Back to Journals » Drug Design, Development and Therapy » Volume 11
Theophylline action on primary human bronchial epithelial cells under proinflammatory stimuli and steroidal drugs: a therapeutic rationale approach
Authors Gallelli L ![]() , Falcone D, Cannataro R, Perri M, Serra R, Pelaia G
, Falcone D, Cannataro R, Perri M, Serra R, Pelaia G ![]() , Maselli R, Savino R, Spaziano G, D’Agostino B
, Maselli R, Savino R, Spaziano G, D’Agostino B ![]()
Received 30 July 2016
Accepted for publication 13 September 2016
Published 23 January 2017 Volume 2017:11 Pages 265—272
DOI https://doi.org/10.2147/DDDT.S118485
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Luca Gallelli,1,* Daniela Falcone,1,* Roberto Cannataro,2 Mariarita Perri,2 Raffaele Serra,3 Girolamo Pelaia,3 Rosario Maselli,3 Rocco Savino,1 Giuseppe Spaziano,4 Bruno D’Agostino4
1Department of Health Science, University of Catanzaro, Catanzaro, 2Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, Rende, 3Department of Medical and Surgical Sciences, University of Catanzaro, Catanzaro, 4Department of Experimental Medicine, School of Medicine, Section of Pharmacology, Second University of Naples, Naples, Italy
*These authors contributed equally to this work
Abstract: Theophylline is a natural compound present in tea. Because of its property to relax smooth muscle it is used in pharmacology for the treatment of airway diseases (ie, chronic obstructive pulmonary disease, asthma). However, this effect on smooth muscle is dose dependent and it is related to the development of side effects. Recently, an increasing body of evidence suggests that theophylline, at low concentrations, also has anti-inflammatory effects related to the activation of histone deacetylases. In this study, we evaluated the effects of theophylline alone and in combination with corticosteroids on human bronchial epithelial cells under inflammatory stimuli. Theophylline administrated alone was not able to reduce growth-stimulating signaling via extracellular signal-regulated kinases activation and matrix metalloproteases release, whereas it strongly counteracts this biochemical behavior when administered in the presence of corticosteroids. These data provide scientific evidence for supporting the rationale for the pharmacological use of theophylline and corticosteroid combined drug.
Keywords: human bronchial epithelial cells, theophylline, corticosteroids, signal transduction
Introduction
Asthma is characterized by activated mast cells, increased numbers of eosinophils, increased numbers of T helper 2 and T helper 17 lymphocytes, and increase in sensory neurogenic release.1 Allergens, some pathogens, and some diseases have been implicated in the exacerbation of asthma in both animal models and humans.2–7 Theophylline is a natural compound present in tea. Because of its property to relax smooth muscle, oral theophylline has been administered to improve the bronchoconstriction in patients with severe asthma or chronic obstructive pulmonary disease (COPD). Theophylline has dose-dependent effects, and the therapeutic index of “high dose” theophylline is narrow. Therefore, its use at “high dose” (plasma levels 10–20 mg/L) as a bronchodilator, frequently, is related with the development of side effects.8
Recently, however, preclinical studies have demonstrated that theophylline at “low dose” (plasma levels 1–5 mg/L) has anti-inflammatory effects not related to the adenosine receptor antagonism or phosphodiesterase (PDE) inhibition, which requires high dose.9 In fact, at “low dose”, theophylline inhibits phosphoinositide 3-kinase (PI3K);10 PI3K generates lipid second messengers involved in airways inflammation.11 Moreover, To et al12 documented that low concentrations of theophylline restore corticosteroid sensitivity by the enhancement of histone deacetylase (HDAC)-2 activity through inhibition of the d-isoform of PI3K and that this is independent from PDE inhibition and adenosine antagonism. In agreement with these data, Sun et al,13 in an experimental study documented that a treatment with theophylline at low dose might improve the anti-inflammatory effects of steroids by increasing HDAC-2 activity.
In patients with airways diseases, particularly with COPD, tumor necrosis factor-alpha (TNF-alpha) and interleukin (IL)-1 blood levels correlate with COPD severity (the higher the levels of these inflammatory stimuli the more severe is COPD expression).14
Montaño et al15 documented increased plasma levels of matrix metalloproteinases (MMP-1, MMP-7, and MMP-9) in patients with COPD, compared to healthy subjects. In agreement, more recently, Ostridge et al16 evaluating inflammatory cytokines in bronchoalveolar lavage from 24 COPD and eight control subjects documented higher levels of MMPs in lungs of subjects with COPD, suggesting that MMPs play a pivotal role in the development of COPD. Taken together, these studies documented that MMPs, ILs, and TNF-alpha may all be involved in the inflammatory pathways of COPD.
Moreover, inflammatory and structural cells, including neutrophils, macrophages, and epithelial cells, which are activated in the airways of patients with COPD release the oxidative stress that activates some transcription factors, for example, nuclear factor-κB (NF-κB) which switches on multiple inflammatory genes, resulting in amplification of the inflammatory response.17 NF-κB is regulated by complex signal transduction pathways mediated by mitogen-activated protein kinases (MAPKs). The ERK1/2 subgroup of MAPKs is activated by an MAPK named Raf (most commonly Raf-1), whose activation in turn requires the guanosine-5′-triphosphate-bound form of Ras family proteins.18 Once activated, Raf-1 phosphorylates the MAPKs MEK1 and MEK2, that finally stimulate ERK1 and ERK2. Airborne pollutants and cigarette smoke can induce the bronchial epithelium to acquire a proinflammatory phenotype, characterized by an increased production of autacoids, cytokines, and chemokines.19 Oxidant-induced phenotypic changes may thus significantly contribute to the key pathogenic role played by bronchial epithelial cells in inflammatory airway disorders (eg, asthma and COPD).
