Back to Journals » Cancer Management and Research » Volume 13
Therapeutic Options for Patients with TP53 Deficient Chronic Lymphocytic Leukemia: Narrative Review
Authors Stefaniuk P ![]() , Onyszczuk J, Szymczyk A
, Onyszczuk J, Szymczyk A ![]() , Podhorecka M
, Podhorecka M
Received 25 September 2020
Accepted for publication 20 November 2020
Published 12 February 2021 Volume 2021:13 Pages 1459—1476
DOI https://doi.org/10.2147/CMAR.S283903
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Kenan Onel
Paulina Stefaniuk,1 Julia Onyszczuk,2 Agnieszka Szymczyk,3 Monika Podhorecka1
1Department of Haematooncology and Bone Marrow Transplantation, Medical University of Lublin, Lublin, Poland; 2Students Scientific Association, Department of Haematooncology and Bone Marrow Transplantation, Medical University of Lublin, Lublin, Poland; 3Department of Clinical Transplantology, Medical University of Lublin, Lublin, Poland
Correspondence: Agnieszka Szymczyk Department of Clinical Transplantology
Medical University of Lublin, Chodźki 7 Street, Lublin, 20-093, Poland
Tel +48-81 534 54 96
Email [email protected]
Abstract: Chronic lymphocytic leukemia (CLL), which is the most common type of leukemia in western countries in adults, is characterized by heterogeneity in clinical course, prognosis and response to the treatment. Although, in recent years a number of factors with probable prognostic value in CLL have been identified (eg NOTCH1, SF3B1 and BIRC-3 mutations, or evaluation of microRNA expression), TP53 aberrations are still the most important single factors of poor prognosis. It was found that approximately 30% of all TP53 defects are mutations lacking 17p13 deletion, whereas sole 17p13 deletion with the absence of TP53 mutation consists of 10% of all TP53 defects. The detection of del(17)(p13) and/or TP53 mutation is not a criterion itself for starting antileukemic therapy, but it is associated with an aggressive course of the disease and poor response to the standard chemoimmunotherapy. Treatment of patients with CLL harbouring TP53-deficiency requires drugs that promote cell death independently of TP53. Novel and smarter therapies revolutionize the treatment of del(17p) and/or aberrant TP53 CLL, but development of alternative therapeutic approaches still remains an issue of critical importance.
Keywords: chronic lymphocytic leukemia, p53 protein, molecular aberrations, drug resistance
Introduction
The World Health Organisation defines chronic lymphocytic leukaemia (CLL) as low-grade lymphoproliferative neoplasm with ≥5×109/L clonal B-cells in peripheral circulation.1,2 Small, mature-appearing CD5, CD19, dimCD20, and CD23 positive B-lymphocytes accumulate in blood, bone marrow, lymph nodes, and spleen.3 The most common presentation of the disease nowadays is asymptomatic peripheral blood lymphocytosis. Patients may also present with lymphadenopathy, hepatomegaly, splenomegaly, bone marrow failure, recurrent infections, autoimmune hemolytic anemia, or autoimmune thrombocytopenia.4
CLL is the most common type of leukaemia in western countries, accounting for approximately 25% of adult leukaemias.5,6 In the United States of America, the median age at diagnosis of CLL is 71 years and the age adjusted incidence is 4.5 cases per 100,000 inhabitants. 5-year survival (calculated in 2005–2011) was estimated as 81.7%.4 Five year survival was calculated as 67.5% in 1975 and increased to 87.9% in 2007.4 It illustrates how novel treatment regimens have revolutionized the management of CLL. As the incidence rate rises with age, the prevalence of CLL is likely to increase due to the demographic changes in western countries. As a consequence of more frequent blood testing, the proportion of younger patients with early stage CLL and no symptoms is also likely to increase.7 CLL is most common in Caucasians and rarely observed in Asians, even those who migrated to western countries.8,9 The incidence of CLL varies between geographical regions, ranging from <0.01% in eastern Asia to 0.06% in Europe and the United States.10
CLL Prognostic Factors
The clinical course of CLL is highly heterogenous: some patients require aggressive treatment at the time of the diagnosis, others do not require treatment for many years, if at all. Numerous prognostic factors have been proposed to evaluate the outcome in CLL. Multivariable models, prognostic indexes and nomograms have been developed in order to predict clinical outcome accurately.8 Rai’s and Binet’s classifications,11 although widely used in clinical routine, are not sufficient to determine if the patient will present a rapidly-progressive disease or an indolent one. Nowadays, genetic, epigenetic and molecular markers are the main focus of attention in prognostication in CLL.12 It is estimated that the typical CLL genome carries approximately 2000 molecular lesions, however only approximately 5 of these are gross structural abnormalities. More than 40 mutated driver genes have been identified in CLL.13–15
Generally, prognostic factors in CLL can be divided into two groups: markers of good and poor prognosis. 13q14 deletion and hypermutation of IGVH are associated with good prognosis, whereas del(11q), del(17p); unmutated immunoglobulin heavy-chain (IGHV); high expression of ZAP70, CD38, CD49d; and mutations in: NOTCH1, SF3B1 and BIRC3 are poor prognostic factors.13
The most frequent genetic aberration in CLL is deletion of 13q14, occurring in 50% to 60% of patients.13,16 The deleted region contains the miR15A and miR16A microRNAs, which inhibit the expression of key regulators of apoptosis and cell cycle.13,17 Levels of the miR15A and miR16A microRNAs negatively correlate with the expression of the antiapoptotic gene BCL2, which codes for an antiapoptotic protein and is one of the genes upregulated in CLL.13 Elevated expression of the antiapoptotic protein BCL2 makes CLL cells resistant to apoptosis, which results in clonal lymphocytes accumulation.18 In Rossi et al's study19 CLL patients harboring solely 13q14 deletion had an expected survival only slightly, not significantly, poorer than that of the healthy population. The progression rate was very slow (approximately 4% per year) and transformation rate low.19 The deletion of chromosome 11q22-q23 is observed in 10–20% of CLL cases.20 It always includes the ATM gene (ataxia telangiectasia mutated). The ATM gene regulates the cell-cycle arrest at G1/S and G2/M to prevent processing of damaged DNA and in case of DNA damage, it alternatively activates the DNA repair pathways or induces apoptosis. ATM deletion at the time of the diagnosis identifies an intermediate-risk group of patients. The presence of 11q22-23 deletion is associated with poor response to chemotherapy.19,21 Trisomy 12 is observed in 10% to 20% of cases of CLL.5 It is the second most common cytogenetic abnormality in patients with CLL.16 The genes involved in the pathogenesis of CLL with a trisomy 12 are unknown. The prognostic significance of this aberration is not completely understood.5,22 Döhner et al considered trisomy 12 an intermediate prognostic risk factor, with a median time to first treatment of 33 months and a median overall survival of 114 months.16 In contrast, Rossi et al qualified +12 patients as low-risked.18 +12 is particularly frequent in patients who develop Richter syndrome, especially in patients harbouring NOTCH1 mutations.23,24 Complex karyotype defined as three or more karyotype aberrations, is also considered an independent marker of poor prognosis.12,25
The unmutated immunoglobulin heavy-chain (IGHV) genes is one of the most important single factors of poor outcome.19 Almost all of IGHV unmutated patients are expected to progress after chemoimmunotherapy. However, progression-free survival (PFS) of IGHV-unmutated patients is similar to that of IGHV-mutated patients in the case of treatment with novel drugs.13 Condoluci et al conducted a study on a group of 4933 asymptomatic, early-stage, CLL patients, aiming to develop a prognostic score to predict time to first treatment (TTFT). Out of all genetic and molecular factors, taken into account in the study, the IGHV unmutated status had the strongest effect on TTFT prognostication. Unmutated IGHV along with absolute lymphocyte count higher than 153 109/L and presence of palpable lymph nodes became components of International Prognostic Score for Early-stage CLL (IPS-E). The IPS-E is a simple prognostic model: each of the three covariates, present in the patient is scored 1 point. Score 0 predicts low-risk (5-year cumulative risk for treatment start was 8.4%), score 1 predicts intermediate-risk (5-year cumulative risk for treatment start was 28.4%), score 2–3 predicts high-risk (cumulative risk for treatment start was 61.2%).26 On the contrary, hypermutation of the rearranged IGHV genes is considered a good prognostic factor. Hamblin et al27 observed that CLL patients carrying mutated IGHV genes experience an indolent course of the disease in comparison with patients harbouring unmutated IGHV genes. Median survival for stage A patients with unmutated IGHV genes was 95 months compared with 293 months for patients with mutated IGHV genes (p = 0.0008).27
