Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Therapeutic Approaches for Nonalcoholic Fatty Liver Disease: Established Targets and Drugs
Authors Huang X, Chen H ![]() , Wen S
, Wen S ![]() , Dong M
, Dong M ![]() , Zhou L, Yuan X
, Zhou L, Yuan X
Received 6 March 2023
Accepted for publication 16 June 2023
Published 21 June 2023 Volume 2023:16 Pages 1809—1819
DOI https://doi.org/10.2147/DMSO.S411400
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Xiaojing Huang,1,2 Huiling Chen,2 Song Wen,2 Meiyuan Dong,2 Ligang Zhou,2 Xinlu Yuan2
1Graduate School of Fudan University, Shanghai, People’s Republic of China; 2Department of Endocrinology, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, People’s Republic of China
Correspondence: Xinlu Yuan, Department of Endocrinology and Metabolic Diseases, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, People’s Republic of China, Email [email protected]
Abstract: Nonalcoholic fatty liver disease (NAFLD), as a multisystemic disease, is the most prevalent chronic liver disease characterized by extremely complex pathogenic mechanisms and multifactorial etiology, which often develops as a consequence of obesity, metabolic syndrome. Pathophysiological mechanisms involved in the development of NAFLD include diet, obesity, insulin resistance (IR), genetic and epigenetic determinants, intestinal dysbiosis, oxidative/nitrosative stress, autophagy dysregulation, hepatic inflammation, gut-liver axis, gut microbes, impaired mitochondrial metabolism and regulation of hepatic lipid metabolism. Some of the new drugs for the treatment of NAFLD are introduced here. All of them achieve therapeutic objectives by interfering with certain pathophysiological pathways of NAFLD, including fibroblast growth factors (FGF) analogues, peroxisome proliferator-activated receptors (PPARs) agonists, glucagon-like peptide-1 (GLP-1) agonists, G protein-coupled receptors (GPCRs), sodium-glucose cotransporter-2 inhibitors (SGLT-2i), farnesoid X receptor (FXR), fatty acid synthase inhibitor (FASNi), antioxidants, etc. This review describes some pathophysiological mechanisms of NAFLD and established targets and drugs.
Keywords: non-alcoholic fatty liver disease, oxidative stress, lipotoxicity, organ dysfunction, dysbiosis, new treatment modalities
Introduction
The global prevalence of nonalcoholic fatty liver disease (NAFLD) is 25% and is the leading cause of cirrhosis and hepatocellular carcinoma.1 NAFLD is defined as the presence of steatosis and metabolic risk factors (especially obesity and type 2 diabetes) in more than 5% of liver cells and the absence of excessive alcohol consumption (≥ 30 g per day for men and ≥ 20 g per day for women) or other chronic liver diseases.2 NAFLD is a series of histopathological changes. It can be divided into nonalcoholic simple fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH). NAFL can be considered present when there is more than 5% steatosis in the liver. The presence of steatosis, hepatic ballooning, and hepatic lobular inflammation can be considered NASH. NASH is characterized by necrotic inflammation and faster fibrotic progression than nonalcoholic fatty liver disease. Both two of these stages have the potential to progress to cirrhosis.3 NAFLD is bidirectionally associated with components of metabolic syndrome.4 Type 2 diabetes increases the risk of cirrhosis and related complications. Although the leading causes of death in patients with NAFLD are cardiovascular disease and extrahepatic malignancy, advanced liver fibrosis is a key prognostic indicator for liver-related outcomes and overall mortality.5 The degree of fibrosis can be assessed by a variety of non-invasive tests. Although several drugs to treat NAFLD are currently in the advanced stages of development, there are currently no approved NAFLD treatments. Due to the complex pathophysiology and heterogeneity of the disease phenotype, many patients with NAFLD may require combination therapy. A healthy lifestyle and weight loss remain important in the prevention and treatment of NAFLD.6
In this review, we discuss NAFLD, the underlying molecular mechanisms of NASH pathogenesis and the development of novel drugs associated with it.
Pathogenesis of Nonalcoholic Fatty Liver Disease
NAFLD Development Mechanism
The pathogenic mechanism of NAFLD’s development and progression to NASH is quite complex and has not been fully understood by scientific research over the past decade. Once the two-factor theory is replaced by the multifactorial hypothesis, it will show that more than just one or two mechanisms can explain the pathogenesis of NAFLD and progression to NASH. It is more likely that multiple factors play a role in the same time.7
The main feature of hepatic steatosis is excessive fat accumulation in liver cells, which is formed due to the breakdown of the liver’s homeostasis between lipid acquisition and breakdown. There are two main sources of liver lipids in the form of free fatty acids (FFA), namely lipolysis of triglycerides (TG) in adipose tissue and synthesis by de novo lipogenesis (DNL) pathways. Lipids are broken down by mitochondrial fatty acid oxidation (FAO) and production of very low-density lipoprotein (VLDL). If the injury occurs somewhere on the way, it can lead to the production of toxic lipid metabolites that further induce hepatocellular stress, in other words, lipotoxicity. This is the first step in the pathway from simple steatosis to NASH.8
Multiple parallel hypotheses promote several factors that collectively contribute to the development of NAFLD, such as diet, obesity, insulin resistance (IR), genetic and epigenetic determinants and intestinal dysbiosis. The key factor in this theory may be IR. IR dysregulation causes fat breakdown in adipose tissue, thereby increasing the accumulation of FFAs in the liver. On the other hand, in obese patients, adipose tissue dysfunction eventually occurs, which also leads to increased uptake of FFAs by the liver, inducing lipid synthesis and gluconeogenesis, which further promotes insulin resistance in the liver.9 Animal studies have shown that obesity in mice stimulate the secrets of dipeptidyl peptidase 4 (DPP4), which promotes insulin resistance and liver inflammation.10 Studies have shown higher levels of advanced glycation end products (AGEs) in NASH compared with healthy controls or patients with simple steatosis.11 These findings suggest that AGEs are associated with inflammation. Whether IR was before or after NAFLD occurred is difficult to say, but the link between them is undeniable. Recently, studies have shown that eradication of HCV infection (with direct-acting antivirals) improves IR and thereby reduces the chance of developing diabetes and cardiovascular disease.12–14 This new observation indirectly confirms the role of IR in the relationship between NAFLD and atherosclerotic disease. In summary, IR is the most associated common causative feature of NAFLD and metabolic syndrome, as well as a common clinical feature of these disorders.14,15 In addition, results reported by Masarone suggest that liver injury is associated with progressive clinical manifestations of IR, and the histological features of NASH are observed on liver biopsy in 98% of T2DM patients, suggesting that NASH may be one of the early complications of T2DM, as they are both pathophysiologic associated with IR.16
