")
Back to Archived Journals » Hypoxia » Volume 3
Therapeutic inhibition of prolyl hydroxylase domain-containing enzymes in surgery: putative applications and challenges
Authors Harnoss JM, Strowitzki MJ, Radhakrishnan P, Platzer L, Harnoss JC, Hank T, Cai J, Ulrich A, Schneider M
Received 25 October 2014
Accepted for publication 13 November 2014
Published 30 January 2015 Volume 2015:3 Pages 1—14
DOI https://doi.org/10.2147/HP.S60872
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Jonathan Michael Harnoss, Moritz Johannes Strowitzki, Praveen Radhakrishnan, Lisa Katharina Platzer, Julian Camill Harnoss, Thomas Hank, Jun Cai, Alexis Ulrich, Martin Schneider
Department of General, Visceral and Transplantation Surgery, University of Heidelberg, Heidelberg, Germany
Abstract: Oxygen is essential for metazoans to generate energy. Upon oxygen deprivation adaptive and protective pathways are induced, mediated by hypoxia-inducible factors (HIFs) and prolyl hydroxylase domain-containing enzymes (PHDs). Both play a pivotal role in various conditions associated with prolonged ischemia and inflammation, and are promising targets for therapeutic intervention. This review focuses on aspects of therapeutic PHD modulation in surgically relevant disease conditions such as hepatic and intestinal disorders, wound healing, innate immune responses, and tumorigenesis, and discusses the therapeutic potential and challenges of PHD inhibition in surgical patients.
Keywords: hypoxia, HIF, PHD, PHI, surgery
Introduction
Since oxygen is indispensable for life, insufficient oxygen availability can lead to cellular dysfunction and ultimately cell death. However, metazoans are able to sense and respond to altered oxygen tension in several physiological and pathological settings. Hypoxia-inducible factors (HIFs) play a central role in this adaption process and have therefore been the subject of intensive and rapidly expanding research in the past two decades.1,2–4 The HIF-mediated adaptive response is orchestrated by HIF prolyl hydroxylase domain-containing enzymes (PHDs), which fulfill specific functions in multiple physiological and pathophysiological processes. Pharmacological manipulation of PHDs therefore represents an interesting therapeutic opportunity in various disorders with high relevance in surgery.
This article reviews the current evidence on PHD function in various visceral organs and diseases, which has emerged from studies applying preclinical disease models in gene-deficient mice or on the pharmacological inhibition of PHDs (prolyl hydroxylase domain-containing enzyme inhibitors [PHI]), as well as from expressional studies in human disease. We do not provide a comprehensive literature survey, but focus on the clinical and translational aspects of PHD modulation by inhibitors (PHI) in visceral surgery and discuss the putative applications and challenges of this novel therapeutic approach.
Hypoxia-inducible factors and prolyl hydroxylase domain-containing enzymes
Three HIF isoforms are known (HIF1, -2, and -3), but HIF1 and -2 have been the most extensively studied. While HIF1 is ubiquitously expressed, HIF2 is more restricted.5 HIFs are transcriptional regulatory proteins, which are composed as heterodimers consisting of two subunits: an alpha (α)-subunit, regulated by hypoxia, and an oxygen-independent beta (β)-subunit, which is constitutively expressed. In normoxia the α-subunit is hydroxylated, which generates a binding site for von Hippel–Lindau tumor-suppressor proteins of the E3 ubiquitin proteasome ligase complex, resulting in polyubiquitination and subsequent proteasomal degradation.6 An autosomal recessive mutation in the von Hippel–Lindau (VHL) gene (Chuvash syndrome) leads to chronically elevated HIF1α levels and is associated with polycythemia, peripheral thrombosis, vascular hypertension, vascular abnormalities, and premature mortality.7 Congenital autosomal dominant mutations in the VHL gene (von Hippel–Lindau disease) predispose to cerebral and retinal hemangioblastoma and renal cell carcinoma.8 In hypoxia, the HIF α-subunits accumulate and dimerize with the β-subunits configuring active HIF. The heterodimers are translocated from the cytoplasm to the nucleus, where they bind to hypoxia response elements in the promoter region of downstream target genes, thus modulating the adaptive cellular response. More than 150 HIF-target genes have been identified, including those regulating angiogenesis, cell proliferation, metabolism, and apoptosis.5 This multifold response indicates the great potential for therapeutic manipulation of the HIF pathway.
PHDs function as oxygen sensors, because they require oxygen (besides iron, 2-oxoglutarate [2OG] and antioxidants like ascorbate or glutathione [GSH]) as an essential co-substrate for the hydroxylation of the HIFα-subunit.9 PHDs are non-heme iron containing 2OG-dependent dioxygenases, and belong to the family of prolyl 4-hydroxylases (P4Hs). The P4H enzyme family consists of collagen- and HIF-P4Hs, which are members of a class of over 60 2OG-dependent dioxygenases.5,10 The group of HIF-PHDs comprises four members: PHD1, PHD2, PHD3, the factor-inhibiting hypoxia-inducible factor (FIH), all of which display a 42%–59% sequence similarity.11 All PHDs are able to hydroxylate HIFα in vitro, but it remains unclear in what proportional contribution.5
In normoxia and mild hypoxia PHD2 is the main regulator of HIF1α due to its relatively abundant frequency in most cells.12,13 In severe and prolonged hypoxia PHD3 regulates HIF2α more efficiently.12 Knockout of PHD2 leads to stabilization of HIF1α, not HIF2α.14,15 In contrast, PHD1 and PHD3 double knockout results in accumulation of HIF2α, not HIF1α.15
PHDs are ubiquitously expressed, however, the PHD homologs display particular, partly overlapping tissue- and subcellular-specific RNA and protein-expression patterns.11,12,16 PHD1 is highly expressed in the testis and liver. PHD2, the most abundant homolog, is expressed in all organs. PHD3 is mainly expressed in the heart.12,16 On the subcellular level, PHD1 is present in the cell nucleus, PHD2 mainly in the cytoplasm, and PHD3 equally in both.12,16,17 Nevertheless, subsequent studies using monoclonal antibodies have indicated that all PHDs are mostly located in the cytoplasm.