High expression of ZAP70, CD38, and CD49d are well-known adverse prognostic factors.28,29 CD38 is a transmembrane type 2 glycoprotein, expressed not only in hematological cells, but also in the brain, the eye, bone tissue, smooth muscles and pancreatic isle cells.30 It plays a critical role in regulation of calcium signalling and hence regulates the insulin secretion, oxytocin production, bone tissue metabolism. It also takes part in the immune response: T cell activation, neutrophil chemotaxis, dendritic cell migration, monocyte chemokine production.28 CD38 is also a lymphocyte differentiation antigen, which interacts with: TCR in T-lymphocytes, BCR in B-lymphocytes, CD16/CD81 complex in NK cells. It induces proliferation in mature B cells and blocks the immature B-cell haematopoiesis.28,31 In the Damle et al32 study, CD38 expressed in more than 30% of CLL cells predicted shorter overall survival.32 Not withstanding, some authors propose 20% or 7% cut-off levels.28,33,34 CD49d is an alpha-4-integrin, which binds either with beta 7 integrin in order to mediate lymphocyte adhesion or with beta 1 integrin (CD29). The combination CD49d/CD29 forms very late antigen-4 (VLA-4), expressed on leukocytes CD34-positive hematopoietic stem/progenitor cells.28 VLA-4 plays an important role in fetal and adult haematopoiesis.28,35,36 Germline CD49d expression superior to the 30% level is associated with shorter OS and PFS.37 It seems that high expression of CD38 and CD49d prolongs CLL cells survival by enhancing the stimulatory signals from microenvironment and blocking apoptotic signals.28 ZAP70 is a protein kinase expressed by T-lymphocytes and NK cells, but also by normal B-cells.38,39 Scielzo et al analyzed ZAP expression on different human normal B-lymphocyte subpopulations, including: naïve, germinal center and memory B cells from tonsils, CD19+ CD5+ cells from cord blood and CD19+ lymphocytes from peripheral blood. ZAP-70 protein was expressed in all cells, although at different levels, probably dependent on stimulation via the B-cell receptor.38 In CLL, patients with ZAP70 positivity show shortened treatment free survival and shorter OS. In contrast, ZAP70 negative patients demonstrate slow progression rate.40
The next-generation sequencing technologies led to identification of additional genetic aberrations in CLL, such as mutations in NOTCH1, SF3B1, and BIRC3, which can be considered as prognostic and predictive biomarkers.41 The NOTCH genes encode transmembrane proteins NOTCH1 to NOTCH4, that function as transcription factors, modifying the expression of numerous target genes, such as MYC and NF-kB signalling components.21 NOTCH1 mutations occur in 4% to 11% of CLL patients and are frequently correlated with trisomy 12 and unmutated IGHV genes.12,15,42 80% of NOTCH1 mutations are represented by c.7544_7545delCT deletion.15 NOTCH1 mutations in CLL result in NOTCH1 impaired degradation and deregulated NOTCH signalling.21 Multivariate analyses has showed that NOTCH1 is an independent poor prognostic factor, especially in patients treated with chemoimmunotherapy.43 Moreover, patients carrying NOTCH mutation have higher risk of Richter transformation.12 NOTCH1 mutations can also be viewed as biomarkers of resistance to the anti-CD20 monoclonal antibodies rituximab and ofatumumab in CLL.13,44 The lower sensitivity to anti-CD20 treatment in CLL harbouring NOTCH1 mutations can be associated with dysregulation of epigenetic repression of CD20 expression, when mutations in exon 34 takes place.45 SF3B1 is a component of the mRNA splicing machinery. SF3B1 mutations, which result in unaltered DNA damage response, and occur with a prevalence from 7 to 10% of CLL patients. SF3B1 mutations frequently accompany other alterations: unmutated IGHV genes and ATM deletion or mutations. The presence of SF3B1 mutations at the time of CLL presentation identifies a group of patients with intermediate-risk disease.21 BIRC3 gene (the Baculoviral IAP repeat containing 3) is mutated in approximately 2% of CLL patients.19 BIRC3 mutations disrupt the C-terminal RING domain, which leads to constitutive NF-κB activation. NF-κB activation in CLL is considered responsible for resistance to conventional chemotherapy. BIRC3 mutations identify a subgroup of patients with poor prognosis.13,19 Although numerous other gene mutations recurring at lower frequency have been indicated for their association with CLL outcome, including SAMHD1, RPS15, NFKBIE, EGR2, KRAS and POT1, their potential role as prognostic biomarkers needs to be further investigated.46
Although factors mentioned above are considered helpful in CLL prognostication, so far it is the TP53 aberrations that seem the most important prognostic and predictive factor in CLL (Table 1).
 |
Table 1 Genetic and Molecular Prognostic Factors in CLL |
P53 Protein – Unveiling the “Guardian of the Genome”
TP53 is a gene, located on chromosome 17, that encodes a tumor suppression protein called p53.47 The tp53 protein has two N-terminal transactivation domains, a central DNA binding domain (encoded by exons 4–8 of the TP53 gene), C-terminus involved in downregulation of DNA binding of the central domain and an oligomerization domain, which is crucial for the transcriptional activity. Regulators, for example a p53 ubiquitin ligase MDM2, keep p53 protein at low levels in normal cells.48,49 In response to cellular stress, including DNA damage and replication stress, p53 is activated to promote the elimination or repair of damaged cells, in order to reduce the risk of propagating mutations.47,48 It allows the ability to decrease the deleterious consequences of mutations in the human genome. It seems that p53 also suppresses chromothripsis - a type of chromosome shattering and rearrangement.48 Due to shelterin defects and incomplete telomeric DNA replication, telomere shortening takes place, which can lead to chromosomal and genomic instability. As cells with dysfunctional telomeres are removed in processes regulated by the p53 protein, telomere dysfunctions promote oncogenesis.50 For the reasons mentioned above, p53 is widely known as the “guardian of the genome”.48 TP53 is mutated or inactivated in approximately 60% of malignant neoplasms.47 Interestingly, other “non–canonical” functions of p53, contributing to its effects, are still being discovered. One of the protection functions of p53 is modulation of autophagy, an essential process to maintain homeostasis and protein control. Autophagy suppresses p53, which is crucial for tumor promotion.51 It was proved that p53 can influence mitochondrial functions as well. When stress stimuli occurs, SCO2 gene (encoding cytochrome C oxidase 2) overexpresses, leading to ROS (reactive oxygen species) generation. Then, p53 can transactivate SCO2, to maintain mitochondrial respiration. In cases of irreversible stress or damage, p53 translocate to mitochondria, leading to apoptosis or necrosis.52 p53 also suppress pluripotency and cellular plasticity, induces an iron-dependent form of cell death known as ferroptosis, increases glutamine catabolism, supports anti-oxidant activity, downregulates lipid synthesis, increases fatty acid oxidation, and stimulates gluconeogenesis. It can have opposing effects on the same metabolic processes, depending on the cell type.48
TP53 aberrations arise alternatively through deletion of the TP53 locus on chromosome 17 or TP53 gene mutations. Gene mutations are predominately concentrated in the DNA- binding domain, encoded by exons 4–8 and comprise of missense mutations, insertions, deletions, nonsense mutations or splice-site mutations. 75% of mutations are missense mutations. Codons 175, 245, 248, 249, 273, and 28 are “hotspots”, the regions affected in particular by mutations.25,53 Figure 1 schematically shows the p53 structure and functions.
 |
Figure 1 p53 structure and functions. The tp53 protein has two N-terminal transactivation domains, a central DNA binding domain, C-terminus involved in down regulation of DNA binding of the central domain and an oligomerization domain, which is essential for transcriptional activity.48,49 In response to cellular stress p53 is activated which leads to promotion of the elimination or repair of damaged cells.47,48 Interestingly, other “non–canonical” functions of p53, contributing to its effects, are still being discovered.48,52,53 |
TP53 Gene Aberrations in CLL
In CLL, TP53 aberrations, associated with either the deletion of chromosome 17p or TP53 gene mutations, are the most important single poor prognostic factors.12 The determination of the outcome of patients with del(17p) had become the aim of numerous clinical trials, in which the median OS oscillated between 2 to 3 years from the time of first-line treatment. Moreover, TP53 abnormality is also clearly associated with poor response to classical chemoimmunotherapy.54 In CLL the most frequent aberration affecting the TP53 gene is del(17p) accompanied by TP53 mutations in the second allele.25 Zenz et al55 performed the analysis of the pattern of TP53 mutations in 254 patients with CLL. It has been proved that the most common are missense mutations (74%), mainly located in the DNA-binding domain. 90% of the mutations were located in exons 5–8. Also the most frequently mutated amino acids were located at positions 175, 179, 248 and 273, which is also observed in all TP53 mutations in general.55 Interestingly, it was suggested in a few studies, that CLL patients with del(17p), show excessive telomere shortening.50 Guièze et al50 evaluated the association between TP53 gene disruptions and impaired telomeres in CLL. TP53 turned out to be the strongest determinant of telomere status in CLL. Patients with del(17p) had shortened telomeres and higher hTERT (human telomerase reverse transcriptase) (hTERT is a component of telomerase, that prevents the shortening of telomeres).50
The issue that also should be raised in the context of TP53 aberrations in CLL is clonal evolution.25 Clonal evolution can be defined as the process of acquisition of vertically transmittable genetic/epigenetic lesions.56 Neoplasm cells are genetically heterogeneous and consist of clonal and subclonal populations, which can either remain in balance or some clones can emerge as dominant. Genetically diverse clones compete and interact with each other, which leads to cancer progression and relapse.25 In CLL clonal evolution is a common phenomenon. TP53 aberrant subclones are more vulnerable to clonal evolution, especially under chemoimmunotherapy pressure. During the course of the disease, TP53 mutations can be acquired or selected.25 While at diagnosis, the TP53 aberrations are observed in 4% to 8% of CLL cases, at the time of first-line treatment the incidence rises to 10%. It is estimated 30−40% in relapsed/refractory CLL and 50–60% in Richter syndrome.13 Rossi et al proved that not only TP53 clonal mutations (represented in >20% leukemic cells), but also the presence of very small TP53 mutated subclones, predict poor prognosis. In patients with TP53 mutated subclones OS was significantly shorter (5-year OS: 46.3%; P = 0.0042) than in individuals with an unmutated TP53 gene (5-year OS: 75.1%) and similar to OS in patients with clonal TP53 mutations (5-year OS: 34.6%; P = 0.6926). Thus, the identification of patients with TP53 subclones might be advisable in order to manage them as high-risk CLL.57
