")
Back to Journals » Drug Design, Development and Therapy » Volume 17
The Role of Traditional Chinese Medicine Natural Products in β-Amyloid Deposition and Tau Protein Hyperphosphorylation in Alzheimer’s Disease
Received 14 August 2023
Accepted for publication 2 November 2023
Published 13 November 2023 Volume 2023:17 Pages 3295—3323
DOI https://doi.org/10.2147/DDDT.S380612
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Huiying Yan,1 Lina Feng,2 Mingquan Li1
1Department of Neurology, the Third Affiliated Clinical Hospital of the Changchun University of Chinese Medicine, Changchun, Jilin Province, People’s Republic of China; 2Shandong Key Laboratory of TCM Multi-Targets Intervention and Disease Control, the Second Affiliated Hospital of Shandong First Medical University, Taian, Shandong Province, People’s Republic of China
Correspondence: Lina Feng, The Second Affiliated Hospital of Shandong First Medical University, Yingsheng Road 2, Taian, Shandong Province, 271000, People’s Republic of China, Tel +86-15543120222, Email [email protected] Mingquan Li, The Third Affiliated Clinical Hospital of the Changchun University of Chinese Medicine, Boshuo Road, Changchun, Jilin, 130117, People’s Republic of China, Tel +86-15948000577, Email [email protected]
Abstract: Alzheimer’s disease is a prevalent form of dementia among elderly individuals and is characterized by irreversible neurodegeneration. Despite extensive research, the exact causes of this complex disease remain unclear. Currently available drugs for Alzheimer’s disease treatment are limited in their effectiveness, often targeting a single aspect of the disease and causing significant adverse effects. Moreover, these medications are expensive, placing a heavy burden on patients’ families and society as a whole. Natural compounds and extracts offer several advantages, including the ability to target multiple pathways and exhibit high efficiency with minimal toxicity. These attributes make them promising candidates for the prevention and treatment of Alzheimer’s disease. In this paper, we provide a summary of the common natural products used in Chinese medicine for different pathogeneses of AD. Our aim is to offer new insights and ideas for the further development of natural products in Chinese medicine and the treatment of AD.
Keywords: Alzheimer’s disease, β-amyloid deposition, tau-protein hyperphosphorylation, natural products, Chinese herbal medicine, review
Introduction
Alzheimer’s disease (AD) is the most prevalent degenerative disease of the central nervous system in elderly individuals. It has a gradual and progressive onset, leading to cognitive decline, mental and behavioral abnormalities, and a reduced ability to perform daily activities.1 As the population ages, dementia has become a widespread condition among older individuals, with AD dementia accounting for 60% to 80% of cases. It is also the primary cause of disability and mortality in elderly individuals.2,3
China currently has the highest number of dementia patients worldwide.4 Epidemiological surveys indicate that there are approximately 15.07 million individuals over the age of 60 with dementia in China, with approximately 9.83 million of them diagnosed with AD.5 This places a significant burden on both the families of the patients and society as a whole. The primary pathological characteristics of AD include the formation of senile plaques (SPs) through the accumulation of extracellular amyloid β-protein (Aβ), the presence of intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated Tau proteins, and synaptic loss.6 Current treatment for cognitive symptoms primarily involves the use of cholinesterase inhibitors (donepezil, carboplatin, and galantamine) and glutamate receptor antagonists (memantine and memantine combined with cholinesterase inhibitors).7 However, it is important to note that cholinesterase inhibitors may lead to increased adverse effects, such as skin irritation, weight loss, nausea, and vomiting.7 On the other hand, glutamate receptor antagonists, particularly memantine, have shown limited effectiveness in the early stages of AD.7 Atypical antipsychotics can help alleviate psychiatric and behavioral symptoms associated with AD, but there is a risk of exacerbating cognitive impairment, among other concerns.7 Currently, there are no therapeutic drugs available that can effectively halt the occurrence or slow down the progression of AD. As a result, researchers both domestically and internationally continue to actively explore alternative treatment methods for AD.
The literature on dementia treatment predates the first reported case of AD in the world by 283 years.8 In Chinese medicine, AD is categorized as a type of dementia, with kidney deficiency as the core issue. The basic pathological mechanism involves marrow reduction, brain depletion, and loss of use of the mental mechanism, with phlegm and blood stasis as the main pathological products. Clinical medications often focus on tonifying the kidney and filling the marrow while also addressing symptoms such as depression, phlegm expulsion, blood invigoration, and orifice opening.9 According to the 2020 edition of the Chinese Alzheimer’s disease dementia treatment guidelines, the treatment of AD dementia with traditional Chinese medicine (TCM) is based on the progression of symptoms. The guidelines suggest that AD treatment should focus on ‘tonifying the kidneys’ in the early stage, “resolving phlegm, activating blood, and dipping fire” in the middle stage, and “detoxifying and fixing the detoxification” in the late stage.10 In the treatment of patients with mild AD for 12–24 weeks, there were better cognitive (p = 0.002) and functional improvements compared to placebo (p = 0.002; n = 2291).7 The cognitive improvement of Qinggong Shoutao pills (14 g/d) was superior to that of ginkgo biloba extract EGb761 (p<0.04) and placebo (p<0.001) in the treatment of prodromal AD patients for 52 weeks (n = 350).11 Blood activating EGb761 (240 mg/d, 160 mg/d) in the treatment of mild/moderate AD patients for 22–24 weeks showed better cognition (p<0.00001; n = 723), behavior (p = 0.00001; n = 547), function (p<0.0001; n = 547), and overall improvement (p<0.0006; n = 542) compared to placebo.12 In the treatment of dementia patients for 12 weeks, Huanglian Jiedu decoction showed a higher clinical improvement rate (p = 0.004) and better functional level compared to placebo (p<0.00001; n = 672).7 However, overall, there is significant heterogeneity among the studies, which requires further verification. In conclusion, Clinical studies have provided initial evidence that combining conventional Western medicine with TCM has a synergistic effect, improving cognitive function and slowing deterioration. This combination treatment appears to be more beneficial in the early stages of AD than in the middle and late stages.13 In recent years, there has been growing interest in the potential efficacy of Chinese herbal medicines and their extracts in preventing and treating AD. Research on this topic has received increasing attention, as natural products derived from Chinese herbal medicines offer several advantages, such as multiple targets, fewer toxic side effects, and cost-effectiveness.14 Consequently, more scholars are dedicating their efforts to studying the natural products and active ingredients of Chinese herbal medicines for AD treatment. This paper aims to review the recent research progress on natural products of Chinese medicine in the context of AD, both domestically and internationally. The findings of this review are expected to provide a theoretical and experimental basis for the clinical treatment of AD.