Genetic deletion of PHD1 in mice does not cause any phenotypical effects in healthy conditions, but induces remarkable tolerance to muscle ischemia and reduced exercise endurance.18 Prenatal PHD2 deficiency is embryonically lethal due to placentation defects. Postnatal PHD2 deficiency promotes angiogenesis, polycythemia, and congestive heart failure.14,19 PHD3 deficiency results in a hypofunctional sympathoadrenal system and reduced blood pressure.20,21
Prolyl hydroxylase domain-containing enzyme inhibitors
PHDs are increasingly considered promising therapeutic targets for pharmacological modulation in various clinical settings involving acute or chronic hypoxia. The biochemistry of PHDs and PHI has been previously reviewed.4 In general, PHI interfere with PHD activity either nonselectively by replacing their essential co-substrates (iron and 2OG) or directly blocking the enzymes’ catalytic site. The PHI deferoxamine, an iron chelator, and cobalt chloride (CoCl2), a competitive iron inhibitor, compete for endogenous iron, and therefore can have systemic side effects. Pan-inhibitors, such as L-mimosine, dimethyloxalylglycine (DMOG), and ethyl-3,4-dihydroxybenzoate (EDHB), inhibit PHD function by mimicking 2OG, an intermediate of the tricarboxylic-acid cycle.5,22 However, several other tricarboxylic-acid-cycle intermediates such as citrate, isocitrate, succinate, fumarate, malate, oxaloacetate, and pyruvate also compete for binding to the active site and thus function as PHI.23,24 Moreover, reactive oxygen species (ROS) and nitric oxide (NO) can act as potent inhibitors of PHD activity [ie, by converting Fe(II) to Fe(III) and by chelating Fe(II), respectively] or via nitric oxide (by chelating Fe[II]), emphasizing the crucial effects of oxidative stress on the PHD–HIF axis.25,26
More recently developed PHI preferentially target protein–protein interactions, PHDs’ amino- or carboxyl terminal ends (eg, FK506-binding protein 38 [FKBP38]) or their active site (eg, TM6008 and TM6089).27–29 However, the PHDs’ catalytic site is highly conserved, thus hampering the development of isoform-specific PHI.30 Present research increasingly focuses on the development of small-molecule inhibitors of PHDs like JNJ-42041935, FG-4497, TRC160334, and AKB-4924.31–34 The use of small interfering ribonucleic acids (siRNAs) as PHI has also been considered.35,36
The greatest challenges remain:
- First, the enormous complexity within the PHD-HIF pathway, which regulates multiple genes, while at the same time interacting with multiple other signaling pathways (eg, the nuclear factor kappa-light-chain-enhancer of activated B cells [NF-κB] pathway, which links hypoxia to inflammation).37
- Second, the selectivity of PHI regarding HIF-PHDs: in order to prevent considerable adverse effects, PHIs should not only be selective for HIF-PHD (instead of targeting multiple other 2OG-dependent dioxgenases), but also for different HIF-PHD homologs. However, crystallographic and sequence analyses revealed that the active site is highly conserved among PHDs and FIH, thus hampering the development of isoform-specific PHI.30
- Third, the identification of the ideal therapeutic niche, which involves not only careful selection of clinical settings, but also the appropriate timing and duration of PHD inhibitor administration. Direct HIF-independent effects of PHI might have a more rapid onset since they occur posttranslationally, whereas downstream effects of HIF stabilization might be delayed.38 In this context, it will be essential to define the cut-off between alleviation and aggravation of symptoms, and to balance the benefits and systemic side effects of PHD inhibitor therapy. Moreover, application routes to enable organ-specific treatment would be desirable. Given the well-known clinical symptoms of Chuvash polycythemia and von Hippel–Lindau disease, long-term studies are required to elucidate the effects of permanent PHD inhibition associated with continuous HIF activation. In this context, a combination therapy of PHIs with additional drugs should also be considered to fine-tune therapeutic responses.5
- Ultimately, there is an urgent need for clinical studies. Clinical studies about PHD inhibition are scarce, albeit that the first Phase II and III clinical trials testing PHI for anemia in chronic kidney disease are ongoing and appear to be encouraging. Peer-reviewed results, however, are pending.39
The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors in the liver
Liver dysfunction following acute or prolonged hepatic ischemia, which might occur after major liver resection or liver transplantation, represents a major challenge in hepatobiliary surgery.40 Ischemia/Reperfusion (I/R) injury is considered the major contributor to primary allograft failure secondary to transplantation.41 Moreover, hepatic I/R injury promotes remote organ inflammation, which may ultimately lead to sepsis and multiorgan failure, emphasizing the key role of organ ischemia in the pathogenesis of severe, systemic complications.42 Intriguingly, recent insights from preclinical studies suggest that PHD inhibition might exert beneficial effects on liver function in various post-surgical conditions, which are outlined following.
Liver ischemia and ischemia/reperfusion damage
Hepatocytes are heavily dependent on mitochondrial adenosine triphosphate (ATP) production. In I/R conditions, however, mitochondrial redox processes promote mitochondrial dysfunction, cellular energy deficiency, oxidative stress, and uncontrolled production of ROS. The resultant oxidative stress is further aggravated by the recruitment of local and circulating inflammatory cells, altogether causing irreversible cell damage and postischemic liver failure.43,44 Preclinical studies in rodents revealed that PHIs are hepatoprotective in the setting of warm hepatic I/R, an effect that is that is at least partially mediated by antioxidant effects of the HIF1-target gene for heme oxygenase 1 (HO-1).45 Interestingly, the combination of I/R injury followed by resection of the nonischemic liver remnant is lethal in up to two-thirds of wild-type mice, but survived by 100% of PHD1-deficient mice. These effects may be attributable to the fact that PHD1 deficiency provides hypoxia tolerance to hepatocytes via stabilization of HIF2α and reprogramming of basal-cell metabolism (Figure 1A): loss of PHD1 function upregulates basal expression of the pyruvate dehydrogenase kinase isozyme 1 (PDK1), leading to reduced oxidative metabolism and less ROS production in ischemic conditions.36 These effects seem to be comparable to previously described effects of PHD1 deficiency on the energy metabolism in skeletal myofibers, which are partly mediated by the master regulator of energy metabolism, peroxisome proliferator-activated receptor alpha (PPARα), ultimately leading to increased expression of catalase and pyruvate dehydrogenase kinase 4 (PDK4). While catalase contributes to the detoxification of ROS, PDK4 inactivates the enzyme pyruvate dehydrogenase (PDH), causing a shift of glucose metabolism from oxidative to more anaerobic ATP production.18 Further, direct effects of HIF are increased glucose uptake via glucose transporter 1 (GLUT1), conversion of pyruvate to lactate catalyzed by lactate dehydrogenase, and clearance of lactate via the monocarboxylate transporter 4 (MCT4). Hence, the reduction of hepatic mitochondrial dysfunction and oxidative stress by inhibition of PHDs (PHD1 in particular) can be considered a promising therapeutic strategy for reduction of liver dysfunction following liver ischemia.
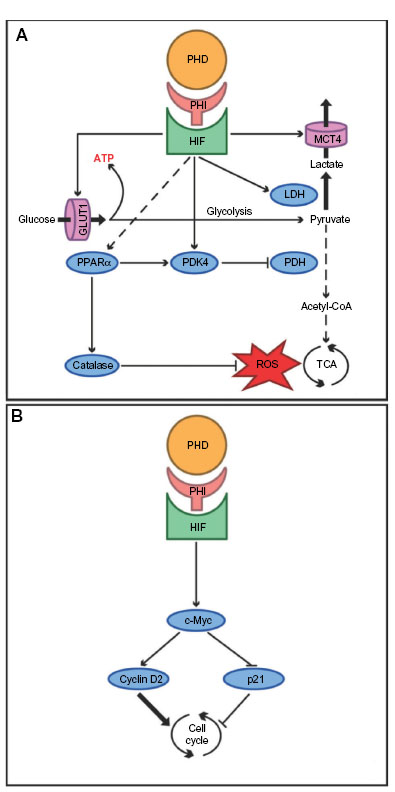 | Figure 1 The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors (PHI) in the liver. |
In the clinical setting, these effects could be of particular interest in liver transplantation, which remains the sole treatment option for acute liver failure and chronic end-stage liver disease. PHI treatment might facilitate the use of marginal donor organs to a greater extent, and thus represent an interesting opportunity to face the constant shortage of adequate organs. Indeed, results from a small, randomized, controlled clinical trial including 60 liver donors, suggest that induction of HIF1α stabilization prior to organ retrieval can attenuate graft injury and improve clinical outcomes.46 As proof of principle it has been demonstrated that in rodent kidney transplantation one single application of the PHD inhibitor FG-4497 6 hours prior to donor organ retrieval is sufficient to stabilize HIF1 and to significantly improve organ function and long-term outcome.33
Liver regeneration
Liver resection is the only potentially curative therapy for primary hepatic malignancies and metastatic liver disease.47 The extent of resection is, however, limited by the function and volume of the prospective remnant liver.48 About 13% of patients undergoing extended liver resection develop liver failure, which is associated with high morbidity and mortality.49 Current research approaches therefore aim at enhancing the regenerative capacity of the liver after resection. Due to altered hepatic hemodynamics, hallmarked by increased portal venous inflow and decreased arterial blood supply, post-resectional regeneration of the liver occurs in a hypoxic environment.50 Not surprisingly, therefore, HIFs and their hepatic target genes are overexpressed in hepatocytes during liver regeneration.51–53 Genetic deficiency of PHD1 enhances liver regeneration in preclinical mouse models via increased hepatocyte proliferation, which is induced by enhanced expression of the cell-cycle promoter cyclin D2 and decreased expression of cell-cycle inhibitor p21 in a c-Myc-dependent fashion (Figure 1B). These effects of PHD1 deficiency in the regenerating liver are likely induced by increased stabilization of HIF2α.54 Collectively, these results support a potential therapeutic role of PHI for liver regeneration after surgical liver resection.