In clinical practice, del(17) in CLL is assessed by the FISH technique and TP53 mutations are evaluated by Sanger sequencing and next-generation sequencing.25 Approximately 30% of all TP53 defects are mutations lacking 17p13 deletion, whereas sole 17p13 deletion with the absence of a TP53 mutation consisting of 10% of all TP53 defects. It is believed that the high proportion of TP53 mutations with no 17p13 deletion may be caused by the presence of two TP53 mutations on individual alleles in CLL cells.21 That is why it is highly recommended to evaluate both the presence of chromosome 17p13 deletion and TP53 mutations.58 In order to unify the methods of detection of TP53 mutation/del(17)(p13), in 2012 the European Research Initiative on CLL (ERIC) published recommendations (available at http://www.ericll.org/tp53_aberrations/) on several methodologies suitable for TP53 analysis. Principal procedures suggested by ERIC for TP53 mutation detection are Sanger Sequencing and Next Generation Sequencing (NGS).49 TP53 sequencing should at very least cover exons 4–10, including splicing sites. As it has been proved that variants can occur in exons outside the DNA binding domain (although it is very rare), the optimal range recommended goes from the second to the eleventh exon49,54,58. It is crucial to test all patients for del(17)(p13) and TP53 mutation before treatment initiation and at every relapse.54 Testing for those mutations at every relapse is recommended due to the ability of leukaemic clones to evolve.21 The detection of del(17)(p13) and TP53 mutation is not a criterion itself for starting therapy. Treatment in patients with CLL who have TP53 mutation should be started according to the International Workshop on Chronic Lymphocytic Leukemia criteria, identically like in patients without aggravating mutations.54,59 Some authors are of the opinion that determination of TP53 status should not be performed before the patient meets criteria for starting therapy.21,25
Numerous studies have been performed, proving that del(17p) and/or TP53 mutations are associated with an aggressive course of the disease and poor response to the standard chemoimmunotherapy.25 One of the most frequently cited studies considering the issue of genetic aberration, including TP53 mutations as predictive factor in CLL, was performed by S. Stilgenbauer et al60 and is called “CLL8 trial”.60 The outcome of the first-line treatment with fludarabine+cyclophosphamide (FC) or fludarabine+cyclophosphamide with rituximab (FCR), has been evaluated in 817 patients with different genetical lesions. TP53 was mutated in 11.5% of patients and interestingly, TP53 mutated was found in 16.1% NOTCH1 mutated patients and in 14.0% of SF3B1 mutated patients. Mutation in TP53 was the most frequent missense mutation and it was situated in the DNA-binding domain. TP53 mutation was positively associated with a CIRS (Cumulative Illness Rating Scale) higher than 1. Patients with TP53 mutated had significantly lower clinical response rates both with FC and FCR treatment. Minimal Residual Disease (MRD) negativity was significantly less frequent in patients with TP53 mutated treated with both regimens. The presence of TP53 mutation was significantly related with shorter PFS (FC: HR 4.295, P<0.001; FCR: HR 3.173, P<0.001) and OS (FC: HR 4.642, P<0.001; FCR: HR 4.447, P<0.001). Only 7 out of 46 patients with del(17) had PFS longer than 24 months. In conclusion, del(17) marked the worst prognosis among subgroups with different genetic aberrations.60
Rossi et al19 reported that median OS in patients harbouring a TP53 mutation was estimated 3 to 5 years. Only 30% of patients were alive at 10 years observation. This subgroup has 70% reduction of the expected survival in comparison with the general population.19 It should be highlighted that this data had been collected before the introduction of novel therapies. Parikh et al61 performed the meta-analysis evaluating the prognostic significance of IGHV mutation status and genetic abnormalities determined by FISH in patients with CLL. Patients with high-risk FISH (del17p13 and del11q23) had shorter median PFS and OS than patients with low or intermediate-risk. High-risk FISH has been proved to be an independent predictor of PFS in 8 of 17 studies analyzed.61
It has also been proved, that among patients with TP53 abnormalities, the risk of Richter transformation is higher than in the general population of patients with CLL.13 Richter transformation, first described by Dr. Maurice Richter in 1928, is defined as the transformation of CLL to an aggressive lymphoma.62,63 Most commonly, CLL transforms to the diffuse large B cell lymphoma (DLBCL).64 A cumulative incidence of Richter transformation in the natural history of CLL is estimated as 2–10%.63,65,66 The factors that increase the probability of the Richter transformation include: advance staged CLL, lymph nodes > 3 cm, unmutated IGHV, del(17p), TP53 mutation, NOTCH1 mutated, and stereotyped B-cell receptor (BCR).63 In around 60% of patients, inactivation of the TP53 gene can be observed.64 In the Fabbri et al study64 del(17p), occurring in 40% of patients has been the most frequent aberration among patients with Richter transformation. Moreover, four TP53 altered cases displayed intrachromosomal rearrangements, which suggests chromothripsis.64 In the Wang et al study63 34.4% of patients with Richter transformation had TP53 disruption. Patients with TP53 aberration had a worse OS (median OS 8.3 vs 12.8 months).
Management of Patients with TP53 Deficient CLL in the Era of Novel Agents
On the basis of research described above and clinicians’ observations, nowadays chemoimmunotherapy is not considered standard management in patients with TP53 aberrations.25 Treatment of patients with CLL harbouring TP53-deficiency requires drugs that promote cell death independently of TP53 (Table 2).
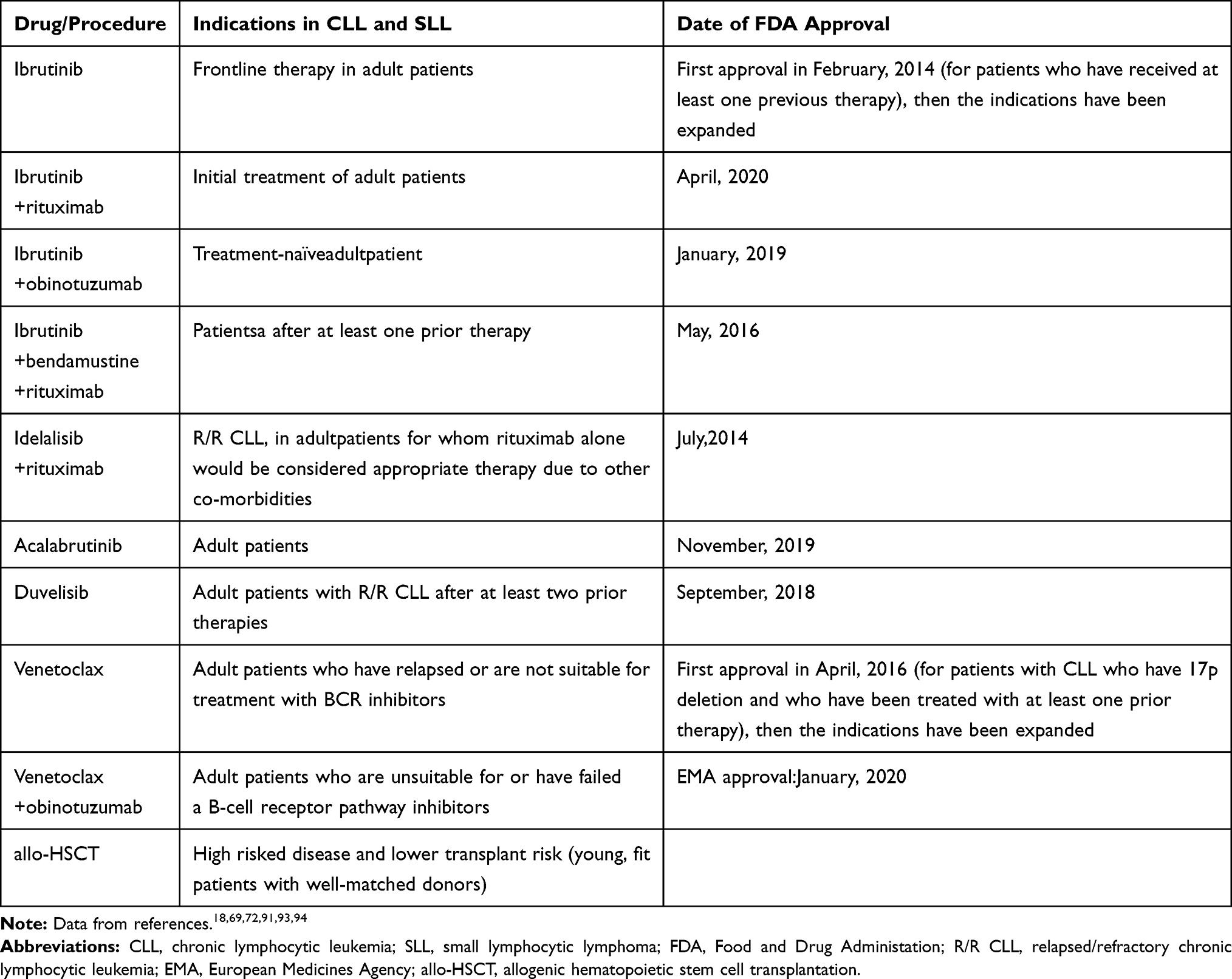 |
Table 2 Current Treatment Options for Patients with CLL with TP53 Disrupted (Outside the Context of Clinical Trials) |
Alemtuzumab
Alemtuzumab is a recombinant, fully humanized, monoclonal antibody against CD52.55 The moderate effectiveness of alemtuzumab in patients with high-risk genetic markers such as del(17p) and TP53 mutations has been proven.5 In a randomized Phase 3 trial (CAM307), de novo patients treated with alemtuzumab demonstrated a 3-fold improvement in overall response rate (64% vs 20%, p = 0.08), compared with patients treated with chlorambucil. The median PFS has also been improved (10.7 months vs 2.2 months, p = 0.41).67 The license of alemtuzumab was withdrawn in 2012 due to a decision by Sanofi. At present alemtuzumab is only available through an international compassionate use program.5 The usage of alemtuzumab in CLL has also been limited due to significant toxicities, most of all high incidence of neutropenia and cytomegalovirus reactivation. Alemtuzumab had been replaced as the treatment of choice for patients with TP53 aberrations, by new agents with better effectiveness.68
Drugs Targeting BCR Signaling
A better insight in CLL pathophysiology, which led to the development of novel targeted approaches, revolutionized the treatment of CLL in TP-53 deficient patients.54 Constitutive BCR signaling is of great significance for the pathogenesis of CLL, as it enhances the proliferation and survival of B lymphocytes.69,70 Upon stimuli of the BCR, protein tyrosine kinases, such as spleen tyrosine kinase (SYK) and LYN kinase become activated. SYK and LYN activate the Bruton tyrosine kinase (BTK) and phosphoinositol-3-kinases (PI3Ks), which stimulate downstream cascades resulting in activation of protein kinase B (AKT), extracellular signal-regulated kinases ERK1 and 2, nuclear factor (NF)-kB, and nuclear factor of activated T-cells (NFAT).69 Inhibition of kinases, involved in BCR signaling became an attractive potentiality in the treatment of CLL.70
Ibrutinib
Ibrutinib is a small molecule BTK inhibitor.4 It binds covalently and irreversibly to cysteine 481 within the ATP binding site. It also cross-reacts with other TEC kinases.4,69 In 2014, on the basis of surprisingly good results of a phase Ib/II study (PCYC-1102), FDA and EMA approved ibrutinib for previously treated CLL patients. The study has been conducted on 85 patients with relapsed or refractory (R/R) CLL. 28 patients had del17p. Patients were given ibrutinib orally, either 420 mg or 840 mg per day. The ORR was 71% in the 420-mg cohort and also 71% in the 840-mg cohort. The 26-month PFS was estimated at 75%, and OS was 83%. Moreover, PFS was elongated, even despite high-risk genomic features, including 17p13.1 deletion. The estimated 26-month PFS was 57% and the OS: 70%. Toxic effects included grade 1 or 2 diarrhea, fatigue, and upper respiratory tract infection.71 A latter analysis, on the extended cohort of PCYC-1102 and additional patients, has been performed by O’Brien et al.72 Continuous treatment with ibrutinib demonstrated efficiency and tolerability. The ORR has been reached in 87% of treatment-naïve (TN) patients and 89% of R/R patients after 5-years follow-up. Treatment with ibrutinib has been proved relatively efficient in patients from high-risked subgroups, including patients with del(17p). The ORR for R/R patients with TP53 aberrations was 79%. The median PFS in patients with del(17p) was 26 months and OS was 57 months, both were the shortest compared with other high-risk groups.
