")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Therapeutic antitumor efficacy of tumor-derived autophagosome (DRibble) vaccine on head and neck cancer
Authors Su H, Luo Q, Xie H, Huang X, Ni Y, Mou Y, Hu Q
Received 1 December 2014
Accepted for publication 23 January 2015
Published 10 March 2015 Volume 2015:10(1) Pages 1921—1930
DOI https://doi.org/10.2147/IJN.S74204
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Hang Su,1,* Qiong Luo,2,* Hao Xie,3 Xiaofeng Huang,1 Yanhong Ni,1 Yongbin Mou,1 Qingang Hu1,4
1Center Laboratory of Stomatology, Stomatological Hospital Affiliated Medical School, 2State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, 3Institute of Life Sciences, Key Laboratory of Developmental Genes and Human Disease, Southeast University, Nanjing, People’s Republic of China; 4Leeds Dental Institute, Faculty of Medicine and Health, University of Leeds, Leeds, UK
*These authors contributed equally to this work
Purpose: Vaccines play important roles in antitumor biotherapy. Autophagy in tumor cells plays a critical role in depredating proteins, including tumor-specific antigens and tumor-associated antigens. We aimed to induce and collect tumor-derived autophagosomes (DRibbles) from tumor cells as a novel antitumor vaccine by inhibiting the functions of proteasomes and lysosomes.
Materials and methods: DRibbles were prepared and their morphological and autophagic properties characterized. Dendritic cells (DCs) generated from the bone marrow monocytes of mice were cocultured with DRibbles, then surface molecules of DCs and B cells, as well as apoptosis of DCs, were determined by flow cytometry. Meanwhile, functional properties of the DRibble-DCs were examined by mixed lymphocyte reactions and animal experiments.
Results: The diameter of autophagic nanoparticles with spherical and double-membrane structure was between 200 nm and 500 nm. DRibbles resulted in the upregulation of costimulatory molecules CD40 and CD86 as well as major histocompatibility complex (MHC)-I molecules on DCs, but not MHC-II. The expressions of CD40, CD80, and CD86 and that of MHC-II molecules on B cells were also upregulated. Moreover, suppression of tumor growth and lifetime prolongation was observed in DRibble-DC-vaccinated tumor-bearing mice.
Conclusion: Our results demonstrate that naïve T cells can be activated effectively by DC cross-presenting antigens on upregulated MHC-I, suggesting that DRibbles be deployed as an effective antitumor vaccine for head and neck cancer immunotherapy in clinical trials.
Keywords: autophagy, nanoparticles, dendritic cells, antitumor immunity, head and neck cancer
Introduction
Cross-presentation is the ability of host professional antigen-presenting cells (APCs) to capture, process, and present exogenous antigens to T cells.1 Antigen cross-presentation is pivotal for the initiation of T-cell immune responses, and is necessary for the elimination of many pathogens.2,3 Evidence in the literature suggests that cross-presentation of melanoma antigens during vaccination was essential for an effective anti-tumor therapy. These data indicate that targeting cross-presentation may be a unique approach for immunotherapy for cancer.4 Therefore, the first critical component of successful therapeutic cancer vaccines is to maximize the efficiency of cross-presentation.5
Autophagy is a basic cellular process in which unnecessary or dysfunctional cellular components are sequestered by autophagosomes and delivered to lysosomes for degradation.6,7 As we know, autophagy in tumor-targeted therapy is thought to be a “double-edged sword”, which plays a prodeath or prosurvival role.8–10 On the other hand, it has been reported that autophagy can be induced by metabolic stress and antitumor therapies.11,12 Although the role of autophagy in tumor therapy is complicated and not yet fully understood, it has been considered an attractive approach for anticancer therapy.
Accumulating evidences suggest that autophagy plays an important role in both innate and adaptive immunity.13,14 Moreover, it has been shown that autophagy in tumor cells is essential for cross-presentation of tumor antigens and subsequent induction of tumor immunity.15 As we know, proteins in tumor cells are degraded either by the autophagy–lysosome pathway or the ubiquitin–proteasome system. Though little about the transition between the two pathways is known, it is believed that short-lived proteins, including defective ribosomal products (DRiPs) are ubiquitinated and degraded by proteasomes and long-lived proteins are separated into autophagosomes for lysosomal degradation. Autophagosomes have been identified as critical tumor-antigen carriers for cross-presentation.16 Recently, some results showed the enhanced antigen presentation was related to autophagy.17,18 In this study, autophagosomes from SCC7, a murine head and neck cancer cell line, were induced by rapamycin (autophagy revulsive), bortezomib (proteasome inhibitor), and ammonium chloride (lysosomotropic agent), by which the fusion of lysosomes and phagosomes would be prevented according to previous reports.16,19
Autophagosomes of tumor cells were isolated from both the cells and the culture medium, and these DRiPs containing autophagosome-rich blebs were termed “DRibbles”. Recently, it has been reported that DRibbles act as a potent antigen source and induce enhanced immune responses in vaccine studies.19 Our present study was to induce and characterize DRibbles from the SCC7 cell line, and further to elucidate its possible therapeutic antitumor efficacy on mice bearing head and neck cancer.
Materials and methods
Mice
Specific pathogen-free, 8-week-old female mice were purchased from the Model Animal Research Center of Nanjing University. Animal welfare and experimental procedures were carried out strictly in accordance with the protocols approved by the Animal Care and Use Committee of the Medical School, Nanjing University.