Hepatic steatosis and fibrosis
Fatty liver disease (FLD) caused by abnormal hepatic triglyceride accumulation has an increasing prevalence in Western industrialized countries, and is associated with alcoholic liver disease or metabolic syndrome.55 Severe steatohepatitis ultimately causes liver fibrosis and cirrhosis.55
Both clinical observations and preclinical studies in gene-deficient mice suggest that HIF pathways are significantly involved in the pathogenesis of FLD. For instance, chronic intermittent hypoxia induced by obstructive sleep apnea syndrome is associated with FLD, promoted by activation of the HIF- and NF-κB pathways.56,57
In mice, chronic stabilization of HIF2α stimulates excessive hepatic lipid accumulation, along with impaired β-oxidation of fatty acids and increased hepatocellular lipid storage capacity, as well as increased inflammation and hepatic fibrosis.52,58 HIF1α expression likewise correlates with the severity of fibrosis in rodents, and HIF1α-deficient mice display reduced fibrosis.59,60 Consistent with these effects in HIF1α-deficient mice, animals deficient in both PHD2 and PHD3 display severe hepatomegaly and hepatic steatosis.61 Conversely, HIF1 may also exert protective effects in alcoholic liver disease: ethanol-induced hepatic lipid accumulation is significantly increased in HIF1α-deficient mice, and DMOG ameliorates steatosis in wild-type, but not in HIF1α-null mice.62
While beneficial effects of PHI in settings of liver ischemia and resection have been well documented in preclinical studies, less is known about the significance of PHD inhibition in the setting of hepatic steatosis and fibrosis. Given the evidence outlined, putative effects of PHI on hepatic steatosis and fibrosis remain ambiguous and most likely depend on the underlying pathogenesis. Thus, while inhibition of collagen-P4Hs reduces collagen formation and prevents liver fibrosis, specific effects of HIF-P4Hs on steatohepatitis and liver fibrosis are subject to further investigation.63,64
The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors in the intestine
Inflammatory bowel disease
The two main types of inflammatory bowel disease (IBD), Crohn’s disease and ulcerative colitis (UC), are distinct in their pathogenesis, and their patterns of manifestation within the gastrointestinal tract: while Crohn’s disease may affect all bowel segments, UC is restricted to the colon. Therefore, surgery can offer a cure for UC: the chronic form, which is resistant to conservative, anti-inflammatory treatment, or fulminant UC are indications for restorative proctocolectomy.65 Further indications for colectomy comprise bacterial-induced colitis and ischemic colitis.66,67
Severe hypoxia of the intestinal mucosa (inflammatory hypoxia) is a hallmark of IBD. At baseline physiological conditions, the intestinal mucosa already has a hypoxic environment, which is potentiated in IBD through a mismatch of localized vascular damage and increased oxygen demand.68,69 It has been shown that HIF1α is significantly overexpressed in human UC.70 In mice, inactivation of epithelial HIF1α in the colon aggravates the susceptibility to mucosal inflammation due to downregulation of barrier-protective HIF-target genes (for eg, intestinal trefoil factor 1 [ITF1], multidrug resistance 1 [MDR1], CD73, and adenosine A2B receptor [A2BAR]). Collectively, this results in diminished mucin production, attenuated xenobiotic defense mechanisms, and impaired nucleotide metabolism and signaling (Figure 2A).71–74 Consistently, the stabilization of HIF1α by PHI has a protective effect:68,69 Several PHI, such as DMOG, FG-4497, TRC160334, and AKB-4924, attenuate the severity of colitis in a variety of preclinical mouse models by, first, augmentation of the epithelial barrier via NF-kB and intestinal barrier genes; second, promotion of neutrophil apoptosis; third, lowering the levels of proinflammatory cytokines such as interleukin (IL)-1b, IL-6, and tumor necrosis factor alpha ([TNFα] a stimulator of NF-κB); and, finally, accelerated initiation of mucosal restitution through fibroblast integrin beta 1 (ITGB1).31,34,75–77 The iron chelator quercetin promotes similar effects in a rat model of colitis.78 Among the individual PHDs, PHD1 appears to be a predominant effector of mucosal protection: PHD1 deficiency leads to increased enterocyte density and intestinal barrier function, and decreased apoptosis of colonic mucosal cells.79 In patients suffering active UC, epithelial expression of PHD1 protein is significantly increased, highlighting these findings’ relevance in human IBD.79,80 Furthermore, loss of PHD3 shortens the lifespan of inflammatory neutrophils by upregulation of the proapoptotic mediator Siva1 and loss of its binding protein, B-cell lymphoma-extra large (Bcl-xL), thus limiting the severity of mucosal inflammation.81 In addition, HIF stabilization supports the immune function of dendritic and mast cells in mice.82,83
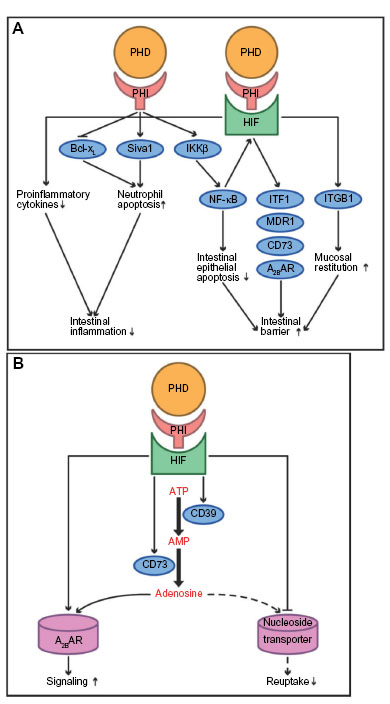 | Figure 2 The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors (PHI) in the intestine. |
Collectively, these observations indicate that PHIs represent a promising tool for the clinical treatment of IBD. Of note, it was likewise observed that high doses of FG-4497 might cause vascular occlusion within the intestine, most likely secondary to elevated hematocrit levels, thus underscoring the challenges of dose finding for therapeutic HIF stabilization.84 Hitherto, research has focused on the significance of PHIs in acute colitis, however, their potential in the treatment of chronic colitis and in the maintenance of remission remains to be addressed. Remarkably, it has been recently shown in rodents that treatment with DMOG 24 hours after radiation-induced enteritis is still gut protective, emphasizing the imperative to further explore the perfect timing of PHD inhibitor administration in the setting of IBD.85
Intestinal ischemia and reperfusion
Bowel damage caused by intestinal I/R represents a frequent challenge in gastrointestinal surgery. While early symptoms of intestinal ischemia are mostly nonspecific, the clinical suspicion can suffice as an indication for surgical exploration and either revascularization or resection. Common reasons for intestinal I/R are strangulated hernias, mesenteric artery occlusion (due to thrombosis or embolism), or circulatory collapse (eg, in septic conditions).86 Current research on novel treatment strategies to alleviate intestinal ischemia injury is therefore highly warranted.