A phase 3 randomized trial, called RESONATE, compared the efficacy of ibrutinib to anti-CD20 antibody ofatumumab in patients with R/R CLL or SLL. 32% of patients in the ibrutinib arm and 33% of patients in the ofatumumab arm had del(17p). In the group treated with ibrutinib, PFS was significantly improved (HR = 0.25, 95% CI: 0.14–0.45). The median duration of PFS was 5.8 months in the ofatumumab group and was not reached in the ibrutinib group. At 6 months follow-up, 83% of patients in the ibrutinib arm with del(17p) were alive with no disease progression compared with 49% in the ofatumumab group.
The results of Farooqui et al73 study, evaluating the efficiency of single-agent treatment with ibrutinib in patients with TP53 aberrations, are also encouraging. 51 patients with active CLL have been enrolled in the study, 47 of which had deletion 17p13.1 and four carried solely a TP53 mutation. 35 patients had not ever been treated and 16 had R/R CLL. An objective response was achieved by 97% of previously untreated patients (95% CI 86–100) and by 80% of patients with R/R CLL (95% CI 52–96). 18% of patients discontinued treatment: 10% due to disease progression and 6% accounted for by patient’s deaths. Richter’s transformation occurred in three patients and prolymphocytic transformation in 2 patients, who discontinued treatment because of disease progression. Median time to transformation was 5.5 months.73 To compare, responses in patients treated with fludarabine, cyclophosphamide, and rituximab were observed in approximately 68% of patients and median PFS was less than 12 months.74 The most frequent adverse events in patients treated with ibrutinib were of grade 1 or 2 and included were arthralgia, diarrhoea, rash, nail ridging, bruising, and muscle spasms or cramps. 24% of patients had grade 3 or worse neutropenia, 14% of patients had anemia, 10% of patients had thrombocytopenia. Cytopenia mostly occurred during the first months of treatment, then improved and were not complicated with infections or bleeding. As regards non-hematological treatment-associated adverse effects, three patients had grade 3 pneumonia, one patient had grade 3 rash.73
Treatment with ibrutinib is generally well tolerated. Common adverse effects include: nausea, diarrhea, fatigue, muscle and bone pain, febrile neutropenia, infections, hypertension, atrial fibrillation and flutter and bleeding problems, especially in patients taking anticoagulants. Some authors claim that in a “real-world setting”, adverse events during therapy with ibrutinib are more common than in clinical trials and the discontinuation of therapy occurs in more than 20% of patients.75 During the treatment with drugs targeting B-cell receptor signaling, including ibrutinib, a transient increase in lymphocytosis in the peripheral blood is frequently observed. Interestingly, lymphocytosis occurs along with the reduction in lymph node and spleen size. It has been proved that BTK inhibitors might mobilize CLL cells from the bone marrow, lymph node, or spleen to peripheral blood.76,77 Burger et al confirmed that treatment with ibrutinib causes CLL cell redistribution from tissues into the blood. After starting ibrutinib therapy, a continuous improvement in bone marrow infiltration by CLL cells was observed. Moreover, authors proved that in patients with unmutated IGHV CLL (which is a poor prognostic factor), the rates of tissue cell death are higher and there is less redistribution after initiation of therapy, as well as faster bone marrow clearance of leukemia cells, compared with patients with mutated IGHV.76 Ponader et al demonstrated in vitro and in vivo in mice that ibrutinib mobilize CLL cells from the tissues. To evaluate the effects of ibrutinib on the number of circulating and tissue-based lymphocytes, an EμTCL1 adoptive transfer mouse model resembling CLL in patients was used. TCL1 leukemia cells were transferred in SCID mice, having led to accelerated disease. Mice were treated with suboptimal (2.5 mg/kg/d) and optimal (25 mg/kg/d) doses of ibrutinib for three weeks. Mice treated with 25 mg/kg/d ibrutinib had significantly smaller livers and spleens with markedly reduced leukemic infiltration, compared with control mice. The crucial mechanism of ibrutinib, leading to the early redistribution of CLL cells from the tissues into the blood is the inhibition of CLL cells chemotaxis toward the chemokines CXCL12 and CXCL13, probably along with inhibition of adhesion molecule function/signaling.77
Lymphocytosis usually normalize while continuing the treatment with ibrutinib. What is important is not to misinterpret elevated lymphocyte counts a sign of progression. For patients meeting the criteria of partial remission except lymphocytosis, the category “partial remission with lymphocytosis” has been established.54
Idelalisib
Idelalisib (previously GS-1101 or CAL-101) is a small-molecule inhibitor of PI3K Delta isoform of the catalytic subunit of PI3K, toward which idelalisib is highly selective, and is expressed in hematopoietic cells, particularly in leukocytes.54,69 In 2014, FDA and EMA approved idelalisib in combination with rituximab for patients with R/R CLL or for those who have CLL and harbour del(17p) or TP53 mutated.69 The decision was a consequence of encouraging results of studies by Furman et al78 and O’Brien et al.79 Furman et al78 evaluated the efficacy and safety of idelalisib in combination with rituximab in comparison with rituximab plus placebo. The study was conducted on 220 patients with relapsed CLL and comorbidities: decreased renal function, myelosuppression, or other major coexisting conditions. Approximately 80% of the patients had unmutated IGHV and more than 40% had 17p deletion or TP53 mutations. The median PFS in patients treated with idelalisib was not reached (HR 0.15; P<0.001) compared with 5.5 months in the placebo group. OR was 81% in the idelalisib group vs 13% in the placebo group (OR 29.92; P<0.001). OS at 12 months was 92% in the idelalisib group vs 80% in the placebo group (p = 0.02). 40% of patients from the idelalisib group and 35% of patients from the placebo group experienced serious adverse effects, the most frequent being: pneumonia, pyrexia, and febrile neutropenia. In 8% of patients in the idelalisib group and 10% in the placebo group adverse events led to treatment discontinuation. Although idelalisib, like ibrutinib, causes lymphocytosis, the addition of rituximab blunted and shortened the duration of the elevation of peripheral lymphocyte counts.78 O’Brien et al79 assessed idelalisib+rituximab as initial therapy in 64 previously untreated elderly patients with CLL or small lymphocytic leukemia. Treatment resulted in durable disease control: the ORR was 97% (100% in 2 patients with del(17p)/TP53 mutations), PFS at 36 months was 83% and OS at 36 months was 90%. All patients had at least one adverse effect and 89.1% experienced grade ≥3 adverse effects. The most common adverse events included diarrhea, rash, pyrexia, nausea, chills, cough and fatigue.79 Idelalisib/ofatumumab combo also proved to be effective in CLL. Median PFS was 16.3 months in the idelalisib plus ofatumumab group (95% CI 13.6–17.8) and 8.0 months (5.7–8.2) in the ofatumumab group (adjusted HR 0.27, 95% CI 0.19–0.39, p<0.0001). In the subgroup with del(17p) OS was 25.8 months in the idelalisib/ofatumumab vs 19.3 months in placebo group. However, in idelalisib+ofatumumab group serious infections, including pneumonia and sepsis, were more common. Out of 261 patients, 22 patients in the idelalisib+ofatumumab group and 6 patients in the placebo group, died due to treatment-related adverse events.80
The most alarming treatment-associated adverse events, fatal and/or severe are diarrhea, colitis, hepatotoxicity, pneumonitis (especially opportunistic Pneumocystis jirovecii pneumonia) and intestinal perforation. Idelalisib should not be used along with other hepatotoxic drugs; strong cytochrome P4503A (CYP3A) inducers, eg rifampicin, and CYP3A substrates, eg midazolam. During idelalisib treatment serum transaminase levels should be assessed on a regular basis and the patient should be monitored for pneumonitis.81
In addition to drugs already widely used in treatment of CLL, like ibrutinib and idelalisib, several second-generation BTK inhibitors and PI3K inhibitors are currently being tested in Phase I and II clinical trials.