Dysfunction of the endocannabinoid system (ES) has been found to be associated with metabolic disorders related to nonalcoholic fatty liver disease (NAFLD). Studies show both of ES and cannabinoid receptor type 1 (CB1) are associated with oxidative/nitrosative stress and may also play a role in modulating liver inflammation, ultimately promoting disease progression toward steatohepatitis.17,18 Studies have shown that endocannabinoids can contribute to oxidative stress in NAFLD through CB1 receptors. Oxidative stress and inflammation are linked bidirectionally. Additionally, liver cell injury and apoptosis can be promoted by lipid peroxidation, increased production of cytokines and ROS, as well as induction of Fas ligand, ultimately accelerating the transition to NASH.17 CB1 receptors are expressed in low amounts in normal liver, but they are significantly upregulated in stellate cells, hepatic vascular endothelial cells, and monocytes during hepatic pathology due to endocannabinoid system activation. Animal studies showed that high fat diet (HFD) increases hepatic levels of CB1 density which prove CB1 might lead to hepatic steatosis when linked to a high fat diet, obesity, and alcohol.19 In relation to, hepatic fat production can be reduced by lowering CB1 receptors in the liver, thereby improving dyslipidemia and slowing down the progression of liver cell damage.20 In this approach, CB1 receptor antagonists may be effective in the treatment of NAFLD. CB1 receptor antagonists have been found to possibly improve hepatocellular damage to iron-related oxidative stress by improving disorders of iron metabolism.17
Autophagy dysregulation is related to the progression of NASH, with studies showing that upregulation of hepatic TXNIP (thioredoxin interacting protein) genes in both NAFLD patients and mice fed MCD diets is positively associated with autophagy dysregulation. Mechanistically, TXNIP regulates the inactivation of mechanistic target of rapamycin kinase complex 1 (MTORC1), thereby promoting autophagy. Inhibition of MTORC1 and induction of autophagy by rapamycin has been shown to attenuate diet-induced steatosis, inflammation, and fibrosis in MCD. Therefore, targeting TXNIP may be a potential approach for treating NASH.21
Nonalcoholic fatty liver disease (NAFLD) associated with type 2 diabetes mellitus (T2DM) is frequently characterized by decreasing gut barrier function and a disturbed gut-liver axis, perhaps due to exposure to hazardous chemicals such as antigens, metabolites, and gut microorganisms. This may worsen hepatic inflammation, eventually leading to severe liver disease characterized by cirrhosis and portal hypertension.22,23 Bacterial endotoxin-induced metabolic endotoxemia can result in intestinal barrier dysfunction, which has been identified as a cause of systemic inflammation.24 Self-maintaining macrophages, immune cells that help maintain intestinal homeostasis and regulate intestinal motility and secretion, rely heavily on the presence of intestinal microbiota.25,26 Research has demonstrated that the gut microbiota can contribute to diet-induced obesity by affecting FXR signaling and bile acid profiles, ultimately promoting hepatic steatosis.27
Progression to NASH-Lipotoxicity, Organelle Distress, and Inflammasome Activation
There is a strong correlation at the cellular and organ levels in the pathogenesis of NASH, especially given the fat-liver and gut-liver axis relationships. Once toxic lipids are metabolized, hepatocytes enter different types of states, which then trigger cascading processes that lead to the development of NASH. In addition to saturated fatty acids such as palmitate and stearate, these toxic lipid molecules include diacylglycerol, ceramides, lysophosphatidylcholine (LPC), and free cholesterol. At the molecular level, lipotoxicity leads to organelle stress and dysfunction, inflammasome activation, and ultimately cell death and activation of inflammatory responses.3,28 Hepatic lipotoxicity may occur because the accumulated liver lipids stimulate the secretion and increase the activity of IL-11 protein, which upregulate NOX4 expression to increase the production of reactive oxygen species. Hepatocytes mitochondria and fatty acid metabolism could be damaged by reactive oxygen species, which accelerate the progression of hepatic steatosis. IL-11 can also promotes fibrosis in hepatic stellate cells.29 Given the major role of hepatic lipid balance in the pathogenesis of NASH, treatment strategies can be considered in terms of reducing fat inflow into the liver or enhancing fat metabolism in the liver. This can be achieved, for example, by increasing fatty acid oxidation (FAO), inhibiting de novo lipogenesis (DNL), increasing fatty acid desaturation, and improving IR. Hepatocyte injury will lead to macrophage-mediated inflammation and hepatic stellate cell activation, which constitute a key triad for the development of NASH. Lipotoxic substances may activate the mechanism of apoptosis in hepatocytes through intrinsic or extrinsic (death receptor) pathways. Major molecular signaling occurs via c-Jun-N terminal kinase (JNK).30 The JNK pathway is known to be stimulated by oxidative stress (OS) and endoplasmic reticulum (ER) stress. At the organelle level, FFA and its derivatives induce mitochondrial hyperfunction, followed by mitochondrial dysfunction, leading to elevated levels of reactive oxygen radicals (ROS), the culprit of oxidative stress.31 Saturated FFAs, especially lipoprotein complexes (LPCs) derived from them, can also accumulate in the endoplasmic reticulum (ER) and trigger ER stress, which stimulates an unfolded protein response (UPR), an attempt by ER to re-establish its homeostasis. However, prolonged activation of UPR leads to activation of JNK, glycogen synthase kinase 3 (GSK3), and transcription factor CCAAT/enhancer-binding homologous protein (CHOP), all of which upregulate pro-death proteins such as p53 up-regulation of apoptosis regulators (PUMA) and extrinsic pathways including tumor necrosis factor (TNF)-associated apoptosis-inducing ligands (TRAIL) to induce cell death through transcriptional upregulation of TRAIL receptor 2 (TRAIL-R2) expression. When the outer mitochondrial membrane is permeabilized, both pathways converge at the mitochondrial level, resulting in cytochrome C release, caspase activation, and apoptosis32 (Figure 1).