DC culture and tumor-cell line
Dendritic cells (DCs) were generated in accordance with our previous work.20 Briefly, bone marrow mononuclear cells were obtained from bone marrow cells after removal of the femurs and tibias of female C3H/HeJ mice. Bone marrow mononuclear cells (1×106/well) were cultured in complete Roswell Park Memorial Institute 1640 medium with 10% fetal bovine serum (Life Technologies, Breda, Netherlands), 10 ng/mL murine granulocyte-macrophage colony-stimulating factor (GM-CSF; eBioscience, San Diego, CA, USA), and 1 ng/mL murine IL-4 (eBioscience) for 5 days. Half the medium with GM-CSF and IL-4 was gently replaced on days 2 and day 4. SCC7, a murine head and neck carcinoma cell line, was cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 units/mL penicillin, and 100 mg/mL streptomycin (Invitrogen, Grand Island, NY, USA).
Preparation of DRibbles and tumor-cell lysate
DRibbles were prepared as described previously.21 Briefly, tumor cells were treated with rapamycin (100 nM; Enzo Life Sciences, Farmingdale, NY, USA), bortezomib (100 nM; Millennium Pharmaceuticals, Cambridge, MA, USA), and ammonium chloride (10 mM, Sigma-Aldrich, St Louis, MO, USA) in complete medium for 24 hours in a 5% CO2 incubator at 37°C. Cells and large-cell debris were removed by centrifugation at 300 g for 5 minutes. Thereafter, the supernatant was centrifuged for 15 minutes at 12,000 g, and DRibbles secreted by tumor cells were collected and stored in phosphate-buffered saline (PBS) at −80°C for further study.
SCC7 cells (1×107 cells in 500 μL PBS) were lysed by three cycles of repetitive rapid freezing in liquid nitrogen and thawing in a 37°C water bath. Cellular debris was spun down at a speed of 300 g for 5 minutes, and the supernatants were used to pulse the DCs.
Transmission electron microscopy
As mentioned earlier, SCC7 cells were treated for 24 hours to collect DRibbles. The autophagosome-enriched DRibble suspension was collected in 1.5 mL Eppendorf tubes and centrifuged at 12,000 g for 15 minutes. The supernatant was removed carefully, and 2.5% cold glutaraldehyde was added to fix samples at 4°C overnight. The next day, the samples were fixed in 1% osmium tetroxide for 1 hour after being rinsed, dehydrated in gradient acetone, replaced with 1:1 acetone:Epon 812 resin overnight, infiltrated with 100% Epon 812 resin (SPI Supplies, West Chester, PA, USA) for 1 hour, and embedded in Epon 812 resin. After polymerization for 24 hours at 60°C, ultrathin sections (70 nm) were cut with an LKB-V ultramicrotome (LKB, Bromma, Sweden) and stained with uranyl acetate and lead citrate. Then, the sections were observed with a JEM-1011 transmission electron microscope (JEOL, Tokyo, Japan), and images were acquired with a charge-coupled-device CCD camera (SIS, Münster, Germany).
Scanning electron microscopy
The prepared DRibbles suspension was centrifuged at 12,000 g for 15 minutes, and then 4% paraformaldehyde was added to fix the sample at 4°C overnight after removing the supernatant. Thereafter, the sample was dripped on a small piece of aluminum foil, which was used as the counterelectrode, and dried in air. The surface morphology of the sample was observed with an Ultra Plus Field Emission scanning electron microscope (Zeiss, Oberkochen, Germany) operating at 1.00 kV, and images were acquired with a CCD camera (Evolution MP 5.0; Media Cybernetics, Rockville, MD, USA).
Western blot
SCC7 cells were treated with 100 nM rapamycin, 100 nM bortezomib, and 10 mM ammonium chloride in complete medium for 12, 24, and 36 hours, respectively. Meanwhile, the untreated SCC7 cells served as control. Proteins were extracted in lysis buffer (30 mM Tris, pH 7.5, 150 mM sodium chloride, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 1% Nonidet P-40, 10% glycerol, 1 mM phosphatase inhibitors, and 1 mM protease inhibitors). The extracted proteins were separated by 4%–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then electrophoretically transferred onto polyvinylidene fluoride membranes. The membrane was blocked with buffer containing 5% skim milk and probed with LC3 antibodies (Santa Cruz Biotechnology, Dallas, TX, USA) overnight at 4°C, and then incubated with horseradish peroxidase-coupled secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA). The protein bands were detected by enhanced chemiluminescence plus Western blotting-detection reagents (Millipore, Bedford, MA, USA) and analyzed by Gel-Pro32 software.
Flow cytometry
For analysis of surface markers, DCs were incubated with DRibbles (0, 5, 25, and 50 μg/mL) for 24 hours. Then, cells were harvested, thoroughly washed, and incubated with fluorochrome-conjugated specific antibodies for CD11c, CD40, CD86, major histocompatibility complex (MHC)-II, and MHC-I (eBioscience) at recommended dilutions for 30 minutes at 4°C. Meanwhile, mononuclear cells obtained from lymph nodes and spleens were loaded with DRibbles (25 μg/mL) for 72 hours and incubated with fluorochrome-conjugated specific antibodies for B220, CD40, CD80, CD86, and MHC-II (eBioscience) at the same condition. Furthermore, DCs loaded with DRibbles (25 μg/mL) or lysate (25 μg/mL) were collected and stained with annexin V-FITC and propidium iodide for determination of apoptosis rate. The surface-marker and apoptotic cell analysis was performed by BD FACSCalibur (BD Biosciences, San Jose, CA, USA) and results were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Mixed lymphocyte reaction
Magnetic bead separation was used to purify the lymph nodes and spleen-derived T cells needed in mixed lymphocyte reaction (MLR). Briefly, inguinal lymph nodes, mesenteric lymph nodes, and spleen were separated from C3H/HeJ mice after the mice were killed. Thereafter, single-cell suspensions were prepared from the lymph nodes and spleen and T cells were obtained by using a mouse Pan T Cell Isolation Kit II (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the supplier’s protocol.
