")
Back to Journals » Biologics: Targets and Therapy » Volume 10
Therapeutic efficacy of monthly subcutaneous injection of daclizumab in relapsing multiple sclerosis
Authors Cohan S
Received 26 February 2016
Accepted for publication 18 May 2016
Published 12 September 2016 Volume 2016:10 Pages 119—138
DOI https://doi.org/10.2147/BTT.S89218
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Stanley Cohan1,2,3
1Providence Multiple Sclerosis Center, 2Providence Brain and Spine Institute, 3Providence Health & Services, Portland, OR, USA
Abstract: Despite the availability of multiple disease-modifying therapies for relapsing multiple sclerosis (MS), there remains a need for highly efficacious targeted therapy with a favorable benefit–risk profile and attributes that encourage a high level of treatment adherence. Daclizumab is a humanized monoclonal antibody directed against CD25, the α subunit of the high-affinity interleukin 2 (IL-2) receptor, that reversibly modulates IL-2 signaling. Daclizumab treatment leads to antagonism of proinflammatory, activated T lymphocyte function and expansion of immunoregulatory CD56bright natural killer cells, and has the potential to, at least in part, rectify the imbalance between immune tolerance and autoimmunity in relapsing MS. The clinical pharmacology, efficacy, and safety of subcutaneous daclizumab have been evaluated extensively in a large clinical study program. In pivotal studies, daclizumab demonstrated superior efficacy in reducing clinical and radiologic measures of MS disease activity compared with placebo or intramuscular interferon beta-1a, a standard-of-care therapy for relapsing MS. The risk of hepatic disorders, cutaneous events, and infections was modestly increased. The monthly subcutaneous self-injection dosing regimen of daclizumab may be advantageous in maintaining patient adherence to treatment, which is important for optimal outcomes with MS disease-modifying therapy. Daclizumab has been approved in the US and in the European Union and represents an effective new treatment option for patients with relapsing forms of MS, and is currently under review by other regulatory agencies.
Keywords: daclizumab, disease-modifying therapy, humanized monoclonal antibody, relapsing multiple sclerosis, therapeutic use
Introduction
Multiple sclerosis (MS) is a chronic, autoimmune, inflammatory, and degenerative disorder of the brain, optic nerves, and spinal cord.1 MS affects an estimated 2.3 million people worldwide, with a global median prevalence of 33 cases per 100,000 people.2 The prevalence is highest in North America and Europe.2 MS is most commonly diagnosed between the ages of 20 and 40 years, affects approximately two to three times as many women as men, and is the most frequent chronic disabling neurologic disease in young adults in the industrialized world.2,3
In the relapsing form of MS (RMS), autoreactive lymphoid cells directed against central nervous system (CNS) antigens are activated in the periphery and subsequently migrate into the CNS, producing multifocal inflammatory lesions that result in demyelination and axonal transection, thereby giving rise to the characteristic acute or subacute neurologic symptoms of a relapse.1 Relapses are transient and followed by complete or incomplete clinical recovery, which may result from resolution of inflammatory activity and a variable degree of remyelination.1,4 The use of corticosteroids during relapses may shorten the duration and/or the intensity of inflammation.3,5 If left untreated, recurring relapses and residual damage can result in worsening sustained neurological disability as a consequence of irreversible axonal damage and neurodegeneration, as well as significant reduction in health-related quality of life3,6,7 and shortened life expectancy.8
The interferon beta products (eg, Betaseron®/Betaferon® [Bayer HealthCare Pharmaceuticals Inc., Montville, NJ, USA/Bayer Pharma AG, Berlin, Germany], Avonex® [Biogen, Cambridge, MA, USA], Rebif® [EMD Serono, Rockland, MA, USA], Extavia® [Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA]) and glatiramer acetate (Copaxone® [Teva Pharmaceuticals, North Wales, PA, USA]), which were approved by the US Food and Drug Administration (US FDA) or the European Medicines Agency (EMA) between 1993 and 2002, reduced the frequency of relapses in clinical studies by ~30% compared with placebo over a 2- to 3-year period of observation.9–12 Interferon beta-1a (Avonex and Rebif) also was shown to reduce the risk of confirmed disability worsening.9,11 These products, which appear to have broad-spectrum immune-modulating properties,13 were the first proven disease-modifying therapies (DMTs) for the chronic treatment of RMS. Mitoxantrone (Novantrone®; EMD Serono), an antineoplastic agent that damages and prevents the repair of DNA and interferes with RNA function, was approved for the treatment of aggressive relapsing–remitting, secondary-progressive, and progressive-relapsing MS in the early 2000s, but it has largely fallen out of use because of limitations in dosage and duration of use due to its potential for serious cardiac toxicity and a heightened risk of acute myeloid leukemia.14
The use of more targeted therapies that regulate immune pathways mediating CNS damage in RMS was first established with the approval of natalizumab (Tysabri®; Biogen), a humanized monoclonal antibody that selectively inhibits inflammatory cell migration into the CNS.15 In clinical studies, natalizumab resulted in a robust therapeutic response, significantly reducing relapse risk, magnetic resonance imaging (MRI) measures of disease activity, and risk of confirmed disability worsening.16,17 Natalizumab also was effective on composite measures of disease activity; in a retrospective analysis of the AFFIRM study, more than 30% of natalizumab-treated patients showed no evidence of radiologic or clinical disease activity (no evidence of disease activity [NEDA]) during 2 years of treatment.18
Three oral agents were subsequently approved by the FDA and EMA as MS DMTs. The first oral agent approved was fingolimod (Gilenya® ; Novartis AG, Basel, Switzerland), a sphingosine-1-phosphate receptor modulator. A putative mechanism of action in MS is retention of reactive memory lymphocytes in lymphoid organs.19 This was followed by delayed-release dimethyl fumarate (Tecfidera® ; Biogen), which in RMS may increase tolerance to the oxidative stress of inflammation by promoting de novo synthesis of oxidative quenching species.20 Teriflunomide (Aubagio® ; Genzyme Corporation, Cambridge, MA, USA) is reported to reduce the population of activated lymphocytes by blocking mitochondrial pyrimidine synthesis.21 Each of these oral agents reduces risk of relapse, disease activity as observed on MRI, and risk of confirmed disability worsening.22–28
A more recently approved MS DMT is alemtuzumab (Lemtrada®; Genzyme Corporation), an anti-CD52 monoclonal antibody that results in the sustained depletion of circulating T and B lymphocytes.29 Alemtuzumab results in reduced risk of relapse activity, disease activity on MRI, and risk of confirmed disability worsening.30,31 Ocrelizumab (Roche, Basel, Switzerland) is unique among the MS DMTs because it has shown significant treatment benefits in Phase III clinical studies in both RMS versus an active comparator and in primary-progressive MS versus placebo.32 Ocrelizumab is an anti-CD20 monoclonal antibody that causes selective and sustained depletion of CD20 B lymphocytes and has been submitted for regulatory approval as an MS DMT.32,33
While many of the recent therapeutic developments for MS were driven by a greater understanding of immune system function, such as the regulatory role of the α 4-integrin receptor subunit (blocked by natalizumab) on the ingress of activated peripheral lymphocytes and monocytes into the CNS34 and the sequestration of activated lymphocytes in peripheral lymphoid organs (enhanced by fingolimod),19 these agents also are nonselective in the sense that they exploit the inhibition of aspects of overall general immune responsivity as a mechanism to block autoimmunity. One potential consequence of broad inhibition of immune system response is increased risk of infection, including opportunistic infection, which has been encountered with some MS DMTs.35–38
An alternative therapeutic approach is to attempt to reestablish equilibrium between autoimmunity and self-tolerance as a means of controlling the abnormal autoreactive state driving MS. Daclizumab, a humanized monoclonal antibody that blocks the α subunit (CD25) of the interleukin 2 (IL-2) receptor (IL-2R), has the potential to restore the homeostasis between autoimmunity and autotolerance that is characteristic of the nondisease state.39,40 Daclizumab has been shown to reduce the expansion of proinflammatory, actived T lymphocytes, and it may simultaneously promote autotolerance through the expansion of CD56bright natural killer (NK) cells, and at least partially preserve the regulatory T cell (TREG) population and function.40 Daclizumab high-yield process (daclizumab [Zinbryta™ [Biogen, Cambridge, MA, USA and AbbVie Inc., North Chicago, IL, USA]]) was approved for noninvestigational use in the US and in the European Union as a once-monthly subcutaneous (SC) injection for the treatment of RMS in May and July 2016, respectively. As described in more detail in the following section, daclizumab reduced relapse risk, disease activity as monitored by MRI, and risk of confirmed disability worsening in pivotal controlled clinical studies.41,42
Proof of concept and the clinical development program
A previous form of daclizumab was developed as an intravenous (IV) therapy (Zenapax®; Hoffmann-La Roche Ltd, Nutley, NJ, USA) and was used as part of a prophylactic drug regimen in patients with a renal transplant to prevent acute organ rejection and for prevention of severe sight-threatening intraocular inflammation in patients with uveitis.39 Because RMS and solid-organ graft rejection are both associated with pathologically heightened effector T lymphocyte activity, it was hypothesized that daclizumab could be of clinical benefit in patients with active RMS.39,43 Subsequent small-scale open-label clinical studies showed promising results.43–46 Daclizumab was reformulated as an SC injection and tested as an adjunct to interferon beta in the Phase II, multicenter, randomized, double-blind CHOICE study, which assessed its efficacy and safety in patients with active RMS.47 CHOICE demonstrated that daclizumab as an add-on therapy with interferon beta reduced disease activity on MRI compared with interferon beta alone.47
Based on these positive results, daclizumab high-yield process was subsequently developed. Daclizumab high-yield process has an identical amino acid sequence and the same antigen-binding domain as earlier forms of daclizumab, but differs in its glycosylation profile, leading to altered binding at the Fc receptor. This new form of daclizumab has been associated with less antibody-dependent cell-mediated cytotoxicity compared with earlier forms of daclizumab.48 In the multicenter, randomized, double-blind, placebo-controlled (SELECT) and active-comparator (DECIDE) clinical studies, daclizumab monotherapy, administered SC every 4 weeks, resulted in statistically significant reductions in both clinical and MRI indicators of disease activity in patients with RMS.41,42 These studies are described in detail in a later section of this review.