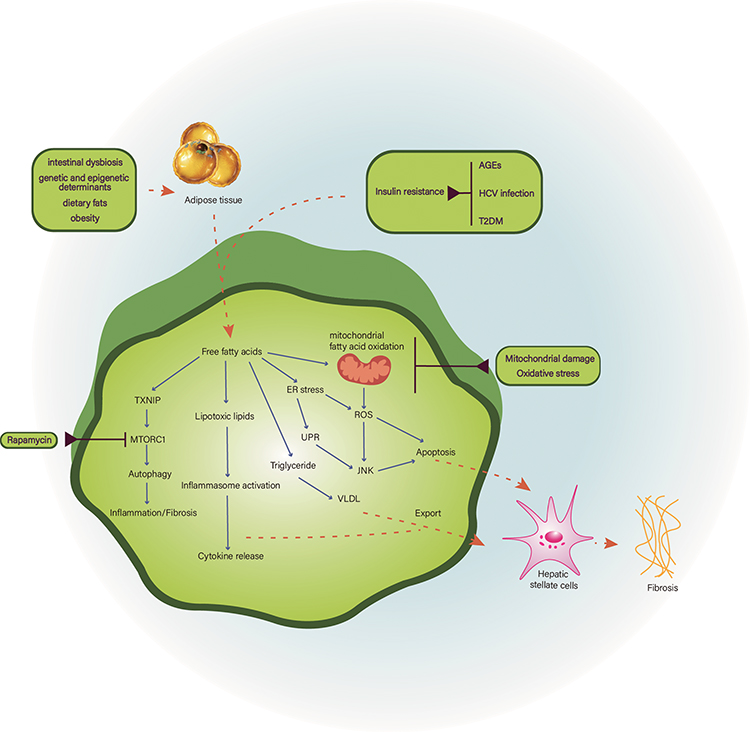 |
Figure 1 Pathogenesis of nonalcoholic fatty liver disease. The pathogenesis of NAFLD is complex and currently considered to be associated with insulin resistance, mitochondrial damage, oxidative stress and intestinal dysbiosis, etc. Abbreviations: AGEs, advanced glycation end products; ER, endoplasmic reticulum; HCV, hepatitis C virus; JNK, c-Jun N-terminal kinase; MTORC1, mechanistic target of rapamycin kinase complex 1; ROS, reactive oxygen species; TXNIP, thioredoxin interacting protein; UPR, unfolded protein response; VLDL, very-low-density lipoprotein. |
The Latest Therapeutic Targets and Representative Drugs of NAFLD
FGF Analogues
FGF21
FGF21 is one of the members of the FGF family. Because of its insulin-sensitizing and hepatoprotective properties, it has become one of the therapeutic hormones for NAFLD.33 In addition, FGF21 has shown its triglyceride-lowering effect in animal models as well as human studies.34 The latter increases the morbidity and mortality of NASH by increasing cardiovascular events. In addition, FGF21 increases the level of adiponectin, which is a kind of adipokine with insulin sensitization, anti-fat, anti-inflammatory and anti-fibrotic effects.35,36
Pegbelfermin, the FGF21 analog, demonstrated by a recent Phase IIa clinical trial, that it can reduce hepatic fat content in patients with NASH. However, in chronic treatments with pegbelfermin, there is a high chance that anti-pegbelfermin and anti-FGF21 antibodies will form, which have been raised concerns about immunogenicity issues.24 Besides, NGMBio is developing a once-monthly formulation, the antibody NGM313, which reduces liver fat content, HbA1c, and transaminases in non-diabetic NAFLD patients by activating the β-Klotho-FGFR1c complex.
Finally, some safety issues for long-term treatment of FGF21 analogues, such as cardiovascular side effects and bone loss, remain unresolved.37
FGF19
FGF19 is a regulator of bile acid synthesis produced by intestinal cells after meals in response to farnesoid X receptor (FXR) stimulation. Because this nutritionally regulated hormone is positively correlated with food intake and there is insufficient evidence that this effect still works in the fasted state, its target population is the NAFLD patients with metabolic syndrome or insulin resistance.38 In addition, FGF19 also has the effect of regulating energy homeostasis, stimulating sugar production, and inhibiting gluconeogenesis. It also reduces lipogenesis by blocking insulin signaling, thereby reducing fat accumulation in the liver.39,40
Recent FGF19-targeted drug developments related to NAFLD include the FGF19 analogue NGM282 and FXR agonists. Obeticholic acid (OCA) is the most reported one as a member of FXR agonists.41
PPARs
There are three different isoforms of peroxisome proliferator-activated receptors (PPARs), namely α, β/δ, and γ. They have the effect of regulating lipid and carbohydrate metabolism.
In NASH, PPARα not only improves lipid metabolism by controlling lipid flux, regulating fatty acid transport, and β oxidation,42 but also reduces liver inflammation by reducing visceral inflammation and intestinal permeability. In the case of cirrhosis, PPARα can reduce portal pressure glucose and lipoprotein metabolism and insulin resistance in skeletal muscle.
PPARβ/δ is highly expressed in human sinusoidal endothelial cells and hepatic macrophages, regulating the expression of key genes involved in innate immunity and inflammation.43
PPARγ regulates insulin sensitivity in adipose tissue, prevents activation of hepatic stellate cells, and thus reduces the incidence of liver fibrosis.44 In addition, against the background of cirrhosis, PPARγ reduces portal venous pressure, visceral inflammation, angiogenesis, and portosystemic shunting.