DCs (2×104/well) were loaded with either DRibbles (25 μg/mL) or lysate (25 μg/mL) for 12 hours with or without cytokine cocktails, which were TNFα (10 ng/mL), IL-1β (10 ng/mL), IL-6 (10 ng/mL) (Peprotech, Rocky Hills, NJ, USA), and prostaglandin E2 (1 μg/mL, Sigma-Aldrich). Then, DCs were cocultured with T cells (2×105/well). After being incubated for 72 hours, the activation and proliferation of T cells were assayed using a Cell Counting Kit (CCK)-8 (Sigma-Aldrich), and absorbances were read at 450 nm using a microplate reader (Synergy HT; BioTek, Winooski, VT, USA) according to the manufacturer’s instructions.
Animal experiments
C3H/HeJ mice were used to establish a murine carcinoma model. These mice were divided into three groups: eight mice each group. For therapeutic experiments, C3H/HeJ mice received subcutaneous injections of 2×106 SCC7 cells in the right flank on day 0. Tumor size was measured with a digital caliper, and the tumor volumes were calculated according to the following formula: tumor volume V (mm3) =1/2×a×b2, where a is the largest diameter (length) and b is the smallest diameter (width) of the tumor. Seven days later, when the average volume of tumors had reached about 80 mm3, tumor-bearing mice were divided randomly into three groups with eight mice each and vaccinated with DCs (1×106) loaded with either DRibbles (25 μg/mL) or tumor lysate (25 μg/mL) in a total volume of 40 μL PBS by subcutaneous injection in both footpads. Meanwhile, those treated with PBS served as controls. On day 14, both sides of inguinal lymph nodes were injected with the same vaccines. During the whole observation, the tumor volume of tumor-bearing mice was monitored every 3 days.
Statistics
Data are expressed as means ± standard error of the mean. Statistical analyses were performed using one-way analysis of variance or Student’s two-tailed t-test. Values of P<0.05 were considered statistically significant. All statistical analysis was performed by GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA).
Results
Ultrastructure and surface morphology of DRibbles
The surface morphology of DRibbles deriving from SCC7 cells by scanning electron microscopy is shown in Figure 1A. The diameters of these particles, with a relatively smooth surface and spherical structure, were in the range of 200–500 nm. The ultrastructure of these particles under transmission electron microscopy is shown in Figure 1B. Numerous vesicles with a double-membrane structure huddled together, and their dimensions also ranged between 200 nm and 500 nm.
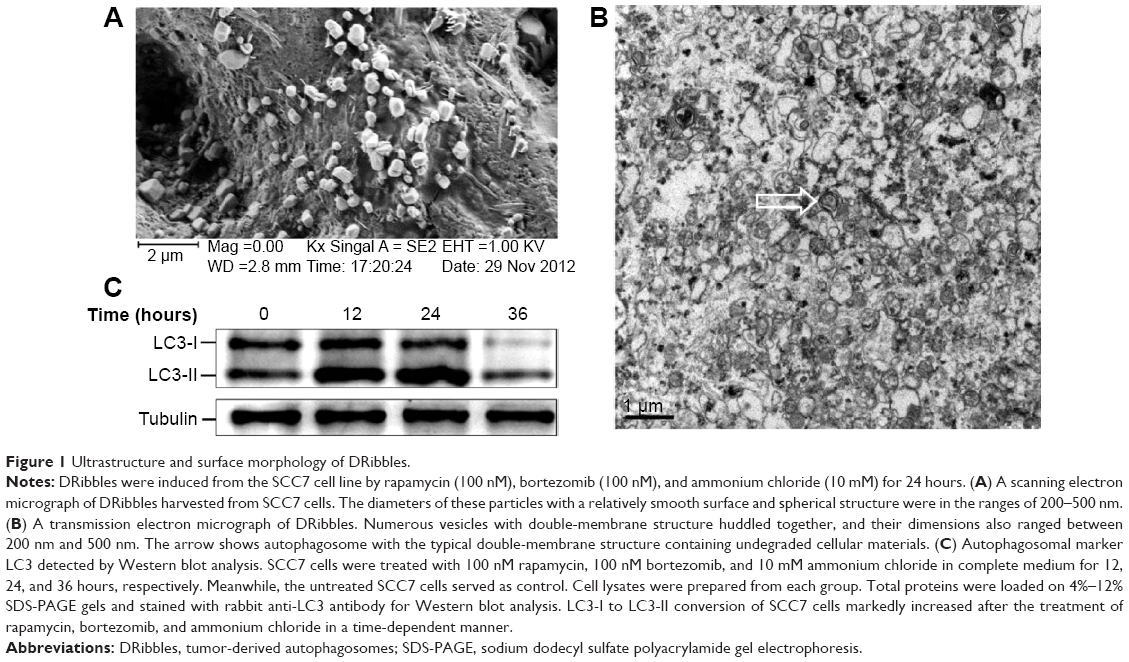 | Figure 1 Ultrastructure and surface morphology of DRibbles. |
Western blot of LC3-II
To further demonstrate the induced DRibbles, expression of LC3, a general autophagosomal marker of autophagosomes,22 was examined by Western blot. The results showed that LC3-I to LC3-II conversion of SCC7 cells markedly increased after treatment with rapamycin, bortezomib, and ammonium chloride in a time-dependent manner (Figure 1C).