Acalabrutinib
Acalabrutinib (ACP-196), a second-generation BTK inhibitor, has been developed to overcome limiting toxicities observed during the treatment with ibrutinib, while reaching similar outcomes in patients with CLL. It could be achieved due to the higher selectivity of acalabrutinib, which limits off-target effects on other kinases such as TEC, EGFR, and ITK. In 2019 the FDA approved acalabrutinib for treatment of TN and relapsed refractory CLL or small lymphocytic lymphoma (SLL).70 The approval was based on the results of two clinical trials: ELEVATE-TN82 and ASCEND.83 In the phase 3 ASCEND trial, conducted on 310 patients with R/R CLL, acalabrutinib monotherapy has been compared to idelalisib+rituximab or bendamustine+rituximab therapies. 16% of patients had del17p. OS at 12 months was 94% for the acalabrutinib group and 91% idelalisib+rituximab or bendamustine+rituximab groups. PFS at 12 months were 88% for acalabrutinib and 68% for idelalisib+rituximab or bendamustine+rituximab. It has been proved that acalabrutinib is better tolerated than thanidelalisib+rituximab. 49% of patients in the idelalisib+rituximab group had to discontinue the treatment compared to 11% of patients treated with acalabrutinib. During the treatment with acalabrutinib, grade ≥3 adverse events included neutropenia (16%), anemia (12%) and pneumonia (5%). Other adverse effects were: headache and cough.83 In phase 3 ELEVATE-TN study82 535 treatment naïve patients with CLL were treated either with acalabrutinib alone or with obinutuzumab or obinutuzumab+chlorambucil. At 28 month follow up, PFS was prolonged in acalabrutinib + obinutuzumab group as well as in in acalabrutinib in monotherapy group vs obinutuzumab+chlorambucil. 30-months PFS rates were estimated 90%, 82%, and 34% respectively, with acalabrutinib+obinutuzumab, acalabrutinib in monotherapy, and obinutuzumab+chlorambucil. PFS improvement with acalabrutinib+obinutuzumab or acalabrutinib in comparison with obinutuzumab+chlorambucil was also observed in the del(17p) subgroup (HR [95% CI]; 0.13 [0.04–0.46]; 0.20 [0.06–0.64]). Median OS has not been reached in any group. The ORR for acalabrutinib+obinutuzumab, for acalabrutinib in monotherapy and for obinutuzumab+chlorambucil was 94%, 85%, and 79%, respectively. Adverse events in acalabrutinib - containing schemes included: headache, diarrhea, neutropenia, atrial fibrillation, bleeding events, hypertension. Adverse events were the reason of treatment discontinuation in 11% of patients on acalabrutinib+obinutuzumab, 9% of patients on acalabrutinib, and 14% of patients on obinutuzumab+chlorambucil.82
Zanubrutinib
Zanubrutinib (BGB-3111) is another second-generation, irreversible BTK inhibitor.70 It has greater selectivity compared with ibrutinib and what seems even more important it has a longer half-life in comparison with acalabrutinib and ibrutinib, which results in longer exposure of CLL cells to BTK inhibition.84 In November, 2019 zanubrutinib received approval for treatment of R/R mantle cell lymphoma.85 Studies evaluating the drug’s efficacy and toxicities in CLL are ongoing. In the Phase 1 study, ORR was 96.2% (95% CI, 89.2–99.2), at a median follow-up of 13.7 months. ORR in all 16 patients with del(17p) or TP53 mutation was 100% (95% CI, 79.4–100).86
Duvelisib
Duvelisib (IPI-145) also appears an interesting alternative for other drugs targeting BCR signaling. It is expected to have better efficiency as it inhibits both p110d and p110g isoforms of PI3K.69 Duvelisib demonstrated better efficiency than ofatumumab in a phase 3 clinical trial conducted on 160 R/R CLL patients. ORR in the duvelisib group was 74% vs 45% with ofatumumab. PFS in duvelisib group was of 13.3 months vs 9.9 months in ofatumumab group. Duvelisib was also superior to ofatumumab in p53-deficient patients. Their median PFS was 12.7 months vs 9 months for patients treated with ofatumumab.87 In 2018, duvelisib has been approved in the treatment of R/R CLL.
New BCR Signaling Inhibitors Under Investigation
Results from clinical trials of multiple drugs targeting BCR signaling are eagerly awaited. New PI3K inhibitors and new BTK inhibitors, especially worth tracking include: umbralisib (TGR-1202), acalisib (GS-9820), copanlisib (BAY 80–6946), bimiralisib (PQR309), tirabrutinib (ONO/GS-4059), vecabrutinib (SNS-062), and fenebrutinib (GDC-0853).54,69 Currently, the data considering the effectiveness of most of these drugs in patients with TP53 mutated is scant.
Apart from BTK inhibitors and PI3K inhibitors, SYK (spleen tyrosine kinase) inhibitors is another promising class of drugs that inhibit BCR signaling.69 Entospletinib (GS-9973), selective inhibitor of SYK has been evaluated in combination with idelelisib in Phase 2 study, conducted on patients with R/R CLL.88 Although it turned out to be effective (ORR was 60%), the study had to be terminated early due to treatment-emergent pneumonitis in 18% of patients (severe in 11 of 12 cases, 2 fatalities).88 Entospletinib and idelalisib combo probably caused excessive mTOR inhibition, which led to immune cell recruitment, T helper 1–type response and consequently, noninfectious pneumonitis. It is suggested, that isolated SYK inhibition may not lead to similar inflammatory effects.69 The trial investigating entospletinib and obinutuzumab combination is ongoing, no results have been posted yet.89 Cerdulatinib (PRT062070), a dual inhibitor of SYK and JAK1/3 and TAK-659, a dual SYK and FLT3 inhibitor are in phase 2a and 1 clinical trials, respectively.88
Inhibiting Bcl-2 with BH3-Mimetics
In numerous studies it has been reported that CLL cells overexpress antiapoptotic protein Bcl2, which results in clonal lymphocytes accumulation that characterize CLL.18,69,90 Based on this observation, BH3-mimetic drugs, a new class of agents triggering cancer cells apoptosis, have been developed.18,69
Venetoclax
Venetoclax (ABT-199) is a highly BCL-2-selective, orally available BH3-mimetic.69,84 In 2016, it was approved by the EMA as a monotherapy for CLL patients with del(17p) or TP53 mutation who have relapsed or are not suitable for treatment with BCR inhibitors, and also for patients without del (17p) or TP53 mutation and who are refractory to chemoimmunotherapy and BCR inhibitors. The decision followed the encouraging results of phase 2 M13-98291 and M14-03292 trials in R/RCLL.69 Phase 2 M13-982 study91 has been conducted on 107 patients with del(17p) from USA, Canada, UK, Germany, Poland, and Australia. At a median follow-up of 12.1 months OR was achieved in 79.4% of patients (95% CI 70.5–86.6). Serious adverse events occurred in 55% of patients and included: pyrexia, autoimmune haemolytic anaemia, pneumonia and febrile neutropenia. Neutropenia, infection, anemia and thrombocytopenia were the most common grade 3–4 adverse events. Seven patients died due to disease progression and four from adverse events, none considered as treatment associated.91
In 2019, venetoclax in combination with obinutuzumab has been approved for TN patients with CLL and SLL.93 The decision was based on the outcome of a CLL14 (NCT02242942) trial of venetoclax+obinutuzumab vs obinutuzumab+chlorambucil in 432 patients with TN CLL with comorbidities. In total, 13.8% of patients had TP53 deletion, mutation, or both. venetoclax+obinutuzumab was associated with longer PFS at 24 months than chlorambucil–obinutuzumab 88.2% (95% CI, 83.7 to 92.6) vs 64.1% (95% CI, 57.4 to 70.8). This benefit was also observed in patients with TP53 deletion, mutation, or both and in patients with unmutated IGHV. Adverse events occurred in 94.3% patients treated with venetoclax–obinutuzumab and in 99.5% of patients in the chlorambucil–obinutuzumab group. The treatment had to be discontinued due to adverse events in 16.0% and 15.4% of patients, respectively. The most frequent adverse effect of grade 3 or 4 were neutropenia, occurring in 52.8% of patients in the venetoclax–obinutuzumab group and in 48.1% of patients in the chlorambucil–obinutuzumab group and infections occurring in 17.5% and 15.0%, respectively. 9.3% of patients died in the venetoclax–obinutuzumab group and 7.9% died in the chlorambucil–obinutuzumab group.94 Seymur et al95 revealed the results of randomized, open-label, phase 3 trial in which 389 patients with R/R CLL treated with venetoclax+rituximab or bendamustine+rituximab took part (MURANO study). After a median follow-up of 23.8 months, PFS was significantly higher in the venetoclax+rituximab group than in the bendamustine+rituximab group. The 2-year rates of PFS were 84.9% and 36.3%, respectively (HR for progression or death, 0.17; 95% confidence interval [CI], 0.11–0.25; P<0.001). The benefit was also observed in patients with TP53 mutated. In the subgroup of patients with chromosome 17p deletion, the 2-year rate of PFS was 81.5% in the venetoclax+rituximab group compared to 27.8% in the bendamustine+rituximab group (HR 0.13; 95% CI, 0.05 to 0.29). The most frequently observed adverse effects grade 3 or 4 was neutropenia in both groups, with higher prevalence in the venetoclax+rituximab group (67% vs 55.3% respectively).95 In 4-year follow-up Kater et al96 reported the long-term outcomes and the predictive value of molecular and genetic characteristics. Four-year PFS and OS were higher in venetoclax+rituximab group compared to the bendamustine+rituximab group. Undetectable MRD at the end of therapy was associated with higher PFS compared with both low and high MRD positivity. In patients with mutated TP53, NOTCH1, XPO1 and BRAF, treated with venetoclax+rituximab, lower undetectable MRD rates were reported.96 Roberts et al97 also evaluated the efficacy of venetoclax and identified the treatment effect modifiers in patients with CLL and SLL. Objective responses were achieved in 75% of all patients, including 22% complete remissions. Chromosome 17p deletion and/or TP53 mutation and NOTCH1 mutation were consistently associated with shorter duration of response, but not the probability of response.97
Levin et al,98 in HOVON 141/VISION trial, are exploring the effects of the combination treatment with ibrutinib+venetoclax in RR/CLL. After a short induction with ibrutinib, patients with RR/CLL are treated with 12 cycles of venetoclax. Those who reach undetectable MRD after 12 cycles are randomised to continue ibrutinib or treatment discontinuation. PFS after 12 months follow-up is to be the primary endpoint of the study, which has not been completed yet.98