The clinical pharmacology, efficacy, and safety of SC daclizumab has been evaluated extensively in Phase I pharmacokinetic studies;48 an open-label safety, immunogenicity, pharmacokinetic, and pharmacodynamic study;49,50 and randomized, double-blind, placebo- and active-controlled studies, and their extension phases (Table 1 and Figure 1).41,42,51–53
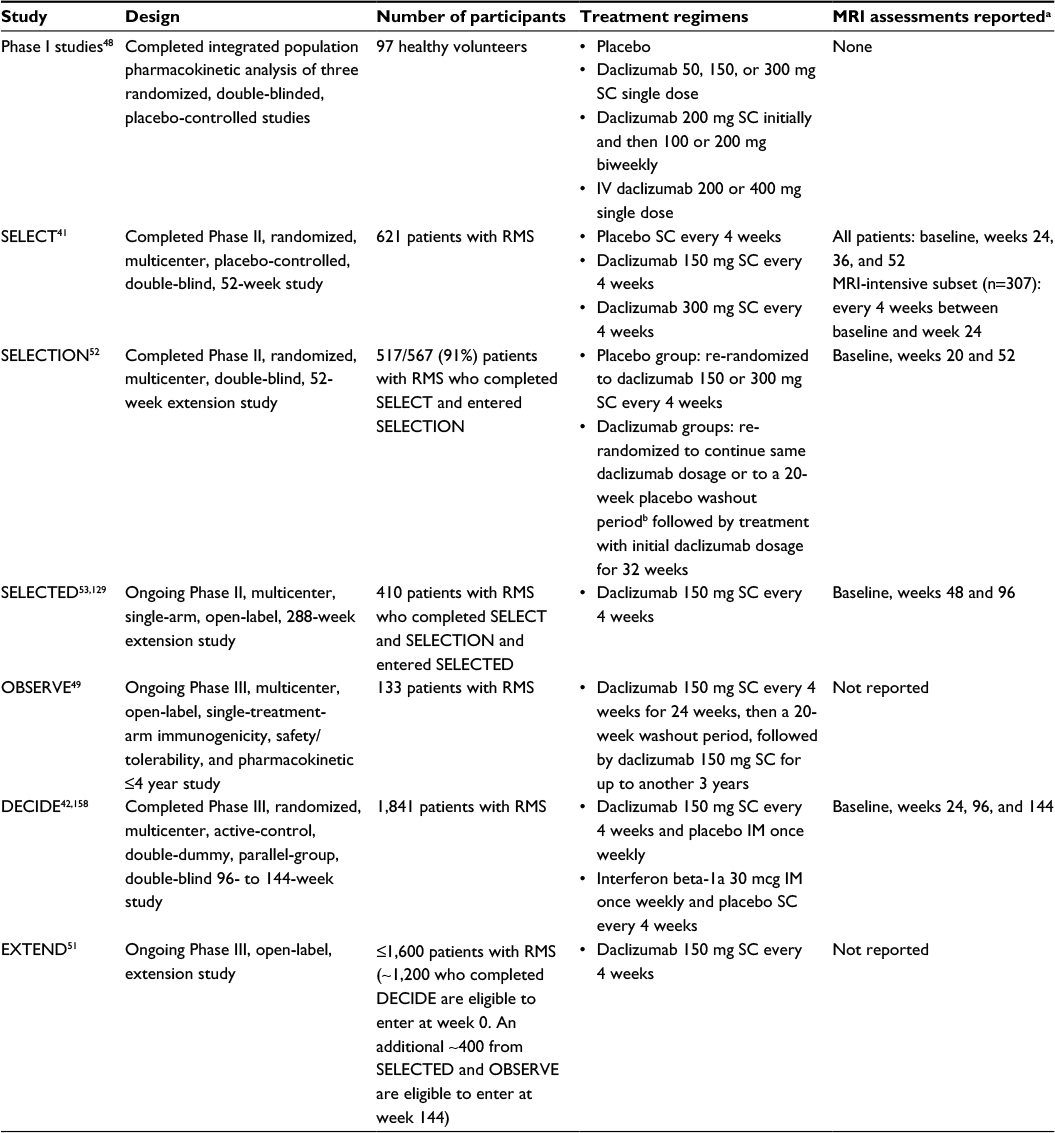 | Table 1 Overview of completed and ongoing daclizumab clinical studies Notes: aTiming of MRI assessments for each study that have been reported in the literature. bIn the placebo washout group, the total time between the last dose of daclizumab in SELECT and reinitiation in SELECTION was 24 weeks. Abbreviations: MRI, magnetic resonance imaging; SC, subcutaneous; IV, intravenous; RMS, relapsing multiple sclerosis; IM, intramuscular. |
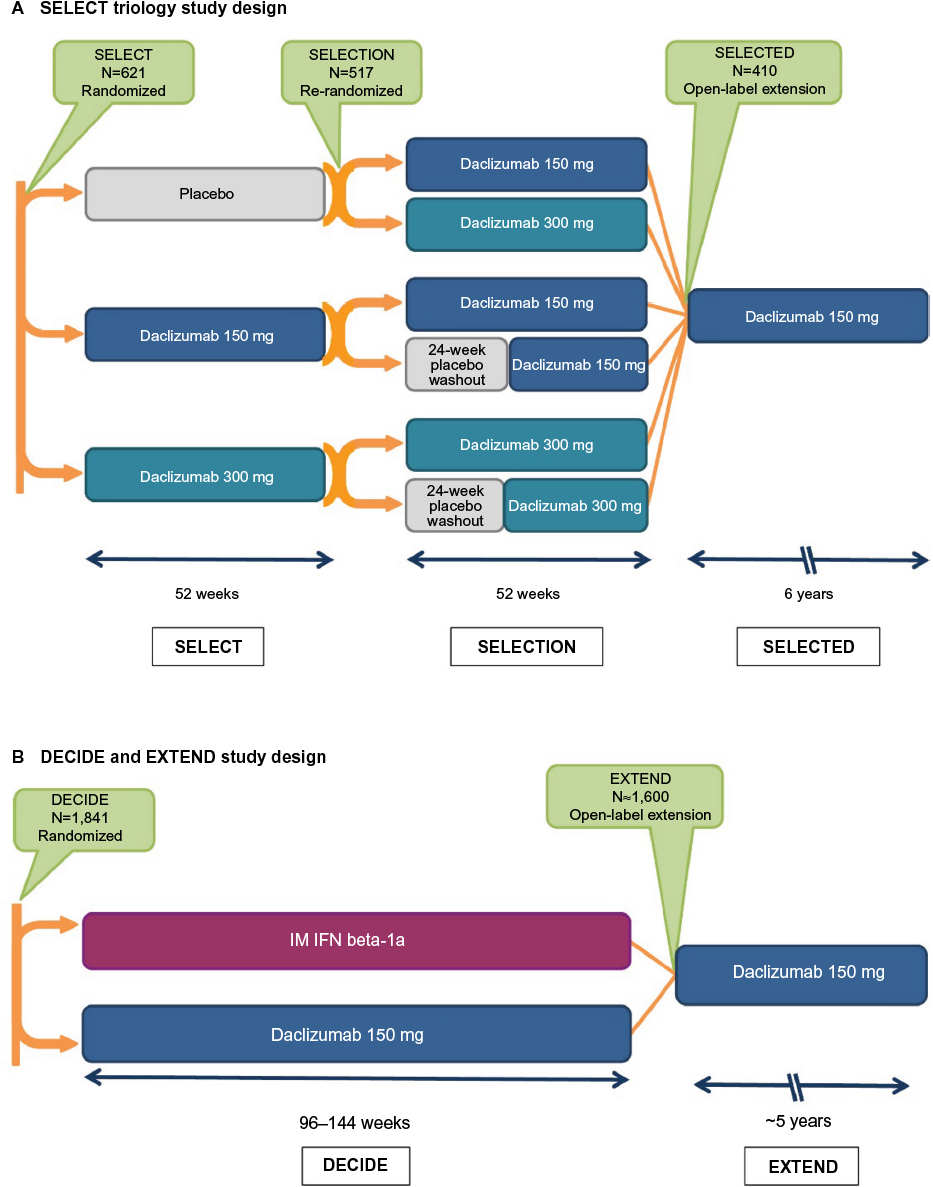 | Figure 1 The SELECT and DECIDE studies and their extension studies.41,42,51–53,129 Notes: (A) In SELECT, patients with RMS were randomized to receive daclizumab 150 or 300 mg subcutaneously every 4 weeks or placebo for 52 weeks.41 Patients who completed SELECT were eligible to enroll in the SELECTION extension study. In SELECTION, patients from the placebo group in SELECT were re-randomized to receive daclizumab 150 or 300 mg subcutaneously every 4 weeks for 52 weeks; patients from the daclizumab 150 and 300 mg treatment arms in SELECT were re-randomized to continue their assigned dosage of daclizumab or to 20 weeks of placebo treatment followed by reinitiation of their previously assigned dosage of daclizumab for 32 weeks.52 In the placebo washout group, the period of time between the last daclizumab dose in SELECT and reinitiation of daclizumab in SELECTION was a total of 24 weeks.52 Patients who completed SELECTION were eligible to enroll in the ongoing open-label SELECTED study in which all patients received daclizumab 150 mg for up to 6 years.53,129 (B) In DECIDE, patients were randomized to receive daclizumab 150 mg subcutaneously every 4 weeks or IFN beta-1a 30 mcg IM once weekly for a minimum of 96 weeks up to a maximum of 144 weeks; the study ended when the last enrolled patient completed the week 96 visit.42 Patients who completed DECIDE were eligible to enroll in the ongoing EXTEND safety extension study in which all patients received daclizumab 150 mg for up to 6 years of total time on treatment.51 Abbreviations: IM, intramuscular; IFN, interferon; RMS, relapsing multiple sclerosis. |
Pharmacology
Mechanism of action
The putative mechanism of action for daclizumab is unique compared with other approved DMTs for the treatment of RMS. As described earlier, daclizumab is a monoclonal antibody that binds to the α subunit (CD25) of the IL-2R expressed on activated T lymphocytes, thereby preventing interaction of IL-2 with its high-affinity receptor and inhibiting the activation of CNS antigen-experienced, proinflammatory, activated T lymphocytes,39,54–57 one of the main drivers of MS pathogenesis.1
The IL-2/IL-2R pathway also has an important role in maintaining the homeostatic balance between proinflammatory autoimmunity and anti-inflammatory autotolerance.40 The cytokine IL-2 is a lymphocyte growth factor produced primarily by activated CD4+ T lymphocytes, and to a lesser degree by CD8+ T lymphocytes, dendritic cells, and NK cells.58 Antigen presentation to T lymphocytes promotes both IL-2 production and secretion by these antigen-activated T lymphocytes and also upregulates IL-2R expression on T lymphocytes.59–61 The IL-2/IL-2R interaction drives T lymphocyte clonal expansion when an immune response is mounted, as is the case in autoimmune conditions such as MS.62 Production of IL-2 is generally short lived, and IL-2 levels are regulated by an autoinhibitory feedback loop such that IL-2 binding to IL-2R activates secondary intracellular messenger systems to repress the IL2 gene.63–65 This process serves to reduce or prevent prolonged T lymphocyte–driven inflammatory responses. IL-2 signaling through IL-2R also is necessary for the support and function of TREG cells, which are required for the maintenance of autotolerance.66–72 Thus, the IL-2/IL-2R system provides a mechanism for both upregulation of proinflammatory responses and maintenance of immune homeostasis.
