")
Back to Journals » The Application of Clinical Genetics » Volume 16
The Unique Spectrum of MUTYH Germline Mutations in Colombian Patients with Extracolonic Carcinomas
Authors Rodriguez-Rojas LX, Candelo E, Pachajoa H , Garcia-Robledo JE , Nastasi-Catanese JA , Olave-Rodriguez JA, Zambrano AR
Received 12 April 2022
Accepted for publication 10 March 2023
Published 18 April 2023 Volume 2023:16 Pages 53—62
DOI https://doi.org/10.2147/TACG.S370416
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Lisa Ximena Rodriguez-Rojas,1,2 Estephania Candelo,3,4 Harry Pachajoa,1,2,4 Juan Esteban Garcia-Robledo,3 Jose Antonio Nastasi-Catanese,1,2 Jorge Andres Olave-Rodriguez,2 Angela R Zambrano5
1Department of Human Genetics, Fundación Valle del Lili, Cali, Colombia; 2Faculty of Health Sciences, Universidad Icesi, Cali, Colombia; 3Fundación Valle del Lili, Centro de Investigaciones Clínicas, Cali, Colombia; 4Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras, Universidad Icesi, Cali, Colombia; 5Department of Hematology/Oncology, Fundación Valle del Lili, Cali, Colombia
Correspondence: Lisa Ximena Rodriguez-Rojas, Department of Human Genetics, Fundación Valle del Lili, Cali, 760032, Colombia, Email [email protected]
Background: Protein MUTYH, encoded by the gene MUTYH, is an important mismatch repair enzyme in the base-excision repair pathway of DNA repair. When genetically altered, different neoplastic conditions can arise. One of the widely known syndromes associated with MUTYH mutations is MUTYH-associated polyposis, a form of familial colorectal cancer syndrome. MUTYH may also be a driver in other familial cancer syndromes, as well as breast cancer and spontaneous cancer cases. However, some controversies about the role of these alterations in oncogenesis remain, especially when affected in a heterozygous way. Most available data on MUTYH mutations are on Caucasian patients.
Material and Methods: We analyzed a small cohort of non-Caucasian, Colombian cancer patients with MUTYH germline heterozygous mutations, clinical features suggestive of familial cancer, and extensive genetic studies with no other mutations and without MUTYH-associated polyposis.
Conclusion: With this case series, we intended to provide important data for the understanding of MUTYH as a possible driver of familial cancer, even when only heterozygous mutations are found.
Keywords: MUTYH mutations, familial cancer, heterozygote MUTYH mutation, Colombia
Background
Oncogenesis develops as a result of multiple disruptions in DNA, epigenetic changes, gene amplifications and other genetic aberrations1 that behave as tumor suppressor genes,2 and human DNA repair genes.3 The DNA repair genes function in a diverse set of pathways that involve the recognition and removal of DNA lesions, tolerance to DNA damage, and protection from errors of incorporation made during DNA replication or DNA repair. Additional defects affect DNA repair indirectly by regulating the cell cycle and providing an opportunity to repair or direct apoptosis. Consequently, dysregulation of gene repair has significant detrimental effects on health, including an increased prevalence of birth defects, accelerated aging rate, and increased cancer risk.3
Mutations are early events in carcinogenesis, and defective DNA repair is a risk factor for the development of many types of cancer. The MUTYH gene (MutY human homolog; MIM 604933) is involved in DNA repair.3–6 It is located at 1p34.2–1p33 and is a member of the base-excision repair (BER) pathway.7 MUTYH encodes for MUTYH glycosylase that participates in BER by repairing oxidative DNA damage. The BER pathway plays a significant role in repairing mutations caused by reactive oxygen species that are generated during aerobic metabolism. BER is a multi-step process that involves the sequential activity of several proteins. MUTYH protein especially corrects oxidative damage on guanine to 8-oxo-7,8-dihy-dro-2-deoxyguanosine (8-oxoG), which leads to an increase in G:C to T:A transversion in tumor suppressor genes such as KRAS and APC, playing an essential role in cellular proliferation in the colorectum.8 Oxidative damage repair is initiated by the DNA glycosylase pathway, which recognizes and removes an improper base by hydrolyzing the N-glycosidic bond. To complete the repair process, the apurinic/apyrimidinic (AP) site is further processed by an incision step, DNA synthesis, an excision step, and DNA ligation through either the short- or long-patch BER pathway. Inherited deficiencies involving components of the nucleotide excision repair, mismatch repair, and recombinational repair pathways have all been linked to specific human genetic disorders. However, in 2003, biallelic mutations in the BER DNA glycosylase MUTYH were found to cause the development of an autosomal recessive syndrome of adenomatous colorectal polyposis and very high colorectal cancer risk.9,10 MUTYH germline mutations have been found in some cases of familial colorectal cancer syndromes and have been related to other cancer types.11