Pioglitazone is a PPAR gamma agonist (thiazolidinedione) that has been shown to reduce the occurrence of hepatic steatosis and lobular inflammation45 and has cardiometabolic benefits.46,47 However, long-term use has been reported to alter bone metabolism and increase fracture risk48 and increase mortality in patients with heart failure due to its fluid retention effects.49
GLP-1
Glucagon-like peptide-1 (GLP-1) is a naturally occurring hormone secreted by enteroendocrine cells (EEC) of the gastrointestinal tract (GIT) and affected by dietary intake. It can regulate glucose metabolism and energy homeostasis by regulating the secretion of islet hormones and glucagon, and can also achieve weight loss by delaying gastric emptying and inhibiting small intestinal peristalsis leading to decreased appetite and delayed food absorption.50 GLP-1R analogues mimic the effects of glucagon-like peptide-1 (GLP-1)51 GLP-1 also regulates the immune system by suppressing inflammation.52 This suggests that GLP-1 analogues can improve insulin sensitivity in NAFLD patients.
Studies50,53 have shown GLP-1 analogues, liraglutide, and exenatide, to reduce the levels of liver enzymes in the serum of NAFLD patients.54 It has also been shown to reduce liver enzymes in patients with diabetes and obesity.55 Not only that, but they have also been found to improve the histological characteristics of the liver by promoting the oxidation of fatty acids,56 steatosis of the liver,57 significantly reducing liver fat content (LFC),58 effectively regulating inflammation caused by fat accumulation in the liver.54 Finally, exenatide treatment appears to be more effective than liraglutide treatment in improving liver enzymes and weight loss.59
GPCRs
G protein-coupled receptors (GPCRs) are an umbrella term for a large class of membrane proteins that mediate a variety of extracellular ligands, including hormones, neurotransmitters, and chemokines.60
Many GPCRs affect NAFLD metabolism by being activated by short-chain fatty acids (SCFAs) and bile acids (BAs). SCFAs activate intestinal cells and regulate intestinal immune responses by activating GPCRs (for example, GPR41/FFAR3 and GPR43/FFAR2) and inhibiting histone deacetylase.61 In addition, GPCRs regulate SCFA-mediated inflammatory responses. SCFAs have also been associated with fructo-oligosaccharides for the improvement of steatohepatitis and chronic inflammation.62 BAs may reduce the incidence of liver and gastrointestinal inflammation by activating nuclear FXR and GPCR signaling.63,64 GPCRs are also members of the intestinal-hepatic axis and regulate the secretion of intestinal hormones (for example, GLP-1) to improve lipid accumulation and fibrosis.65,66
Compared with other NAFLD therapies, targeted GPCRs have outstanding advantages in the treatment of NAFLD. Functional selection of GPCR ligands helps reduce side effects of treatment. GPCRs act on the entire process of NAFLD development, such as lipid accumulation, inflammation and fibrosis. Therefore, targeted GPCRs can be applied to different stages of NAFLD treatment, such as steatosis, NASH, liver fibrosis, and liver cancer.
SGLT-2i
Sodium-glucose co-transporter-2 (SGLT-2) is highly expressed in the proximal tubular cells of the kidneys and is responsible for filtering glucose reabsorption.67
In animal study, Nasiri-Ansari et al found empagliflozin, SGLT-2 inhibitor can attenuate the progress of NAFLD in apoE((-/-)) mice by activating autophagy and reducing ER stress and apoptosis.68
SGLT2i lowers blood glucose by inhibiting glucose reabsorption and increasing the excretion of glucose in the urine. In addition, they can reduce blood volume, reduce body weight and fat mass, and lower blood pressure through osmotic diuresis.61,69,70 They have also been shown to have cardioprotective properties for the heart and kidneys.71–73 And it is currently approved for use in patients with T2DM.
More than half of patients with T2DM also have NAFLD. This seems to prove a bidirectional pathological relationship between type 2 diabetes and NAFLD. In other words, having one disease increases the probability of the other.74 This may be due to their pathophysiologic association with insulin resistance.
Several studies have shown that SGLT2i can reduce the incidence of NAFLD in patients with T2DM, improve liver aminotransferases, blood lipids, blood glucose, insulin resistance and reduce body weight.75,76 And it further attenuates liver fat deposition, liver inflammation and liver fibrosis.65,77 However, YAN H reported that it shows no apparent effect in improving IR in NAFLD compared with GLP-1 RAs.78
FXR
The farnesoid X receptor (FXR) belongs to the gene family of nuclear receptors and is highly expressed in the gastrointestinal tract. As an endogenous ligand for bile acids, FXR regulates bile acid production and integrates and regulates glucose, lipid, and energy metabolism.79,80 The FXR nuclear receptor plays a key role in preserving hepatic homeostasis. By blocking de novo lipogenesis via SREBP-1c, this receptor is engaged to lessen lipotoxicity while also boosting mitochondrial β-oxidation and cholesterol excretion. These activities slow the growth of fibrosis, inflammation, and insulin resistance. In addition, Bile acids can stimulate the production of antimicrobial peptides by binding to FXR. These peptides can inhibit the growth of intestinal probiotics and disrupt intestinal barrier function.81
Obeticholic acid (OCA), the natural agonist of the farnesoid X receptor, could decreases insulin resistance and hepatic steatosis in preclinical research. In a Phase 2 trial, patients with type 2 diabetes mellitus and nonalcoholic fatty liver disease were randomly assigned to given placebo, 25 or 50 mg OCA for 6 weeks, it shows increased insulin sensitivity (by 28.0% from baseline with 25 mg OCA (P=0.019) and 20.1% from baseline with 50 mg OCA (P=0.060)), and decreased biological indicators of liver inflammation and fibrosis (NCT00501592).82
In a multicentre, randomised trial, 282 patients with biopsy evidence of non-alcoholic steatohepatitis, 25 mg OCA or placebo was randomly assigned to patients once a day. Patients given obeticholic acid showed improvement in fibrosis, hepatocellular ballooning, steatosis, lobular inflammation, the mean change in the NAFLD activity score (change from baseline=–1·7 vs –0·7; p<0.0001) and biochemical markers for the liver, like serum alanine aminotransferase and aspartate aminotransferase, when compared to placebo. Although there have been improvements in the individual histological features of nonalcoholic steatohepatitis (NASH), the proportion of patients with resolution of NASH remained unchanged. Pruritus was the only adverse event occurring more frequently with obeticholic acid than with placebo. Overall, the drug was well-received with no other significant adverse events reported (NCT01265498).83
FASN Inhibitor
Fatty acid synthase (FASN) is a multi-enzyme protein that catalyzes the synthesis of palmitate from acetyl-CoA and malonyl-CoA in the de novo lipogenesis (DNL) pathway. There are studies confirmed that those with NAFLD have higher levels of FFA due to hepatic DNL which promotes liver fibrosis in various ways.84 Additionally, it was confirmed that the level of FASN gene is significantly higher in the NAFLD patients compared with healthy individuals.85 Therefore, FASN may be a potential therapeutic target for NAFLD treatment.