Evidence suggests that MS may result, in part, from a breakdown in immune self-tolerance due to abnormalities in the IL-2/IL-2R signaling pathway, resulting in an excess of peripherally activated CNS antigen-primed T lymphocytes.40 For example, polymorphisms in the CD25 gene are associated with MS susceptibility, directly implicating IL-2 signaling in MS pathogenesis.73,74 These polymorphisms also are associated with increased CD25 expression on naive CD4+ T lymphocytes.75 Levels of soluble CD25, a marker of IL- 2-driven T lymphocyte proliferation,76 also are elevated in MS,77 and have been linked with MS severity.78
IL-2 signaling is mediated through two IL-2R isoforms, which differ in their affinity for IL-2 and expression across different immune cell types.79 On the surface of activated T lymphocytes and TREG cells, IL-2 signaling is mediated via the high-affinity isoform that is composed of the IL-2-capturing α subunit (CD25), which is expressed transiently following antigen activation in T lymphocytes and constitutively in TREG cells, and two signaling subunits, CD132 and CD122 (the γ and β chains, respectively; Figure 2).58,70,79–81 IL-2 binding to CD25 promotes the association with CD122 and CD132, resulting in the formation of the quaternary high-affinity receptor complex.79,82 CD25 functions solely to increase the affinity of IL-2R for IL-2 and has no known signaling function.58,79 Thus, the intracellular transmission of the IL-2 signal is dependent on the cytoplasmic tails of CD122 and CD132.83 In contrast, the intermediate-affinity IL-2R, composed only of CD122 and CD132, is found on resting T and B lymphocytes and cytotoxic immunoregulatory CD56bright NK cells, and binds IL-2 with an affinity significantly lower than the high-affinity CD25-containing IL-2R (Figure 2).79
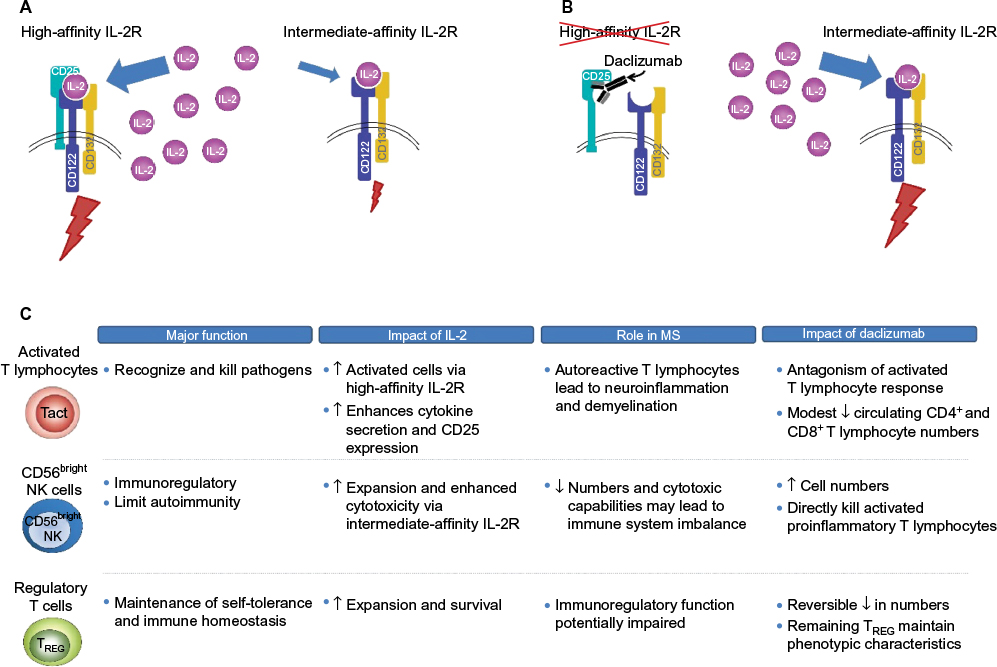 | Figure 2 Proposed mechanism of action of daclizumab and effects on key immune cell populations. Notes: (A) IL-2 binds with higher affinity to the heterotrimeric IL-2R complex composed of CD25, CD122, and CD132 than to the heterodimeric intermediate-affinity IL-2R composed of the CD122 and CD132 subunits.79 (B) Daclizumab binds to CD25 and prevents formation of the high-affinity receptor complex, which increases the availability of IL-2 to bind and signal through the intermediate-affinity receptor complex.55,84,85 (C) Blockade of CD25 has effects on several different immune cell populations in patients with MS, most notably proinflammatory, activated T lymphocytes, CD56bright NK cells, and TREG cells.40,41,54,86–89,107,159 Abbreviations: IL-2R, interleukin 2 receptor; IL-2, interleukin 2; MS, multiple sclerosis; Tact, activated T cell; NK, natural killer; TREG, regulatory T cell. |
Daclizumab binds selectively and with greater affinity than IL-2 to CD25 expressed on effector T lymphocytes (dissociation constants, 0.27 and 10 nM, respectively), thus blocking the assembly of the high-affinity IL-2R and preventing IL-2 signal transmission via this route (Figure 2).84,85 Daclizumab does not block IL-2 signaling via the intermediate-affinity IL-2R on resting T lymphocytes and other immune cells.84,86 The primary overall effects of CD25 blockade by daclizumab in patients with RMS are antagonism of proinflammatory, activated T lymphocytes, expansion of CD56bright NK cells, and a reversible reduction in TREG cell numbers (Figure 2).54,86–89
Daclizumab, by inhibiting the formation of the high-affinity IL-2R, results in an increase in the amount of IL-2 available for interaction with the intermediate-affinity IL-2R expressed on CD56bright NK cells,55,79 to which daclizumab cannot bind due to the absence of CD25.39,79 Unimpeded activation of the intermediate-affinity IL-2R activates and induces the expansion of CD56bright NK cells,41,44,54–56,90 which can penetrate the blood–brain barrier where they recognize, establish direct contact with, and cause lysis of proinflammatory, activated T lymphocytes, while leaving resting T lymphocytes intact.54,90,91 CD25 blockade also may have indirect effects on proinflammatory T lymphocyte responses by inhibiting trans-presentation of CD25 by dendritic cells to the IL-2R on resting T lymphocytes, further preventing the formation of the high-affinity IL-2R.57 The effects of daclizumab on activated T lymphocytes and CD56bright NK cells are fully reversible after treatment is stopped.54,87,89
As noted earlier, TREG cells express the high-affinity CD25-containing IL-2R, and the function of TREG cells is believed to be dependent upon IL-2/IL-2R interaction.66–69,71 If this is the case, one might anticipate potential worsening of autoimmunity upon CD25 blockade, but just the opposite is observed.40 Daclizumab has demonstrated efficacy in reducing MS disease activity, even though treatment with daclizumab results in a reversible reduction in TREG cell numbers of ~50%.86,89 Several lines of evidence may provide at least partial answers to this seeming paradox. Expression of the TREG cell phenotype is largely defined by the presence of FOXP3+ expression in CD4+ T lymphocytes.92,93 The signal that activates the FOXP3 gene is transmitted via the γ (CD132) chain of IL-2R.68 Thus, FOXP3 gene activation by IL-2 via the intermediate-affinity receptor may serve to maintain and support the TREG population.94,95 In addition, the CD132 γ chain, which is found in multiple cytokine receptors, also may be stimulated by other cytokines, such as IL-4, IL-7, IL-9, and IL-21.94 Daclizumab does not result in TREG cell depletion, and evidence suggests that the TREG cells that remain preserve the normal TREG lineage and phenotype.86 In this scenario, daclizumab reduces activated T lymphocyte support by IL-2 by blocking CD25 bioavailability, while increasing NK cell activation and maintaining TREG cell function and phenotype via the still-available intermediate- and low-affinity receptor subunits of IL-2R and other cytokine receptors.55,86
Lymphoid tissue inducer (LTi) cells are a proinflammatory subset of lymphocytes that appear to originate from hematopoetic CD34+ precursor cells.96,97 Among the proposed actions of LTi cells is the promotion of ectopic lymphoid follicles, as are found in the meninges of patients with MS.97–99 The findings of two studies suggest that daclizumab therapy in patients with RMS was associated with a decrease in circulating LTi cells and a decrease in the cerebrospinal fluid content of CXCL13, a chemokine produced by CNS lymphoid aggregates.97,100 The reported decrease in LTi cells was hypothesized to be a consequence of increased stimulation of the intermediate-affinity IL-2R due to CD25 blockade by daclizumab, driving its progenitor cells toward the formation of CD56bright NK cells at the expense of the LTi lineage.97,100 In the past few years, studies have shown that innate lymphoid cells are more diverse than previously appreciated.101 More recently published data do not confirm the previously reported reduction in LTi cells by daclizumab, and may be a result of different analytic methods used in these studies to identify innate lymphoid cell subsets.102 The findings from this recent study confirmed that circulating CD56bright NK cells were increased in patients with RMS treated with daclizumab, but there was no apparent effect of daclizumab on circulating innate lymphoid cell subpopulations, including LTi cells.102
Pharmacokinetics
Four studies evaluated the pharmacokinetics of IV daclizumab and SC daclizumab in healthy individuals and patients with RMS.48,50 In general, there were no clinically meaningful differences between the two populations in the pharmacokinetic profile of daclizumab, which was characterized by slow clearance, linear pharmacokinetics (doses ≥100 mg), high SC bioavailability (>80%), and an elimination half-life suitable for monthly administration (~3 weeks).48,50 Daclizumab had a low volume of distribution of 6.4 L in healthy volunteers, which is to be expected for an immunoglobulin G1 monoclonal antibody that is primarily confined to the vascular and interstitial spaces.48
Daclizumab best fits a two-compartment pharmacokinetic model with first-order absorption and elimination, and this finding was used to predict its steady-state pharmacokinetic profile in Phase III studies.48 Steady-state serum concentrations of daclizumab were attained by week 16 in 26 patients with RMS who received daclizumab 150 mg SC every 4 weeks for 20 weeks (total of six doses) in the Phase III OBSERVE study (NCT01462318).50 Maximum observed concentration (Cmax) was achieved after a median of 7 and 5 days for doses 1 and 6, respectively, with a mean steady-state serum peak-to-trough concentration ratio of ~2 (Table 2).50 There are no known adverse drug–drug interactions in patients treated with daclizumab. In a recently reported study of the effects of daclizumab on the pharmacokinetics of drugs metabolized by a range of hepatic cytochrome P450 isoenzymes, no interaction was seen, co-administration neither inhibiting nor inducing enzymatic activities, and the serum kinetics of medications characteristically metabolized by these isoenzymes were not affected.103
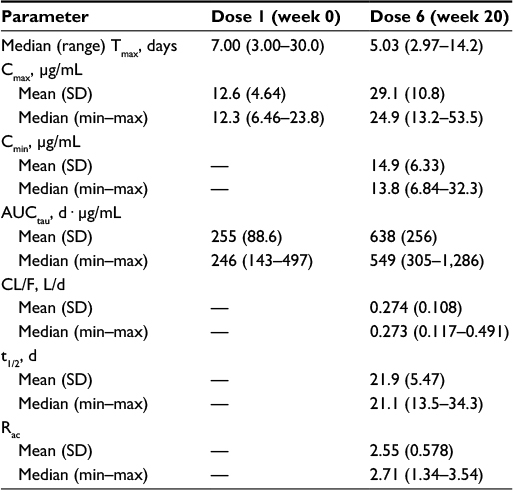 | Table 2 Pharmacokinetic parameters of daclizumab after administration of daclizumab 150 mg subcutaneously every 4 weeks in patients with RMS50 Notes: Reprinted from Tran et al,50 copyright (2016), with permission from Dove Medical Press Ltd. Abbreviations: RMS, relapsing multiple sclerosis; Tmax, time to maximum observed concentration; Cmax, maximum observed concentration; SD, standard deviation; min, minimum; max, maximum; Cmin, minimum observed concentration; AUCtau, area under the concentration–time curve within a dosing interval; CL/F, apparent clearance; L, liter; t1/2, elimination half-life; Rac, accumulation ratio. |
Pharmacodynamics
Daclizumab has a well-defined pharmacodynamic profile, demonstrating rapid saturation of CD25 on peripheral blood T lymphocytes within a few hours after SC administration that persisted throughout the 4-week dosing interval, and more slowly reversed following treatment discontinuation.104 Unbound CD25 returned to pretreatment levels within 4–6 months after the last daclizumab dose due to its long half-life of elimination.104 The longevity of this effect suggests that delayed or isolated missed doses of daclizumab due to patient nonadherence may have minimal to no adverse impact upon clinical efficacy.
Total CD4+ and CD8+ T lymphocyte counts were modestly reduced in daclizumab-treated patients in the SELECT (mean reductions of 7%–10% over 52 weeks) and DECIDE (median reductions of 15%–18% over 96 weeks) studies.41,105 Despite reductions in each of these T lymphocyte populations, the ratio of the number of CD4+ T lymphocytes to CD8+ T lymphocytes remained stable during daclizumab treatment in both studies.41,105 Furthermore, no association was observed between CD4+ or CD8+ T lymphocyte counts and the occurrence of infections in daclizumab-treated patients in DECIDE.105 In contrast, activated effector CD4+ T lymphocytes that express HLA-DR2, a haplotype associated with susceptibility to MS,106 were reduced by ~25% during the treatment period in daclizumab-treated patients with active RMS in CHOICE,107 suggesting a selective reduction in activated CD4+ T lymphocytes.
Serum IL-2 levels increased approximately twofold within 4 weeks of daclizumab administration and stabilized thereafter with continued treatment.89 Accompanying the increased IL-2 levels was a rapid, up to fivefold, increase in the number of CD56bright NK cells, reaching a plateau within 52 weeks that was sustained with continued treatment54,87,89 and subsequently decreased during a placebo washout period.89 A significant increase in the number of CD56bright NK cells also was observed in cerebrospinal fluid after IV daclizumab administration.90 Notably, higher CD56bright NK cell counts at week 4 were associated with lower numbers of new or newly enlarging T2 hyperintense lesions over 52 weeks (P<0.001).87 During the first year of treatment, both the lowest and highest quartiles of peripheral blood CD56bright NK counts at week 8 were associated with fewer new or newly enlarging T2 hyperintense lesions between weeks 24 and 52 (63% and 86%, respectively) compared with placebo (P<0.0001 for each quartile comparison).87 The observed reduction in MRI measures of disease activity may be the direct result of the cytotoxic effects of CD56bright NK cells upon activated T lymphocytes.108 However, even in daclizumab-treated patients who experienced no expansion of CD56bright NK cell numbers, there was a 54% reduction in new or newly enlarging T2 hyperintense lesions over 52 weeks versus the placebo group, emphasizing that mechanisms other than the expansion of CD56bright NK cells also contribute to the reduction in MS disease activity.87 The predictive value of CD56bright NK cell counts measured during the first 4 or 8 weeks after daclizumab initiation upon disease activity during the first 52 weeks of observation was not seen in the second year of daclizumab therapy.87
In addition, TREG cells express CD25, and reductions in TREG cell counts of ~50% that persisted throughout the duration of treatment have been observed in daclizumab-treated patients.86,88 The effect of daclizumab on TREG cell counts is reversible on treatment discontinuation and nondepleting because remaining TREG cells maintain the TREG function and phenotype (ie, do not acquire effector CD4+ T lymphocyte characteristics).86,89 No association has been found to date between daclizumab-mediated declines in TREG cell numbers and clinical efficacy during treatment.86 Daclizumab shows continued efficacy in RMS despite reducing the number of TREG cells.86 This reduction in TREG cell numbers may in part be compensated for by daclizumab-induced expansion of CD56bright NK cells, the latter playing a significant role in restoring immune tolerance through their destruction of proinflammatory T cells,54,91 and thus may be a major mediator of the effects of daclizumab on RMS disease activity.