It has been demonstrated that adenosine and its receptors are protective in intestinal I/R injury. Extracellular adenosine is generated from ATP or adenosine diphosphate, which are converted into adenosine monophosphate (AMP) in a process driven by the ectopyrase CD39. The subsequent conversion of AMP to adenosine is catalyzed by CD73. Both CD39 and CD73 are induced by hypoxia (Figure 2B).72,87 In mice, CD39 deficiency causes increased severity of intestinal inflammation and CD73 deficiency amplifies intestinal injury and organ failure in response to I/R.88–90 The effects of adenosine in the setting of intestinal ischemia are importantly mediated by A2BAR, which is likewise induced in hypoxia via HIF.91 While conditional deletion of intestinal epithelial HIF1α aggravates intestinal I/R damage in a mouse model of short superior mesenteric artery occlusion (SMAO; 15 minutes), DMOG significantly attenuates I/R injury by increased extracellular adenosine generation involving CD73, and signaling via A2BAR.91,92 Furthermore, HIF amplifies extracellular adenosine concentrations by repressing equilibrative nucleoside transporters (thus inhibiting its reuptake), and by repressing its further metabolism by the adenosine kinase, which reconverts adenosine into AMP.93,94
While these insights indicate a putative benefit of PHIs in the setting of intestinal I/R, conflicting data likewise exist: in a mouse model of severe SMAO (90 minutes), partial deficiency of HIF1 seems to be protective against intestinal I/R injury, emphasizing that duration and severity of I/R injury determine the role of HIF in this setting.95 However, in other rodent models of severe SMAO (60 minutes), induction of the HIF-target gene for HO-1 causes a significant reduction of intestinal I/R injury.96
In summary, PHI may represent an interesting novel treatment opportunity in clinical settings of intestinal ischemia, despite these apparently paradoxical preclinical data. However, it remains a major challenge to determine the therapeutic niche for PHD inhibitor treatment in this setting, and more preclinical studies are required to identify the perioperative timing of PHD inhibitor application, in order to balance the beneficial and deleterious effects of HIF stabilization.
The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors in wound healing
Wound healing is of greatest interest to the surgeon, especially if this complex process – in which inflammation, (neo-)angiogenesis, and reepithelialization play pivotal roles – is impaired.97 Like the intestinal mucosa, the skin is constitutively hypoxic even under baseline conditions, resulting in continuous HIF1α stabilization within the basal epidermal layer.98,99 Skin injury further enhances hypoxia and epidermal HIF1α stabilization, which represents a major stimulus for wound healing. Subsequently, several angiogenic factors are released, and circulating endothelial precursor cells are recruited to initiate revascularization (Figure 3).100,101 Accordingly, heterozygous loss of HIF1α impairs wound healing in mice, and dermal suppression of HIF1α and its target genes negatively affects the wound-healing process in diabetic mice.102,103 HIF1α stabilization by DMOG crucially improves wound closure via enhanced angiogenesis, which is stimulated by increased levels of vascular endothelial growth factor and enhanced proliferation of endothelial precursor cells, a process that appears to be specifically mediated by inhibition of PHD2.103–106 Specific depletion of PHD2 in epidermal keratinocytes accelerates reepithelialization in a murine model of skin wound healing: this effect is promoted by reduced activity of the canonical transforming growth factor beta (TGFβ) 2-pathway and increased HIF1α-dependent expression of β3-integrin, thus enhancing the migration and proliferation of keratinocytes.107 Consistently, HIF1α deficiency in keratinocytes delays wound healing in aged mice.108 In contrast, in dermal fibroblasts HIF stabilization by DMOG and FG-4497 induces the transcription of ITGB1.76 Integrin signaling through α3β1-integrin inhibits SMAD7, likewise a HIF-responsive gene and potent inhibitor of TGFβ1, and thus enhances cutaneous reepithelialization in a murine model of skin wound healing.109,110
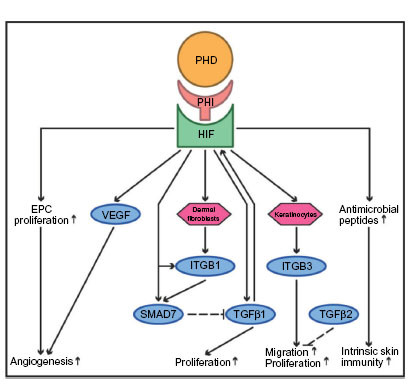 | Figure 3 The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors (PHI) in wound healing. |
While hypoxia increases TGFβ1 in human dermal fibroblasts in vitro, TGFβ1 likewise stabilizes HIF1α through selective downregulation of PHD2, thus underscoring the extensive crosstalk between the HIF- and TGFβ-pathways in wound healing.111,112 Furthermore, in mice HIF1α directly stimulates the production of antimicrobial peptides in keratinocytes, including cathelicidin, thus strengthening the intrinsic immunity of the skin layer.113 In a mouse model of skin abscess, topical treatment of keratinocytes with the HIF1-stabilizing PHD inhibitor AKB-4924 leads to increased bactericidal capacity against skin pathogens.114 In contrast to these reported effects of HIF1α, keratinocyte-specific deletion of HIF2α accelerates wound closure by increasing keratinocyte migration, and reducing the bacterial load in the acute inflammatory phase.115
Despite these adverse effects caused by HIF2α, these preclinical data collectively indicate that PHIs might represent an interesting therapeutic tool in skin wound healing, especially in the restoration of complicated wounds such as in diabetic patients or in ischemic flaps, or as an adjunctive therapy in antibiotic-resistant bacterial wound infections.103–106 A major advantage of PHI application in this setting might be the opportunity for topical administration, which would reduce systemic side effects.
The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors in the innate immune response
From a surgical perspective, abdominal sepsis is highly relevant since it is associated with a lethality of 25–30%, and is one of the leading causes for mortality on surgical intensive care units.116 Sepsis represents a severe systemic inflammatory response syndrome due to bacterial pathogens, which can ultimately lead to multiorgan failure. Clinical sepsis symptoms and septic multiorgan failure are induced by an overwhelming innate immune response, which is importantly stimulated by hypoxia in a HIF-dependent manner (Figure 4).117,118 The intertwined relationships between hypoxia, inflammation, and innate immunity have been well-documented:68,119 tissue hypoxia causes accumulation of inflammatory cells, and leakage across epithelial and vascular barriers.120,121 Remarkably, inflammation potentiates tissue hypoxia due to an imbalance of decreased oxygen supply (caused by vascular thrombosis) and increased oxygen demand (due to enhanced consumption by both resident and inflammatory cells).84,92,122 At the cellular and subcellular level, HIF1α enhances the adhesion and migration of hypoxic myeloid cells through β2-integrin transcription, and is indispensable for anaerobic ATP-generation and myeloid cell survival in inflamed tissues, as well as for their ability to aggregate and invade.117,123 HIF1α likewise stimulates bactericidal effectors of the innate immune response such as TNFα, inducible nitric oxide synthase and subsequently nitric oxide, and promotes neutrophil survival via NF-κB.124–127
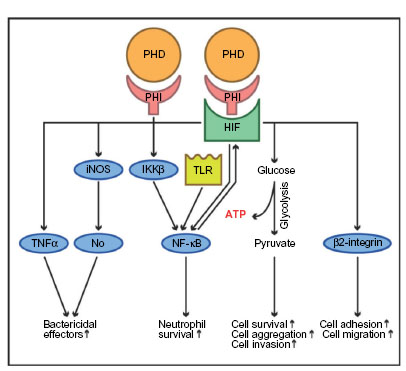 | Figure 4 The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors (PHI) in innate immune response. |
From a clinical perspective, the significance of HIF in the innate immune response appears to be double-edged. Depending on the type and extent of the underlying inflammatory disease, both adverse and beneficial effects have been described: while HIF-dependent effects in myeloid cells importantly support the ability of the immune system to resolve localized bacterial invasion and tissue inflammation, HIF1α can play an adverse role in sepsis. In murine sepsis models, macrophage-specific deletion of HIF1α is protective against lipopolysaccharide-induced mortality by reducing proinflammatory cytokines such as IL-1β, IL-6, or TNFα.128,129 Furthermore, HIF1α amplifies the proinflammatory NF-κB pathway directly by upregulation of NF-κB, as well as indirectly, through induction of toll-like receptors (TLR).127,130 In a reciprocal self-reinforcing process, NF-κB likewise stimulates HIF1α transcription.131
Of note, PHDs regulate both pathways and are, therefore, important mediators of the innate immune response.84 PHD1 represses NF-κB in normoxia by hydroxylation and consecutive inactivation of IkB kinase beta (IKKβ), a promoter of NF-κB.132 PHD3 interacts with IKKβ independently of its hydroxylase function by blocking the interaction of IKKβ with its chaperone heat shock protein 90 (Hsp90), which is required for IKKβ phosphorylation and release of NF-kB.133 Accordingly, global or myeloid cell-specific deficiency of PHD3 aggravates clinical symptoms and the lethality of abdominal sepsis in preclinical mouse models via stabilization of HIF1α and increased activity of NF-κB.134
Finally, apart from their multifold effects on innate immune functions, HIF and PHDs likewise affect adaptive immunity. For instance, specific stabilization of HIF1α in T-cells impairs the survival of septic mice due to reduced T-cell proliferation and proinflammatory cytokine secretion.135
From a surgical, as well as from an anesthesiologic standpoint, these insights, which were mostly generated in preclinical mouse models, are highly relevant concerning putative applications of PHI in critically ill or septic patients. For instance, boosted activation of the innate immune response via PHI could be an interesting therapeutic option in bacterial infections of immunodeprived patients, but it may likewise have severe side effects. More experimental evidence is required to evaluate the therapeutic implications of PHIs for the modulation of the innate immune response in specific inflammatory disease settings.