DRibbles induced the expression of surface molecules on DCs
To investigate the role of DRibbles in modulating DC function, we evaluated the effects of DRibbles on the expressions of CD40, CD86, MHC-II, and MHC-I on DCs. As shown in Figure 2A and B, DRibbles significantly induced the expression of CD40 and CD86 on DCs in a dose-dependent manner. Moreover, the expression of MHC-I was also increased by DRibbles (Figure 2D), but there were no obvious changes in the expression of MHC-II (Figure 2C).
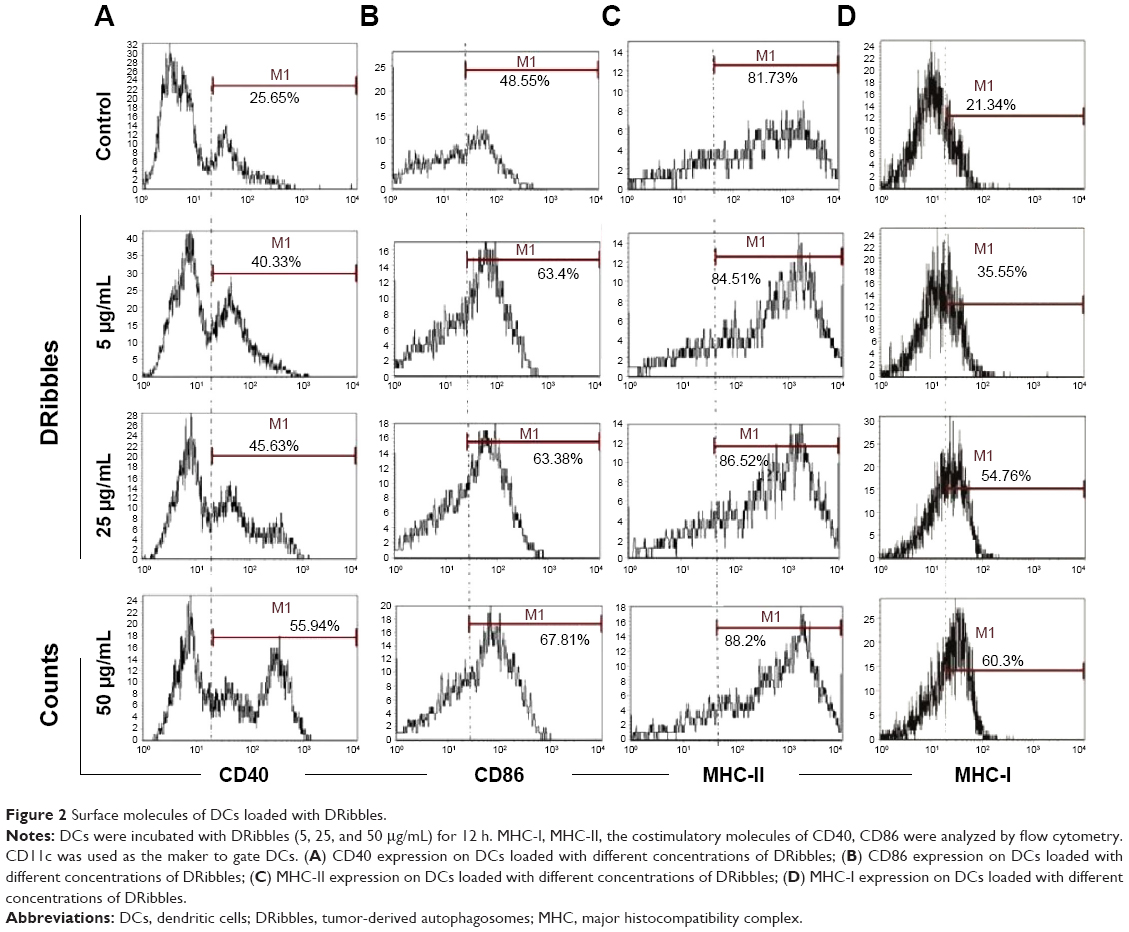 | Figure 2 Surface molecules of DCs loaded with DRibbles. |
DRibbles induced the expression of surface molecules on B cells
To investigate the effects of DRibbles on B-cell responses, we evaluated the expressions of CD40, CD80, CD86, and MHC-II on the surface of B cells. As shown in Figure 3, DRibbles significantly induced the expression of CD40 on B cells derived from lymph nodes or spleen. Interestingly, the expressions of CD80, CD86, and MHC-II were increased by DRibbles as well (Figure 3).