Efficacy
Pivotal studies
Two large double-blind controlled studies compared daclizumab 150 mg SC every 4 weeks with placebo over 52 weeks (SELECT)41 and with intramuscular (IM) interferon beta-1a 30 mcg once weekly for a minimum of 96 weeks and up to a maximum of 144 weeks (DECIDE; Table 1 and Figure 1).42 Eligible patients were adults (18–55 years of age) with documented relapsing–remitting MS according to the 2005 McDonald criteria.109 The primary endpoint in both SELECT and DECIDE was the annualized relapse rate (ARR), where relapse was defined as new or recurrent neurological symptoms not associated with fever or infection, lasting 24 hours or more, and accompanied by new or worsened objective neurological findings at assessment.41,42 Secondary and tertiary outcomes included the impact of daclizumab on confirmed disability worsening, MRI lesion burden and activity, patient-reported outcomes, safety, and immune system activity markers. SELECT (and its extension study, SELECTION; Figure 1) also included a daclizumab 300 mg treatment group, but because efficacy outcomes were similar between the 150 and 300 mg dose groups, only the 150 mg dose was evaluated in subsequent pivotal studies in MS.110 Thus, this review focuses on results from treatment with daclizumab 150 mg.
The primary endpoint was met in both SELECT and DECIDE; ARR was reduced significantly by daclizumab 150 mg compared with placebo over 52 weeks in SELECT (54% reduction; P<0.0001) and compared with IM interferon beta-1a over 144 weeks in DECIDE (45% reduction; P<0.001; Table 3).41,42 In DECIDE, daclizumab also significantly reduced the annualized rate of severe/serious relapses (38% reduction; P=0.002).42 In SELECT, there was a 57% reduction in 12-week confirmed disability worsening with daclizumab (P=0.021).41 In DECIDE, there was a 16% difference in the percentage of patients with 12-week confirmed disability worsening that favored daclizumab, but the effect was not statistically significant (P=0.16).42 However, there was an imbalance between treatment arms in the number of patients censored who experienced disability worsening, but left the study before disability worsening could be confirmed 12 weeks later, which was considered to have impacted the test for statistical significance. Specifically, the primary analysis assumed that none of the censored patients had 12-week confirmed disability worsening. However, when all censored patients were instead assumed to have 12-week confirmed disability worsening, a 24% reduction in 12-week confirmed disability worsening was observed for daclizumab versus IM interferon beta-1a (P=0.016).42 When censored patients were assumed as either confirmed or not confirmed based on an imputation of the observed rate, a 21% reduction in 12-week confirmed disability worsening was observed that favored daclizumab (P=0.047).42 DECIDE also included assessment of 24-week confirmed disability worsening – considered by the EMA to be a more robust measure than 12-week confirmed disability worsening111 – as a preplanned tertiary endpoint using the imputed methodological approach.42 Daclizumab significantly reduced 24-week confirmed disability worsening by 27% compared with IM interferon beta-1a (P=0.03).42
 | Table 3 Clinical efficacy of daclizumab 150 mg given subcutaneously every 4 weeks in multicenter, randomized, double-blind, comparative studies of patients with RMS41,42 Notes: aResults are presented for efficacy analysis populations. Results from the daclizumab 300 mg treatment arm have been reported,41 but are not included in this table comparing efficacy outcomes of the 150 mg dose. In both the DECIDE and SELECT studies, between treatment group differences in ARR were evaluated using negative binomial regression models. Times to first relapse or to confirmed disability worsening and proportion of patients with relapse or confirmed disability worsening were estimated based on Kaplan–Meier survival distribution and between treatment-group differences were evaluated using Cox proportional-hazards models. Each of the models included adjustments for baseline factors. In the analysis of 24-week confirmed disability worsening in DECIDE, patients with a missing confirmatory assessment after a tentative progression were imputed using multiple imputation based on risk factors at time of tentative progression. The statistical models are detailed in the original articles.41,42 SELECT data adapted with permission from Elsevier (The Lancet, 2013, 381, 2167–75). Abbreviations: RMS, relapsing multiple sclerosis; ARR, annualized relapse rate; CI, confidence interval; NE, not evaluated; HR, hazard ratio; IM, intramuscular. |
Consistent with the effects of daclizumab on clinical endpoints, statistically significant effects were observed on MRI measures of disease activity in both SELECT and DECIDE (Table 4).41,42 At week 52 in SELECT and week 96 in DECIDE, daclizumab 150 mg significantly reduced the number of gadolinium-enhancing (Gd+) lesions (new Gd+ lesions in SELECT, P<0.0001, and all Gd+ lesions in DECIDE, P<0.001, respectively) and new or newly enlarging T2 hyperintense lesions (P<0.0001 and P<0.001, respectively).41,42 In DECIDE, the effects on both of these MRI lesion parameters were observed as early as the first MRI assessment at week 24 (P<0.001 for both measures),42 while in SELECT, the effect on new Gd+ lesions was seen as early as 4 weeks after initiating treatment, which was the time of the first post-baseline MRI.41 The mean percentage of brain volume loss, which may be indicative of neurodegeneration and tissue damage,112 was not significantly different between treatment groups after 1 year of treatment in SELECT, but was significantly different between the daclizumab and IM interferon beta-1a arms after 2 years of treatment in DECIDE (P<0.001; Table 4).41,42 A benefit for daclizumab over IM interferon beta-1a on brain volume loss also was observed during the intervals of baseline to week 24 (P=0.03) and weeks 24–96 (P<0.001).42 The effects of daclizumab across all MRI lesion outcomes were maintained in the subgroup of patients treated for 144 weeks.113,114
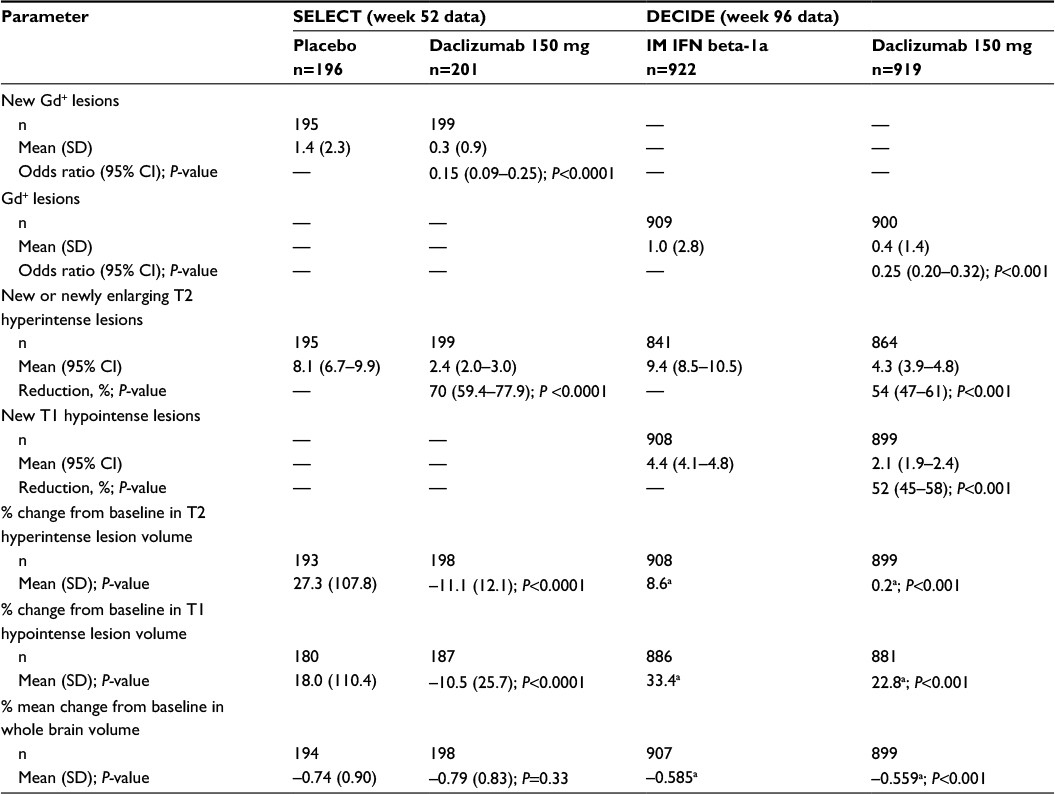 | Table 4 Neuroradiological efficacy of daclizumab 150 mg given subcutaneously every 4 weeks in multicenter, randomized, double-blind, comparative studies of patients with RMS41,42 Notes: Results are presented for patients in the efficacy analysis population who had a post-baseline scan.41,42 Results from the daclizumab 300 mg treatment arm have been reported,41x but are not included in this table comparing efficacy outcomes of the 150 mg dose. aSD not reported. In both the DECIDE and SELECT studies, between treatment group differences were evaluated using ordinal logistic regression models (new Gd+/Gd+ lesions), negative binomial regression models (new or newly enlarging T2 lesions, new T1 hypointense lesions), or analysis of covariance model based on ranks (T2 hyperintense and T1 hypointense lesion volume, whole brain volume). Each of the models included adjustments for baseline factors. The statistical models are detailed in the original articles.41,42 SELECT data adapted with permission from Elsevier (The Lancet, 2013, 381, 2167–75). Abbreviations: RMS, relapsing multiple sclerosis; IM, intramuscular; IFN, interferon; Gd+, gadolinium-enhancing; SD, standard deviation; CI, confidence interval. |
A post hoc subgroup analysis of SELECT revealed statistically significant reductions in ARR, new or newly enlarging T2 hyperintense lesions, and new Gd+ lesions with daclizumab compared with placebo in patients with highly active RMS, defined as at least two relapses in the year before randomization and at least one Gd+ lesion at baseline.115A benefit for daclizumab versus IM interferon beta-1a on relapse and MRI lesion activity in patients with highly active RMS also has been reported in DECIDE.116 In DECIDE, the clinical and MRI benefits of daclizumab were consistent across a number of predefined patient subgroups stratified by demographics and baseline MS disease characteristics, including patients with or without prior use of interferon beta.117
Additional post hoc analyses examined the proportion of patients with NEDA, defined as no relapses, no confirmed disability worsening, no new Gd+ lesions (SELECT) or no Gd+ lesions (DECIDE), and no new or newly enlarging T2 hyperintense lesions on brain MRI.18,118 In SELECT, a significantly higher percentage of patients receiving daclizumab 150 mg compared with placebo exhibited NEDA at week 52 (36% versus 11%; adjusted odds ratio 6.31; 95% confidence interval [CI] 3.59–11.11; P<0.0001).119 Similarly, in DECIDE, a significantly higher percentage of patients receiving daclizumab 150 mg compared with IM interferon beta-1a exhibited NEDA at week 96 (25% versus 14%; odds ratio 2.06; 95% CI 1.59–2.66; P<0.0001).118
Extension studies
Both SELECT and DECIDE are being followed up with extension phases, enabling evaluation of the long-term safety profile and persistence of daclizumab efficacy (Figure 1).51–53,120 Final results are available for SELECTION, a 1-year extension of SELECT,52 and interim 3-year results for SELECTED, an ongoing, long-term, open-label extension of SELECTION.53 In SELECTION, half of the patients who received daclizumab 150 mg SC in SELECT were randomized to continue daclizumab 150 mg SC for an additional year (Table 1 and Figure 1). Patients who completed SELECTION were then eligible to enroll in the SELECTED long-term extension study (Figure 1).53 For patients who received daclizumab 150 mg for 1 year in SELECT and continued to receive daclizumab 150 mg for an additional year in SELECTION, the ARR (0.148 and 0.165), the proportion of patients with relapse (14.7% and 13.6%), and the proportion of patients with 12-week confirmed disability worsening (6% and 5%) were similar in years 1 and 2 of treatment, respectively.52 In the subset of patients (n=94) in SELECTED who had received continuous daclizumab 150 mg since their initial enrollment in SELECT, efficacy outcomes were sustained in the third year of treatment.53 EXTEND is an ongoing open-label extension study of DECIDE, from which no interim data have yet been reported, that will provide additional information on the long-term safety and efficacy of daclizumab for up to 6 years of treatment.51
Patient-centered outcomes