Culpitt et al20 in an in vitro study documented that the release of TNF-alpha and MMP-9 from macrophages taken from healthy subjects and normal smokers is inhibited by corticosteroids, whereas they are relatively ineffective in macrophages from patients with COPD. The reasons for resistance to corticosteroids in patients with COPD might be the marked reduction in activity of HDAC-2, which is recruited to activated inflammatory genes by glucocorticoid receptors to switch off inflammatory genes.21 The decreased activity of HDAC-2 is related to both increased secretion of cytokines (eg, TNF-alpha) and reduced response to corticosteroids.17 However, a recent finding highlights that the theophylline by itself has the capability to restore corticosteroid sensitivity.22
With this knowledge, in this study, we evaluated the effects of theophylline alone and theophylline in combination with corticosteroids on primary cultures of human bronchial epithelial cells (HBECs) under inflammatory stimuli.
Methods
Reagents
Anti-phospho-ERK1/2, Anti-phospho-NF-κB, and monoclonal antibodies were purchased from New England Biolabs (Beverly, MA, USA); an anti-(total)-ERK1/2 polyclonal antibody was commercially provided by Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). All reagents and drugs needed for cell culture, protein extraction, and Western blotting were purchased from Sigma (St Louis, MO, USA). The drugs were obtained from Sigma and then dissolved in agreement with the datasheet of each drug, in saline (0.9%) or in light-protected dimethyl sulfoxide (DMSO) as a stock solution (stored at −20°C). Stock solution was then further diluted in cell culture medium to create working concentrations. The maximum final concentration of DMSO was <0.1%.
Recombinant TNF-alpha was obtained from PeproTech, Inc. (Rocky Hill, NJ, USA). Enzyme-linked immunosorbent assay (ELISA) for MMP-2 and MMP-9 monoclonal antibody (Biotrak Human ELISA System, Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, UK) was performed according to manufacturer’s protocol.
Primary cultures of HBECs
Adjacent normal lung tissue from a surgical specimen of lung tumor was obtained with the written informed consent of the patients. The study was approved by the Research Ethics Committee (EUDRACT number 2010019530-27), and was carried out in accordance with the Declaration of Helsinki and Guidelines for Good Clinical Practice. To avoid contamination of tumor tissue, the normal tissue was obtained far from the tumor lesion. Therefore, HBECs were obtained from fresh surgical specimens derived from patients who underwent either lobectomy or pneumectomy, in agreement with our previous papers.23–27
The cells were then harvested and cultured in bronchial epithelial growth medium (BEGM; Clonetics, Sand Diego, CA, USA) in the presence of antibiotics (100 U/mL of penicillin and 100 μg/mL of streptomycin; Sigma) and fungizone (1 μg/mL; Gibco BRL, Gaithersburg, MD, USA). Cells were maintained at 37°C in a humidified incubator, with 5% CO2. The cell line generated by primary epithelial culture medium was referred to as HBECs.
Experimental protocol
For assays, cells at passage 3 or 4 (4 days for each passage) were seeded into 24-well cell culture plates (1 mL BEGM/well containing 5×104 cells/mL). When cells reached 70%–80% confluence, they were then treated for 24 h with TNF-alpha (50 ng/mL) or IL-1 (1 ng/mL) in the presence or absence of an overnight treatment with methylprednisone (10−5 and 10−10 M), hydrocortisone (10−5 and 10−10 M), and theophylline (10−5 M). The medium was not changed after the treatment. The solvent employed to dissolve these drugs was used as control. After this period, the medium was collected for ELISA determination and cells were processed for protein extraction and immunoblotting.
Protein extraction and immunoblot analysis
Following treatment with TNF-alpha or IL-1, cells were lysed for Western blotting in radioimmunoprecipitation assay buffer as previously reported.28–32
Briefly, protein extracts were then separated on 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Amersham Pharmacia Biotech). Immunoblotting was performed using an anti-phospho-ERK1/2 monoclonal antibody. Membranes were reprobed with a polyclonal antibody against total (phosphorylated and unphosphorylated) protein. Antibody binding was visualized by enhanced chemiluminescence-Plus (Amersham Pharmacia Biotech); intensities of experimental bands were analyzed by computer-assisted densitometry (ImageJ software for Windows) and expressed as arbitrary units (control levels set at 100). These experiments were performed in triplicate.
Enzyme-linked immunosorbent assay
A commercially available sandwich ELISA kit for MMP-2 and MMP-9 determination based on monoclonal antibody (Human Biotrak ELISA System, Amersham Pharmacia Biotech) was used to determine MMP levels, in agreement, with our previous papers.33–35 These experiments were performed in triplicate.
Statistical analysis
All data are expressed as mean ± standard error of the mean. Statistical evaluation of the results was performed by analysis of variance (ANOVA). Differences identified by ANOVA were pinpointed by paired Student’s t-test. The threshold of statistical significance was set at P<0.05.