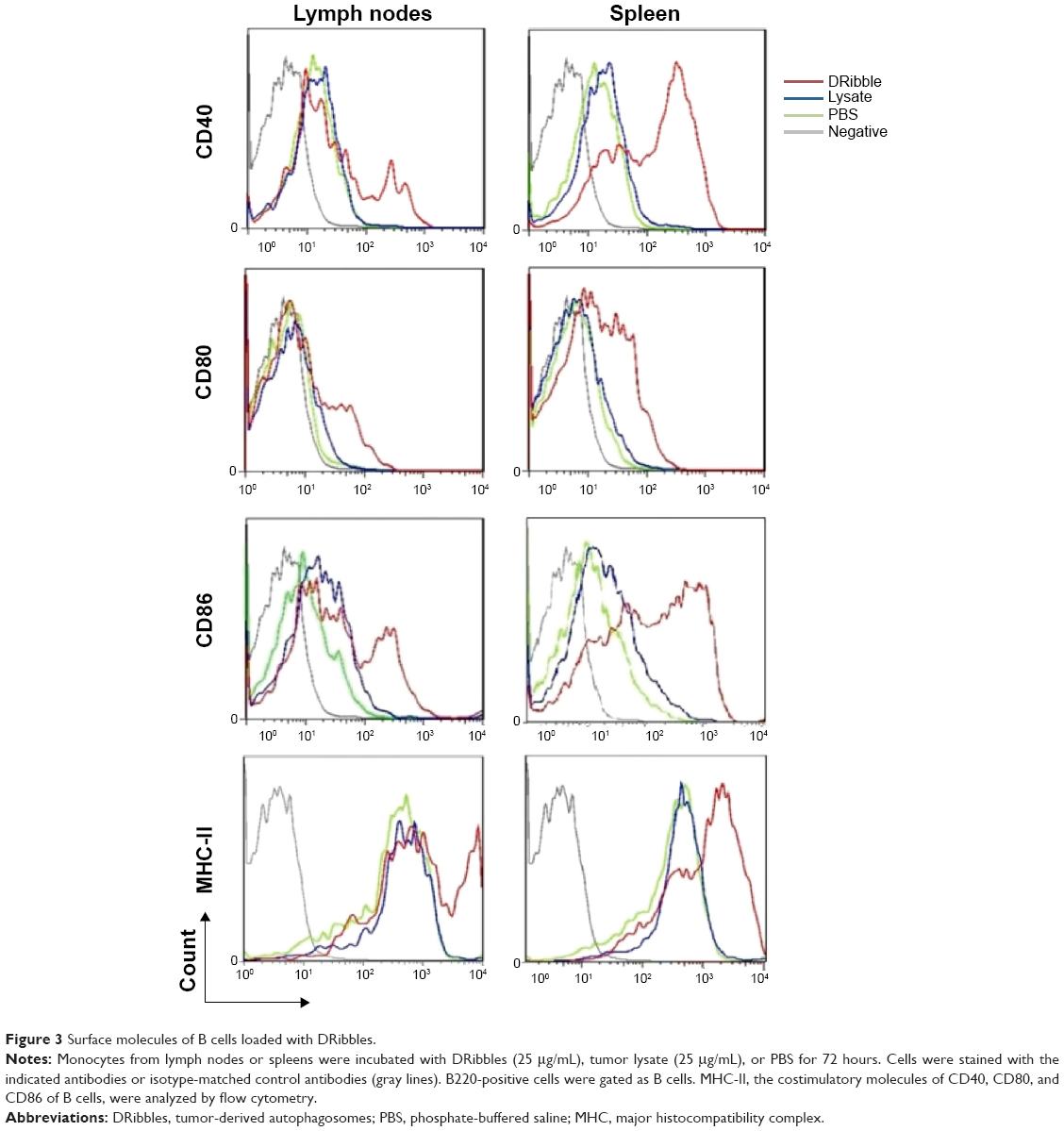 | Figure 3 Surface molecules of B cells loaded with DRibbles. |
DCs treated with DRibbles facilitated proliferative responses of T cells
Before measurement of DC function, we first determined whether DRibbles or lysate could induce apoptosis of DCs after DCs had been incubated with either lysate or DRibbles from SCC7 cells for 24 hours. DRibbles did not influence the apoptosis rate when compared to the untreated group (Figure 4A) (P>0.05).
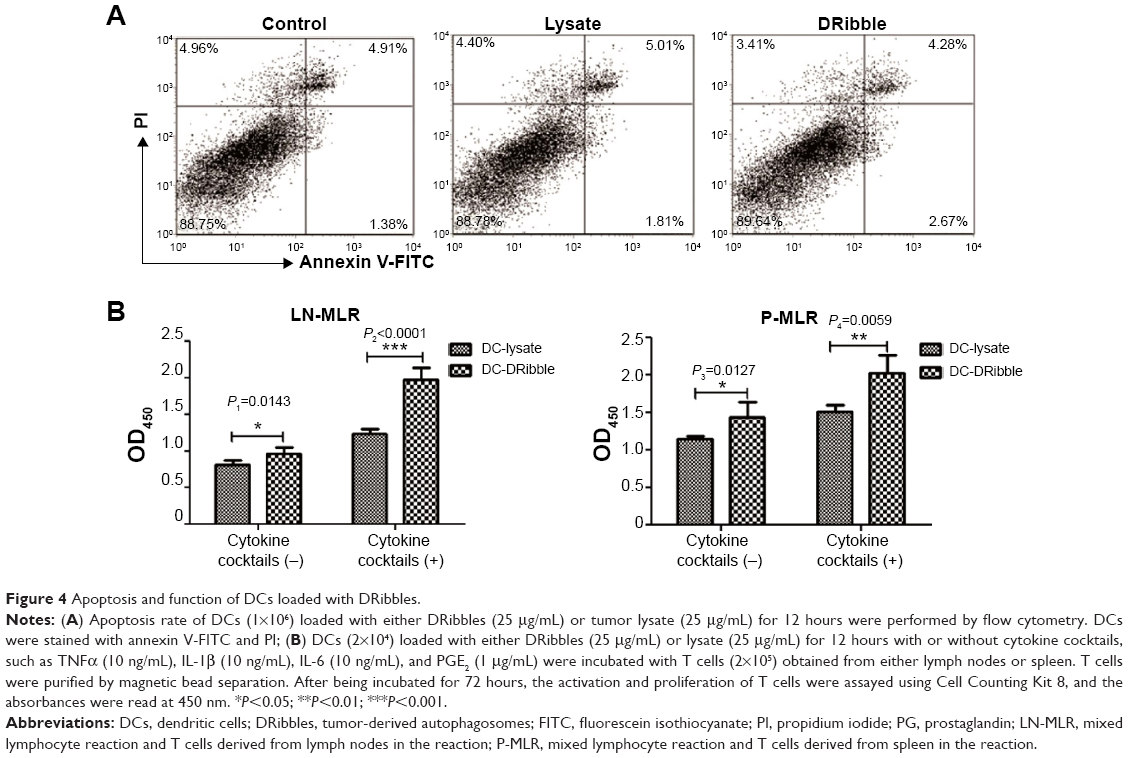 | Figure 4 Apoptosis and function of DCs loaded with DRibbles. |
Functional activity of DCs was determined by the primary MLR assay. DCs treated with DRibbles or lysate were subsequently cocultured with T cells from lymph nodes or spleen of C3H/HeJ mice for 72 hours. As shown in Figure 4B, there was a difference between the proliferation of T cells stimulated by DRibble-loaded DCs and those stimulated by lysate-loaded DCs without cytokine cocktails (P<0.05). DCs in the DRibble-treatment group induced significantly the proliferation of T cells after being stimulated with cytokine cocktails compared to lysate-loaded DCs (P<0.01).