Consistent with the effects of daclizumab on primary and secondary clinical and MRI endpoints in SELECT and DECIDE, significant benefits of daclizumab were observed on health-related quality-of-life measures.41,42 Patient-reported outcomes in both studies included the 29-item Multiple Sclerosis Impact Scale (MSIS-29), which is used to assess the impact of MS on physical (physical impact subscale [PHYS]) and psychological (psychological impact subscale) functioning,121 as well as the EuroQol 5-Dimensions (EQ-5D), which is used to assess general health status.122 The EQ-5D includes a descriptive component in which patients self-report their current level of difficulty on five dimensions of health (index score) and a valuation component in which patients self-rate their general health status on a visual analog scale (VAS).122
In SELECT, a statistically significant benefit for daclizumab 150 mg compared with placebo was observed on MSIS-29 PHYS subscale (P=0.00082), ED-5D index (P=0.0091), and EQ-5D VAS (P<0.0001) scores (Figure 3).41 In DECIDE, significantly greater mean improvements from baseline were observed at week 96 with daclizumab 150 mg compared with IM interferon beta-1a on MSIS-29 PHYS subscale (P<0.001), MSIS-29 psychological impact subscale (P=0.04), EQ-5D index (P=0.005), and EQ-5D VAS (P<0.001) scores (Figure 3).42 Clinically meaningful worsening on the MSIS PHYS subscale has been defined as a ≥7.5-point change from baseline,110 and this was evaluated post hoc in SELECT and as a secondary outcome in DECIDE.42,110 In SELECT, significantly fewer patients treated with daclizumab 150 mg compared with placebo exhibited clinically meaningful worsening on the MSIS-29 PHYS subscale at week 52 (20% versus 28%, respectively; P<0.01).110 In DECIDE, significantly fewer patients treated with daclizumab 150 mg compared with IM interferon beta-1a exhibited clinically meaningful worsening on the MSIS-29 PHYS subscale at week 96 (19% versus 23%, respectively), representing a 24% relative reduction in the odds for worsening between treatment groups (95% CI 5−40; P=0.0176).42,123
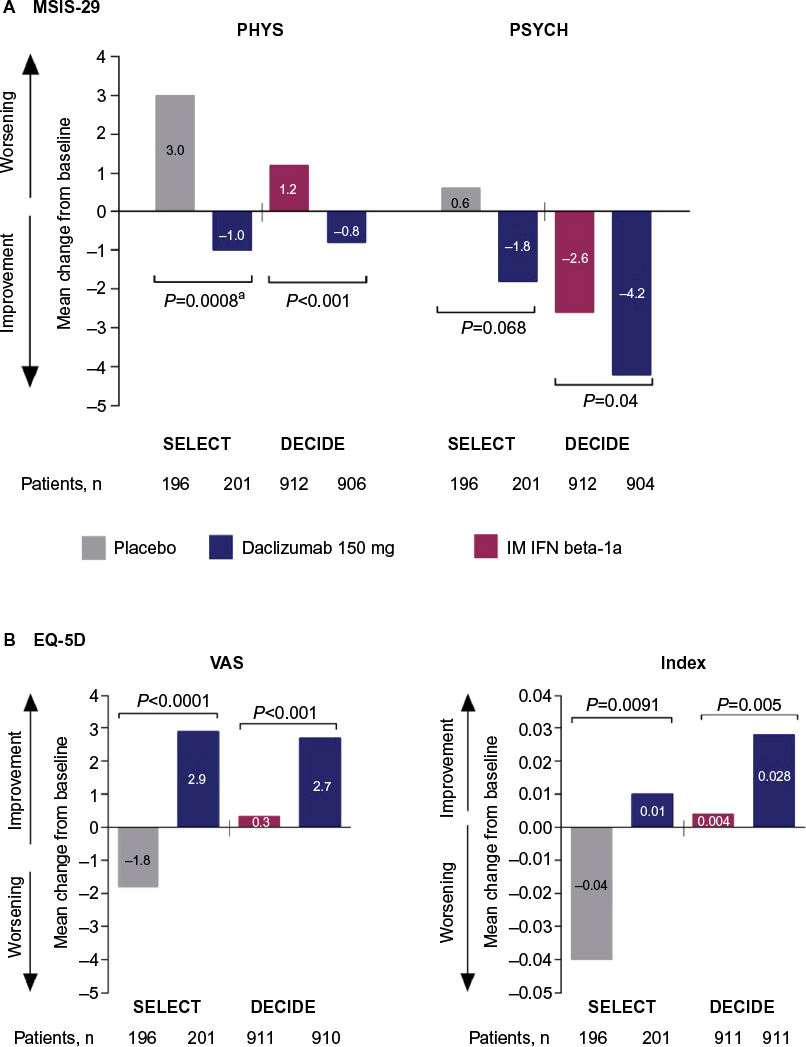 | Figure 3 Changes from baseline to weeks 52 and 96 in health-related quality-of-life endpoints in SELECT and DECIDE.41,42 Notes: aNominal P-value. Comparison was not considered statistically significant within the sequential closed testing procedure for secondary endpoints.41 (A) Change from baseline to week 52 or 96 in MSIS-29 PHYS and PSYCH subscale scores. (B) Change from baseline to week 52 or 96 in EQ-5D VAS and Index scores. In both the DECIDE and SELECT studies, between treatment group differences in MSIS-29 PHYS and PSYCH scores and EQ-5D VAS and index scores were evaluated using analysis of covariance models adjusting for baseline factors. The statistical models are detailed in the original articles.41,42 SELECT data adapted with permission from Elsevier (The Lancet, 2013, 381, 2167–75). Abbreviations: MSIS-29, 29-item Multiple Sclerosis Impact Scale; PHYS, physical impact subscale; PSYCH, psychological impact subscale; IM, intramuscular; IFN, interferon; EQ-5D, EuroQol 5-Dimensions; VAS, visual analog scale. |
The Multiple Sclerosis Functional Composite (MSFC) was administered in DECIDE. The MSFC is a quantitative measure with three components assessing lower limb function/walking ability (Timed 25-Foot Walk [T25FW]), upper limb function (9-Hole Peg Test [9HPT]), and cognitive function (3-second Paced Auditory Serial Addition Test [PASAT-3]).124 At week 96 of DECIDE, daclizumab 150 mg was associated with a greater improvement baseline in MSFC composite score compared with IM interferon beta-1a (0.091 versus 0.055, respectively; P<0.001).42 Significant benefits of daclizumab treatment versus IM interferon beta-1a also were seen on all three separate components that comprise the MSFC at week 96 (T25FW [P=0.006], 9HPT [P=0.002], and PASAT-3 [P=0.04]).42 A benefit for daclizumab on cognitive outcomes also was observed on the Symbol Digit Modalities Test at week 96 with greater mean improvements from baseline observed for daclizumab (4.1±12.4) versus IM interferon beta-1a (2.9±12.7; P=0.03).42
Safety and tolerability
Two decades of clinical experience with interferon beta and glatiramer acetate have shown that these DMTs are relatively safe, although serious adverse events (AEs) do infrequently occur.125,126 Newer DMTs, including natalizumab, fingolimod, delayed-release dimethyl fumarate, teriflunomide, and alemtuzumab, have improved on the modest efficacy outcomes achieved with interferon beta and glatiramer acetate; however, each is associated with a greater degree of concern regarding safety or tolerability.37,125,127,128 It is important to have a clear understanding of the safety profile of each DMT in order to weigh potential risks against potential benefits.
Safety data have been reported for SELECT, SELECTION, and DECIDE (Table 5),41,42,52 and for the 3-year interim analysis of SELECTED.129 The overall incidence of AEs was similar between daclizumab and placebo in SELECT and between daclizumab and IM interferon beta-1a in DECIDE (Table 5).41,42 The incidence of serious AEs, excluding MS relapse, was similar in the daclizumab and placebo groups after 1 year of treatment in SELECT,41 but was higher in daclizumab-treated patients than in IM interferon beta-1a-treated patients after 2–3 years of treatment in DECIDE42 (Table 5). Most treatment-emergent AEs in patients receiving daclizumab were mild or moderate in severity, and the incidence of these AEs did not increase with increasing duration of treatment.42,129 Daclizumab treatment was associated with a higher incidence of AEs leading to treatment discontinuation compared with both placebo (3% versus <1%) and IM interferon beta-1a (15% versus 12%), respectively.42,129 There was no evidence of an increased risk of malignancies in either SELECT or DECIDE, and rates were similar between comparator arms (Table 5).42,129 Five deaths were reported in patients treated with daclizumab during the clinical studies; in two of them (ischemic colitis as a complication of psoas abscess and autoimmune hepatitis in a patient treated with daclizumab 300 mg), a contributory role for daclizumab could not be ruled out, while the other three deaths (acute exacerbation of MS [n=2]; subarachnoid hemorrhage due to a fall [n=1]) were considered by the investigators to be unrelated to treatment.41,42,52,120 Daclizumab does not appear to be associated with tolerability issues that are common among the interferon betas, such as influenza-like symptoms and injection site reactions (Table 5).41,42,130 The safety profile for patients in SELECTION in year 2 of daclizumab 150 mg treatment was similar to that observed in patients in SELECT and SELECTION after 1 year of treatment (Table 5).52 The incidence of AEs in daclizumab-treated patients did not increase over time in a 3-year interim analysis of SELECTED; the safety profile of daclizumab after extended treatment was similar to that observed in SELECT and SELECTION.129
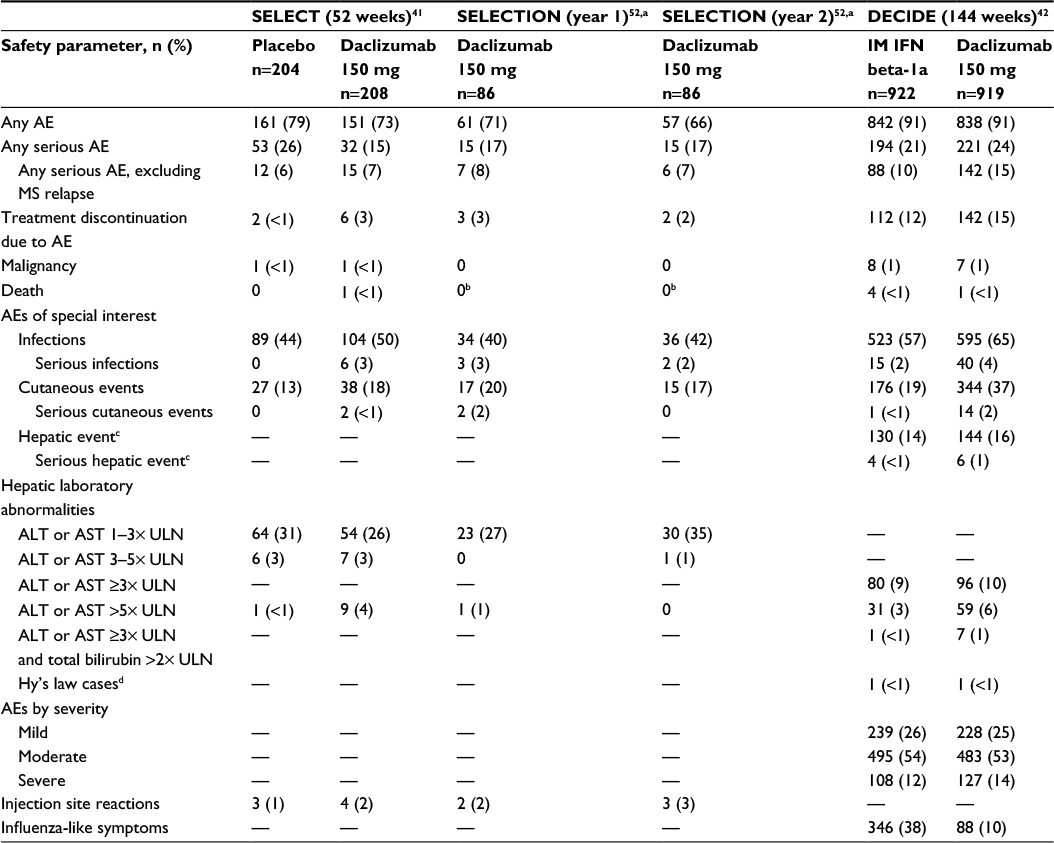 | Table 5 Incidence of AEs in daclizumab 150 mg-treated patients in SELECT, SELECTION, and DECIDE41,42,52 Notes: Results from the daclizumab 300 mg treatment arms in SELECT and SELECTION and the daclizumab 150 mg washout and reinitiation arm in SELECTION have been reported,41,52 but are not included in this table showing the safety profile of continuous treatment with the 150-mg dose. Dashes are used where data were not reported for the parameter. aYear 1 represents patients in the placebo group in SELECT who were randomized to initiate treatment with daclizumab 150 mg in SELECTION, and year 2 represents patients in the daclizumab 150 mg group in SELECT who were randomized to continue daclizumab 150 mg treatment in SELECTION.52 bOne death (autoimmune hepatitis) was reported in SELECTION in a patient randomized to washout for 24 weeks followed by reinitiation of daclizumab 300 mg. cDefined using the standardized Medical Dictionary for Regulatory Activities query for drug-related hepatic disorders. dOne patient in each treatment group was assessed by an independent panel to meet the criteria of Hy’s law based on a causality score of probable or higher.131,132 There were no cases assessed as having a causality score of definitive. SELECT and SELECTION data adapted with permission from Elsevier (The Lancet, 2013, 381, 2167–75) and (Lancet Neurology, 2014, 13, 472–81), respectively. Abbreviations: AE, adverse event; IM, intramuscular; IFN, interferon; MS, multiple sclerosis; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ULN, upper limit of normal. |
Hepatic AEs, including liver enzyme elevations, have been observed in daclizumab treatment groups, but the incidence of treatment-limiting events is low (Table 5).41,42,52 Similar percentages of patients had elevations of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) 1–3× or 3–5× the upper limit of normal (ULN) in the daclizumab and placebo groups after 1 year of treatment in SELECT (Table 5).41 The incidence of elevations of ALT or AST 1–3× ULN was slightly higher in patients in year 2 of treatment than in year 1 of treatment in SELECTION; however, the incidence of ALT or AST elevations >5× ULN was similar in years 1 and 2 of treatment (Table 5).52 Elevations of ALT or AST >5× ULN were more common with daclizumab 150 mg than with placebo (4% versus <1%) or IM interferon beta-1a (6% versus 3%), respectively (Table 5).41,42 Elevated serum transaminases were observed throughout the treatment period in the daclizumab group, but occurred predominantly in the early phases of treatment in the IM interferon beta-1a group.42 In DECIDE, drug-related hepatic events, as defined by a Medical Dictionary of Regulatory Activities query, were observed in 16% of daclizumab-treated patients and 14% of IM interferon beta-1a-treated patients, with serious hepatic events occurring in 1% and <1% of patients, respectively.42 Seven patients receiving daclizumab and one patient receiving IM interferon beta-1a experienced ALT or AST ≥3× ULN concurrent with total bilirubin >2× ULN.42 However, the independent hepatic safety assessment concluded that only one patient in each treatment group that met the criteria for Hy’s law131 had a causality score of probable or higher132 (Table 5).