Results
ERKs phosphorylation and NF-κB modulation under proinflammatory stimuli and steroid drugs
The exposure of HBECs for 24 h to TNF-alpha (50 ng/mL) induced a significant increase in the ERK1/2 phosphorylation (pERKs; P<0.01; Figure 1A, C, lane 2). In agreement with pERK, an increase in the activation of NF-κB (P<0.01) was recorded (Figure 1B, C, lane 2). The effect of TNF-alpha on both pERKs and NF-κB was significantly inhibited (P<0.01) by a 24-h treatment with methylprednisone and hydrocortisone at high dosage (10−5) (Figure 1A–C, lanes 3, 4). In contrast, corticosteroids at low dosage (10−10) did not modify the effects of TNF-alpha on both pERKs and NF-κB (Figures 1A–C, lanes 5, 6).
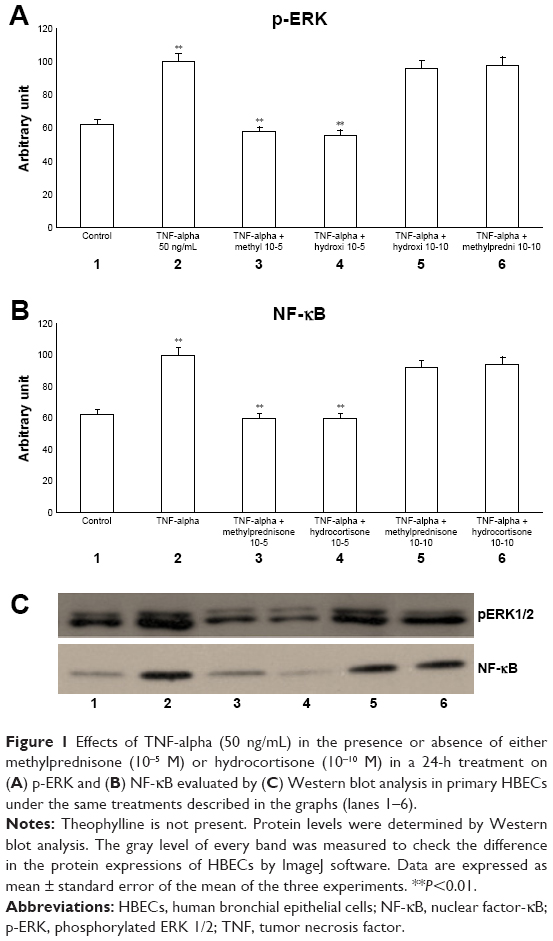 | Figure 1 Effects of TNF-alpha (50 ng/mL) in the presence or absence of either methylprednisone (10−5 M) or hydrocortisone (10−10 M) in a 24-h treatment on (A) p-ERK and (B) NF-κB evaluated by (C) Western blot analysis in primary HBECs under the same treatments described in the graphs (lanes 1–6). |
The exposure of HBECs for 24 h to IL-1 (1 ng/mL) significantly increased (P<0.01) both pERK1/2 and NF-κB (Figure 2A–C, lane 2). These effects were significantly inhibited (P<0.01) by a 24-h treatment with methylprednisone and hydrocortisone at high concentrations (10−5) (Figure 2A–C, lanes 3, 4). In contrast, a 24-h treatment with methylprednisone and hydrocortisone at low concentrations (10−10) did not modify the effects of IL-1 on pERK and NF-κB (Figures 2A–C, lanes 5, 6). The treatment with theophylline (10−5) did not modify the effects of both TNF-alpha and IL-1 on p-ERK and NF-κB (Figures 3A–C, lane 2; 4A–C, lane 2), but potentiated the inhibitory effects of methylprednisone and hydrocortisone on both TNF-alpha and IL-1 (Figures 3A–C, lanes 5, 6; 4A–C, lanes 5, 6). Both TNF-alpha and IL-1 exerted their effects uniquely on phosphorylation-dependent activation of ERK1/2, without affecting its total expression (data not shown).