What needs to be remembered during managing CLL patients with venetoclax is the possibility of tumor lysis syndrome (TLS).54 Roberts et al.18 and Seymour et al99 reported cases of TLS with fatal outcome during therapy with venetoclax. Before starting the therapy, to prevent TLS, a risk assessment for the development of this adverse effect should be performed, based on the patient’s renal function and tumour cell load. A stepwise ramp-up of dosage should be introduced (20 mg for a start, then weekly increase to a final dose of 400 mg). Patients should be closely monitored during the treatment in search of TLS.54
Chimeric Antigen Receptor—T Cell Therapy in the Treatment of CLL
Chimeric antigen receptor—T cell therapy (CAR-T) is a very recent treatment in CLL, attracting more and more attention.4 Genetically modified T cells express chimeric antigen receptor (CAR) targeting specific antigen, which is, in CLL, CD19.54,100 Preclinical and clinical trials prove that CAR-T therapy is a promising treatment in R/R CLL. Turtle et al101 evaluated the safety and effectiveness of CAR-T cell therapy in patients with CLL, previously treated with ibrutinib. 23 out of 24 patients had either complex karyotype or del(17p). Four weeks after CAR-T cell infusion, ORR was 71%. 83% of patients developed cytokine release syndrome, 33% developed neurotoxicity which was the reason of one fatal outcome. In patients who achieved the clinical response, 88% had no minimal residual disease (MRD) evaluated by flow cytometry, and 58% had no detectable disease evaluated by deep IGH sequencing. Then, patients who respond to CAR-T therapy may achieve a deep remission without MRD.101
Common adverse events of CAR-T cell therapy in CLL include cytokine release syndrome (CRS), B cell aplasia, neurotoxicity, and infections.100 CRS occurs due to the sudden activation of CAR-T cells, after encountering target cells. The cytokines released include most of all IL-6, IL-10, TNF-α, and IFN-γ. Inflammatory factors can be also produced by macrophages, monocytes, natural killer cells, and dendritic cells in the response for stimulation of CAR-T cells, target cells, and inflammatory mediators.100 CRS occurs in nearly 83% of CLL patients, with a higher frequency in patients who were previously treated with ibrutinib.100 Symptoms of CRS range from fever and hypoxia to renal failure and, rarely, death. Treatment with tocilizumab (IL-6 receptor antagonist) and corticosteroids can be a CRS management strategy.102 The limitations of CAR-T consist the complexity of the approach, expenses and toxicities.4
Allogenic Stem-Cell Transplantation in High Risk CLL Patients
Despite novel treatment possibilities, allogenic stem-cell transplantation (alloPBSCT) still remains an option for patients with CLL.54 However, the cohort of patients that should be qualified to the procedure seem to change. Therapy with novel drugs gives a chance of durable remission in patients with del(17), so one must be careful not to overweight the risks of alloPBSCT over the benefits of therapy with new agents. OS at 6 years from alloPBSCT is 50–60%. Treatment related mortality in CLL was estimated 16–25% and graft vs host disease was observed in 50–55% of patients. As alloPBSCT is the only treatment that offers a full recovery, it could be relevant for young, high-risk patients, without comorbidities, with a perfectly matched donor.54,84
Resistance to Novel Therapies
Although novel agents, such as ibrutinib, idelalisib and venetoclax, are significantly efficient in CLL, some patients develop resistance to these drugs and progress.103 Sedlarikova et al103 provided an overview of the acquired resistance to novel agents. 80% of patients who failed on ibrutinib, had acquired mutations in BTK and PLCG2 genes. In idelalisib failure, no resistance-associated mutations or deregulated signalling were reported. In patients who had failed on venetoclax, acquired mutations in the BCL2 gene were found. Resistance-associated mutations in patients who have progressed on ibrutinib and venetoclax are often presented at low allelic frequencies.103 One of the studies analysed by Sedlarikova et al was by Woyach et al,104 who performed the whole-exome sequencing of six patients with R/R CLL, previously treated with ibrutinib. In 5 patients acysteine-to-serine (C481S) mutation in BTK at the binding site of ibrutinib was found and in 3 patients mutations in PLCγ2 were identified.104 Acysteine-to-serine mutation of BTK results in solely transient inhibition of BTK, which results in poor therapeutic outcome. Mutations in PLCγ2 possibly lead to autonomous BCR activity, due to gain-of-function.71 Although acquired resistance to ibrutinib occurs in a small proportion of patients, patients with del(17p13.1), del(11q22.3) or a complex karyotype seem to be at greater risk of relapse. Gángó et al105 reported that in patients with CLL treated with ibrutinib, with persisting TP53 mutated subclones, no BTK mutations were detected. Conversely, an association between reduction or elimination of TP53 mutated clones and the presence of BTK mutations has been proved. The authors hypothesize that the treatment with ibrutinib may create conditions, in which the survival of subclones with BTK mutations is facilitated, by eliminating subclones with TP53 mutations or that the elimination of TP53 subclones allow the expansion of subclones harboring BTK mutations.103,105 As the mutations in BTK, PLCG2 and BCL2 leading to the treatment failure, are possible to detect a few months before clinical relapse, in patients receiving long-term treatment with BCR and BCL-2 inhibitors, regular genetic testing should be performed, preferably using highly sensitive ultradeep NGS approaches.103
New Perspectives in Treatment of CLL with del(17p) and/or TP53 Mutation – A Straight Way from Biology to Therapy
Improvements in understanding the biology of CLL keeps resulting in the development of new treatment strategies, which are being evaluated in pre-clinical trials. Here we present a few of the approaches that, in our opinion, seem most promising.
Boudny at al41 hypothesized that checkpoint kinase 1 (CHK1), which takes part in DNA replication and repair, could be a suitable target in the treatment of CLL. CHK1 is an important component of the DNA damage response (DDR) pathway, which serves as an anti-cancer barrier.106 The newly developed agent MU380 is a selective inhibitor of CHK1. In CLL-derived cell lines: TP53-mut MEC-1 and MEC-2, but also in TP53-wild type OSU-CLL, MU380 altered cell cycle and induced apoptosis. MU380 reduced the viability of CLL cells, both unstimulated (even though their CHK1 level is low) and induced to proliferate by anti-CD40/IL-4 stimuli. Effects were comparable in samples harbouring p53 pathway dysfunction and samples without such alterations. Only a few samples were resistant to the inhibitor, only 5 of 96 showed viability ≥ 80% after 72 h treatment. It is worth adding that non-cancerous cells were much less sensitive to MU380. Thus, in the future MU380 may represent an attractive option for high-risk CLL patients.41
Another potential target in the therapy of CLL is USP7-PTEN network. USP7 is an de-ubiquitinase, that is over-expressed in about 70% of CLL CD19+ cells, both at the mRNA and protein levels. It is of great significance in CLL pathogenesis, as it inactivates three major tumor suppressors: p53, PTEN and FoxO.106 Carra et al107 demonstrated that in CLL cell lines USP7 can be effectively targeted by the USP7 inhibitor P5091, which results in reactivation of tumour suppressive functions and hence promotes the induction of cell growth arrest and apoptosis. These effects were observed both in TP53-wild type and TP53-null environment. It is very probable that, P5091 effects are p53-independent, as P5091 promoted apoptosis in TP53-mutated MEC-1 cells.107
One of the features of CLL cells is their high intrinsic reactive oxygen species (ROS) stress, which is even higher when p53 function is lost.108 Liu et al108 proved that CLL cells with loss of p53 are sensitive to ROS-mediated cell killing, even when CLL cells are under the protection of bone marrow stromal cells.103 The ROS-modulating factor used by the authors was phenethyl isothiocyanate (PEITC), a natural substance, that can be found in cruciferous vegetables.108,109 In mouse model, the usage of PEITC resulted in massive cell death in primary 17p-CLL cells and p53-null leukemia cells. PEITC at a concentration of 5 µM killed 17p-CLL cells, with only 17% viable cells remained at 24 h after drug incubation. 17p-CLL cells were resistant to standard anti-CLL agents such as F-ara-A and Oxaliplatin. Survival 48 h after treatment with F-ara-A and Oxaliplatin were 53% and 42% respectively. The overall survival in mice treated with PEITC was 7.8 months vs 3.5 months for untreated mice.108
Ciardullo et al110 examined the effect of the second-generation MDM2 inhibitor, RG7388, on the induction of p53-transcriptional targets in CLL cells and normal cells. MDM2 is an ubiquitin ligase, which controls the levels and activity of p53 via ubiquitin-dependent proteasomal degradation. RG7388 is a non-genotoxic compound, that binds to MDM2 in the p53-binding pocket and can release p53, leading to effective stabilization of the p53 pathway. Ciardullo et al110 proved that RG7388 induces a pro-apoptotic p53 gene signature in CLL, but not in normal blood and bone marrow cells.110
Conclusions
In recent years we witnessed novel and smarter therapies revolutionise the treatment of del(17p) and/or aberrant TP53 CLL. As a consequence of the wide availability of ibrutinib, idelalisib, venetoclax and other targeted therapies, nowadays the prognosis of those patients has improved significantly.68 However, TP53 aberrations are still the most important single factors of poor prognosis in CLL.12 That is due to the early progression of the disease in patients with TP53 mutations, high risk of Richter transformation, adverse effects mandating treatment discontinuation, acquired resistance to BTK inhibitors. Hence, the development of alternative therapeutic approaches still remains an issue of critical importance.101 Efforts should be focused both on the improvement of the already approved approaches, but also on the development of new strategies. CAR-T cells therapy is one of the most promising advances, that, wide spread, could be another milestone in the treatment of CLL. New technologies, combined with ever-increasing understanding of CLL pathogenesis justify our hopes for new therapies for TP53-mutated CLL patients.