Pathogenic MUTYH variants cause colorectal familial adenomatous polyposis autosomal recessive (FAP2; MIM 608456).12 This entity is also called MUTYH-associated polyposis (MAP). Patients with MAP have an 18 to 100-fold increased risk of colorectal cancer (CC) compared to the general population.13 Other neoplasias have been associated with somatic MUTYH mutations in gastric cancer (GC; MIM 613659).14 However, some reports have found germline mutations on the MUTYH gene in other carcinomas including breast, ovarian, endometrial, bladder, and skin cancers.15 Most of the MUTYH-associated carcinomas have biallelic alterations, either homozygous or compound heterozygous, exemplifying the common behavior of a tumor suppressor gene.16,17
Monoallelic MUTYH mutations, occurring in 1–2% of the Caucasian population, are associated with a moderately increased risk of colorectal cancer.18 Previous studies have reported an increased risk of gastric, liver, endometrial,19 and breast cancers15,20,21 for monoallelic mutation carriers, whereas other studies did not find statistically significant evidence for an increased risk of breast cancer.22 Clarity on these cancer risks is important for the clinical management of MUTYH mutation carriers. The two most common mutations are p.Y179C and p.G396D, which are present in around 70–80% of MAP in European families identified with a MUTYH germline mutation.8 In a Brazilian cohort of 60 probands, 6.6% of patients with hereditary CC presented with biallelic germline MUTYH mutations in the two most frequent hotspots.8 While data have been reported in Latin American population, the majority of case series reported on MUTYH mutations are in Caucasian populations.17,23 To our knowledge, in Colombia, no previous similar studies have been published. Here, we present a case series report of 11 cancer patients, of which nine had breast cancer, one had vulvar cancer, and one had a malignant solitary fibrous tumor. All of them presented with monoallelic MUTYH mutations and a strong familial history of cancer including breast, prostate, colon, gastric, and lung cancers.
Materials and Methods
Participants
A retrospective study on the clinical and molecular characteristics of 500 patients of the oncogenetic consultation registry from Fundación Valle del Lili, a reference service for southwestern Colombia, located in the city of Cali (Colombia, South America), with a population of 2.2 million inhabitants. The rare disease cohort (Protocol “1504”) was approved by the institutional ethical committee. Data concerning the histological type and cancer diagnosis were provided by medical pathology reports in diagnostic core biopsies or tumor resections. All patients were screened for germline gene mutation predisposition to cancer.
Sample Collection and Next-Generation Sequencing Analysis
Peripheral blood samples were obtained from participants after signing the informed consent. Data were collected by interview and review of medical records from patients attended at our institution. Genomic DNA was isolated using the DNeasy® Blood Kit (QIAGEN, Hilden, Germany). After the extraction phase, DNA has been quantified by Qubit®3.0 fluorometer (Thermofisher Scientific, Waltham, MA, USA). For parallel next-generation sequencing (NGS), the genomic library preparation was performed using the Sistemas Genomicos Library Preparation kit (Valencia, Spain). The regions of interest were selected by using a hybridization probe, which included intronic and exonic regions adjacent to the genes included in the corresponding panel. Following that, clonal amplification and sequencing of the selected regions were done using the Illumina platform MiSeq (San Diego, USA) following the paired-ends strategy of the GeneSystem platform. The median coverage of this sample was 278X for the total of sequencing samples with a range of 250X to 310X. Bioinformatic analysis was performed in alignment with the reference genome (GRCh38). In this analysis, a variant was defined as alterations in DNA sequence found in the general population at a frequency less than 1%. The percentages of sequence reads observed matching to a specific DNA variant was divided by overall coverage at the locus, as NGS provides a random sample, variant allele frequency is surrogate by the proportion of DNA molecules in the original specimen. The studies covered the following genes: APC, ATM, BARD1, BMPR1A, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, TP53, BRCA1, BRCA2, POLD1, POLE, GREM1, HOXB13, AXIN2, GALNT12, RPS20, RNF43, NTHL1, and MSH3. Pathogenic or likely pathogenic variants found by NGS were classified using the AAMG guidelines and then confirmed with Sanger sequencing, and variants that were not identified as disease-causing were not confirmed by this methodology. Additionally, in silico studies were conducted for the latter variants using bioinformatic tools such as Varsome and ClinVar (Table 1). Finally, copy number variation tests were performed with all patients.