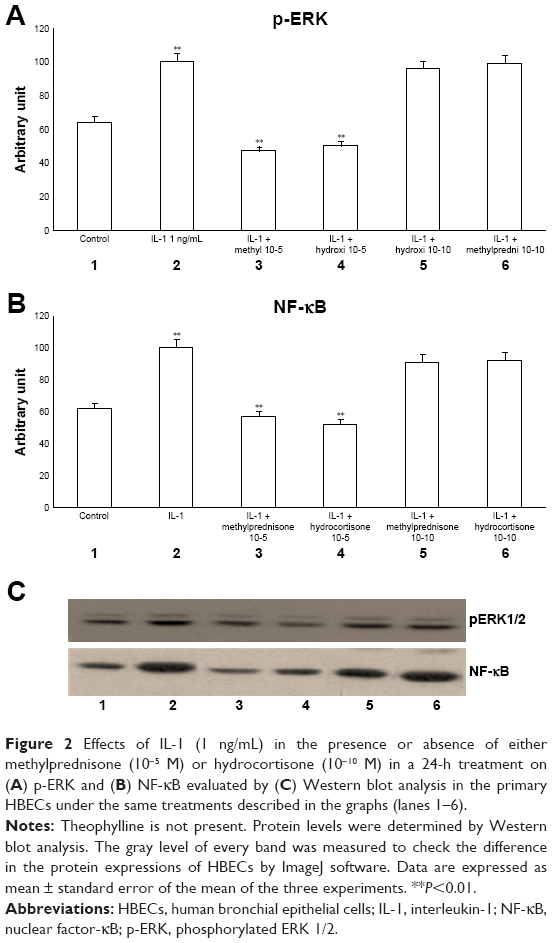 | Figure 2 Effects of IL-1 (1 ng/mL) in the presence or absence of either methylprednisone (10−5 M) or hydrocortisone (10−10 M) in a 24-h treatment on (A) p-ERK and (B) NF-κB evaluated by (C) Western blot analysis in the primary HBECs under the same treatments described in the graphs (lanes 1–6). |
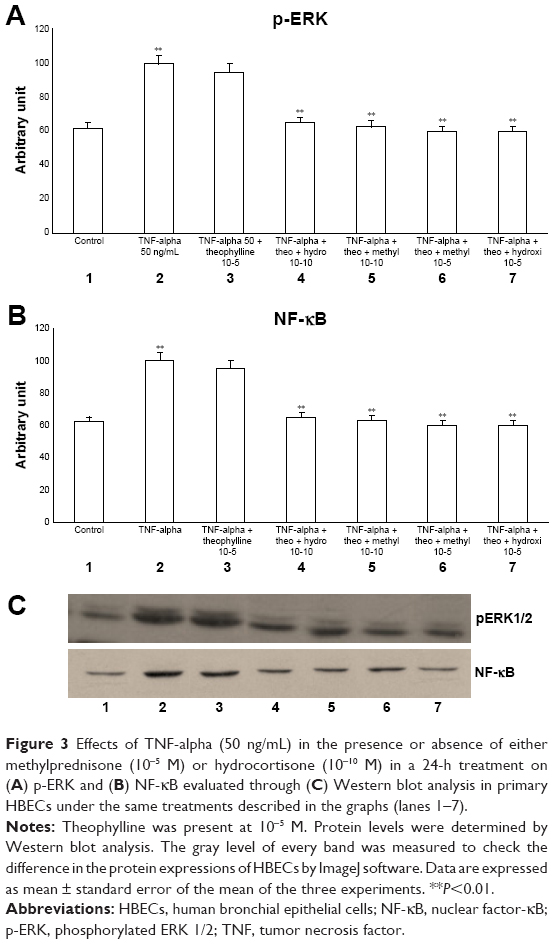 | Figure 3 Effects of TNF-alpha (50 ng/mL) in the presence or absence of either methylprednisone (10−5 M) or hydrocortisone (10−10 M) in a 24-h treatment on (A) p-ERK and (B) NF-κB evaluated through (C) Western blot analysis in primary HBECs under the same treatments described in the graphs (lanes 1–7). |
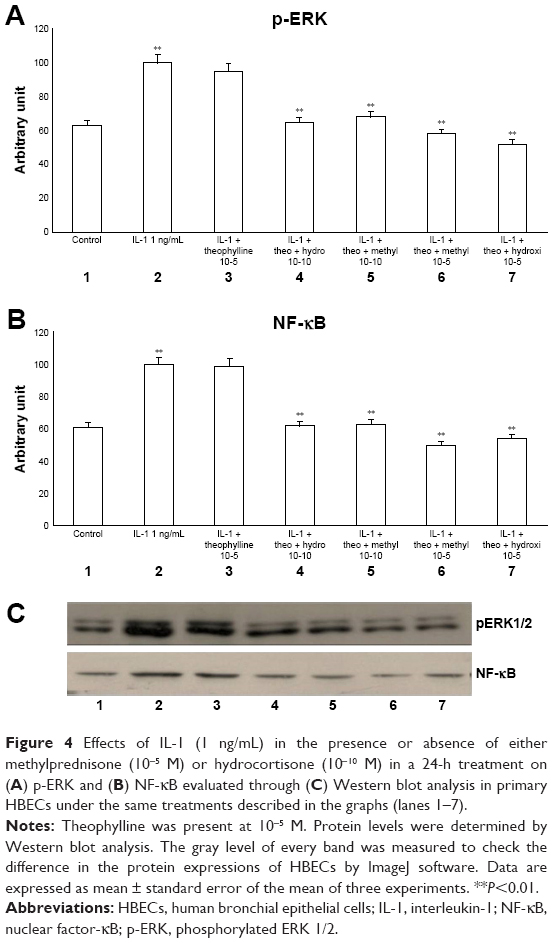 | Figure 4 Effects of IL-1 (1 ng/mL) in the presence or absence of either methylprednisone (10−5 M) or hydrocortisone (10−10 M) in a 24-h treatment on (A) p-ERK and (B) NF-κB evaluated through (C) Western blot analysis in primary HBECs under the same treatments described in the graphs (lanes 1–7). |
Matrix metalloproteinases expression
The exposure of HBECs for 24 h to TNF-alpha (50 ng/mL) induced a significant increase (P<0.01) in MMP-2 and MMP-9 expression. These effects were reversed by a 24-h treatment with methylprednisone and hydrocortisone at high dosage (10−5); in contrast, the treatment with corticosteroids at low dosage (10−10) did not modify the effects of TNF-alpha on MMPs expression (Figure 5A and B). Similarly, IL-1 (1 ng/mL) increased the expression of MMPs and this effect was significantly reversed (P<0.01) by a 24-h treatment with corticosteroids at high dosage (Figure 6A and B). Theophylline (10−5) potentiated the effects of methylprednisone and hydrocortisone on MMP-2 and MMP-9 expression (Figures 5C and D, 6C and D).