Funding
This study was funded by the research grant of Medical University of Lublin [DS176 and DS 175].
Disclosure
The authors report no conflicts of interest or competing financial interests in this work.
References
1. Parikh SA. Chronic lymphocytic leukemia treatment algorithm 2018. Blood Cancer J. 2018;8:93. doi:10.1038/s41408-018-0131-2
2. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–2390. doi:10.1182/blood-2016-01-643569
3. Rozman C, Montserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333:1052–1057. doi:10.1056/NEJM199510193331606
4. Rai KR, Jain P. Chronic lymphocytic leukemia (CLL) - then and now. Am J Hematol. 2016;91:330–340. doi:10.1002/ajh.24282
5. Hallek M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2017;92:946–965. doi:10.1002/ajh.24826
6. Filip AA, Grenda A, Popek S, et al. Expression of circulating miRNAs associated with lymphocyte differentiation and activation in CLL-another piece in the puzzle. Ann Hematol. 2017;96(1):33–50. doi:10.1007/s00277-016-2840-6
7. Mauro FR, Foa R, Giannarelli D, et al. Clinical characteristics and outcome of young chronic lymphocytic leukemia patients: a single institution study of 204 cases. Blood. 1999;94:448–454. doi:10.1182/blood.V94.2.448
8. Kipps TJ, Stevenson FK, Wu CJ, et al. Chronic lymphocytic leukaemia. Nat Rev Dis Prim. 2017;3:17008. doi:10.1038/nrdp.2017.8
9. Pan JW, Cook LS, Schwartz SM, Weis NS. Incidence of leukemia in Asian migrants to the United States and their descendants. Cancer Causes Control. 2002;13:791–795. doi:10.1023/A:1020608328969
10. Nabhan C, Aschebrook-Kilfoy B, Chiu BC, et al. The impact of race, ethnicity, age and sex on clinical outcome in chronic lymphocytic leukemia: a comprehensive surveillance, epidemiology, and end results analysis in the modern era. Leuk Lymphoma. 2014;55:2778–2784. doi:10.3109/10428194.2014.898758
11. Eichhorst B, Robak T, Montserrat E, et al.; ESMO Guidelines Committee. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26:v50–54. doi:10.1093/annonc/mdv303
12. Eichhorst B, Hallek M. Prognostication of chronic lymphocytic Leukemia in the era of new agents. Hematology Am Soc Hematol Educ Program. 2016;2016:149–155. doi:10.1182/asheducation-2016.1.149
13. Gaidano G, Rossi D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematology Am Soc Hematol Educ Program. 2017;2017:329–337. doi:10.1182/asheducation-2017.1.329
14. Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi:10.1038/nature15395
15. Puente XS, Beà S, Valdés-Mas R, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–524. doi:10.1038/nature14666
16. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–1916. doi:10.1056/NEJM200012283432602
17. Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi:10.1016/j.ccr.2009.11.019
18. Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–322. doi:10.1056/NEJMoa1513257
19. Rossi D, Rasi S, Spina V, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013;121:1403–1412. doi:10.1182/blood-2012-09-458265
20. Guarini A, Marinelli M, Tavolaro S, et al. ATM gene alterations in chronic lymphocytic leukemia patients induce a distinct gene expression profile and predict disease progression. Haematologica. 2012;97:47–55. doi:10.3324/haematol.2011.049270
21. Rossi D, Gaidano G. The clinical implications of gene mutations in chronic lymphocytic leukaemia. Br J Cancer. 2016;114:849–854. doi:10.1038/bjc.2016.78
22. Seiffert M, Dietrich S, Jethwa A, Glimm H, Lichter P, Zenz T. Exploiting biological diversity and genomic aberrations in chronic lymphocytic leukemia. Leuk Lymphoma. 2012;53:1023–1031. doi:10.3109/10428194.2011.631638
23. Rossi D, Cerri M, Capello D, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2008;142:202–215. doi:10.1111/j.1365-2141.2008.07166.x
24. Autore F, Strati P, Laurenti L, Ferrajoli A. Morphological, immunophenotypic, and genetic features of chronic lymphocytic leukemia with trisomy 12: a comprehensive review. Haematologica. 2018;106:931–938. doi:10.3324/haematol.2017.186684
25. Campo E, Cymbalista F, Ghia P, et al. TP53 aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica. 2018;103:1956–1968. doi:10.3324/haematol.2018.187583
26. Condoluci A, Terzi Di Bergamo L, Langerbeins P. International prognostic score for asymptomatic early-stage chronic lymphocytic leukemia. Blood. 2020;135(21):1859–1869. doi:10.1182/blood.2019003453
27. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated IgV(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–1854. doi:10.1182/blood.V94.6.1848
28. Brachtl G, Piñón Hofbauer J, Greil R, Hartmann TN. The pathogenic relevance of the prognostic markers CD38 and CD49d in chronic lymphocytic leukemia. Ann Hematol. 2014;93:361–374. doi:10.1007/s00277-013-1967-y
29. D’Arena G, Tarnani M, Rumi C, et al. Prognostic significance of combined analysis of ZAP-70 and CD38 in chronic lymphocytic leukemia. Am J Hematol. 2007;82:787–791. doi:10.1002/ajh.20936
30. Malavasi F, Deaglio S, Funaro A, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841–886. doi:10.1152/physrev.00035.2007
31. Kumagai M, Coustan-Smith E, Murray DJ, et al. Ligation of CD38 suppresses human B lymphopoiesis. J Exp Med. 1995;181:1101–1110. doi:10.1084/jem.181.3.1101
32. Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–1847. doi:10.1182/blood.V94.6.1840
33. Dürig J, Naschar M, Schmücker U, et al. CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia. 2002;16:30–35. doi:10.1038/sj.leu.2402339
34. Krober A, Seiler T, Benner A, et al. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood. 2002;100:1410–1416. doi:10.1182/blood.V100.4.1410.h81602001410_1410_1416
35. Rose DM, Han J, Ginsberg MH. Alpha4 integrins and the immune response. Immunol Rev. 2002;186:118–124. doi:10.1034/j.1600-065X.2002.18611.x
36. Rettig MP, Ansstas G, DiPersio JF. Mobilization ofhematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia. 2012;26:34–53.
37. Gattei V, Bulian P, Del Principe MI, et al. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood. 2008;111:865–873. doi:10.1182/blood-2007-05-092486
38. Scielzo C, Camporeale A, Geuna M, et al. ZAP-70 is expressed by normal and malignant human B-cell subsets of different maturational stage. Leukemia. 2006;20:689–695. doi:10.1038/sj.leu.2404138
39. Wang YH, Fan L, Xu W, Li JY. Detection methods of ZAP-70 in chronic lymphocytic leukemia. Clin Exp Med. 2012;12:69–77. doi:10.1007/s10238-011-0148-3
40. Shanafelt TD. Predicting clinical outcome in CLL: how and why. Hematology Am Soc Hematol Educ Program. 2009;421–429. doi:10.1182/asheducation-2009.1.421
41. Boudny M, Zemanova J, Khirsariya P, et al. Novel CHK1 inhibitor MU380 exhibits significant single-agent activity in TP53-mutated chronic lymphocytic leukemia cells. Haematologica. 2019;104:2443–2455. doi:10.3324/haematol.2018.203430
42. Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119:521–529. doi:10.1182/blood-2011-09-379966
43. Cortese D, Sutton LA, Cahill N, et al. On the way towards a ‘CLL prognostic index’: focus on TP53, BIRC3, SF3B1, NOTCH1 and MYD88 in a population-based cohort. Leukemia. 2014;28:710–713. doi:10.1038/leu.2013.333
44. Tausch E, Beck P, Schlenk RF, et al. NOTCH1 mutation and treatment outcome in CLL patients treated with chlorambucil (Chl) or ofatumumab-Chl (O-Chl): results from the Phase III study complement 1 (OMB110911). Blood. 2013;122:527.
45. Pozzo F, Bittolo T, Arruga F, et al. NOTCH1 mutations. associate with low CD20 level in chronic lymphocytic leukemia: evidence for a NOTCH1 mutation- driven epigenetic dysregulation. Leukemi. 2015;30:182–189. doi:10.1038/leu.2015.182
46. Amin NA, Malek SN. Gene mutations in chronic lymphocytic leukemia. Semin Oncol. 2016;43:215–221. doi:10.1053/j.seminoncol.2016.02.002
47. Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=TP53.