DRibbles improved survival rate and inhibited tumor growth in mice
To investigate the efficacy of DRibbles in vivo, we subcutaneously injected 2×106 SCC7 cells into mice on day 0. Seven days later, we distributed the mice into three groups (n=8). Vaccines of DCs loaded with either DRibbles or tumor lysates were administered into the mice footpads subcutaneously on day 7 and the lymph nodes on day 14 (Figure 5A). At the same time, we measured tumor volumes until day 24. DRibbles significantly improved survival rate (Figure 5C) and markedly inhibited the growth of tumors (Figure 5C) compared with tumor lysates and PBS.
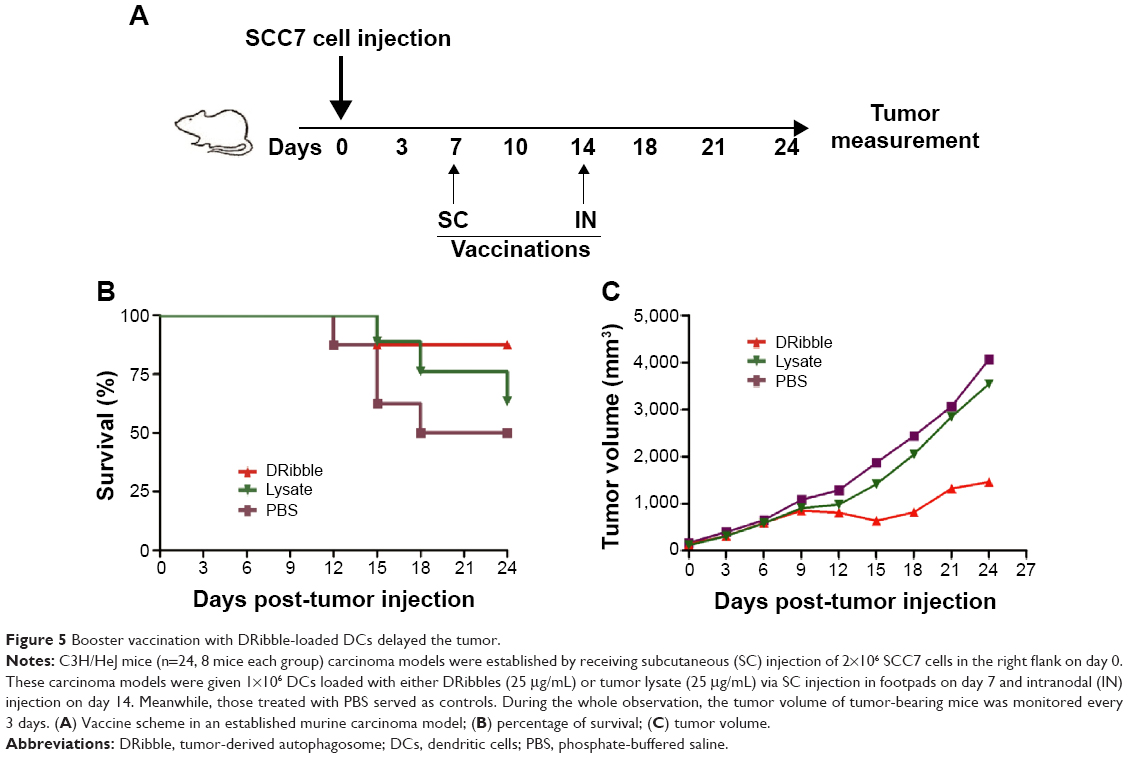 | Figure 5 Booster vaccination with DRibble-loaded DCs delayed the tumor. |
Discussion
Squamous cell carcinoma (SCC) is a highly malignant tumor of squamous epithelial cells.23 The incidence of SCC has been increasing faster and faster.24 In the US, there were 53,640 new cases and 11,520 deaths due to SCC of the head and neck in 2013.25 Moreover, 139,000 new cases of SCC of the head and neck are reported yearly in Europe.26 Although many kinds of antitumor strategies have been developed, SCC is still treated traditionally with surgery, radiotherapy, and chemotherapy.27 The limitations in treatment efficacy of current therapeutic modalities point to the urgent need for new efficient therapeutic strategies to decrease the incidence and mortality of SCC. In this case, immunotherapy becomes a fourth treatment option for SCC. In past years, a variety of cancer vaccines have been used in immunotherapy trials.21,28 These vaccines highlight new therapeutic approaches to enhance the patients’ own immunity. Therefore, tumor-antigen inclusion and efficient cross-presentation are important elements for successful treatment of cancer vaccines.21 In this study, we demonstrated that DRibbles deriving from the SCC7 cell line could enhance costimulatory molecule expression on DCs and B cells and strengthen the immunostimulatory function of DCs, and we explored, for the first time, the efficacy of DRibbles in an in vivo model for head and neck SCC.