The putative effects of prolyl hydroxylase domain-containing enzyme inhibitors in cancer
Hypoxia occurs in virtually all human tumors because expanding cancer cells rapidly outgrow the development of nourishing blood vessels, and because tumor vessels are poorly functional and chaotic, altogether leading to insufficient oxygen supply.136,137 Accordingly, HIF is upregulated in a variety of tumor entities, and has been demonstrated to enhance tumor aggressiveness, associated with poor prognosis and resistance against anticancer therapy.24,138–140 HIFs importantly influence all major aspects of cancer biology, including cell survival, resistance to apoptosis, angiogenesis, invasion, and metastasis.141 On one hand, clinical disorders such as the hereditary von Hippel–Lindau disease underscore the tumor-promoting role of HIF2α.8 On the other hand, tumor-suppressive functions of HIF have likewise been demonstrated, and the effects of HIF1 and HIF2 on tumor growth can be intriguingly different:142 for instance, HIF1α (but not HIF2α) decreases the activity of the cell-cycle regulator Myc and may have antiproliferative functions. Overexpression of HIF2α in rat glioma tumors enhances angiogenesis, but reduces tumor growth by increasing tumor-cell apoptosis.143–145
The impact of PHI on occult or dormant tumor disease may lead to significant side effects in patients undergoing treatment with PHI, and putative applications of PHI in gastrointestinal surgery (such as protection of liver function following major resections) might specifically apply for cancer patients. From a clinical standpoint, it will therefore become crucial to sort out the specific effects of PHI on tumor growth and metastasis. Although immunohistochemical analyses of human cancers have predominantly revealed increased expression of PHDs, conflicting evidence exists about how they specifically influence tumorigenesis.11,146 In fact, the effects of the individual PHDs on tumor growth appear to be heterogeneous, and the impact of pharmacological pan-inhibition of PHDs on tumor growth is poorly defined. In the following, we summarize hitherto available evidence regarding specific effects of PHD1, PHD2, and PHD3 on tumor growth, which has mostly been generated by functional genomic studies.
PHD1 overexpression in tumor cells can inhibit tumor growth in mice.147 Conversely, downregulation of PHD1 in vitro impairs cyclin D1, thus suppressing the proliferation of various cancer cell lines.148
Broad, but partly conflicting, evidence has been generated concerning the tumor-specific effects of PHD2. While overexpression of PHD2 in pancreatic cancer cells appears to impair tumor growth, downregulation of PHD2 in pancreatic-, colorectal-, and breast-cancer cells stimulates tumor growth in immunodeficient mice, altogether suggesting tumor-suppressive functions of PHD2.149–151 Moreover, PHD2 haplodeficiency stimulates hepatocarcinogenesis in mice.152 In human patients, PHD2 expression is positively correlated with gastric-cancer survival, and may serve as a prognostic marker.152,153 PHD2 deficiency is likewise associated with the immortality of human endometrial cancer cells, while the reintroduction of PHD2 induces senescence.154 In the literature one case is reported describing a patient with a PHD2 mutation, which was associated with multiple tumors.155 Conversely, in osteosarcoma cells PHD2 deficiency significantly alleviates tumor growth by the matrix metalloproteinase-induced transformation of TGFβ into a tumor suppressor.156,157 This is remarkable in the context of the mentioned potential positive feedback loop between TGFβ and HIF1α.111 However, in breast-cancer cells, TGFβ seems to be a tumor promoter.158 In rodents, haplodeficiency of PHD2 reduces tumor-cell invasion, intravasation, and metastasis without affecting primary tumor size, and improves the delivery of chemotherapy and accompanied tumor response to chemotherapy by normalizing the tumor vasculature.159,160 At the same time, stabilization of HIF1α may counteract the efficiency of chemotherapy via increased expression of MDR1.74 PHD2 can also contribute to tumorigenesis independently of HIF: inactivation of PHD2 in colon carcinoma cells stimulates tumor growth, mediated by NF-κB-dependent expression of IL-8 and angiogenin, thereby stimulating angiogenesis and vasculogenesis through the recruitment of bone-marrow-derived vascular modulatory cells.150
PHD3 expression is decreased in human colorectal-cancer tissue and associated with higher tumor grade and metastasis. In this context, PHD3 seems to be a HIF-independent tumor suppressor acting via NF-κB.133 Moreover, PHD3 is essential for the apoptosis of sympathetic neuron precursor cells in vitro. Thus, loss of PHD3 contributes to the development of pheochromocytomas.21
In summary, the reviewed conflicting data on HIF and PHD in tumorigenesis might be in part due to the following: first, different experimental setups in vivo or in vitro with inactivation of PHDs only in tumor cells, in tumor micro-environments (fibroblasts, endothelial, epithelial and immune cells), or in both; and, second, the HIF-dependent or -independent functions of PHDs, such as on NF-κB or TGFβ.120,138,157 Obviously, modulation of PHDs might enhance tumor growth, which may limit the clinical use of PHIs in any therapeutic approach. More preclinical studies elucidating the precise cancer- and stromal-cell-specific functions of PHDs in tumorigenesis are warranted.
Conclusion
The possible applications of PHI in various diseases are multifold, and open a broad perspective for clinical implications in surgery. Although several ongoing clinical trials are encouraging,39 translation to the patient’s bedside mandates more preclinical and clinical studies, which need to settle several problems: first, PHI with certain selectivity for the individual PHDs, as well as for different tissues, are required; second, the timeframe of PHD inhibitor application in preconditioning and treatment has to be defined to balance the benefits and potential side effects of constant HIF stabilization; third, alternative PHD inhibitor application pathways such as local application in wound healing or enteral application in IBD are desirable to minimize systemic side effects; and, finally, the significance of PHI in tumorigenesis (either tumor suppressive as anticancer therapeutics, or cancer promoting as a severe systemic side effect) remains elusive and needs to be clarified.
Acknowledgment
Supported in the framework of the Clinical Research Group KFO 227 by the Deutsche Forschungsgemeinschaft, Germany.
Disclosure
The authors declare no conflicts of interest in this work.
