negative p38α. Exp Eye Res. 2012;100:101–108. doi:10.1016/j.exer.2012.03.011
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.