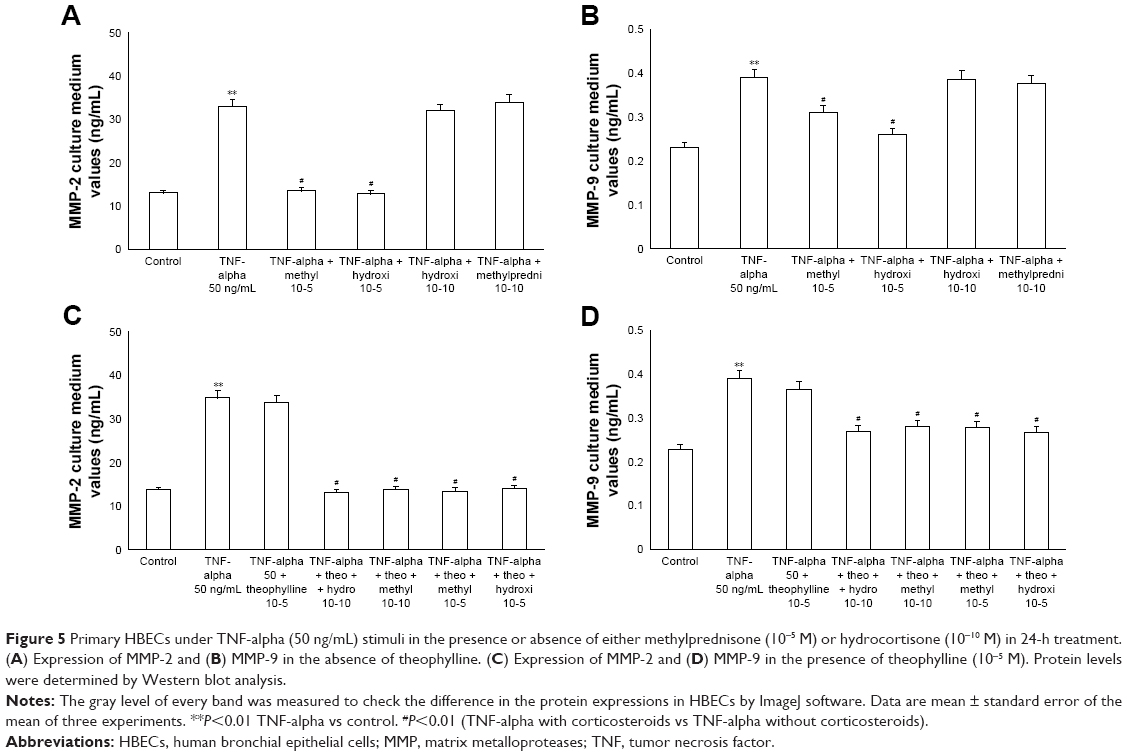 | Figure 5 Primary HBECs under TNF-alpha (50 ng/mL) stimuli in the presence or absence of either methylprednisone (10−5 M) or hydrocortisone (10−10 M) in 24-h treatment. (A) Expression of MMP-2 and (B) MMP-9 in the absence of theophylline. (C) Expression of MMP-2 and (D) MMP-9 in the presence of theophylline (10−5 M). Protein levels were determined by Western blot analysis. |
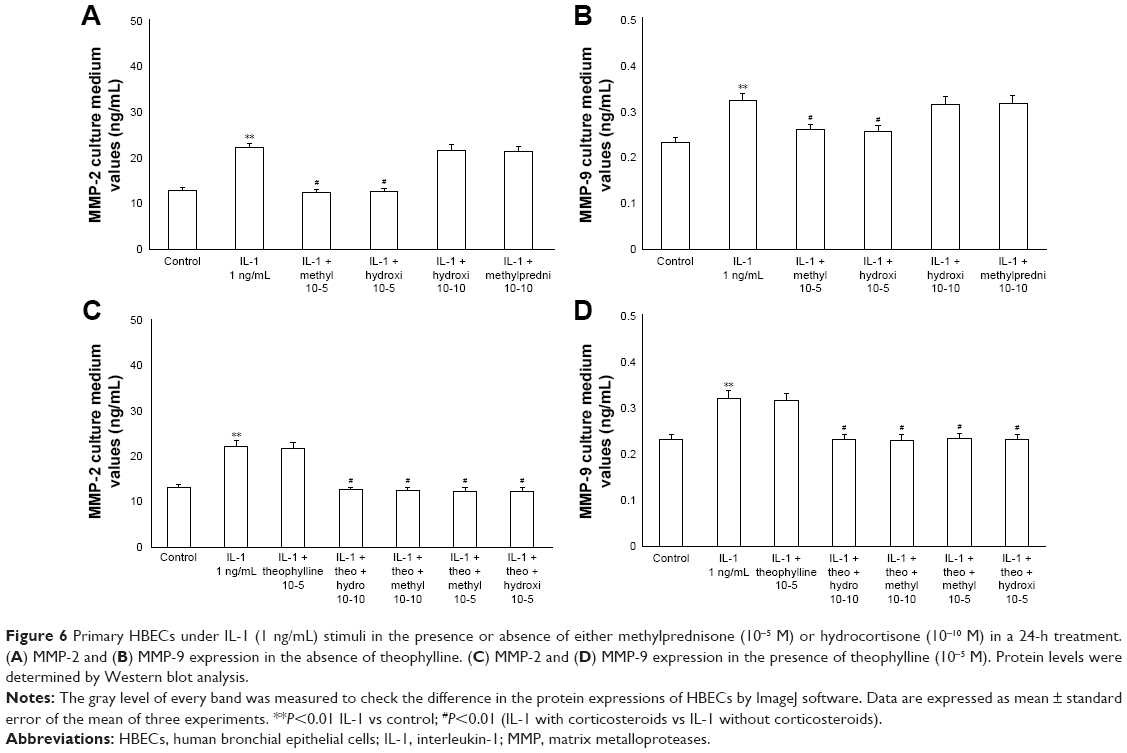 | Figure 6 Primary HBECs under IL-1 (1 ng/mL) stimuli in the presence or absence of either methylprednisone (10−5 M) or hydrocortisone (10−10 M) in a 24-h treatment. (A) MMP-2 and (B) MMP-9 expression in the absence of theophylline. (C) MMP-2 and (D) MMP-9 expression in the presence of theophylline (10−5 M). Protein levels were determined by Western blot analysis. |
Discussion
TNF-alpha plays an important role in defense of the body against foreign pathogens such as viruses, bacteria, and fungi. TNF-alpha production within the lung (from several cells, ie, neutrophils, T cells, macrophages, monocytes, epithelial cells, fibroblasts, and smooth muscle cells) plays a central role in both inducing the expression of adhesion molecules (eg, intercellular adhesion molecule [ICAM]-1, vascular cell adhesion molecule-1, and E-selectin) and cytokines (eg, IL-1, IL-6, IL-8, platelet-derived growth factor, granulocyte macrophage colony stimulating factor, monocyte chemoattractant protein-1, and macrophage inflammatory protein 2).36
Increased levels of chemokines, cytokines, and adhesion molecules induce both the activation and recruitment of neutrophils and macrophages to the lung and lead to tissue destruction.37 In agreement, it has been documented that blood levels of TNF-alpha and IL-1 correlate with COPD severity.38,39 In this view, we treated primary HBECs with TNF-alpha and IL-1 to mimic organ conditions in COPD. In our study, both TNF-alpha and IL-1 induced a significant increase in pERK and NF-κB suggesting that these proinflammatory molecules play a role in the inflammatory process. These data are in agreement with other authors who documented that in human lung cells, TNF-alpha and IL-1beta induce the activation of NF-κB and MAPK signaling pathways.40,41
Previously in HBECs, we documented that TNF-alpha stimulates, via activation of p38 MAPK signaling pathway, IL-8 release and airway epithelial cell apoptosis, an effect that was inhibited by budesonide.42
In agreement, Ito et al43 showed both in vitro and in vivo that low-dose theophylline enhances HDAC activity in epithelial cells and macrophages via p38 MAPK activation and this effect increases the activity of corticosteroids.
Now, we have documented the involvement of ERK pathway in this mechanism. In fact in our study both methylprednisone and hydrocortisone reduced the effects of both TNF-alpha and IL-1 on both pERK and NF-κB, suggesting a suppressive effect at high concentration. In contrast, low concentration of theophylline did not counteract this effect, but significantly inhibited these mechanisms in the presence of both corticosteroids at low dosage, suggesting a synergic mechanism.