References
Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8(5):588–594. | |
Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8(2):139–152. | |
Kiss J, Kirchberg J, Schneider M. Molecular oxygen sensing: implications for visceral surgery. Langenbecks Arch Surg. 2012;397(4): 603–610. | |
Nagel S, Talbot NP, Mecinovic J, Smith TG, Buchan AM, Schofield CJ. Therapeutic manipulation of the HIF hydroxylases. Antioxid Redox Signal. 2010;12(4):481–501. | |
Selvaraju V, Parinandi NL, Adluri RS, et al. Molecular mechanisms of action and therapeutic uses of pharmacological inhibitors of HIF-prolyl 4-hydroxylases for treatment of ischemic diseases. Antioxid Redox Signal. 2014;20(16):2631–2665. | |
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | |
Ang SO, Chen H, Hirota K, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32(4):614–621. | |
Shen C, Kaelin WG Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23(1):18–25. | |
Nytko KJ, Maeda N, Schlafli P, Spielmann P, Wenger RH, Stiehl DP. Vitamin C is dispensable for oxygen sensing in vivo. Blood. 2011; 117(20):5485–5493. | |
Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008;4(3):152–156. | |
Jokilehto T, Jaakkola PM. The role of HIF prolyl hydroxylases in tumour growth. J Cell Mol Med. 2010;14(4):758–770. | |
Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. | |
Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22(16): 4082–4090. | |
Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116(7):774–781. | |
Takeda K, Aguila HL, Parikh NS, et al. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008; 111(6):3229–3235. | |
Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80(4):421–426. | |
Metzen E, Berchner-Pfannschmidt U, Stengel P, et al. Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci. 2003;116(Pt 7):1319–1326. | |
Aragonés J, Schneider M, Van Geyte K, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40(2):170–180. | |
Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008; 111(6):3236–3244. | |
Bishop T, Gallagher D, Pascual A, et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3-/- mice. Mol Cell Biol. 2008;28(10):3386–3400. | |
Lee S, Nakamura E, Yang H, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8(2):155–167. | |
Kasiganesan H, Sridharan V, Wright G. Prolyl hydroxylase inhibitor treatment confers whole-animal hypoxia tolerance. Acta Physiol (Oxf). 2007;190(2):163–169. | |
Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. | |
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4): 393–402. | |
Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14(8):3470–3481. | |
Pan Y, Mansfield KD, Bertozzi CC, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27(3):912–925. | |
Barth S, Nesper J, Hasgall PA, et al. The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol Cell Biol. 2007;27(10):3758–3768. | |
Nangaku M, Izuhara Y, Takizawa S, et al. A novel class of prolyl hydroxylase inhibitors induces angiogenesis and exerts organ protection against ischemia. Arterioscler Thromb Vasc Biol. 2007;27(12):2548–2554. | |
Wenger RH, Camenisch G, Stiehl DP, Katschinski DM. HIF prolyl-4-hydroxylase interacting proteins: consequences for drug targeting. Curr Pharm Des. 2009;15(33):3886–3894. | |
McDonough MA, Li V, Flashman E, et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci U S A. 2006;103(26):9814–9819. | |
Keely S, Campbell EL, Baird AW, et al. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol. 2014;7(1):114–123. | |
Barrett TD, Palomino HL, Brondstetter TI, et al. Pharmacological characterization of 1-(5-chloro-6-(trifluoromethoxy)-1H-benzoimidazol-2-yl)-1H-pyrazole-4-carboxylic acid (JNJ-42041935), a potent and selective hypoxia-inducible factor prolyl hydroxylase inhibitor. Mol Pharmacol. 2011;79(6):910–920. | |
Bernhardt WM, Gottmann U, Doyon F, et al. Donor treatment with a PHD-inhibitor activating HIFs prevents graft injury and prolongs survival in an allogenic kidney transplant model. Proc Natl Acad Sci U S A. 2009;106(50):21276–21281. | |
Gupta R, Chaudhary AR, Shah BN, et al. Therapeutic treatment with a novel hypoxia-inducible factor hydroxylase inhibitor (TRC160334) ameliorates murine colitis. Clin Exp Gastroenterol. 2014;7:13–23. | |
Huang M, Chan DA, Jia F, et al. Short hairpin RNA interference therapy for ischemic heart disease. Circulation. 2008;118(14 Suppl): S226–S233. | |
Schneider M, Van Geyte K, Fraisl P, et al. Loss or silencing of the PHD1 prolyl hydroxylase protects livers of mice against ischemia/reperfusion injury. Gastroenterology. 2010;138(3):1143–1154. | |
Scholz CC, Cavadas MA, Tambuwala MM, et al. Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc Natl Acad Sci U S A. 2013;110(46):18490–18495. | |
Howell NJ, Tennant DA. The role of HIFs in ischemia-reperfusion injury. Hypoxia. 2014;2:107–115. | |
Flight MH. Deal watch: AstraZeneca bets on FibroGen’s anaemia drug. Nat Rev Drug Discov. 2013;12(10):730. | |
Benzoni E, Lorenzin D, Favero A, et al. Liver resection for hepatocellular carcinoma: a multivariate analysis of factors associated with improved prognosis. The role of clinical, pathological and surgical related factors. Tumori. 2007;93(3):264–268. | |
Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation – from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10(2):79–89. | |
Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Paneth cell-derived interleukin-17A causes multiorgan dysfunction after hepatic ischemia and reperfusion injury. Hepatology. 2011;53(5):1662–1675. | |
Jaeschke H, Woolbright BL. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant Rev (Orlando). 2012;26(2):103–114. | |
Weigand K, Brost S, Steinebrunner N, Buchler M, Schemmer P, Müller M. Ischemia/Reperfusion injury in liver surgery and transplantation: pathophysiology. HPB Surg. 2012;2012:176723. | |
Zhong Z, Ramshesh VK, Rehman H, et al. Activation of the oxygen-sensing signal cascade prevents mitochondrial injury after mouse liver ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol. 2008;295(4):G823–G832. | |
Amador A, Grande L, Martí J, et al. Ischemic pre-conditioning in deceased donor liver transplantation: a prospective randomized clinical trial. Am J Transplant. 2007;7(9):2180–2189. | |
Reissfelder C, Rahbari NN, Koch M, et al. Postoperative course and clinical significance of biochemical blood tests following hepatic resection. Br J Surg. 2011;98(6):836–844. | |
Rahbari NN, Reissfelder C, Koch M, et al. The predictive value of postoperative clinical risk scores for outcome after hepatic resection: a validation analysis in 807 patients. Ann Surg Oncol. 2011;18(13):3640–3649. | |
Hammond JS, Guha IN, Beckingham IJ, Lobo DN. Prediction, prevention and management of postresection liver failure. Br J Surg. 2011;98(9):1188–1200. | |
Abshagen K, Eipel C, Vollmar B. A critical appraisal of the hemodynamic signal driving liver regeneration. Langenbecks Arch Surg. 2012;397(4):579–590. | |
Maeno H, Ono T, Dhar DK, Sato T, Yamanoi A, Nagasue N. Expression of hypoxia inducible factor-1alpha during liver regeneration induced by partial hepatectomy in rats. Liver Int. 2005;25(5):1002–1009. | |
Rankin EB, Rha J, Selak MA, et al. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol. 2009;29(16):4527–4538. | |
Tajima T, Goda N, Fujiki N, et al. HIF-1alpha is necessary to support gluconeogenesis during liver regeneration. Biochem Biophys Res Commun. 2009;387(4):789–794. | |
Mollenhauer M, Kiss J, Dudda J, et al. Deficiency of the oxygen sensor PHD1 augments liver regeneration after partial hepatectomy. Langenbecks Arch Surg. 2012;397(8):1313–1322. | |