There is a a randomized, placebo-controlled trial, showing TVB-2640, a fatty acid synthase inhibitor, can significantly reduce excessive hepatic fat production, reduce insulin resistance, inflammation and reactive oxygen species production, and improve liver fibrosis in a dose-dependent manner (NCT03938246).86
Antioxidant Treatment for NAFDL
The antioxidant activity of vitamins C and E reduces liver cell damage, and changes in serum vitamin D, vitamin B12, and folate levels are strongly associated with the severity of NAFLD.
Vitamin E
Vitamin E is a fat-soluble and powerful chain-breaking antioxidant in the human body that has the most significant evidence of therapeutic benefits in liver disease.87 Vitamin E has the ability to reduce oxidative stress and slow the onset of NASH.88 Vitamin E reduces the inflammatory response by increasing the expression of adiponectin and inhibits the expression of a variety of cytokines, including tumor necrosis factor-α (TNF-α), IL-1, IL-2, IL-4, IL-6, and IL-8. It also acts as a scavenger of hydroxyl, peroxyl, and superoxide radicals and stimulates superoxide dismutase (SOD) production. The proportional relationship between BCL2-associated with X (BAX) and Bcl-2 proteins is a key factor in determining the strength of inhibition of apoptosis. Vitamin E exerts an anti-apoptotic effect by enhancing levels of the anti-apoptotic protein, BCL-2, and reducing BCL2-associated with X (BAX) protein and p53 BCL-2. Although there is no standard regimen for the treatment of NAFLD/NASH, patients with NAFLD often require vitamin E supplementation.89
Vitamin C
Vitamin C (ascorbic acid) is a powerful antioxidant that keeps the body healthy, slowing the progression of liver fibrosis by scavenging free oxygen radicals90 to reduce inflammation and activation of hepatic stellate cells (HSCs). Vitamin C treatment reduces oxidative stress in the liver. Ascorbic acid has also been suggested to modulate adiponectin to reduce lipid accumulation in the liver, systemic insulin resistance and inflammation, and to prevent NAFLD.91,92 In addition, ascorbic acid supplementation inhibits hepatic steatosis and stress by increasing mRNA levels in the liver (Figure 2).
 |
Figure 2 The latest therapeutic targets and representative drugs of NAFLD. This article lists a series of pathophysiological pathways of NAFLD, including fibroblast growth factors (FGF) analogues, peroxisome proliferator-activated receptors (PPARs) agonists, glucagon-like peptide-1 (GLP-1) agonists, G protein-coupled receptors (GPCRs), sodium-glucose cotransporter-2 inhibitors (SGLT-2i), farnesoid X receptor (FXR), fatty acid synthase inhibitor (FASNi) and antioxidants. |
Conclusion
NAFLD, the most prevalent chronic liver disease in the world, places a significant burden on the world’s healthcare system because mortality and morbidity are not only connected to the liver but also to a number of extrahepatic organ systems. This article lists a series of pathophysiological pathways of NAFLD, including fibroblast growth factors (FGF) analogues, peroxisome proliferator-activated receptors (PPARs) agonists, glucagon-like peptide-1 (GLP-1) agonists, G protein-coupled receptors (GPCRs), sodium-glucose cotransporter-2 inhibitors (SGLT-2i), farnesoid X receptor (FXR), fatty acid synthase inhibitor (FASNi), antioxidants, etc. Recent studies have continued to provide new evidence strongly indicating that the gut microbiota may be a key link in the pathogenesis of NAFLD and its progression to NASH and cirrhosis. This is in addition to the previously established links between the various signaling pathways involved in the development of NAFLD. New treatments and targets are now being created concurrently with the rapid appearance of new data, which includes certain targets and pathways that are the subject of the aforementioned drug development. A multidisciplinary approach is needed for the evaluation of NAFLD in order to detect and treat this multisystem disease early and lessen the effects of this important public health issue. However, regardless of the progress of drug therapy in the treatment of NAFLD, maintaining a healthy lifestyle and losing weight remains important measures to prevent the onset of NAFLD and slow its progression.