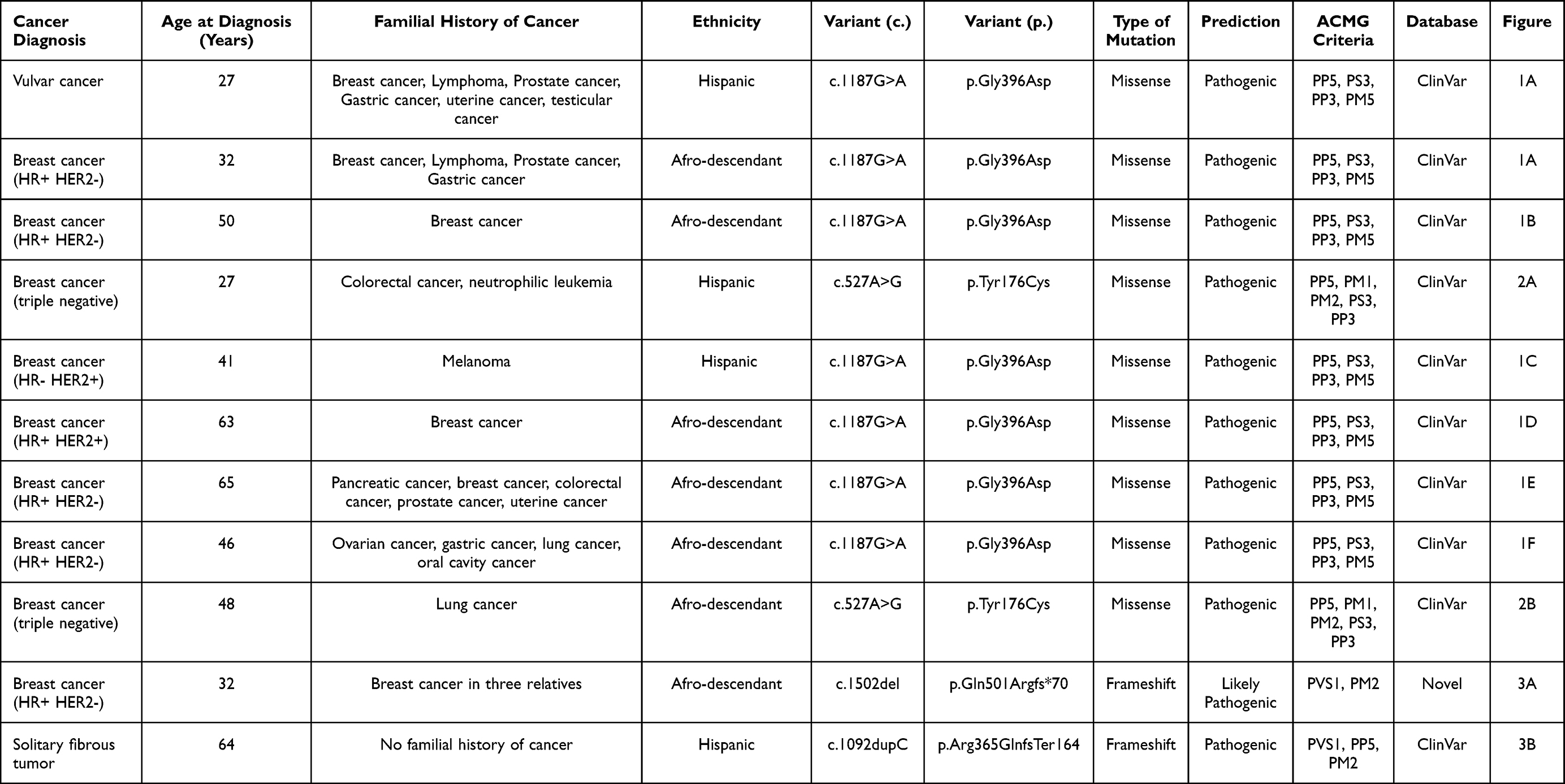 |
Table 1 Demographics, Clinical Presentation and Associated Variants |
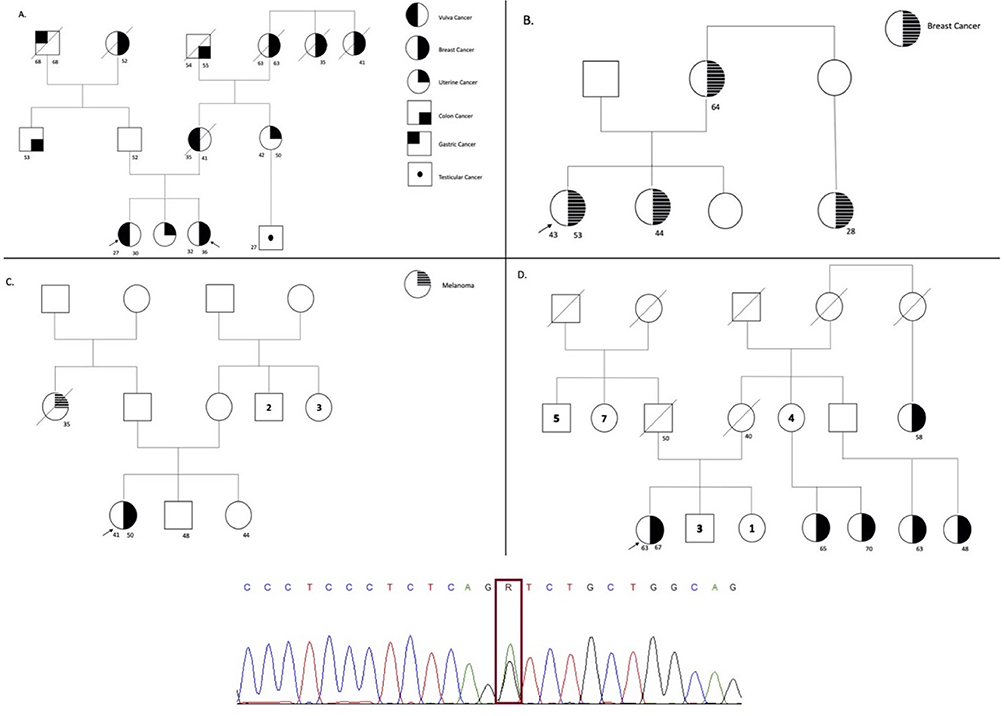 |
Figure 1 The figure represents the pedigree of each participant, which is correlate in Table 1. The symbols for each neoplasia type are stated (A) on the right side for (A–D). The arrow represents the proband, the line crossing the symbol represents the dead status, the unique number represent current age, and symbols with two numbers represent the current age and age of diagnosis. On the bottom of the figure the sanger sequencing showing the variant encoding the MUTYH gene for all the families is stated. |
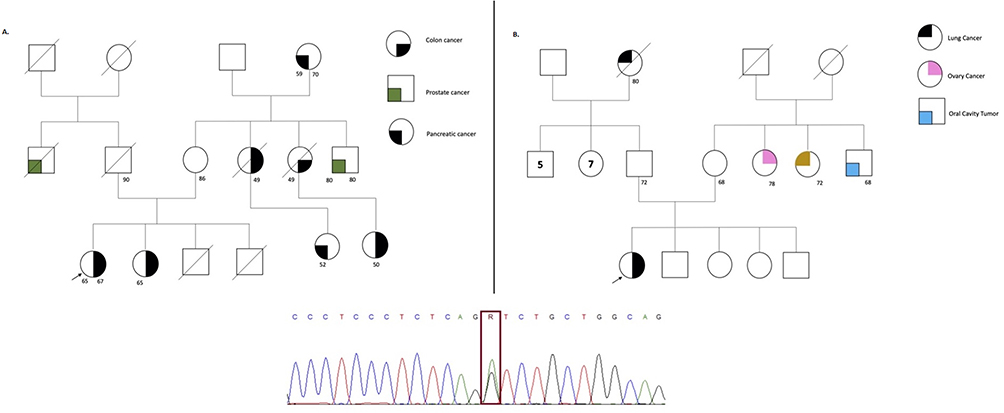 |
Figure 2 The figure represents the pedigree of each participant, which is correlate in Table 1. The symbols for each neoplasia type are stated (A and B) on the right side. The arrow represents the proband, the line crossing the symbol represents the dead status, the unique number represent current age, and symbols with two numbers represent the current age and age of diagnosis. On the bottom of the figure the sanger sequencing showing the variant encoding the MUTYH gene for all the families is stated. |
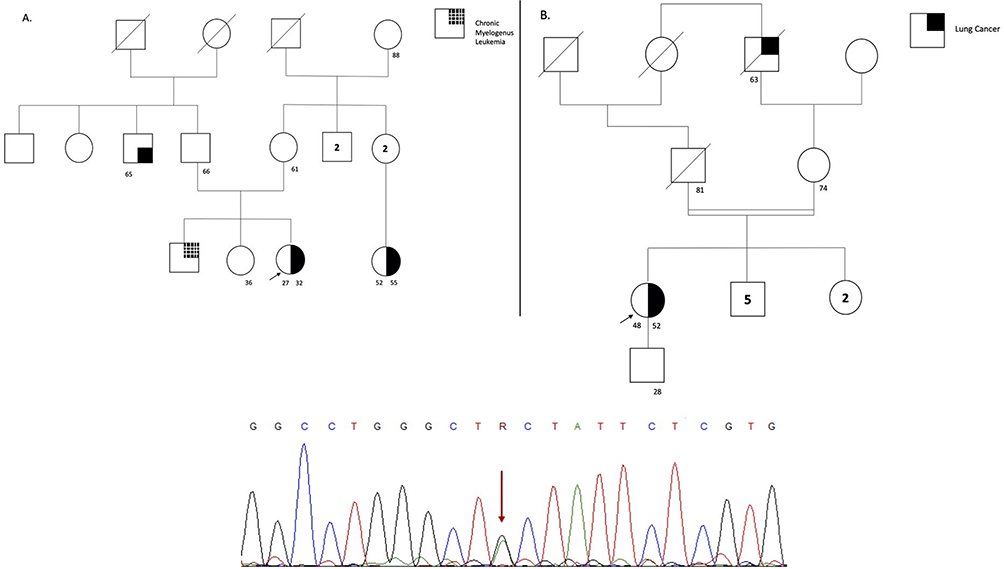 |
Figure 3 The figure represents the pedigree of each participant, which is correlate in Table 1. The symbols for each neoplasia type are stated (B) on the right side for (A and B). The arrow represents the proband, the line crossing the symbol represents the dead status, the unique number represent current age, and symbols with two numbers represent the current age and age of diagnosis. On the bottom of the figure the sanger sequencing showing the variant encoding the MUTYH gene for all the families is stated. |
Bioinformatic Analysis
Genetic variants and all available information about the variants were searched in dbSNP, ClinVar, and PubMed. The effects of missense variants were predicted by SIFT (Sorting Intolerant From Tolerant), PolyPhen-2 (Polymorphism Phenotyping v2), Provean (Protein Variation Effect Analyzer), Mutation Taster, FATHMM (Functional Analysis through Hidden Markov Models), CADD v1.3 (Combined Annotation–Dependent Depletion), and DANN (Deleterious Annotation of genetic variants using Neural Networks). Finally, their effect was predicted using the standard and guidelines for the interpretation of Sequence Variants from the joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG criteria).