Rozman D. From nonalcoholic Fatty liver disease to hepatocellular carcinoma: a systems understanding. Dig Dis Sci. 2014;59(2):238–241. | |
Musso G, Cassader M, Olivetti C, Rosina F, Carbone G, Gambino R. Association of obstructive sleep apnoea with the presence and severity of non-alcoholic fatty liver disease. A systematic review and meta-analysis. Obes Rev. 2013;14(5):417–431. | |
Musso G, Olivetti C, Cassader M, Gambino R. Obstructive sleep apnea-hypopnea syndrome and nonalcoholic fatty liver disease: emerging evidence and mechanisms. Semin Liver Dis. 2012;32(1):49–64. | |
Qu A, Taylor M, Xue X, et al. Hypoxia-inducible transcription factor 2α promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology. 2011;54(2):472–483. | |
Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2009;296(3):G582–G592. | |
Bozova S, Elpek GO. Hypoxia-inducible factor-1alpha expression in experimental cirrhosis: correlation with vascular endothelial growth factor expression and angiogenesis. APMIS. 2007;115(7):795–801. | |
Minamishima YA, Moslehi J, Padera RF, Bronson RT, Liao R, Kaelin WG Jr. A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol Cell Biol. 2009;29(21):5729–5741. | |
Nishiyama Y, Goda N, Kanai M, et al. HIF-1α induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J Hepatol. 2012;56(2):441–447. | |
Bickel M, Baringhaus KH, Gerl M, et al. Selective inhibition of hepatic collagen accumulation in experimental liver fibrosis in rats by a new prolyl 4-hydroxylase inhibitor. Hepatology. 1998;28(2):404–411. | |
Sakaida I, Matsumura Y, Kubota M, Kayano K, Takenaka K, Okita K. The prolyl 4-hydroxylase inhibitor HOE 077 prevents activation of Ito cells, reducing procollagen gene expression in rat liver fibrosis induced by choline-deficient L-amino acid-defined diet. Hepatology. 1996;23(4):755–763. | |
Bennis M, Tiret E. Surgical management of ulcerative colitis. Langenbecks Arch Surg. 2012;397(1):11–17. | |
Rutgeerts P, Vermeire S, Van Assche G. Biological therapies for inflammatory bowel diseases. Gastroenterology. 2009;136(4):1182–1197. | |
Nielsen OH, Vainer B, Rask-Madsen J. Non-IBD and noninfectious colitis. Nat Clin Pract Gastroenterol Hepatol. 2008;5(1):28–39. | |
Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med (Berl). 2007;85(12):1295–1300. | |
Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114(8):1098–1106. | |
Giatromanolaki A, Sivridis E, Maltezos E, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56(3):209–213. | |
Furuta GT, Turner JR, Taylor CT, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. 2001;193(9):1027–1034. | |
Synnestvedt K, Furuta GT, Comerford KM, et al. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110(7):993–1002. | |
Eltzschig HK, Ibla JC, Furuta GT, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198(5):783–796. | |
Comerford KM, Colgan SP. Assessing oxygen sensitivity of the multidrug resistance (MDR) gene. Methods Enzymol. 2004;381:376–387. | |
Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134(1):145–155. | |
Keely S, Glover LE, MacManus CF, et al. Selective induction of integrin beta1 by hypoxia-inducible factor: implications for wound healing. FASEB J. 2009;23(5):1338–1346. | |
Cummins EP, Seeballuck F, Keely SJ, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134(1):156–165. | |
Jeon H, Kim H, Choi D, et al. Quercetin activates an angiogenic pathway, hypoxia inducible factor (HIF)-1-vascular endothelial growth factor, by inhibiting HIF-prolyl hydroxylase: a structural analysis of quercetin for inhibiting HIF-prolyl hydroxylase. Mol Pharmacol. 2007;71(6):1676–1684. | |
Tambuwala MM, Cummins EP, Lenihan CR, et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology. 2010;139(6):2093–2101. | |
Van Welden S, Laukens D, Ferdinande L, De Vos M, Hindryckx P. Differential expression of prolyl hydroxylase 1 in patients with ulcerative colitis versus patients with Crohn’s disease/infectious colitis and healthy controls. J Inflamm (Lond). 2013;10(1):36. | |
Walmsley SR, Chilvers ER, Thompson AA, et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest. 2011;121(3):1053–1063. | |
Jantsch J, Chakravortty D, Turza N, et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180(7):4697–4705. | |
Jeong HJ, Moon PD, Kim SJ, et al. Activation of hypoxia-inducible factor-1 regulates human histidine decarboxylase expression. Cell Mol Life Sci. 2009;66(7):1309–1319. | |
Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7(5):281–287. | |
Taniguchi CM, Miao YR, Diep AN, et al. PHD inhibition mitigates and protects against radiation-induced gastrointestinal toxicity via HIF2. Sci Transl Med. 2014;6(236):236ra264. | |
Mallick IH, Yang W, Winslet MC, Seifalian AM. Ischemia-reperfusion injury of the intestine and protective strategies against injury. Dig Dis Sci. 2004;49(9):1359–1377. | |
Hart ML, Gorzolla IC, Schittenhelm J, Robson SC, Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol. 2010;184(7):4017–4024. | |
Friedman DJ, Künzli BM, A-Rahim YI, et al. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci U S A. 2009;106(39):16788–16793. | |
Hart ML, Henn M, Köhler D, et al. Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 2008;22(8):2784–2797. | |
Louis NA, Robinson AM, MacManus CF, Karhausen J, Scully M, Colgan SP. Control of IFN-alphaA by CD73: implications for mucosal inflammation. J Immunol. 2008;180(6):4246–4255. | |
Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, Eltzschig HK. Hypoxia-inducible factor-1alpha-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5’-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol. 2011;186(7):4367–4374. | |
Grenz A, Clambey E, Eltzschig HK. Hypoxia signaling during intestinal ischemia and inflammation. Curr Opin Crit Care. 2012;18(2): 178–185. | |
Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136(2):607–618. | |
Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008;111(12):5571–5580. | |
Kannan KB, Colorado I, Reino D, et al. Hypoxia-inducible factor plays a gut-injurious role in intestinal ischemia reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G853–G861. | |
Wasserberg N, Pileggi A, Salgar SK, et al. Heme oxygenase-1 upregulation protects against intestinal ischemia/reperfusion injury: a laboratory based study. Int J Surg. 2007;5(4):216–224. | |
Shaw TJ, Martin P. Wound repair at a glance. J Cell Sci. 2009;122(Pt 18):3209–3213. | |
Bedogni B, Welford SM, Cassarino DS, Nickoloff BJ, Giaccia AJ, Powell MB. The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell. 2005;8(6): 443–454. | |
Lokmic Z, Musyoka J, Hewitson TD, Darby IA. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int Rev Cell Mol Biol. 2012;296:139–185. | |
Grunewald M, Avraham I, Dor Y, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124(1):175–189. | |
Jin DK, Shido K, Kopp HG, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12(5):557–567. | |
Zhang X, Liu L, Wei X, et al. Impaired angiogenesis and mobilization of circulating angiogenic cells in HIF-1alpha heterozygous-null mice after burn wounding. Wound Repair Regen. 2010;18(2): 193–201. | |
Botusan IR, Sunkari VG, Savu O, et al. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci U S A. 2008;105(49):19426–19431. | |
Wetterau M, George F, Weinstein A, et al. Topical prolyl hydroxylase domain-2 silencing improves diabetic murine wound closure. Wound Repair Regen. 2011;19(4):481–486. | |
Zimmermann AS, Morrison SD, Hu MS, et al. Epidermal or dermal specific knockout of PHD-2 enhances wound healing and minimizes ischemic injury. PLoS One. 2014;9(4):e93373. | |