48. Kastenhuber E, Lowe S. Putting p53 in context. Cell. 2017;170:1062–1078. doi:10.1016/j.cell.2017.08.028
49. Minervini CF, Cumbo C, Orsini P, et al. TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn Pathol. 2016;11:1–9. doi:10.1186/s13000-016-0550-y
50. Guièze R, Pages M, Véronèse L, et al. Telomere status in chronic lymphocytic leukemia with TP53 disruption. Oncotarget. 2016;7:56976–56985. doi:10.18632/oncotarget.10927
51. White E. Autophagy and p53. Cold Spring Harb Perspect Med. 2016;6(4):a026120. doi:10.1101/cshperspect.a026120
52. Dai CQ, Luo TT, Luo SC, et al. p53 and mitochondrial dysfunction: novel insight of neurodegenerative diseases. J Bioenerg Biomembr. 2016;48(4):337–347. doi:10.1007/s10863-016-9669-5
53. Leroy B, Ballinger ML, Baran-Marszak F, et al. Recommended guidelines for validation, quality control, and reporting of TP53 variants in clinical practice. Cancer Res. 2017;77:1250–1260. doi:10.1158/0008-5472.CAN-16-2179
54. Edelmann J, Gribben JG. Managing patients with TP53-deficient chronic lymphocytic leukemia. J Oncol Pract. 2017;13:371–377. doi:10.1200/JOP.2017.023291
55. Zenz T, Vollmer D, Trbusek M, et al.; European Research Initiative on CLL (ERIC). TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia. 2010;24:2072–2079. doi:10.1038/leu.2010.208
56. Shlush LI, Hershkovitz D. Clonal evolution models of tumor heterogeneity. Am Soc Clin Oncol Educ Book. 2015;e662–5. doi:10.14694/EdBook_AM.2015.35.e662
57. Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123(14):2139–2147. doi:10.1182/blood-2013-11-539726
58. Pospisilova S, Gonzalez D, Malcikova J, et al.; European Research Initiative on CLL (ERIC). TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia. 2012;26:1458–1461. doi:10.1038/leu.2012.25
59. Hallek M, Cheson BD, Catovsky D, et al.; International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–5456. doi:10.1182/blood-2007-06-093906
60. Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123:3247–3255. doi:10.1182/blood-2014-01-546150
61. Parikh SA, Strati P, Tsang M, West CP, Shanafelt TD. Should IGHV status and FISH testing be performed in all CLL patients at diagnosis? A systematic review and meta-analysis. Blood. 2016;127:1752–1761. doi:10.1182/blood-2015-10-620864
62. Richter MN. Generalized reticular cell sarcoma of lymph nodes associated with lymphatic leukemia. Am J Pathol. 1928;4:285–292.
63. Wang Y, Tschautscher MA, Rabe KG, et al. Clinical characteristics and outcomes of Richter transformation: experience of 204 patients from a single center. Haematologica. 2020;105:765–773. doi:10.3324/haematol.2019.224121
64. Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210:2273–2288. doi:10.1084/jem.20131448
65. Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131:2761–2772. doi:10.1182/blood-2018-01-791376
66. Rossi D, Gaidano G. Richter syndrome: pathogenesis and management. Semin Oncol. 2016;43:311–319. doi:10.1053/j.seminoncol.2016.02.012
67. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol. 2007;25:5616–5623. doi:10.1200/JCO.2007.12.9098
68. Rafei H, Kharfan-Dabaja MA. Treatment of Del17p and/or aberrant TP53 chronic lymphocytic leukemia in the era of novel therapies. Hematol Oncol Stem Cell Ther. 2018;11:1–12. doi:10.1016/j.hemonc.2017.04.002
69. Yosifov D, Wolf C, Stilgenbauer S, Mertens D. From biology to therapy: the CLL success story. HemaSphere. 2019;3:e175. doi:10.1097/HS9.0000000000000175
70. Isaac K, Mato AR. Acalabrutinib and its therapeutic potential in the treatment of chronic lymphocytic leukemia: a short review on emerging data. Cancer Manag Res. 2020;12:2079–2085. doi:10.2147/CMAR.S219570
71. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi:10.1056/NEJMoa1215637
72. O’Brien S, Furman RR, Coutre S, et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood. 2018;131:1910–1919. doi:10.1182/blood-2017-10-810044
73. Farooqui MZ, Valdez J, Martyr S, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16:169–176. doi:10.1016/S1470-2045(14)71182-9
74. Hallek M, Fischer K, Fingerle-Rowson G, et al.; International Group of Investigators; German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–1174. doi:10.1016/S0140-6736(10)61381-5
75. Mato AR, Nabhan C, Thompson MC, et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: a real-world analysis. Haematologica. 2018;103(5):874–879. doi:10.3324/haematol.2017.182907
76. Burger JA, Li KW, Keating MJ, et al. Leukemia cell proliferation and death in chronic lymphocytic leukemia patients on therapy with the BTK inhibitor ibrutinib. JCI Insight. 2017;2(2):e89904. doi:10.1172/jci.insight.89904
77. Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–1189. doi:10.1182/blood-2011-10-386417
78. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007. doi:10.1056/NEJMoa1315226
79. O’Brien SM, Lamanna N, Kipps TJ, et al. A phase 2 study of idelalisib plus rituximab in treatment-naive older patients with chronic lymphocytic leukemia. Blood. 2015;126:2686–2694. doi:10.1182/blood-2015-03-630947
80. Jones JA, Robak T, Brown JR, et al. Efficacy and safety of idelalisib in combination with ofatumumab for previously treated chronic lymphocytic leukaemia: an open-label, randomised phase 3 trial. Lancet Haematol. 2017;4:e114–e126. doi:10.1016/S2352-3026(17)30019-4
81. Coutré SE, Barrientos JC, Brown JR, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma. 2015;56:2779–2786. doi:10.3109/10428194.2015.1022770
82. Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395:1278–1291. doi:10.1016/S0140-6736(20)30262-2
83. Ghia P, Pluta A, Wach M, et al. ASCEND: Phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2020;38:2849–2861. doi:10.1200/JCO.19.03355
84. Aitken MJL, Lee HJ, Post SM. Emerging treatment options for patients with p53-pathway-deficient CLL. Ther Adv Hematol. 2019;10:2040620719891356. doi:10.1177/2040620719891356
85. Syed YY. Zanubrutinib: first approval. Drugs. 2020;80:91–97. doi:10.1007/s40265-019-01252-4
86. Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134:851–859. doi:10.1182/blood.2019001160
87. Flinn IW, Hillmen P, Montillo M, et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood. 2018;132:2446–2455. doi:10.1182/blood-2018-05-850461
88. Barr PM, Saylors GB, Spurgeon SE, et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood. 2016;127:2411–2415. doi:10.1182/blood-2015-12-683516
89. Available from: https://clinicaltrials.gov/ct2/show/NCT03010358.
90. Hanada M, Delia D, Aiello A, Stadtmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood. 1993;82:1820–1828. doi:10.1182/blood.V82.6.1820.1820
91. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768–778. doi:10.1016/S1470-2045(16)30019-5
92. Jones JA, Mato AR, Wierda WG, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19:65–75. doi:10.1016/S1470-2045(17)30909-9
93. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-venetoclax-cll-and-sll.
94. Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380:2225–2236. doi:10.1056/NEJMoa1815281
95. Seymour JF, Kipps TJ, Eichhorst B. Venetoclax–rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107–1120. doi:10.1056/NEJMoa1713976
96. Kater AP, Wu JQ, Kipps T, et al. Venetoclax plus rituximab in relapsed chronic lymphocytic leukemia: 4-year results and evaluation of impact of genomic complexity and gene mutations from the MURANO Phase III study. J Clin Oncol. 2020;28:JCO2000948.
97. Roberts AW, Ma S, Kipps TJ, et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood. 2019;134(2):111–122. doi:10.1182/blood.2018882555
98. Levin MD, Kater A, Mattsson M, et al. Protocol description of the HOVON 141/VISION trial: a prospective, multicentre, randomised Phase II trial of ibrutinib plus venetoclax in patients with creatinine clearance ≥30 mL/min who have relapsed or refractory chronic lymphocytic leukaemia (RR-CLL) with or without TP53 aberrations. BMJ Open. 2020;10(10):e039168. doi:10.1136/bmjopen-2020-039168
99. Seymour JF, Ma S, Brander DM, et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol. 2017;18:230–240. doi:10.1016/S1470-2045(17)30012-8
100. Zou Y, Xu W, Li J. Chimeric antigen receptor-modified T cell therapy in chronic lymphocytic leukemia. J Hematol Oncol. 2018;11:130. doi:10.1186/s13045-018-0676-3
101. Turtle CJ, Hay KA, Hanafi LA, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of Ibrutinib. J Clin Oncol. 2017;35:3010–3020. doi:10.1200/JCO.2017.72.8519
102. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi:10.1182/blood-2014-05-552729
103. Sedlarikova L, Petrackova A, Papajik T, et al. Resistance-associated mutations in chronic lymphocytic leukemia patients treated with novel agents. Front Oncol. 2020;10:894. doi:10.3389/fonc.2020.00894
104. Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–2294. doi:10.1056/NEJMoa1400029
105. Gángó A, Alpár D, Galik B, et al. Dissection of subclonal evolution by temporal mutation profiling in chronic lymphocytic leukemia patients treated with ibrutinib. Int J Cancer. 2020;146:85–93. doi:10.1002/ijc.32502
106. Bartkova J, Horejsí Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi:10.1038/nature03482
107. Carrà G, Panuzzo C, Torti D, et al. Therapeutic inhibition of USP7-PTEN network in chronic lymphocytic leukemia: a strategy to overcome TP53 mutated/deleted clones. Oncotarget. 2017;8:35508–35522. doi:10.18632/oncotarget.16348
108. Liu J, Chen G, Pelicano H, et al. Targeting p53-deficient chronic lymphocytic leukemia cells in vitro and in vivo by ROS-mediated mechanism. Oncotarget. 2016;7:71378–71389. doi:10.18632/oncotarget.12110
109. Ranjan A, Ramachandran S, Gupta N, et al. Role of phytochemicals in cancer prevention. Int J Mol Sci. 2019;20:4981. doi:10.3390/ijms20204981
110. Ciardullo C, Aptullahoglu E, Woodhouse L, et al. Non-genotoxic MDM2 inhibition selectively induces a pro-apoptotic p53 gene signature in chronic lymphocytic leukemia cells. Haematologica. 2019;104(12):2429–2442. doi:10.3324/haematol.2018.206631
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.