Daclizumab has been associated with an increased risk of cutaneous AEs when compared with placebo or IM interferon beta-1a (Table 5).41,42 In DECIDE, cutaneous AEs occurred in 37% of daclizumab-treated patients and 19% of IM interferon beta-1a-treated patients over 2–3 years of treatment and resulted in treatment discontinuation in 5% and 1% of patients, respectively.42 The most common cutaneous events were rash (7% versus 3% of patients) and eczema (4% versus 1%) in the daclizumab and IM interferon beta-1a groups, respectively.42 Most patients with cutaneous AEs had mild (55%) or moderate (38%) events, and these events were predominantly treated with topical corticosteroids or did not require any topical or systemic corticosteroid treatment (ie, 81% of mild and 73% of moderate events).133 Severe or serious cutaneous AEs were each reported in 2% of daclizumab-treated patients and most commonly were treated with systemic corticosteroids.133 Among patients with serious cutaneous events in the daclizumab group, dermatitis was reported in three patients and angioedema in two patients; all other serious cutaneous events were reported in one patient each.42 There were no cases of Stevens–Johnson syndrome or toxic epidermal necrolysis reported in DECIDE.42 In SELECTED, one case was reported by the investigator as Stevens–Johnson syndrome, but the diagnosis was not supported by the study’s central independent dermatologist or by the local site dermatologist upon their review of the case.129
Both infections and serious infections have been observed at a higher incidence in daclizumab-treated patients compared with those treated with placebo or IM interferon beta-1a (Table 5).41,42 Most patients with infections remained on daclizumab treatment.42 The most common (incidence ≥10%) infections in DECIDE were nasopharyngitis (25% versus 21%), upper respiratory tract infection (16% versus 13%), and urinary tract infection (10% versus 11%) in the daclizumab and IM interferon beta-1a groups, respectively.42 In SELECT and SELECTION, most patients stayed on daclizumab treatment or restarted daclizumab treatment after resolution of their serious infection.41,52 No cases of progressive multifocal leukoencephalopathy have been reported to date in the daclizumab clinical study program.41,42,129 Interim analyses of safety extension studies showed that the risk of infections did not increase with longer duration of treatment with daclizumab (up to 6 years).120,129
Antidrug antibodies (ADAs) and neutralizing antibodies (NAbs) have been detected in relatively low proportions of daclizumab-treated patients. In patients who initiated daclizumab treatment in SELECTION, 4% of patients were ADA positive and 2% of patients were NAb positive during the 52-week treatment period.52 In these patients, NAbs were not persistent, because patients who tested positive at week 72 were negative for NAbs at week 104.52 In patients continuing daclizumab treatment in SELECTION, one patient was ADA positive, and there were no new cases of NAb positivity in the second year of treatment.52 In DECIDE, ADAs were observed in 19% of evaluable patients treated with daclizumab; in 7% of patients, ADAs were persistent (defined as more than one consecutive positive evaluation ≥74 days apart or a positive evaluation at the final assessment with no further samples available).134 NAbs were observed in 8% of evaluable patients treated with daclizumab; in 2% of patients, NAbs were persistent.134 The majority of ADA reactivity to daclizumab occurred early during treatment (during the first year), and this reactivity was transient.134 ADA titers were generally low in DECIDE, and ADAs and NAbs had no discernible impact on efficacy or safety outcomes associated with daclizumab.134
There was no evidence of increased risk of adverse pregnancy outcomes or fetal abnormalities following gestational exposure to daclizumab during the first trimester.135 Because the number of pregnancies occurring during the daclizumab clinical studies was small (45 pregnancies in 40 patients), no definitive conclusions can be drawn regarding safety during pregnancy.135 There is no information available on the safety of daclizumab in breastfeeding women.
A substudy of daclizumab-treated patients enrolled in SELECTED who received seasonal influenza vaccination achieved acceptable levels of serologic protection against the seasonal influenza and experienced no unique or worsened safety issues associated with vaccination.136
Administration/acceptability
Poor adherence to self-injectable DMT regimens may lead to suboptimal treatment outcomes for patients with MS.137,138 Furthermore, higher frequency of injection has been associated with reduced adherence.139 Daclizumab has several properties that may improve use on a long-term basis. First, self-administration of daclizumab on a once-monthly schedule is likely to be better adhered to by more patients than the daily to twice-monthly administration schedules of other DMTs, because reduced patient adherence to MS medications is clearly related to the frequency of administration.138–141 Furthermore, reducing the dosing frequency of drugs in indications other than MS has been shown to be associated with better treatment adherence, health-related quality of life, patient satisfaction, and reduced costs.142,143 The relatively simple and infrequent procedure of SC administration is unlikely to be a barrier to routine use,141,144 and the favorable pharmacokinetic and pharmacodynamic profile of daclizumab should minimize the risk of breakthrough disease activity resulting from delayed dosing. Second, the superior therapeutic profile of daclizumab compared with platform MS medications such as interferon beta may encourage patients to adhere to the dosing regimen of daclizumab, because some patients report perceived lack of efficacy as contributing to nonadherence.138,140 Finally, the overall tolerability of daclizumab may improve adherence, because some patients report tolerability issues as a reason for nonadherence.138,140 Better adherence to DMTs would be expected to lead to improved clinical outcomes, in addition to lower medical resource utilization and costs.138
Because daclizumab received its first approval for use outside of approved investigational clinical studies in May 2016, patient adherence to daclizumab and its clinical efficacy have not been studied in the overall “real-world” community clinic-based RMS patient population. Additional data, including its use in the noninvestigational clinical practice setting, are needed to confirm the validity of these hypotheses.
Conclusion
Management of RMS can be challenging for clinicians because of highly variable intra- and inter-individual therapeutic responses regarding efficacy and safety.145,146 Only a minority of patients attain the goal of NEDA with current DMTs,18,147,148 while the majority of patients achieve a partial reduction in the frequency of relapses and inflammatory brain lesion activity on MRI, and delayed accumulation of disability.145,149 It is important to note that although patients with residual clinical or radiologic disease activity despite treatment with a first-line DMT may be experiencing some therapeutic benefit, they are commonly at increased risk of disability worsening in the short term150,151 and a lower health-related quality of life.152,153 Apart from a high incidence of breakthrough disease activity associated with first-line DMTs,145,146 many patients experience treatment-related AEs,154 as well as reduced quality of therapeutic response to the extent that clinical and pharmacoeconomic outcomes are compromised because of patient nonadherence to therapy.137,140,155–157 Hence, new therapies are required that have a favorable benefit–risk profile, few tolerability issues, and different attributes to encourage improved levels of adherence. In this regard, the superior efficacy of daclizumab over IM interferon beta-1a, a standard of care in MS, has been demonstrated. The risks associated with daclizumab, including hepatic disorders, cutaneous events, and infections, were typically mild or moderate in severity, manageable with routine medical care, and did not appear to increase with extended treatment. Daclizumab has been approved by the US FDA and EMA for relapsing MS.
Acknowledgments
The daclizumab clinical development program was funded by Biogen and AbbVie Biotherapeutics Inc. Biogen and AbbVie Biotherapeutics Inc. provided funding for medical writing support in the development of this paper; Malcolm Darkes from Excel Scientific Solutions wrote the first draft of the manuscript based on input from Dr Cohan, and Kristen DeYoung from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. Biogen and AbbVie Biotherapeutics Inc. reviewed and provided feedback on the paper to Dr Cohan. Dr Cohan had full editorial control of the paper and provided his final approval of all content.
Disclosure
Stanley Cohan serves on advisory boards for Biogen, Mallinckrodt, Novartis, and Sanofi-Genzyme; has received research support from Biogen, Mallinckrodt, Novartis, Opexa, Roche-Genentech, Sanofi-Genzyme, and Teva; has received speaker honoraria from Acorda, Biogen, Novartis, Roche-Genentech and Sanofi-Genzyme; and has received funds for transportation, meals, and lodging from Acorda, Biogen, Mallinckrodt, Novartis, Roche-Genentech, and Sanofi-Genzyme. The author reports no other conflicts of interest in this work.