Results
Eleven patients with an average age of 45 years (IQR 32.0–56.5). Seventy-two percent of patients were 50 years old or younger, and 18% were under 30 years old, highlighting the possible importance of MUTYH alterations and other BER proteins as drivers of cancer when affected in a germline manner. Eighty-two percent of the patients in this cohort presented breast cancer with a clinical spectrum, which ranged from hormone receptor-negative to Her-2-Neu-positive (78%), and triple negative (22%). The remaining 18% presented vulvar cancer and a malignant solitary fibrous tumor. No alterations in BRCA1 and BRCA2 were found in breast cancer patients. None of the 11 cancer patients had previous or current evidence of colon polyps. A summary of the general characteristics of these patients is shown in Table 1. Seven patients presented with the c.1187G > A (63%) variant, two patients with the c.527A>G (18%) variant, and two patients with deletion and duplication in the corresponding positions (c.1502del and c.1092dupC). Nine of the 11 variants reported in this series were in missense, and two were in frameshift (Figures 1– 4). In the present study, we present the spectrum of genetic variants encoding the MUTYH gene and we did not find any significant changes on the CNV test. The complete coding sequence of MUTYH gene was investigated in 11 index patients and pathogenic variants are present in table for each index patient.
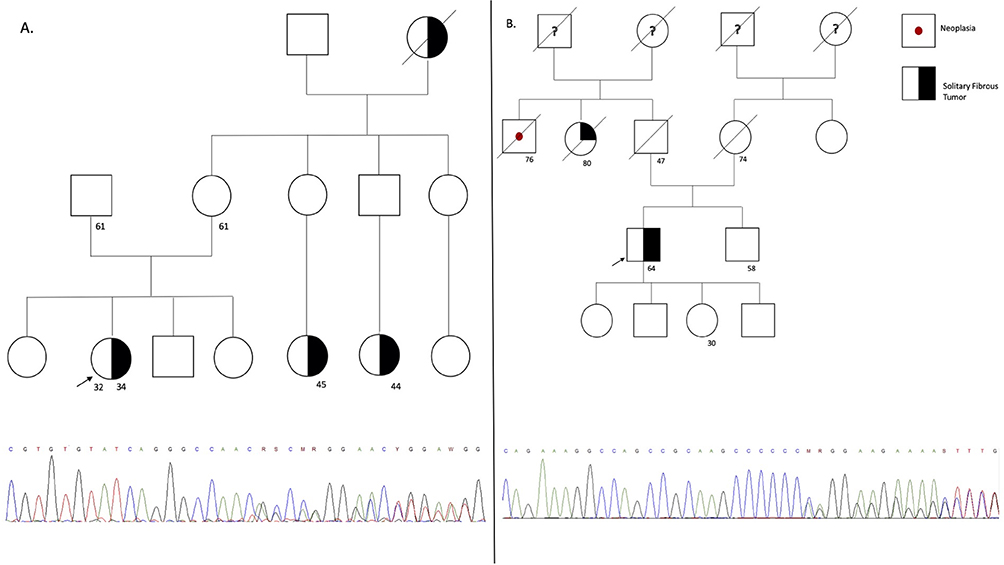 |
Figure 4 The figure represents the pedigree of each participant, which is correlate in Table 1. The symbols for each neoplasia type are stated (B) on the right side for (A and B). The arrow represents the proband, the line crossing the symbol represents the dead status, the unique number represent current age, and symbols with two numbers represent the current age and age of diagnosis. On the bottom of the figure the sanger sequencing showing the variant encoding the MUTYH gene for all the families is stated. |
Variants’ effect was predicted using the standard and guidelines for the interpretation of Sequence Variants from the joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG criteria). Bioinformatic analysis showed the fourth variants encoding the MUTYH gene from likely pathogenic to pathogenic with a maximum ACCM classification score of 18 and minimum of 9, and a wide range of pathogenic bioinformatic predictors such as: SIFT, Polyphen-2, Provean, with uncertain significance for the following bioinformatic predictors; Mutation Taster, FATHMM, DANN. The first variant, NM_001128425.2:c.1187G>A shows to be pathogenic and confirmed by functional studies, and the variant is known to cause disease. The second variant, NM_012222.3:c.527A>G very strong evidence is likely pathogenic according to combined evidence of Varsome, Uniprot and the other predictive markers, the variant affect a hot-spot of length on the 17 amino-acids and 34 missense/in-frame variants are pathogenics on this region. The third variant NM_012222.2:c.1502del and NM_012222.2:c.1092dupC are null variants frame-shift in gene MUTYH predicted to cause loss-of-function as disease mechanisms. The present exon contains 5 pathogenic variants and the truncated region has 12 pathogenic variants and the exon in the duplication variant contains 17 pathogenic variants. The truncated region contains 75 pathogenic variants, respectively.