Funding
This work was supported by Talents Training Program of Pudong Hospital affiliated to Fudan University (PJ202001), National Natural Science Foundation of China (82100850), the Project of Key Medical Specialty of Pudong Hospital of Fudan University (No Tszb2023-14) and Scientific Program of Shanghai Pudong Hospital (YJRCJJ201808).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi:10.1002/hep.28431
2. European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64(6):1388–1402. doi:10.1016/j.jhep.2015.11.004
3. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic Steatohepatitis: a review. JAMA. 2020;323(12):1175–1183. doi:10.1001/jama.2020.2298
4. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. doi:10.1038/nrgastro.2017.109
5. Powell EE, Jonsson JR, Clouston AD. Steatosis: co-factor in other liver diseases. Hepatology. 2005;42(1):5–13. doi:10.1002/hep.20750
6. Wong VWS, Wong GLH, Chan RSM, et al. Beneficial effects of lifestyle intervention in non-obese patients with non-alcoholic fatty liver disease. J Hepatol. 2018;69(6):1349–1356. doi:10.1016/j.jhep.2018.08.011
7. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65(8):1038–1048. doi:10.1016/j.metabol.2015.12.012
8. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–1846. doi:10.1002/hep.24001
9. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–922. doi:10.1038/s41591-018-0104-9
10. Ghorpade DS, Ozcan L, Zheng Z, et al. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature. 2018;555(7698):673–677. doi:10.1038/nature26138
11. Takeuchi M. Toxic AGEs (TAGE) theory: a new concept for preventing the development of diseases related to lifestyle. Diabetol Metab Syndr. 2020;12(1):105. doi:10.1186/s13098-020-00614-3
12. Adinolfi LE, Petta S, Fracanzani AL, et al. Reduced incidence of type 2 diabetes in patients with chronic hepatitis C virus infection cleared by direct-acting antiviral therapy: a prospective study. Diabetes Obes Metab. 2020;22(12):2408–2416. doi:10.1111/dom.14168
13. Adinolfi LE, Petta S, Fracanzani AL, et al. Impact of hepatitis C virus clearance by direct-acting antiviral treatment on the incidence of major cardiovascular events: a prospective multicentre study. Atherosclerosis. 2020;296:40–47. doi:10.1016/j.atherosclerosis.2020.01.010
14. Sasso FC, Pafundi PC, Caturano A, et al. Impact of direct acting antivirals (DAAs) on cardiovascular events in HCV cohort with pre-diabetes. Nutr Metab Cardiovasc Dis. 2021;31(8):2345–2353. doi:10.1016/j.numecd.2021.04.016
15. Rinaldi L, Pafundi PC, Galiero R, et al. Mechanisms of non-alcoholic fatty liver disease in the metabolic syndrome. A narrative review. Antioxidants. 2021;10(2):270. doi:10.3390/antiox10020270
16. Masarone M, Rosato V, Aglitti A, et al. Liver biopsy in type 2 diabetes mellitus: steatohepatitis represents the sole feature of liver damage. PLoS One. 2017;12(6):e0178473. doi:10.1371/journal.pone.0178473
17. Jorgačević B, Mladenović D, Ninković M, et al. Rimonabant improves oxidative/nitrosative stress in mice with nonalcoholic fatty liver disease. Oxid Med Cell Longev. 2015;2015:842108. doi:10.1155/2015/842108
18. Gornicka A, Morris-Stiff G, Thapaliya S, Papouchado BG, Berk M, Feldstein AE. Transcriptional profile of genes involved in oxidative stress and antioxidant defense in a dietary murine model of steatohepatitis. Antioxid Redox Signal. 2011;15(2):437–445. doi:10.1089/ars.2010.3815
19. Alswat KA. The role of endocannabinoids system in fatty liver disease and therapeutic potentials. Saudi J Gastroenterol. 2013;19(4):144–151. doi:10.4103/1319-3767.114505
20. Bartelt A, Orlando P, Mele C, et al. Altered endocannabinoid signalling after a high-fat diet in Apoe(-/-) mice: relevance to adipose tissue inflammation, hepatic steatosis and insulin resistance. Diabetologia. 2011;54(11):2900–2910. doi:10.1007/s00125-011-2274-6
21. Park HS, Song JW, Park JH, et al. TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy. 2021;17(9):2549–2564. doi:10.1080/15548627.2020.1834711
22. Trebicka J, Bork P, Krag A, Arumugam M. Utilizing the gut microbiome in decompensated cirrhosis and acute-on-chronic liver failure. Nat Rev Gastroenterol Hepatol. 2021;18(3):167–180. doi:10.1038/s41575-020-00376-3
23. Trebicka J, Macnaughtan J, Schnabl B, Shawcross DL, Bajaj JS. The microbiota in cirrhosis and its role in hepatic decompensation. J Hepatol. 2021;75(Suppl 1):S67–S81. doi:10.1016/j.jhep.2020.11.013
24. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi:10.2337/db06-1491
25. Honda M, Surewaard BGJ, Watanabe M, et al. Perivascular localization of macrophages in the intestinal mucosa is regulated by Nr4a1 and the microbiome. Nat Commun. 2020;11(1):1329. doi:10.1038/s41467-020-15068-4
26. De Schepper S, Verheijden S, Aguilera-Lizarraga J, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. 2018;175(2):400–415.e13. doi:10.1016/j.cell.2018.07.048
27. Parséus A, Sommer N, Sommer F, et al. Microbiota-induced obesity requires farnesoid X receptor. Gut. 2017;66(3):429–437. doi:10.1136/gutjnl-2015-310283
28. Pierantonelli I, Svegliati-Baroni G. Nonalcoholic fatty liver disease: basic pathogenetic mechanisms in the progression from NAFLD to NASH. Transplantation. 2019;103(1):e1–e13. doi:10.1097/TP.0000000000002480
29. Dong J, Viswanathan S, Adami E, et al. Hepatocyte-specific IL11 cis-signaling drives lipotoxicity and underlies the transition from NAFLD to NASH. Nat Commun. 2021;12(1):66. doi:10.1038/s41467-020-20303-z
30. Parthasarathy G, Revelo X, Malhi H. Pathogenesis of nonalcoholic steatohepatitis: an overview. Hepatol Commun. 2020;4(4):478–492. doi:10.1002/hep4.1479
31. Simões ICM, Fontes A, Pinton P, Zischka H, Wieckowski MR. Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol. 2018;95:93–99. doi:10.1016/j.biocel.2017.12.019
32. Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1049–1061. doi:10.1016/j.metabol.2016.02.014
33. Zarei M, Pizarro-Delgado J, Barroso E, Palomer X, Vázquez-Carrera M. Targeting FGF21 for the treatment of nonalcoholic steatohepatitis. Trends Pharmacol Sci. 2020;41(3):199–208. doi:10.1016/j.tips.2019.12.005
34. Kharitonenkov A, DiMarchi R. Fibroblast growth factor 21 night watch: advances and uncertainties in the field. J Intern Med. 2017;281(3):233–246. doi:10.1111/joim.12580
35. Saxena NK, Anania FA. Adipocytokines and hepatic fibrosis. Trends Endocrinol Metab. 2015;26(3):153–161. doi:10.1016/j.tem.2015.01.002
36. Hui X, Feng T, Liu Q, Gao Y, Xu A. The FGF21-adiponectin axis in controlling energy and vascular homeostasis. J Mol Cell Biol. 2016;8(2):110–119. doi:10.1093/jmcb/mjw013
37. Dushay J, Lai M. Is trimming the fat enough? Fibroblast growth factor 21 as an emerging treatment for nonalcoholic fatty liver disease. Hepatology. 2019;70(5):1860–1862. doi:10.1002/hep.30789
38. Alvarez-Sola G, Uriarte I, Latasa MU, et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut. 2017;66(10):1818–1828. doi:10.1136/gutjnl-2016-312975
39. Cicione C, Degirolamo C, Moschetta A. Emerging role of fibroblast growth factors 15/19 and 21 as metabolic integrators in the liver. Hepatology. 2012;56(6):2404–2411. doi:10.1002/hep.25929
40. Bhatnagar S, Damron HA, Hillgartner FB. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J Biol Chem. 2009;284(15):10023–10033. doi:10.1074/jbc.M808818200
41. Sciarrillo CM, Keirns BH, Koemel NA, Anderson KL, Emerson SR. Fibroblast growth factor 19: potential modulation of hepatic metabolism for the treatment of non-alcoholic fatty liver disease. Liver Int. 2021;41(5):894–904. doi:10.1111/liv.14802
42. Bougarne N, Weyers B, Desmet SJ, et al. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr Rev. 2018;39(5):760–802. doi:10.1210/er.2018-00064
43. Sanderson LM, Boekschoten MV, Desvergne B, Müller M, Kersten S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol Genomics. 2010;41(1):42–52. doi:10.1152/physiolgenomics.00127.2009
44. Zardi EM, Navarini L, Sambataro G, et al. Hepatic PPARs: their role in liver physiology, fibrosis and treatment. Curr Med Chem. 2013;20(27):3370–3396. doi:10.2174/09298673113209990136
45. Brown E, Hydes T, Hamid A, Cuthbertson DJ. Emerging and established therapeutic approaches for nonalcoholic fatty liver disease. Clin Ther. 2021;43(9):1476–1504. doi:10.1016/j.clinthera.2021.07.013
46. Spence JD, Viscoli CM, Inzucchi SE, et al. Pioglitazone therapy in patients with stroke and prediabetes: a post hoc analysis of the IRIS randomized clinical trial. JAMA Neurol. 2019;76(5):526–535. doi:10.1001/jamaneurol.2019.0079
47. DeFronzo RA, Inzucchi S, Abdul-Ghani M, Nissen SE. Pioglitazone: the forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diab Vasc Dis Res. 2019;16(2):133–143. doi:10.1177/1479164118825376
48. Kernan WN, Viscoli CM, Furie KL, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med. 2016;374(14):1321–1331. doi:10.1056/NEJMoa1506930
49. Francque S, Szabo G, Abdelmalek MF, et al. Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors. Nat Rev Gastroenterol Hepatol. 2021;18(1):24–39. doi:10.1038/s41575-020-00366-5
50. Gupta NA, Mells J, Dunham RM, et al. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology. 2010;51(5):1584–1592. doi:10.1002/hep.23569
51. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. doi:10.1016/S0140-6736(06)69705-5
52. Drucker DJ. The cardiovascular biology of glucagon-like peptide-1. Cell Metab. 2016;24(1):15–30. doi:10.1016/j.cmet.2016.06.009
53. Seino Y, Yabe D. Glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1: incretin actions beyond the pancreas. J Diabetes Investig. 2013;4(2):108–130. doi:10.1111/jdi.12065
54. Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387(10019):679–690. doi:10.1016/S0140-6736(15)00803-X
55. Fadini GP, Frison V, Rigato M, et al. Trend 2010–2018 in the clinical use of GLP-1 receptor agonists for the treatment of type 2 diabetes in routine clinical practice: an observational study from Northeast Italy. Acta Diabetol. 2020;57(3):367–375. doi:10.1007/s00592-019-01445-z
56. Svegliati-Baroni G, Saccomanno S, Rychlicki C, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31(9):1285–1297. doi:10.1111/j.1478-3231.2011.02462.x
57. Eguchi Y, Kitajima Y, Hyogo H, et al. Pilot study of liraglutide effects in non-alcoholic steatohepatitis and non-alcoholic fatty liver disease with glucose intolerance in Japanese patients (LEAN-J). Hepatol Res. 2015;45(3):269–278. doi:10.1111/hepr.12351
58. Sathyanarayana P, Jogi M, Muthupillai R, Krishnamurthy R, Samson SL, Bajaj M. Effects of combined exenatide and pioglitazone therapy on hepatic fat content in type 2 diabetes. Obesity. 2011;19(12):2310–2315. doi:10.1038/oby.2011.152
59. Teshome G, Ambachew S, Fasil A, Abebe M. Efficacy of glucagon-like peptide-1 analogs in nonalcoholic fatty liver disease: a systematic review. Hepat Med. 2020;12:139–151. doi:10.2147/HMER.S265631
60. Tripathi A, Debelius J, Brenner DA, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. 2018;15(7):397–411. doi:10.1038/s41575-018-0011-z
61. Vinolo MAR, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3(10):858–876. doi:10.3390/nu3100858
62. Takai A, Kikuchi K, Ichimura M, et al. Fructo-oligosaccharides ameliorate steatohepatitis, visceral adiposity, and associated chronic inflammation via increased production of short-chain fatty acids in a mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol. 2020;20(1):46. doi:10.1186/s12876-020-01194-2