MMP-2 and MMP-9 are members of gelatinases, which start the degradation components of the endothelial basal lamina including type IV collagen, fibronectin, laminin, and heparan sulfate. In a recent clinical trial performed in patients with post-thrombotic syndrome, we documented a correlation between MMPs, TNF-alpha, and ILs suggesting that these molecules are involved in inflammatory pathways.44 Moreover, previous experimental studies documented in cerebral endothelial cells show that TNF-alpha and IL-1 beta were able to increase the MMP-9 levels and this effect was reduced by dexamethasone treatment.45,46 In agreement, in the present study, TNF-alpha and IL-1-induced MMPs release that were inhibited by methylprednisone and hydrocortisone. The effects of these corticosteroids on MMPs secretion, seems to be related to two mechanisms: 1) block of the activator protein (AP)-1 site in the MMP-9 gene, and 2) induction of the expression of tissue inhibitor of metalloproteinase-1 (TIMP-1).46 AP-1 is involved in the coordinated expression of several genes that control inflammation, cell proliferation, and apoptosis, while TIMP-1 is the controller of MMPs.47 An imbalance between MMPs and TIMP is involved in airway diseases.48,49 Yigit et al50 documented in 27 patients with inflammatory nasal polyposis that the treatment with oral corticosteroid reduces the tissutal expression of MMP2, while increases the tissutal expression of TIMP-1. Previously, in patients with COPD as well as in patients with asthma it has been documented that a combination therapy with corticosteroid and low-dose theophylline may reduce airway inflammation, improving clinical symptoms.51,52 An explanation of this synergic mechanism has been postulated by Sun et al,13 that in an experimental study it is documented that a low-dose theophylline might improve the anti-inflammatory effects of steroids by increasing HDAC -2 activity.
In our study, theophylline used at low dosage potentiated the inhibitory effects of methylprednisone and hydrocortisone on MMP-2 and MMP-9, suggesting that theophylline may be able to improve the effects of steroids also through its anti-inflammatory activity.
Our study has several limitations. First, these data must be confirmed in clinical trials. Second, we measured the immunoreactivity of MMPs in the cells but these values are not related to the activity of the enzymes, which would have been better and more reflective of their actual enzymatic functional activity. In fact, although we used antibodies against active MMPs in this study, we did not measure activity or localize the activity. However, this study could increase the knowledge related to the synergic effects of these compounds supporting the rationale for the use of theophylline at low dosage with corticosteroid in respiratory diseases with an inflammatory component. This synergy (theophylline-corticosteroids at low dosages) resulting in improved anti-inflammatory effect, could improve the clinical efficacy of the compounds, reducing their side effects.