Discussion and Conclusion
MAP is a familial genetic disorder in which patients present with colon polyps early in life and over time develop colorectal carcinomas as new mutations emerge due to unresolved DNA damage.10 As a type of familial adenomatous polyposis 1 (FAP1) (MIM 175100) in which APC is affected by germline mutations,24 MAP (FAP2) behaves similarly. Therefore, some MAP cases are very difficult to distinguish clinically from FAP1.7,13,25 Even though MAP is the most well-known syndrome associated with MUTYH mutations, other types of cancer could develop because of this tumor suppressor gene compromise.17 One of the cases in this study presented with solitary fibrous tumor (SFT), a rare mesenchymal tumor that can grow in almost any part of the body.26 To the best of our knowledge, no previous reports on MUTYH variants associated with this type of cancer have been published. This patient was the only one from the whole cohort that did not have any family history of cancer.
Most patients in the cohort had breast cancer (81%). Some researchers have reported cases of breast cancer that were associated with biallelic MUTYH mutations.25,27 However, others have also reported monoallelic cases.15,20 In our cohort, all patients were monoallelic. Out of the breast cancer cases in this study, 77% were aged 50 years or below and had a strong familial history of cancer, including prostate, colorectal, melanoma, breast, uterine, and gastric cancers. The vulvar cancer case was not associated with human papillomavirus infection. Interestingly, this patient and one of the breast cancer patients were siblings, and both presented with the same genetic variant. It is worth saying that statistics about these mutations are mostly reported in Caucasian population, while data from Latin American countries are scarce. Furthermore, most of our patients were Hispanic African-Americans.17
Regarding specific mutations, it has been reported that approximately 50–82% of European Caucasian MAP patients present the p.Tyr179Cys or p.Gly396Asp variant. In our cohort, the p.Gly396Asp variant was the most common, which corresponds to 60% of our patients, all of which were diagnosed with breast cancer (BC), plus the vulvar cancer case. The p.Tyr179Cys mutation was the second most frequent mutation present in two of our patients diagnosed with BC. Thus, 80% of our patients had either of the two mutations, resembling the findings of other studies on Caucasian populations.
One of the BC patients presented with deletion c.1502del. No previous publications have reported the association of this variant with BC. For that reason, we considered the deletion as a new genotype-phenotype association. We strongly associate this rare genetic alteration with the patient’s case, as she was 32 years old and had three relatives with BC history (these relatives could not be tested as their health insurance did not authorize it). Another infrequent genetic alteration found was the c.1092dupC in a patient with an SFT, a very rare sarcoma, with which MUTYH has also not been previously associated. Most SFT cases present with the NAB2-STAT6 fusion gene as an oncogenic hallmark,28 which was not observed in this cohort.
Some controversies still exist in the literature regarding the actual impact of monoallelic mutations in patients with BC. Fulk et al29 found no association between the increased risk of BC and monoallelic MUTYH mutations. Other authors have demonstrated that biallelic mutations may considerably increase the risk of BC in female and male patients, while also showing that this association is slightly stronger for monoallelic mutations.22,29 In the present study, we described a series of Colombian patients with monoallelic MUTYH mutations that developed different tumors, wherein BC was the most prevalent. Most patients developed these tumors at an early age, which supports the hypothesis on germline mutations, as demonstrated by molecular analysis. Two of the patients, who were 27- and 32-year-old siblings, had the same mutation, but one presented with BC, whereas the other with vulvar cancer (Figure 1A and B). Eighty percent of our patients presented with the most common genetic variants usually associated with MAP.
Even though it is a small cohort of patients, it suggests that outcomes of MUTYH alterations may differ between Caucasian and Latin American populations. Considering that the Latin American population has one of the most heterogeneous genetic compositions,30,31 the patterns of disease from a genetic origin in this population can be diverse. An increase in genomic analysis of patients with cancer in the last decade has yielded significant data on the origins of different types of tumors. MUTYH was initially associated with gastrointestinal tumors, but today some data suggest that it may also play a key role as a driver of mutation in other types of cancer. Our cohort provides very important data on MUTYH alterations in Latin American patients with cancer, given the scarce existing information in the literature.