The efficacy of antitumor vaccine was based on whether APCs could cross-present tumor-associated antigens to tumor antigen-specific T cells and prime naïve T cells to develop to effector T cells.29 The normal function of APCs and methods of tumor antigens loading are two principal variables for vaccines.30,31 DCs, the most important APC for innate immunity and adaptive immunity, have impaired function in most head and neck SCC (HNSCC) patients.32,33 To make matters worse, a suitable tumor-associated antigen of HNSCC for a vaccine has not been found. This less optimistic fact might lead to little progress made in vaccines for HNSCC. In recent years, the relationships between autophagy and tumors have been widely explored. Many studies demonstrated that cross-presentation of tumor antigens could be enhanced dramatically by inducing autophagy on tumors.15,18 Combining these findings with the DRiP hypothesis proposed previously,34 tumor antigens that might be cross-presented to induce antitumor immune responses were regarded as encapsulated into autophagosomes instead of being degraded in some conditions.16 These autophagosomes were collected and used as effective vaccines for lung cancer and melanoma.21 Therefore, we aimed to induce autophagosome-based DRibbles from HNSCC cells and use them as vaccines to explore their efficacy.
First, SCC7 cells were treated with rapamycin, bortezomib, and ammonium chloride for 12, 24, and 36 hours. The expression of LC3 was detected. The amount of LC3-II was correlated with the number of autophagosomes.35 The expression of LC3-II at the 24-hour time point was higher than that at other points (Figure 1C). As a result, we inferred the most appropriate point of autophagosomes from SCC7 cells was 24 hours. In our experiment, ammonium chloride acted as a lysosomal lumen alkalizer to inhibit the fusion of lysosomes and autophagosomes, leading to the accumulation of autophagic cargo and LC3-II. Therefore, the LC3 level was greatly affected by lysosomal activity. The mechanism of ammonium chloride inhibiting lysosomal activity was similar to that of chloroquine. As we know, acidic extracellular pH can result in a remarkably reduced cellular uptake of chloroquine. Chloroquine can affect lysosomal activity and induce accumulation of LC3-II only at the physiological pH 7.4, but not at acidic pH conditions.36 Therefore, we speculated that culture medium was at acidic pH when cells had been cultured for more than 36 hours. Ammonium chloride did not inhibit lysosomal activity under acidic extracellular pH. This led to the degradation of LC3-II. Therefore, the expression of LC3-II decreased in SCC7 cells treated for 36 hours (Figure 1C). Then, we developed autophagosome-based DRibbles induced from SCC7 cells with rapamycin, bortezomib, and ammonium chloride for 24 hours. The unique characteristics of DRibbles were determined by transmission electron microscopy and scanning electron microscopy (Figure 1A and B). The features and dimensions of the double-membrane particles shown in Figure 1A and B were in accordance with autophagosomes in previous research.21 Western blot analysis showed that LC3-II, the typical autophagosome marker, was presented in these DRibbles as expected (Figure 1C). These findings indicated that DRibbles might be able to activate immune responses.
Next, we focused on DRibble modulation on APCs, especially on DCs and B cells. Our study showed that DRibbles could induce expression of CD40, CD86, and MHC-I on DCs, but not MHC-II (Figure 2), which meant DRibbles could induce the upregulation of costimulatory molecules on the surface of DCs and antigen cross-presentation by DCs. Similarly, the expressions of CD40, CD80, CD86, and MHC-II on B cells derived from lymph nodes or spleens were also enhanced by DRibbles (Figure 3). These costimulatory molecules were upregulated to engage in the APC–T cell interaction, which were key factors for the activity of APCs and subsequent immune responses. Therefore, APCs loaded with DRibbles might be able to activate naïve T cells through the increased expressions of costimulatory molecules.
To determine whether DRibble DCs could be efficient in stimulating naïve T cells, we cocultured T cells with DRibble-loaded DCs. Subsequently, we examined the proliferation of T cells by CCK-8 analysis. DCs loaded with DRibbles were highly efficient in driving T-cell proliferation (Figure 4B). These results indicated that tumor cell-derived DRibbles serving as a source of tumor antigens were superior in activating antigen-specific T cells when loaded onto DCs. To determine the antitumor effect of DRibbles, we subcutaneously injected SCC7 cells into mice, and vaccinated these tumor-bearing mice with DRibble-loaded DCs. Vaccination with DCs loaded with DRibbles significantly improved the survival rate and inhibited the growth of tumors compared with tumor lysate-loaded DCs (Figure 5).