Acknowledgments
The authors thank Dr Erika Cione and Dr Manuela Sgambato for significant technical help and Dr Domenico Sturino for the careful English review of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
D’Agostino B, Advenier C, de Palma R, et al. The involvement of sensory neuropeptides in airway hyper-responsiveness in rabbits sensitized and challenged to Parietaria judaica. Clin Exp Allergy. 2002;32(3):472–479. | ||
D’Agostino B, Marrocco G, De Nardo M, et al. Activation of the nociceptin/orphanin FQ receptor reduces bronchoconstriction and microvascular leakage in a rabbit model of gastroesophageal reflux. Br J Pharmacol. 2005;144(6):813–820. | ||
D’Agostino B, Orlotti D, Calò G, et al. Nociceptin modulates bronchoconstriction induced by sensory nerve activation in mouse lung. Am J Respir Cell Mol Biol. 2010;42(2):250–254. | ||
Sullo N, Roviezzo F, Matteis M, et al. Nociceptin/orphanin FQ receptor activation decreases the airway hyperresponsiveness induced by allergen in sensitized mice. Am J Physiol Lung Cell Mol Physiol. 2013;304(10):657–664. | ||
Gallelli L, D’Agostino B, Marrocco G, et al. Role of tachykinins in the bronchoconstriction induced by HCl intraesophageal instillation in the rabbit. Life Sci. 2003;72(10):1135–1142. | ||
Miraglia Del Giudice M, Maiello N, Decimo F, et al. Airways allergic inflammation and L. reuterii treatment in asthmatic children. J Biol Regul Homeost Agents. 2012;26 (1 Suppl):S35–S40. | ||
Stoltz DJ, Jackson DJ, Evans MD, et al. Specific patterns of allergic sensitization in early childhood and asthma & rhinitis risk. Clin Exp Allergy. 2013;43(2):233–241. | ||
Gallelli L, Ferreri G, Colosimo M, et al. Retrospective analysis of adverse drug reactions to bronchodilators observed in two pulmonary divisions of Catanzaro, Italy. Pharmacol Res. 2003;47(6):493–499. | ||
Morfín Maciel BM, Castillo Morfín BM. Theophylline, a new look to an old drug. Rev Alerg Mex. 2010;57(4):112–122. | ||
Foukas LC, Daniele N, Ktori C, Anderson KE, Jensen J, Shepherd PR. Direct effects of caffeine and theophylline on p110 delta and other phosphoinositide 3-kinases: differential effects on lipid kinase and protein kinase activities. J Biol Chem. 2002;277(40):37124–37130. | ||
Ito K, Caramori G, Adcock IM. Therapeutic potential of phosphatidylinositol 3-kinase inhibitors in inflammatory respiratory disease. J Pharmacol Exp Ther. 2007;321(1):1–8. | ||
To Y, Ito K, Kizawa Y, et al. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(7):897–904. | ||
Sun X, Li Q, Gong Y, Ren L, Wan H, Deng W. Low-dose theophylline restores corticosteroid responsiveness in rats with smoke-induced airway inflammation. Can J Physiol Pharmacol. 2012;90(7):895–902. | ||
Huang AX, Lu LW, Liu WJ, Huang M. Plasma inflammatory cytokine IL-4, IL-8, IL-10, and TNF-α levels correlate with pulmonary function in patients with asthma-chronic obstructive pulmonary disease (COPD) overlap syndrome. Med Sci Monit. 2016;22:2800. | ||
Montaño M, Sansores RH, Becerril C, et al. FEV1 inversely correlates with metalloproteinases 1, 7, 9 and CRP in COPD by biomass smoke exposure. Respir Res. 2014;15:74. | ||
Ostridge K, Williams N, Kim V, et al. Relationship between pulmonary matrix metalloproteinases and quantitative CT markers of small airways disease and emphysema in COPD. Thorax. 2016;71(2):126–132. | ||
Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. | ||
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. | ||
Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev. 2011;242(1):205–219. | ||
Culpitt SV, Rogers DF, Shah P, et al. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167(1):24–31. | ||
Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol. 2009;71:451–464. | ||
Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636–645. | ||
Pelaia G, Cuda G, Vatrella A, et al. Effects of hydrogen peroxide on MAPK activation, IL-8 production and cell viability in primary cultures of human bronchial epithelial cells. J Cell Biochem. 2004;93(1):142–152. | ||
Cione E, Tucci P, Senatore V, et al. Synthesized esters of ferulic acid induce release of cytochrome c from rat testes mitochondria. J Bioenerg Biomembr. 2008;40(1):19–26. | ||
Cione E, Genchi G. Characterization of rat testes mitochondrial retinoylating system and its partial purification. J Bioenerg Biomembr. 2004;36(2):211–217. | ||
Bonofiglio D, Cione E, Vizza D, et al. Bid as a potential target of apoptotic effects exerted by low doses of PPARγ and RXR ligands in breast cancer cells. Cell Cycle. 2011;10(14):2344–2354. | ||
Kane MA, Folias AE, Pingitore A, et al. CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Mol Cell Biol. 2011;31(16):3277–3285. | ||
Pelaia G, Gallelli L, D’Agostino B, et al. Effects of TGF-beta and glucocorticoids on map kinase phosphorylation, IL-6/IL-11 secretion and cell proliferation in primary cultures of human lung fibroblasts. J Cell Physiol. 2007;210(2):489–497. | ||