Monoallelic MUTYH mutations have been found to increase female BC risk, but this conclusion remains controversial.29 We present a case series of 11 Colombian patients that included nine BC cases, one vulvar cancer case, and one SFT case. Eighty-one percent of the mutations detected were c.1187G>A (63%) and c.536A>G (18%), which have been previously reported as causal mutations for MAP. We concluded that, in our population, the presence of heterozygous MUTYH mutations is present in familial extracolonic cancer cases and may be considered causal. In non-mutated BRCA1 and BRCA2 BC patients, other genes should be evaluated using gene panels where MUTYH is included.32 MAP may not be the only cancer syndrome associated with MUTYH alterations. We encourage all researchers and clinicians with cancer patients that have MUTYH alterations of a possible germline source to publish their results in order to collaborate with familial cancer literature and improve the understanding of these MUTYH-associated syndromes. The study is limited by the small sample size and lack of functional analysis. However, a few studies have documented the variants encoding the MUTYH gene in the South-America region. For that reason, we present a novel study evaluating these variants in our population.
Abbreviations
AXIN2, Axin-related protein; ATM, ATM Serine/Threonine Kinase; APC, Adenomatous polyposis coli; AP, Apurinic/apyrimidinic; BARD1, BRCA1 Associated RING Domain 1; BMPR1A, Bone Morphogenetic Protein Receptor Type 1A; BCRA1, Breast cancer type 1; BCRA2, Breast cancer type 2; BRIP1, BRCA1 Interacting Helicase 1; BER, base-excision repair; BC, Breast Cancer; CC, Colorectal Cancer; CDH1, cadherin 1, type 1, E-cadherin; CDK4, cyclin dependent kinases; CDKN2A, cyclin-dependent kinase inhibitor 2A; CHEK2, Recombinant protein of human CHK2 checkpoint homolog; EPCAM, epithelial cellular adhesion molecule; DNA, Deoxyrobonucleic acid; GC, Gastric Cancer; GALNT12, Polypeptide N-Acetylgalactosaminyltransferase 12; GREM1, Gremlin 1, DAN Family BMP Antagonist; HOXB13, Homeobox B13; IQR, Interquartile range; FAP1, Familial adenomatous polyposis 1; FAP2, Familial adenomatous polyposis autosomal recessive; ROS, Reactive oxygen species; MAP, MUTYH-associated polyposis; MA, Massachusetts; MLH1, MutL Homolog 1; MSH2, MutS homolog 2; MSH3, MutS homolog 3; MSH6, MutS homolog 6; MUTYH, MutY human homolog; NAB2-STATG, Nuclear polyadenylated RNA-binding; NBN, Nibrin; NGS, Next-generation sequencing; NTHL1, Nth Like DNA Glycosylase 1; KRAS, Kristen rat sarcoma virus; PALB2, Partner And Localizer Of BRCA2; PMS2, PMS1 Homolog 2, Mismatch Repair System Component; PTEN, Phosphatase and tensin homolog; POLD, DNA Polymerase Delta 1, Catalytic Subunit; POLE, DNA Polymerase Epsilon, Catalytic Subunit; RAD51C, RAD51 homolog C; RAD51D, RAD51 homolog D; RPS20, Ribosomal Protein S20; RNF43, Ring Finger Protein 43; SFT, Solitary Fibrous Tumor; SMAD4, SMAD Family Member 4; STK11, serine/threonine kinase 11; TP53, Tumor Protein P53; USA, United States of America.
Data Sharing Statement
All data generated or analysed during this study are included in this published article.
Ethics Approval and Consent to Participate
The study was assessed and supported by the biomedical ethics committee at Fundación Valle del Lili. All patients included in this descriptive study fill out the written informed consent. The study was performed according to the Helsinki Declarations.
Consent for Publication
All patients whose clinical or molecular data are used in this manuscript gave consent for publication during their informed consent completion.
Acknowledgments
We thank our patients to participate in the study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding of any kind of funding source was provided during the implementation and execution of this study.
Disclosure
The authors declare that they have no competing interests.
References
1. Vicente-Dueñas C, Romero-Camarero I, Cobaleda C, Sánchez-García I. Function of oncogenes in cancer development: a changing paradigm. EMBO J. 2013;32:1502–1513.
2. Science M, There I, Rna RO. Molecular basis of cancer: oncogenes and tumor suppressor genes. Microbiol Immunol. 1993;37(1):11–22. doi:10.1111/j.1348-0421.1993.tb03173.x
3. Ronen A, Glickman BW. Human DNA repair genes. Environ Mol Mutagen. 2001;37(3):241–283. doi:10.1002/em.1033
4. Loeb KR, Loeb LA. Significance of multiple mutations in cancer. Carcinogenesis. 2000;21(3):379–385. doi:10.1093/carcin/21.3.379
5. Benhamou S, Sarasin A. Variability in nucleotide excision repair and cancer risk: a review. Mutat Res. 2000;462(2–3):149–158. doi:10.1016/S1383-5742(00)00032-6
6. Matta J, Ortiz C, Encarnación J, Dutil J, Suárez E. Variability in DNA repair capacity levels among molecular breast cancer subtypes: triple negative breast cancer shows lowest repair. Int J Mol Sci. 2017;18(7):1505. doi:10.3390/ijms18071505
7. Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004; 427:652–656.