Compared with tumor lysate-pulsed DCs, autophagic nanoparticle-pulsed DCs increased the expressions of costimulatory molecules significantly, and were more effective in activating naïve T cells. Autophagic nanoparticles might contain tumor antigens that could be directly presented by tumor cells. DRibble-pulsed DCs might cross-present tumor antigens to naïve T cells and induce specific immune responses against SCC7 cells. These might lead to suppression of tumor growth and lifetime prolongation in tumor-bearing mice. There are also many different variables in preparing and administering DC vaccines.37 Our preliminary study proved DRibble-loaded DCs were more effective in improving survival rate and delaying tumor progression in tumor-bearing mice than lysate-loaded DCs. However, much remains to be done. We will optimize the protocol of DC vaccine preparation and administration to improve survival benefit in mice in our further studies.
In summary, we identified the unique characteristics and potential antitumor efficacy of autophagic nanoparticles (DRibbles) on the basis of these findings. We provided insights into the mechanism of DRibbles that could be responsible for their efficacy as a novel cancer immunotherapy. The efficacy of DRibbles should be further determined in clinical trials.
Acknowledgments
We acknowledge the kind support of Dr Hongming Hu, Professor Yayi Hou, and Professor Zichun Hua for technical assistance, and Guohua Xia and Tom Luther for language editing. The authors are grateful for grants from the National Natural Sciences Foundation of China (81271698, 81371680), the Project of Natural Science Foundation of Jiangsu Province (BK2012744), the Six Talent Peaks Project in Jiangsu Province (2012-SWYY-002), and the Nanjing Medical Development Foundation (ZKX12034).
Disclosure
The authors report no conflicts of interest in this work.
References
Bevan MJ. Cross-priming. Nat Immunol. 2006;7:363–365. | ||
Bevan MJ. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J Exp Med. 1976;143:1283–1288. | ||
Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. | ||
Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264:961–965. | ||
Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20:89–95. | ||
Lin NY, Beyer C, Giessl A, et al. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann Rheum Dis. 2013;72:761–768. | ||
Abeliovich H, Dunn WA Jr, Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol. 2000;151:1025–1034. | ||
Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res. 2010;70:3431–3434. | ||
Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. | ||
Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther. 2011;131:130–141. | ||
Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–666. | ||
Liu EY, Ryan KM. Autophagy and cancer – issues we need to digest. J Cell Sci. 2012;125:2349–2358. | ||
Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. | ||
Heath RJ, Xavier RJ. Autophagy, immunity and human disease. Curr Opin Gastroenterol. 2009;25:512–520. | ||
Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68:6889–6895. | ||
Li Y, Wang LX, Pang P, et al. Cross-presentation of tumor associated antigens through tumor-derived autophagosomes. Autophagy. 2009;5:576–577. | ||
English L, Chemali M, Duron J, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009;10:480–487. | ||
Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15:267–276. | ||
Twitty CG, Jensen SM, Hu HM, Fox BA. Tumor-derived autophagosome vaccine: induction of cross-protective immune responses against short-lived proteins through a p62-dependent mechanism. Clin Cancer Res. 2011;17:6467–6481. | ||
Mou Y, Chen B, Zhang Y, et al. Influence of synthetic superparamagnetic iron oxide on dendritic cells. Int J Nanomedicine. 2011;6:1779–1786. | ||
Li Y, Wang LX, Pang P, et al. Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin Cancer Res. 2011;17:7047–7057. | ||
Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. | ||
Duray A, Demoulin S, Hubert P, Delvenne P, Saussez S. Immune suppression in head and neck cancers: a review. Clin Dev Immunol. 2010;2010:701657. | ||
Psyrri A, Rampias T, Vermorken JB. The current and future impact of human papillomavirus on treatment of squamous cell carcinoma of the head and neck. Ann Oncol. 2014;25:2101–2115. | ||
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. | ||
Grégoire V, Lefebvre JL, Licitra L, Felip E. Squamous cell carcinoma of the head and neck: EHNS-ESMO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21:v184–v186. | ||
Freiser ME, Serafini P, Weed DT. The immune system and head and neck squamous cell carcinoma: from carcinogenesis to new therapeutic opportunities. Immunol Res. 2013;57:52–69. | ||
Mittendorf EA, Peoples GE, Singletary SE. Breast cancer vaccines: promise for the future or pipe dream? Cancer. 2007;110:1677–1686. | ||
Corundolo V, Hermans IF, Salio M. Dendritic cells: a journey from laboratory to clinic. Nat Immunol. 2004;5:7–10. | ||
Strome SE, Voss S, Wilcox R, et al. Strategies for antigen loading of dendritic cells to enhance the antitumor immune response. Cancer Res. 2002;62:1884–1889. | ||
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. | ||
Gottfried E, Kreutz M, Mackensen A. Tumor-induced modulation of dendritic cell function. Cytokine Growth Factor Revs. 2008;19:65–77. | ||
Wang ZY, Shi PH, Huang XF, et al. Peripheral blood dendritic cells and vascular endothelial growth factor in oral squamous cell carcinoma: correlation analysis and in vitro study. Int J Oral Maxillofac Surg. 2010;39:713–720. | ||
Yewdell JW, Nicchitta CV. The DRiP hypothesis decennial: support, controversy, refinement and extension. Trends Immunol. 2006;27:368–373. | ||
Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. | ||
Pellegrini P, Strambi A, Zipoli C, et al. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy. 2014;10:562–571. | ||
Lesterhuis WJ, Aarntzen E, De Vries IJ, et al. Dendritic cell vaccines in melanoma: from promise to proof? Crit Rev Oncol Hematol. 2008;66:118–134. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.