Takaku M, Tomita S, Kurobe H, et al. Systemic preconditioning by a prolyl hydroxylase inhibitor promotes prevention of skin flap necrosis via HIF-1-induced bone marrow-derived cells. PLoS One. 2012;7(8):e42964. | |
Kalucka J, Ettinger A, Franke K, et al. Loss of epithelial hypoxia-inducible factor prolyl hydroxylase 2 accelerates skin wound healing in mice. Mol Cell Biol. 2013;33(17):3426–3438. | |
Rezvani HR, Ali N, Serrano-Sanchez M, et al. Loss of epidermal hypoxia-inducible factor-1α accelerates epidermal aging and affects re-epithelialization in human and mouse. J Cell Sci. 2011;124(Pt 24): 4172–4183. | |
Reynolds LE, Conti FJ, Silva R, et al. alpha3beta1 integrin-controlled Smad7 regulates reepithelialization during wound healing in mice. J Clin Invest. 2008;118(3):965–974. | |
Heikkinen PT, Nummela M, Jokilehto T, Grenman R, Kähäri VM, Jaakkola PM. Hypoxic conversion of SMAD7 function from an inhibitor into a promoter of cell invasion. Cancer Res. 2010;70(14): 5984–5993. | |
McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006;281(34):24171–24181. | |
Falanga V, Qian SW, Danielpour D, Katz MH, Roberts AB, Sporn MB. Hypoxia upregulates the synthesis of TGF-beta 1 by human dermal fibroblasts. J Invest Dermatol. 1991;97(4):634–637. | |
Peyssonnaux C, Boutin AT, Zinkernagel AS, Datta V, Nizet V, Johnson RS. Critical role of HIF-1alpha in keratinocyte defense against bacterial infection. J Invest Dermatol. 2008;128(8):1964–1968. | |
Okumura CY, Hollands A, Tran DN, et al. A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J Mol Med (Berl). 2012;90(9):1079–1089. | |
Cowburn AS, Alexander LE, Southwood M, Nizet V, Chilvers ER, Johnson RS. Epidermal deletion of HIF-2alpha stimulates wound closure. J Invest Dermatol. 2014;134(3):801–808. | |
Russell JA. Management of sepsis. N Engl J Med. 2006;355(16): 1699–1713. | |
Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. | |
Zinkernagel AS, Johnson RS, Nizet V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J Mol Med (Berl). 2007;85(12):1339–1346. | |
Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586(Pt 17):4055–4059. | |
Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011;17(11):1391–1401. | |
Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–665. | |
Karhausen J, Haase VH, Colgan SP. Inflammatory hypoxia: role of hypoxia-inducible factor. Cell Cycle. 2005;4(2):256–258. | |
Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci U S A. 2004; 101(28):10440–10445. | |
Walmsley SR, Cowburn AS, Clatworthy MR, et al. Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (pVHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood. 2006;108(9):3176–3178. | |
Peyssonnaux C, Datta V, Cramer T, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005; 115(7):1806–1815. | |
Kim HY, Kim YH, Nam BH, et al. HIF-1alpha expression in response to lipopolysaccaride mediates induction of hepatic inflammatory cytokine TNFalpha. Exp Cell Res. 2007;313(9):1866–1876. | |
Walmsley SR, Print C, Farahi N, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201(1):105–115. | |
Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol. 2007;178(12):7516–7519. | |
Harris AJ, Thompson AA, Whyte MK, Walmsley SR. HIF-mediated innate immune responses: cell signaling and therapeutic implications. Hypoxia. 2014;2:47–58. | |
Kuhlicke J, Frick JS, Morote-Garcia JC, Rosenberger P, Eltzschig HK. Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like receptors TLR2 and TLR6 during hypoxia. PLoS One. 2007;2(12): e1364. | |
Rius J, Guma M, Schachtrup C, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807–811. | |
Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103(48): 18154–18159. | |
Xue J, Li X, Jiao S, Wei Y, Wu G, Fang J. Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKbeta independent of hydroxylase activity. Gastroenterology. 2010;138(2):606–615. | |
Kiss J, Mollenhauer M, Walmsley SR, et al. Loss of the oxygen sensor PHD3 enhances the innate immune response to abdominal sepsis. J Immunol. 2012;189(4):1955–1965. | |
Thiel M, Caldwell CC, Kreth S, et al. Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS One. 2007;2(9):e853. | |
Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000;60(5):1388–1393. | |
Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–975. | |
Zhong H, De Marzo AM, Laughner E, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59(22):5830–5835. | |
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. | |
Strofer M, Jelkmann W, Metzen E, Brockmeier U, Dunst J, Depping R. Stabilisation and knockdown of HIF – two distinct ways comparably important in radiotherapy. Cell Physiol Biochem. 2011;28(5): 805–812. | |
Semenza GL. Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med. 2003;54:17–28. | |
Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12(1):9–22. | |
Acker T, Diez-Juan A, Aragones J, et al. Genetic evidence for a tumor suppressor role of HIF-2alpha. Cancer Cell. 2005;8(2):131–141. | |
Henze AT, Acker T. Feedback regulators of hypoxia-inducible factors and their role in cancer biology. Cell Cycle. 2010;9(14):2749–2763. | |
Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23(9):1949–1956. | |
Henze AT, Riedel J, Diem T, et al. Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer Res. 2010;70(1):357–366. | |
Erez N, Milyavsky M, Eilam R, Shats I, Goldfinger N, Rotter V. Expression of prolyl-hydroxylase-1 (PHD1/EGLN2) suppresses hypoxia inducible factor-1alpha activation and inhibits tumor growth. Cancer Res. 2003;63(24):8777–8783. | |
Zhang Q, Gu J, Li L, et al. Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer Cell. 2009;16(5):413–424. | |
Su Y, Loos M, Giese N, et al. Prolyl hydroxylase-2 (PHD2) exerts tumor-suppressive activity in pancreatic cancer. Cancer. 2012;118(4): 960–972. | |
Chan DA, Kawahara TL, Sutphin PD, Chang HY, Chi JT, Giaccia AJ. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell. 2009;15(6): 527–538. | |
Bordoli MR, Stiehl DP, Borsig L, et al. Prolyl-4-hydroxylase PHD2- and hypoxia-inducible factor 2-dependent regulation of amphiregulin contributes to breast tumorigenesis. Oncogene. 2011;30(5): 548–560. | |
Heindryckx F, Kuchnio A, Casteleyn C, et al. Effect of prolyl hydroxylase domain-2 haplodeficiency on the hepatocarcinogenesis in mice. J Hepatol. 2012;57(1):61–68. | |
Kamphues C, Wittschieber D, Klauschen F, et al. Prolyl hydroxylase domain 2 protein is a strong prognostic marker in human gastric cancer. Pathobiology. 2012;79(1):11–17. | |
Kato H, Inoue T, Asanoma K, Nishimura C, Matsuda T, Wake N. Induction of human endometrial cancer cell senescence through modulation of HIF-1alpha activity by EGLN1. Int J Cancer. 2006;118(5): 1144–1153. | |
Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359(25):2685–2692. | |
Klotzsche-von Ameln A, Muschter A, Mamlouk S, et al. Inhibition of HIF prolyl hydroxylase-2 blocks tumor growth in mice through the antiproliferative activity of TGFβ. Cancer Res. 2011;71(9):3306–3316. | |
Klotzsche-von Ameln A, Muschter A, Heimesaat MM, Breier G, Wielockx B. HIF prolyl hydroxylase-2 inhibition diminishes tumor growth through matrix metalloproteinase-induced TGFβ activation. Cancer Biol Ther. 2012;13(4):216–223. | |
Hung SP, Yang MH, Tseng KF, Lee OK. Hypoxia-induced secretion of TGF-β1 in mesenchymal stem cell promotes breast cancer cell progression. Cell Transplant. 2013;22(10):1869–1882. | |
Leite de Oliveira R, Deschoemaeker S, Henze AT, et al. Gene-targeting of Phd2 improves tumor response to chemotherapy and prevents side-toxicity. Cancer Cell. 2012;22(2):263–277. | |
Mazzone M, Dettori D, Leite de Oliveira R, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136(5):839–851. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.