Gallelli L, Falcone D, Pelaia G, et al. Interleukin-6 receptor superantagonist Sant7 inhibits TGF-induced proliferation of human lung fibroblasts. Cell Prolif. 2008;41(3):393–407. | ||
Pelaia G, Gallelli L, Renda T, et al. Effects of statins and farnesyl transferase inhibitors on ERK phosphorylation, apoptosis and cell viability in non small lung cancer cells. Cell Prolif. 2012;45(6):557–565. | ||
Preianò M, Pasqua L, Gallelli L, et al. Simultaneous extraction and rapid visualization of peptidomic & lipidomic body fluids fingerprints by using mesoporous aluminosilicate and MALDI-TOF MS. Proteomics. 2012;12(22):3286–3294. | ||
Roccia F, Campolo B, Gallelli L, et al. Effects of ambrisentan in a patient affected by combined pulmonary fibrosis and emphysema and by severe pulmonary hypertension: clinical, functional and biomolecular findings. Clin Drug Invest. 2013;33(6):451–457. | ||
Serra R, Gallelli L, Conti A, et al. The effects of sulodexide on both clinical and molecular parameters in patients with mixed arterial and venous ulcers of lower limbs. Drug Des Devel Ther. 2014;8:519–527. | ||
Serra R, Buffone G, Falcone D, et al. Chronic venous leg ulcers are associated with high levels of metalloproteinases-9 and neutrophil gelatinase-associated lipocalin. Wound Repair Regen. 2013;21(3):395–401. | ||
Serra R, Grande R, Buffone G, Gallelli L, De Franciscis S. The effects of minocycline on extracellular matrix in patients with chronic venous leg ulcers. Acta Phlebol. 2013;14(3):99–107. | ||
Cromwell O, Hamid Q, Corrigan CJ, et al. Expression and generation of interleukin-8, IL-6 and granulocyte-macrophage colony-stimulating factor by bronchial epithelial cells and enhancement by IL-1 beta and tumour necrosis factor-alpha. Immunology. 1992;77(3):330–337. | ||
Demirjian L, Abboud RT, Li H, Duronio V. Acute effect of cigarette smoke on TNF-alpha release by macrophages mediated through the erk1/2 pathway. Biochim Biophys Acta. 2006;1762(6):592–597. | ||
von Haehling S, Hopkinson NS, Polkey MI, Niethammer M, Anker SD, Genth-Zotz S. Elevated TNFalpha production in whole blood in patients with severe COPD: the potential link to disease severity. Wien Klin Wochenschr. 2009;121(9–10):303–308. | ||
Dina R Hammad, Ahmed G Elgazzar, Tarek S Essawy, Sohair A Abd El Sameie. Evaluation of serum interleukin-1 beta as an inflammatory marker in COPD patients. Egypt J Chest Dis Tuberc. 2015;64:347–352. | ||
Chunlian W, Heyong W, Jia X, Jie H, Xi C, Gentao L. Magnolol inhibits tumor necrosis factor-α-induced ICAM-1 expression via suppressing NF-κB and MAPK signaling pathways in human lung epithelial cells. Inflammation. 2014;37(6):1957–1967. | ||
Lin FS, Lin CC, Chien CS, Luo SF, Yang CM. Involvement of p42/p44 MAPK, JNK, and NF-κB in IL-1β-induced ICAM-1 expression in human pulmonary epithelial cells. J Cell Physiol. 2005;202:464–473. | ||
Gallelli L, Pelaia G, Fratto D, et al. Effects of budesonide on P38 MAPK activation, apoptosis and IL-8 secretion, induced by TNF-alpha and haemophilus influenzae in human bronchial epithelial cells. Int J Immunopathol Pharmacol. 2010;23(2):471–479. | ||
Ito K, Lim S, Caramori G, et al. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc Natl Acad Sci U S A. 2002;99(13):8921–8926. | ||
de Franciscis S, Gallelli L, Amato B, et al. Plasma MMP and TIMP evaluation in patients with deep venous thrombosis: could they have a predictive role in the development of post-thrombotic syndrome? Int Wound J. Epub 2015 Sep 24. | ||
Harkness KA, Adamson P, Sussman JD, Davies-Jones GA, Greenwood J, Woodroofe MN. Dexamethasone regulation of matrix metalloproteinase expression in CNS vascular endothelium. Brain. 2000;123(4):698–709. | ||
Förster C, Kahles T, Kietz S, Drenckhahn D. Dexamethasone induces the expression of metalloproteinase inhibitor TIMP-1 in the murine cerebral vascular endothelial cell line cEND. J Physiol. 2007;580(3):937–949. | ||
Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31(6):1334–1356. | ||
Guan WJ, Gao YH, Xu G, et al. Sputum matrix metalloproteinase-8 and -9 and tissue inhibitor of metalloproteinase-1 in bronchiectasis: clinical correlates and prognostic implications. Respirology. 2015;20(7):1073–1081. | ||
Chaudhuri R, McSharry C, Brady J, et al. Low sputum MMP-9/TIMP ratio is associated with airway narrowing in smokers with asthma. Eur Respir J. 2014;44(4):895–904. | ||
Yigit O, Acioğlu E, Gelişgen R, Server EA, Azizli E, Uzun H. The effect of corticosteroid on metalloproteinase levels of nasal polyposis. Laryngoscope. 2011;121(3):667–673. | ||
Ford PA, Durham AL, Russell RE, Gordon F, Adcock IM, Barnes PJ. Treatment effects of low-dose theophylline combined with an inhaled corticosteroid in COPD. Chest. 2010;137(6):1338–1344. | ||
Markham A, Faulds D. Theophylline. A review of its potential steroid sparing effects in asthma. Drugs. 1998;56(6):1081–1091. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.