8. Pitroski C, Cossio S, Koehler-Santos P, Graudenz M, Prolla J, Ashton-Prolla P. Frequency of the common germline MUTYH mutations p.G396D and p.Y179C in patients diagnosed with colorectal cancer in Southern Brazil. Int J Colorectal Dis. 2011;26:841–846. doi:10.1007/s00384-011-1172-1
9. Cheadle JP, Sampson JR. Exposing the MYtH about base excision repair and human inherited disease. Hum Mol Genet. 2003;12(suppl_2):R159–65. doi:10.1093/hmg/ddg259
10. Poulsen MLM, Bisgaard ML. MUTYH Associated Polyposis (MAP). Curr Genomics. 2008;6:420–435. doi:10.2174/138920208785699562
11. Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet. 2002;30(2):227–232. doi:10.1038/ng828
12. Nielsen M, Joerink - van de Beld MC, Jones N, et al. Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology. 2009;136(2):471–476. doi:10.1053/j.gastro.2008.10.056
13. Nielsen M, Infante E, Brand R. MUTYH Polyposis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, editors. GeneReviews® [Internet]. Seattle: University of Washington; 1993.
14. Kim CJ, Cho YG, Park CH, et al. Genetic alterations of the MYH gene in gastric cancer. Oncogene. 2004;23(40):6820–6822. doi:10.1038/sj.onc.1207574
15. Rennert G, Lejbkowicz F, Cohen I, Pinchev M, Rennert HS, Barnett-Griness O. MutYH mutation carriers have increased breast cancer risk. Cancer. 2012;118(8):1989–1993. doi:10.1002/cncr.26506
16. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
17. Win AK, Reece JC, Dowty JG, et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int J Cancer. 2016;139(7):1557–1563. doi:10.1002/ijc.30197
18. Jones N, Vogt S, Nielsen M, et al. Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology. 2009;137(2):489–94, 494.e1; quiz 725–6. doi:10.1053/j.gastro.2009.04.047
19. Win AK, Cleary SP, Dowty JG, et al. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer. 2011;129(9):2256–2262. doi:10.1002/ijc.25870
20. Wasielewski M, Out AA, Vermeulen J, et al. Increased MUTYH mutation frequency among Dutch families with breast cancer and colorectal cancer. Breast Cancer Res Treat. 2010;124(3):635–641. doi:10.1007/s10549-010-0801-7
21. Zhu M, Chen X, Zhang H, et al. AluYb8 insertion in the MUTYH gene and risk of early-onset breast and gastric cancers in the Chinese population. Asian Pac J Cancer Prev. 2011;12(6):1451–1455.
22. Beiner ME, Zhang WW, Zhang S, Gallinger S, Sun P, Narod SA. Mutations of the MYH gene do not substantially contribute to the risk of breast cancer. Breast Cancer Res Treat. 2009;114(3):575–578. doi:10.1007/s10549-008-0042-1
23. Win AK, Dowty JG, Cleary SP, et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology. 2014;146(5):1208–1211.e5. doi:10.1053/j.gastro.2014.01.022
24. Sehgal R, Sheahan K, O’Connell PR, Hanly AM, Martin ST, Winter DC. Lynch syndrome: an updated review. Genes. 2014;5(3):497–507. doi:10.3390/genes5030497
25. Nielsen M, Franken PF, Reinards THCM, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet. 2005;42(9):e54. doi:10.1136/jmg.2005.033217
26. Vogels RJ, Vlenterie M, Versleijen-Jonkers YMH, et al. Solitary fibrous tumor - clinicopathologic, immunohistochemical and molecular analysis of 28 cases. Diagn Pathol. 2014;9:224. doi:10.1186/s13000-014-0224-6
27. Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology. 2009;137(6):1976–1985.e10. doi:10.1053/j.gastro.2009.08.052
28. Park HK, Yu DB, Sung M, et al. Molecular changes in solitary fibrous tumor progression. J Mol Med. 2019;97(10):1413–1425. doi:10.1007/s00109-019-01815-8
29. Fulk K, LaDuca H, Black MH, et al. Monoallelic MUTYH carrier status is not associated with increased breast cancer risk in a multigene panel cohort. Fam Cancer. 2019;18(2):197–201. doi:10.1007/s10689-018-00114-4
30. Alshawi A, Essa A, Al-Bayatti S, Hanotte O. Genome analysis reveals genetic admixture and signature of selection for productivity and environmental traits in Iraqi cattle. Front Genet. 2019;10:609. doi:10.3389/fgene.2019.00609
31. Wang S, Ray N, Rojas W, et al. Geographic patterns of genome admixture in Latin American Mestizos. PLoS Genet. 2008;4(3):e1000037. doi:10.1371/journal.pgen.1000037
32. Kurian AW, Hare EE, Mills MA, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014;32(19):2001–2009. doi:10.1200/JCO.2013.53.6607
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.