References
Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. | ||
Multiple Sclerosis International Federation. Atlas of MS 2013. Available from: http://www.msif.org/about-us/advocacy/atlas/. Accessed August 26, 2015. | ||
Freedman MS. Disease-modifying drugs for multiple sclerosis: current and future aspects. Expert Opin Pharmacother. 2006;7(Suppl 1):S1–S9. | ||
Lublin FD. The incomplete nature of multiple sclerosis relapse resolution. J Neurol Sci. 2007;256(Suppl 1):S14–S18. | ||
Miller DM, Weinstock-Guttman B, Béthoux F, et al. A meta-analysis of methylprednisolone in recovery from multiple sclerosis exacerbations. Mult Scler. 2000;6(4):267–273. | ||
Buchanan RJ, Huang C, Kaufman M. Health-related quality of life among young adults with multiple sclerosis. Int J MS Care. 2010;12(4):190–199. | ||
Forbes A, While A, Mathes L, Griffiths P. Health problems and health-related quality of life in people with multiple sclerosis. Clin Rehabil. 2006;20(1):67–78. | ||
Capkun G, Dahlke F, Lahoz R, et al. Mortality and comorbidities in patients with multiple sclerosis compared with a population without multiple sclerosis: an observational study using the US Department of Defense administrative claims database. Mult Scler Relat Disord. 2015;4(6):546–554. | ||
Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol.1996;39(3):285–294. | ||
Johnson KP, Brooks BR, Cohen JA, et al; The Copolymer 1 Multiple Sclerosis Study Group. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. Neurology. 1995;45(7):1268–1276. | ||
PRISMS (Prevention of Relapses and Disability by Interferon β-1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352(9139):1498–1504. | ||
The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43(4):655–661. | ||
Liu Z, Pelfrey CM, Cotleur A, Lee JC, Rudick RA. Immunomodulatory effects of interferon beta-1a in multiple sclerosis. J Neuroimmunol. 2001;112(1–2):153–162. | ||
Cocco E, Marrosu MG. The current role of mitoxantrone in the treatment of multiple sclerosis. Expert Rev Neurother. 2014;14(6):607–616. | ||
Aktas O, Kieseier B, Hartung HP. Neuroprotection, regeneration and immunomodulation: broadening the therapeutic repertoire in multiple sclerosis. Trends Neurosci. 2010;33(3):140–152. | ||
Polman CH, O’Connor PW, Havrdova E, et al; AFFIRM Investigators. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. | ||
Rudick RA, Stuart WH, Calabresi PA, et al; SENTINEL Investigators. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–923. | ||
Havrdova E, Galetta S, Hutchinson M, et al. Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the Natalizumab Safety and Efficacy in Relapsing-Remitting Multiple Sclerosis (AFFIRM) study. Lancet Neurol. 2009;8(3):254–260. | ||
Mehling M, Kappos L, Derfuss T. Fingolimod for multiple sclerosis: mechanism of action, clinical outcomes, and future directions. Curr Neurol Neurosci Rep. 2011;11(5):492–497. | ||
Fox RJ, Kita M, Cohan SL, et al. BG-12 (dimethyl fumarate): a review of mechanism of action, efficacy, and safety. Curr Med Res Opin. 2014;30(2):251–262. | ||
Bar-Or A, Pachner A, Menguy-Vacheron F, Kaplan J, Wiendl H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs. 2014;74(6):659–674. | ||
Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–556. | ||
Cohen JA, Barkhof F, Comi G, et al; TRANSFORMS Study Group. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. | ||
Confavreux C, O’Connor P, Comi G, et al; TOWER Trial Group. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(3):247–256. | ||
Fox RJ, Miller DH, Phillips JT, et al; CONFIRM Study Investigators. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. | ||
Gold R, Kappos L, Arnold DL, et al; DEFINE Study Investigators. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | ||
Kappos L, Radue EW, O’Connor P, et al; FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. | ||
O’Connor P, Wolinsky JS, Confavreux C, et al; TEMSO Trial Group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. | ||
Hartung HP, Aktas O, Boyko AN. Alemtuzumab: a new therapy for active relapsing-remitting multiple sclerosis. Mult Scler. 2015;21(1):22–34. | ||
Cohen JA, Coles AJ, Arnold DL, et al; CARE-MS I investigators. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–1828. | ||
Coles AJ, Twyman CL, Arnold DL, et al; CARE-MS II investigators. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829–1839. | ||
Roche. Roche’s ocrelizumab first investigational medicine to show positive pivotal study results in both relapsing and primary progressive forms of multiple sclerosis [news release]. Basel, Switzerland: Roche; October 8, 2015. Available from: http://www.roche.com/media/store/releases/med-cor-2015-10-08.htm. Accessed January 7, 2016. | ||
Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378(9805):1779–1787. | ||
Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4ß1 integrin. Nature. 1992;356(6364):63–66. | ||
Arvin AM, Wolinsky JS, Kappos L, et al. Varicella-zoster virus infections in patients treated with fingolimod: risk assessment and consensus recommendations for management. JAMA Neurol. 2015;72(1):31–39. | ||
Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–1880. | ||
Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476–1478. | ||
Novartis Pharmaceuticals Corporation. Gilenya [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015. | ||
Waldmann TA. Anti-Tac (daclizumab, Zenapax) in the treatment of leukemia, autoimmune diseases, and in the prevention of allograft rejection: a 25-year personal odyssey. J Clin Immunol. 2007;27(1):1–18. | ||
Wiendl H, Gross CC. Modulation of IL-2Rα with daclizumab for treatment of multiple sclerosis. Nat Rev Neurol. 2013;9(7):394–404. | ||
Gold R, Giovannoni G, Selmaj K, et al; SELECT study investigators. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9884):2167–2175. | ||
Kappos L, Wiendl H, Selmaj K, et al. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2015;373(15):1418–1428. | ||
Bielekova B, Richert N, Howard T, et al. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon ß. Proc Natl Acad Sci U S A. 2004;101(23):8705–8708. | ||
Bielekova B, Howard T, Packer AN, et al. Effect of anti-CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch Neurol. 2009;66(4):483–489. | ||
Rose JW, Watt HE, White AT, Carlson NG. Treatment of multiple sclerosis with an anti–interleukin-2 receptor monoclonal antibody. Ann Neurol. 2004;56(6):864–867. | ||
Rose JW, Burns JB, Bjorklund J, Klein J, Watt HE, Carlson NG. Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results. Neurology. 2007;69(8):785–789. | ||
Wynn D, Kaufman M, Montalban X, et al; CHOICE investigators. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010;9(4):381–390. | ||
Othman AA, Tran JQ, Tang MT, Dutta S. Population pharmacokinetics of daclizumab high-yield process in healthy volunteers: integrated analysis of intravenous and subcutaneous, single- and multiple-dose administration. Clin Pharmacokinet. 2014;53(10):907–918. | ||
ClinicalTrials.gov [homepage on the Internet]. An immunogenicity and pharmacokinetics (PK) study of BIIB019 (daclizumab high yield process (DAC HYP)) prefilled syringe in relapsing remitting multiple sclerosis (RRMS) (OBSERVE). Available from: https://clinicaltrials.gov/ct2/show/NCT01462318. Accessed December 7, 2015. | ||
Tran JQ, Othman A, Mikulskis A, Wolstencroft P, Elkins J. Pharmacokinetics of daclizumab high-yield process with repeated administration of the clinical subcutaneous regimen in patients with relapsing-remitting multiple sclerosis. Clin Pharmacol. 2016;8:9–13. | ||
ClinicalTrials.gov [homepage on the Internet]. Long-term extension study in participants with multiple sclerosis who have completed study 205MS301 (NCT01064401) to evaluate the safety and efficacy of BIIB019 (EXTEND). Available from: https://clinicaltrials.gov/ct2/show/NCT01797965. Accessed December 7, 2015. | ||
Giovannoni G, Gold R, Selmaj K, et al; SELECTION Study Investigators. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECTION): a multicentre, randomised, double-blind extension trial. Lancet Neurol. 2014;13(5):472–481. | ||
Radue E-W, Giovannoni G, Gold R, et al. Long-term efficacy of daclizumab HYP in relapsing-remitting multiple sclerosis: 3 year results from the SELECTED extension study. Neurology. 2015;84(Suppl 14):P7.226. | ||
Bielekova B, Catalfamo M, Reichert-Scrivner S, et al. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2Rα-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006;103(15):5941–5946. | ||
Martin JF, Perry JS, Jakhete NR, Wang X, Bielekova B. An IL-2 paradox: blocking CD25 on T cells induces IL-2–driven activation of CD56bright NK cells. J Immunol. 2010;185(2):1311–1320. | ||
Sheridan JP, Zhang Y, Riester K, et al. Intermediate-affinity interleukin-2 receptor expression predicts CD56bright natural killer cell expansion after daclizumab treatment in the CHOICE study of patients with multiple sclerosis. Mult Scler. 2011;17(12):1441–1448. | ||
Wuest SC, Edwan JH, Martin JF, et al. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med. 2011;17(5):604–609. | ||
Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. | ||
Depper JM, Leonard WJ, Drogula C, Krönke M, Waldmann TA, Greene WC. Interleukin 2 (IL-2) augments transcription of the IL-2 receptor gene. Proc Natl Acad Sci U S A. 1985;82(12):4230–4234. | ||
Malek TR, Ashwell JD. Interleukin 2 upregulates expression of its receptor on a T cell clone. J Exp Med. 1985;161(6):1575–1580. | ||
Waldmann TA. The IL-2/IL-2 receptor system: a target for rational immune intervention. Immunol Today. 1993;14(6):264–270. | ||
Smith KA. Interleukin-2: inception, impact, and implications. Science. 1988;240(4856):1169–1176. | ||
Gong D, Malek TR. Cytokine-dependent Blimp-1 expression in activated T cells inhibits IL-2 production. J Immunol. 2007;178(1):242–252. | ||
Shaw J, Meerovitch K, Bleackley RC, Paetkau V. Mechanisms regulating the level of IL-2 mRNA in T lymphocytes. J Immunol. 1988;140(7):2243–2248. | ||
Villarino AV, Tato CM, Stumhofer JS, et al. Helper T cell IL-2 production is limited by negative feedback and STAT-dependent cytokine signals. J Exp Med. 2007;204(1):65–71. | ||
Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2Rα and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169(9):4850–4860. | ||
de la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34(9):2480–2408. | ||
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–1151. | ||
Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rß-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17(2):167–178. | ||
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–1164. | ||
Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201(5):723–735. | ||
Takahashi T, Kuniyasu Y, Toda M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10(12):1969–1980. | ||
Cavanillas ML, Alcina A, Nuñez C, et al. Polymorphisms in the IL2, IL2RA and IL2RB genes in multiple sclerosis risk. Eur J Hum Genet. 2010;18(7):794–799. | ||
Hafler DA, Compston A, Sawcer S, et al; International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357(9):851–862. | ||
Dendrou CA, Plagnol V, Fung E, et al. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat Genet. 2009;41(9):1011–1015. | ||
Rubin LA, Kurman CC, Fritz ME, et al. Soluble interleukin 2 receptors are released from activated human lymphoid cells in vitro. J Immunol. 1985;135(5):3172–3177. | ||
Greenberg SJ, Marcon L, Hurwitz BJ, Waldmann TA, Nelson DL. Elevated levels of soluble interleukin-2 receptors in multiple sclerosis. N Engl J Med. 1988;319(15):1019–1020. | ||
Maier LM, Anderson DE, Severson CA, et al. Soluble IL-2RA levels in multiple sclerosis subjects and the effect of soluble IL-2RA on immune responses. J Immunol. 2009;182(3):1541–1547. | ||
Nelson BH, Willerford DM. Biology of the interleukin-2 receptor. Adv Immunol. 1998;70:1–81. | ||
Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167(3):1245–1253. | ||
Rogers WO, Weaver CT, Kraus LA, Li J, Li L, Bucy RP. Visualization of antigen-specific T cell activation and cytokine expression in vivo. J Immunol. 1997;158(2):649–657. | ||
Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its α, ß, and γc receptors. Science. 2005;310(5751):1159–1163. | ||
Nelson BH, Lord JD, Greenberg PD. Cytoplasmic domains of the interleukin-2 receptor ß and γ chains mediate the signal for T-cell proliferation. Nature. 1994;369(6478):333–336. | ||
Goebel J, Stevens E, Forrest K, Roszman TL. Daclizumab (Zenapax) inhibits early interleukin-2 receptor signal transduction events. Transpl Immunol. 2000;8(3):153–159. | ||
Yang H, Wang J, Du J, et al. Structural basis of immunosuppression by the therapeutic antibody daclizumab. Cell Res. 2010;20(12):1361–1371. | ||
Huss DJ, Mehta DS, Sharma A, et al. In vivo maintenance of human regulatory T cells during CD25 blockade. J Immunol. 2015;194(1):84–92. | ||
Elkins J, Sheridan J, Amaravadi L, et al. CD56bright natural killer cells and response to daclizumab HYP in relapsing-remitting MS. Neurol Neuroimmunol Neuroinflamm. 2015;2(2):e65. | ||
Oh U, Blevins G, Griffith C, et al. Regulatory T cells are reduced during anti-CD25 antibody treatment of multiple sclerosis. Arch Neurol. 2009;66(4):471–479. | ||
Mehta D, Riester K, Sheridan J, Elkins J, Amaravadi L. Rapid, sustained and reversible pharmacodynamics of DAC HYP in MS patients supports mechanism of action via modulation of the IL-2 pathway. Mult Scler. 2014;20(Suppl 1):491–492. | ||