63. Chiang JYL. Bile acid metabolism and signaling. Compr Physiol. 2013;3(3):1191–1212. doi:10.1002/cphy.c120023
64. Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. 2018;15(2):111–128. doi:10.1038/nrgastro.2017.119
65. Hirasawa A, Tsumaya K, Awaji T, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11(1):90–94. doi:10.1038/nm1168
66. Larraufie P, Martin-Gallausiaux C, Lapaque N, et al. SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci Rep. 2018;8(1):74. doi:10.1038/s41598-017-18259-0
67. Sala-Rabanal M, Hirayama BA, Ghezzi C, et al. Revisiting the physiological roles of SGLTs and GLUTs using positron emission tomography in mice. J Physiol. 2016;594(15):4425–4438. doi:10.1113/JP271904
68. Nasiri-Ansari N, Nikolopoulou C, Papoutsi K, et al. Empagliflozin attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) mice by activating autophagy and reducing ER stress and apoptosis. Int J Mol Sci. 2021;22(2):818. doi:10.3390/ijms22020818
69. Inoue M, Hayashi A, Taguchi T, et al. Effects of canagliflozin on body composition and hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease. J Diabetes Investig. 2019;10(4):1004–1011. doi:10.1111/jdi.12980
70. Katsuno K, Fujimori Y, Takemura Y, et al. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J Pharmacol Exp Ther. 2007;320(1):323–330. doi:10.1124/jpet.106.110296
71. Zinman B, Inzucchi SE, Lachin JM, et al. Empagliflozin and cerebrovascular events in patients with type 2 diabetes mellitus at high cardiovascular risk. Stroke. 2017;48(5):1218–1225. doi:10.1161/STROKEAHA.116.015756
72. Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7):644–657. doi:10.1056/NEJMoa1611925
73. Ismail-Beigi F, Moghissi E, Kosiborod M, Inzucchi SE. Shifting paradigms in the medical management of type 2 diabetes: reflections on recent cardiovascular outcome trials. J Gen Intern Med. 2017;32(9):1044–1051. doi:10.1007/s11606-017-4061-7
74. Yokono M, Takasu T, Hayashizaki Y, et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol. 2014;727:66–74. doi:10.1016/j.ejphar.2014.01.040
75. Pradhan R, Yin H, Yu O, Azoulay L. Glucagon-like peptide 1 receptor agonists and sodium-glucose cotransporter 2 inhibitors and risk of nonalcoholic fatty liver disease among patients with type 2 diabetes. Diabetes Care. 2022;45(4):819–829. doi:10.2337/dc21-1953
76. Ding C, Tang Y, Zhu W, et al. Sodium-glucose cotransporter protein-2 inhibitors and glucagon-like peptide-1 receptor agonists versus thiazolidinediones for non-alcoholic fatty liver disease: a network meta-analysis. Acta Diabetol. 2022;59(4):519–533. doi:10.1007/s00592-021-01830-7
77. Raj H, Durgia H, Palui R, et al. SGLT-2 inhibitors in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: a systematic review. World J Diabetes. 2019;10(2):114–132. doi:10.4239/wjd.v10.i2.114
78. Yan H, Huang C, Shen X, Li J, Zhou S, Li W. GLP-1 RAs and SGLT-2 inhibitors for insulin resistance in nonalcoholic fatty liver disease: systematic review and network meta-analysis. Front Endocrinol. 2022;13:923606. doi:10.3389/fendo.2022.923606
79. Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017;1(1):3–9. doi:10.1016/j.livres.2017.05.001
80. Li T, Chiang JYL, Ma Q. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66(4):948–983. doi:10.1124/pr.113.008201
81. Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103(10):3920–3925. doi:10.1073/pnas.0509592103
82. Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145(3):574–582.e1. doi:10.1053/j.gastro.2013.05.042
83. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi:10.1016/S0140-6736(14)61933-4
84. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–735. doi:10.1053/j.gastro.2013.11.049
85. Mitsuyoshi H, Yasui K, Harano Y, et al. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol Res. 2009;39(4):366–373. doi:10.1111/j.1872-034X.2008.00464.x
86. Loomba R, Mohseni R, Lucas KJ, et al. TVB-2640 (FASN inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a randomized, placebo-controlled Phase 2a trial. Gastroenterology. 2021;161(5):1475–1486. doi:10.1053/j.gastro.2021.07.025
87. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55(6):2005–2023. doi:10.1002/hep.25762
88. Oseini AM, Sanyal AJ. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. 2017;37(Suppl 1):97–103. doi:10.1111/liv.13302
89. Sarkhy AA, Al-Hussaini AA, Nobili V. Does vitamin E improve the outcomes of pediatric nonalcoholic fatty liver disease? A systematic review and meta-analysis. Saudi J Gastroenterol. 2014;20(3):143–153. doi:10.4103/1319-3767.132983
90. Moser MA, Chun OK. Vitamin C and heart health: a review based on findings from epidemiologic studies. Int J Mol Sci. 2016;17(8):1328. doi:10.3390/ijms17081328
91. Kim JY, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117(9):2621–2637. doi:10.1172/JCI31021
92. Rose FJ, Webster J, Barry JB, Phillips LK, Richards AA, Whitehead JP. Synergistic effects of ascorbic acid and thiazolidinedione on secretion of high molecular weight adiponectin from human adipocytes. Diabetes Obes Metab. 2010;12(12):1084–1089. doi:10.1111/j.1463-1326.2010.01297.x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Platelet-Activating Factor Promotes the Development of Non-Alcoholic Fatty Liver Disease
Yin H, Shi A, Wu J
Diabetes, Metabolic Syndrome and Obesity 2022, 15:2003-2030
Published Date: 8 July 2022
The Lipid-Oxidative Stress Axis: Novel Therapeutic Targets for Podocytopathy

Liu Y, Zou M, Wang Y
Journal of Inflammation Research 2025, 18:12505-12532
Published Date: 11 September 2025
Metabolic Crosstalk in Diabetic Kidney Disease: Synergistic Effects of Glucotoxicity and Lipotoxicity
Cao J, Dun Z, Tian D, Qiao Z, Wang Y
Diabetes, Metabolic Syndrome and Obesity 2026, 19:580866
Published Date: 23 March 2026