Bielekova B, Richert N, Herman ML, et al. Intrathecal effects of daclizumab treatment of multiple sclerosis. Neurology. 2011;77(21):1877–1886. | ||
Jiang W, Chai NR, Maric D, Bielekova B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J Immunol. 2011;187(2):781–790. | ||
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–336. | ||
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. | ||
Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor ß-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178(1):280–290. | ||
Passerini L, Allan SE, Battaglia M, et al. STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+CD25– effector T cells. Int Immunol. 2008;20(3):421–431. | ||
Spits H, Artis D, Colonna M, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13(2):145–149. | ||
Perry JS, Han S, Xu Q, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med. 2012;4(145):145ra106. | ||
Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(Pt 4):1089–1104. | ||
Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14(2):164–174. | ||
Lin YC, Winokur P, Blake A, Wu T, Romm E, Bielekova B. Daclizumab reverses intrathecal immune cell abnormalities in multiple sclerosis. Ann Clin Transl Neurol. 2015;2(5):445–455. | ||
Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. 2014;41(3):354-65. | ||
Gillard GO, Saenz SA, Huss DJ, Fontenot JD. Circulating innate lymphoid cells are unchanged in response to DAC HYP therapy. J Neuroimmunol. 2016;294:41–45. | ||
Tran JQ, Othman AA, Wolstencroft P, Elkins J. Therapeutic protein-drug interaction assessment for daclizumab high-yield process in patients with multiple sclerosis using a cocktail approach. Br J Clin Pharmacol. 2016;52(1):160–167. | ||
Minocha M, Tran JQ, Sheridan JP, Othman AA. Blockade of the high-affinity interleukin-2 receptors with daclizumab high-yield process: pharmacokinetic/pharmacodynamic analysis of single- and multiple-dose phase I trials. Clin Pharmacokinet. 2015;55(1):121–130. | ||
Wiendl H, Giovannoni G, Castro-Borrero W, et al. Lymphocyte counts in patients receiving daclizumab HYP in DECIDE. Presented at: 31st Congress of the European Committee for Treatment and Research in Multiple Sclerosis; October 7–10, 2015; Barcelona, Spain. | ||
Haines JL, Terwedow HA, Burgess K, et al; The Multiple Sclerosis Genetics Group. Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. Hum Mol Genet. 1998;7(8):1229–1234. | ||
Zhang Y, McClellan M, Efros L, et al. Daclizumab reduces CD25 levels on T cells through monocyte-mediated trogocytosis. Mult Scler. 2014;20(2):156–164. | ||
Xu W, Fazekas G, Hara H, Tabira T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;163(1–2):24–30. | ||
Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58(6):840–846. | ||
Phillips GA, Wyrwich KW, Guo S, et al. Responder definition of the Multiple Sclerosis Impact Scale physical impact subscale for patients with physical worsening. Mult Scler. 2014;20(13):1753–1760. | ||
European Medicines Agency. Guideline on clinical investigation of medicinal products for the treatment of multiple sclerosis. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/03/WC500185161.pdf. Accessed November 3, 2015. | ||
Miller DH, Barkhof F, Frank JA, Parker GJ, Thompson AJ. Measurement of atrophy in multiple sclerosis: pathological basis, methodological aspects and clinical relevance. Brain. 2002;125(Pt 8):1676–1695. | ||
Arnold DL, Kappos L, Khan O, et al. Reduction in brain volume loss in patients receiving daclizumab HYP versus intramuscular interferon beta-1a: results of the DECIDE study. Mult Scler. 2015;21(Suppl 11):257–258. | ||
Arnold DL, Kappos L, Wiendl H, et al. Benefits on brain MRI lesion activity with daclizumab HYP compared with intramuscular interferon beta-1a are maintained through 144 weeks’ treatment: results from the DECIDE study. Mult Scler. 2015;21(Suppl 11):255–256. | ||
Giovannoni G, Radue EW, Havrdova E, et al. Effect of daclizumab high-yield process in patients with highly active relapsing-remitting multiple sclerosis. J Neurol. 2014;261(2):316–323. | ||
Wiendl H, Kappos L, Selmaj K, et al. Daclizumab high-yield process (DAC HYP) vs. interferon beta-1a in patients with highly active disease: DECIDE study results. Eur J Neurol. 2015;22(Suppl 1):50. | ||
Wiendl H, Havrdova E, Rose J, Riester K, Tsao LC, Greenberg S. Daclizumab HYP versus interferon ß-1a across patient demographic and disease activity subgroups in the DECIDE phase 3 study. Neurology.2015;84(Suppl 14):P4.007. | ||
Kappos L, Havrdova E, Giovannoni G, et al. Effect of daclizumab HYP versus intramuscular interferon beta-1a on no evidence of disease activity in patients with relapsing-remitting multiple sclerosis: analysis of the DECIDE study. Presented at: 31st Congress of the European Committee for Treatment and Research in Multiple Sclerosis; October 7–10, 2015; Barcelona, Spain. | ||
Havrdova E, Giovannoni G, Stefoski D, et al. Disease-activity-free status in patients with relapsing-remitting multiple sclerosis treated with daclizumab high-yield process in the SELECT study. Mult Scler. 2014;20(4):464–470. | ||
Giovannoni G, Kappos L, Gold R, et al. Safety and tolerability of daclizumab HYP in patients with relapsing-remitting multiple sclerosis: an integrated analysis of six clinical studies. Presented at: 31st Congress of the European Committee for Treatment and Research in Multiple Sclerosis; October 7–10, 2015; Barcelona, Spain. | ||
Hobart J, Lamping D, Fitzpatrick R, Riazi A, Thompson A. The Multiple Sclerosis Impact Scale (MSIS-29): a new patient-based outcome measure. Brain. 2001;124(Pt 5):962–973. | ||
Rabin R, de Charro F. EQ-5D: a measure of health status from the EuroQol Group. Ann Med. 2001;33(5):337–343. | ||
Kappos L, Selmaj K, Arnold D, et al. Daclizumab HYP versus interferon beta-1a in relapsing-remitting multiple sclerosis: primary results of the DECIDE study. Neurology. 2015;84(14 Suppl):S4.003. | ||
Fischer JS, Rudick RA, Cutter GR, Reingold SC, National MS Society Clinical Outcomes Assessment Task Force. The Multiple Sclerosis Functional Composite Measure (MSFC): an integrated approach to MS clinical outcome assessment. Mult Scler. 1999;5(4):244–250. | ||
Yadav V, Bourdette D. New disease-modifying therapies and new challenges for MS. Curr Neurol Neurosci Rep. 2012;12(5):489–491. | ||
Lublin FD, Cofield SS, Cutter GR, et al. Randomized study combining interferon and glatiramer acetate in multiple sclerosis. Ann Neurol. 2013;73(3):327–340. | ||
Miller AE. Teriflunomide: a once-daily oral medication for the treatment of relapsing forms of multiple sclerosis. Clin Ther. 2015;37(10):2366–2380. | ||
Willis MD, Harding KE, Pickersgill TP, et al. Alemtuzumab for multiple sclerosis: long term follow-up in a multi-centre cohort. Mult Scler.Epub 2015 Oct 29. doi:10.1177/1352458515614092. | ||
Gold R, Giovannoni G, Selmaj K, et al. Long-term safety of daclizumab HYP in patients with relapsing-remitting multiple sclerosis: 3–4 year results from the SELECTED extension study. Presented at: 67th Annual Meeting of the American Academy of Neurology; April 18–25, 2015;Washington, DC. | ||
Tremlett HL, Oger J. Interrupted therapy: stopping and switching of the ß-interferons prescribed for MS. Neurology. 2003;61(4):551–554. | ||
Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241–243. | ||
Rockey DC, Seeff LB, Rochon J, et al. Causality assessment in drug-induced liver injury using a structured expert opinion process: comparison to the Roussel-Uclaf causality assessment method. Hepatology. 2010;51(6):2117–2126. | ||
Kircik L, Krueger J, Lebwohl M, et al. Incidence, severity, duration, and treatment of cutaneous adverse events in the DECIDE study of daclizumab HYP versus intramuscular interferon beta-1a in patients with relapsing-remitting multiple sclerosis. Mult Scler. 2015;21(Suppl 11):251–252. | ||
Amaravadi L, Mikulskis A, Lucas N, Riester K, Sweetser M, Elkins J. Evaluation of immunogenicity in patients with multiple sclerosis treated with daclizumab HYP. Presented at: 67th Annual Meeting of the American Academy of Neurology; April 18–25, 2015; Washington, DC. | ||
Gold R, Stefoski D, Selmaj K, et al. Pregnancy experience: preclinical data and pregnancy outcomes in the daclizumab high-yield process clinical program. Presented at: 31st Congress of the European Committee for Treatment and Research in Multiple Sclerosis; October 7–10, 2015; Barcelona, Spain. | ||
Mehta L, Priestley R, Umans K, et al. Immune response to seasonal influenza vaccine in patients with relapsing-remitting multiple sclerosis on long-term daclizumab HYP treatment. Mult Scler. 2015;21(Suppl 11):234–235. | ||
Steinberg SC, Faris RJ, Chang CF, Chan A, Tankersley MA. Impact of adherence to interferons in the treatment of multiple sclerosis: a non-experimental, retrospective, cohort study. Clin Drug Investig. 2010;30(2):89–100. | ||
Menzin J, Caon C, Nichols C, White LA, Friedman M, Pill MW. Narrative review of the literature on adherence to disease-modifying therapies among patients with multiple sclerosis. J Manag Care Pharm. 2013;19(1 Suppl A):S24–S40. | ||
Chen C, Baraban E, Stuchiner T, Kim E, Suh K, Cohan S. Evaluating medication adherence to disease-modifying therapy (DMT) and associated factors using data from the Pacific Northwest Multiple Sclerosis Registry. Mult Scler. 2013;19(Suppl 11):479. | ||
Devonshire V, Lapierre Y, Macdonell R, et al; GAP Study Group. The Global Adherence Project (GAP): a multicenter observational study on adherence to disease-modifying therapies in patients with relapsing-remitting multiple sclerosis. Eur J Neurol. 2011;18(1):69–77. | ||
Poulos C, Kinter E, Yang J-C, Bridges JFP, Posner J, Reder AT. Patient preferences for injectable treatments for multiple sclerosis in the United States: a discrete-choice experiment. Patient. 2016;9(2):171–180. | ||
Richter A, Anton SF, Koch P, Dennett SL. The impact of reducing dose frequency on health outcomes. Clin Ther. 2003;25(8):2307–2335; discussion 2306. | ||
Cotté FE, Fardellone P, Mercier F, Gaudin AF, Roux C. Adherence to monthly and weekly oral bisphosphonates in women with osteoporosis. Osteoporos Int. 2010;21(1):145–155. | ||
Stoner KL, Harder H, Fallowfield LJ, Jenkins VA. Intravenous versus subcutaneous drug administration. Which do patients prefer? A systematic review. Patient. 2015;8(2):145–153. | ||
Rudick RA, Polman CH. Current approaches to the identification and management of breakthrough disease in patients with multiple sclerosis. Lancet Neurol. 2009;8(6):545–559. | ||
Freedman MS, Cohen B, Dhib-Jalbut S, et al. Recognizing and treating suboptimally controlled multiple sclerosis: steps toward regaining command. Curr Med Res Opin. 2009;25(10):2459–2470. | ||
Arnold DL, Calabresi PA, Kieseier BC, et al. Effect of peginterferon beta-1a on MRI measures and achieving no evidence of disease activity: results from a randomized controlled trial in relapsing-remitting multiple sclerosis. BMC Neurol. 2014;14:240. | ||
Khatri B, Barkhof F, Comi G, Jin J, Francis G, Cohen J. Fingolimod treatment increases the proportion of patients who are free from disease activity in multiple sclerosis compared to IFN-ß1a: results from a phase 3, active-controlled study (TRANSFORMS). Neurology. 2012;78(Meeting Abstracts 1):PD5.006. | ||
Miller AE, Rhoades RW. Treatment of relapsing-remitting multiple sclerosis: current approaches and unmet needs. Curr Opin Neurol. 2012;25(Suppl):S4–S10. | ||
Prosperini L, Gallo V, Petsas N, Borriello G, Pozzilli C. One-year MRI scan predicts clinical response to interferon beta in multiple sclerosis. Eur J Neurol. 2009;16(11):1202–1209. | ||
Rio J, Rovira A, Tintoré M, et al. Relationship between MRI lesion activity and response to IFN-ß in relapsing–remitting multiple sclerosis patients. Mult Scler. 2008;14(4):479–484. | ||
Benito-León J, Mitchell AJ, Rivera-Navarro J, Morales-González JM. Impaired health-related quality of life predicts progression of disability in multiple sclerosis. Eur J Neurol. 2013;20(1):79–86. | ||
Karampampa K, Gustavsson A, Miltenburger C, Eckert B. Treatment experience, burden and unmet needs (TRIBUNE) in MS study: results from five European countries. Mult Scler. 2012;18(Suppl 2):7–15. | ||
Giovannoni G, Southam E, Waubant E. Systematic review of disease-modifying therapies to assess unmet needs in multiple sclerosis: tolerability and adherence. Mult Scler. 2012;18(7):932–946. | ||
Cerghet M, Dobie E, Elston Lafata J, et al. Adherence to disease-modifying agents and association with quality of life among patients with relapsing-remitting multiple sclerosis. Int J MS Care. 2010;12(2):51–58. | ||
Uitdehaag B, Constantinescu C, Cornelisse P, et al. Impact of exposure to interferon beta-1a on outcomes in patients with relapsing–remitting multiple sclerosis: exploratory analyses from the PRISMS long-term follow-up study. Ther Adv Neurol Disord. 2011;4(1):3–14. | ||
Tan H, Cai Q, Agarwal S, Stephenson JJ, Kamat S. Impact of adherence to disease-modifying therapies on clinical and economic outcomes among patients with multiple sclerosis. Adv Ther. 2011;28(1):51–61. | ||
Arnold D, Kappos L, Havrdova E, et al. Brain MRI results of DECIDE: a randomized, double-blind trial of DAC HYP vs. IFN β-1a in RRMS patients. Presented at: Joint ACTRIMS/ECTRIMS Meeting; September 10–13, 2014; Boston, MA. | ||
Duffy SS, Lees JG, Moalem-Taylor G. The contribution of immune and glial cell types in experimental autoimmune encephalomyelitis and multiple sclerosis. Mult Scler Int. 2014;2014:285245. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.