Natural Products of Traditional Chinese Medicine and AD
The pathological changes observed in AD are complex and diverse, and the reasons for the appearance of these changes cannot be fully explained. The pathogenesis of AD remains unclear. The mainstream theories regarding the common pathogenesis of AD include extraneuronal amyloid aggregation, intraneuronal Tau protein hyperphosphorylation, the cholinergic theory, the neuroinflammatory theory, and the oxidative stress response theory.15,16 β-amyloid deposition and Tau protein hyperphosphorylation are well-known pathological markers of AD and play a crucial role in its development.17 These markers are closely associated with oxidative stress, microglial activation, autophagy, and other factors involved in AD. Therefore, this paper aims to review recent experimental studies on the use of natural products in traditional Chinese herbal medicine to inhibit Aβ deposition and Tau protein hyperphosphorylation, focusing on the pathways mentioned above.
Inhibition of Aβ Deposition by Natural TCM Products
The Aβ theory is the prevailing theory of AD development. One of the key pathological features in the development of AD is the formation of SPs, which results from the extracellular aggregation of Aβ in nerve cells. Aβ is produced from amyloid precursor protein (APP) through a series of protein hydrolysis processes. APP has two main metabolic pathways. Under normal physiological conditions, APP is cleaved by α-secretase and γ-secretase to produce the soluble extracellular fragment sAPPα and other polypeptides with neuroprotective effects. This pathway is referred to as the nonamyloid hydrolysis pathway. However, under pathological conditions, the production of Aβ is divided into two types. The first involves the sequential cleavage of APP embedded in the cell membrane by β-APP cleavage enzyme 1 (BACE-1) and the γ-secretase complex,18 resulting in the secretion of Aβ polypeptides outside the cell (polypeptides of different lengths, including Aβ1-40 and Aβ1-42). The second pathway involves the reinternalization of APP followed by cleavage by β-secretase and γ-secretase, leading to the production of intracellular Aβ polypeptides.19–21 This pathway is known as the amyloid hydrolysis pathway. In the pathological process of AD, there is an imbalance in the production and clearance of Aβ. Normally, the production and degradation of Aβ are in equilibrium. However, in AD, there is excessive production of Aβ that cannot be cleared efficiently, leading to its accumulation in large quantities. This accumulation results in the formation of insoluble amyloid, which is deposited in brain tissue and contributes to the formation of SPs. The process of extracellular amyloid beta deposition leading to AD is depicted in Figure 1.
 |
Figure 1 How extracellular β-amyloid deposition occurs. Amyloid precursor protein (APP) is cleaved by β-secretase to produce the transmembrane 99-residue C-terminal fragment (C99). This C99 fragment is then processed by γ-secretase, leading to the release of the amyloid precursor protein intracellular domain (AICD) into the cytoplasm. |
Li et al22 conducted a study using intraperitoneal injection of D-galactose to induce senescence, hippocampal injection of Aβ1-40 compound to develop an AD model, and the middle cerebral artery line embolization reperfusion method to create a vascular dementia (VD) rat model. This study evaluated the effects of tanshinone intervention on learning and memory impairment in both AD and VD rats. The results demonstrated that tanshinone significantly reduced the latency of evasion and the number of misfirings into the blind end in both groups, indicating its therapeutic potential in improving learning and memory impairment in AD and VD rats. Zhuang et al23 conducted a study in which they injected Aβ25-35-induced AD model rats in the bilateral hippocampus and provided them with high, medium and low doses of ginsenoside Rg2 intervention. The study revealed that each dose of ginsenoside Rg2 improved the learning and memory ability of AD model rats. Furthermore, medium and high doses of ginsenoside Rg2 significantly inhibited the formation of SPs caused by Aβ deposition. In a study conducted by Lu et al.24 APPswe, PSEN1dE9 (APP/PS1) transgenic AD model mice were selected. After 28 days of intervention with berberine, the berberine group showed a reduction in the expression level of Aβ42 and the number of SPs compared to the saline group. This suggests that berberine has the potential to significantly enhance learning and memory abilities while also reducing the expression and aggregation of Aβ42, leading to decreased formation of SPs. Niu et al25 conducted a study using 4-month-old 5×FAD (APPSwFlLon, PSEN1*M146L*L286V) transgenic mice to simulate the deposition of SPs in patients with AD. The mice were administered β-asarone ether at a concentration of 10 mg/kg for 90 days through continuous gavage. The findings demonstrated that β-asarone ether effectively decreased the formation of SPs in the hippocampal and cortical tissues of AD mice and improved the characteristic pathological manifestations of AD. Xiao et al26 developed an in vitro cell model by overexpressing Aβ42 in the M146 cell line through stable transfection with the APP/PS1 gene. They conducted an intervention study using various concentrations of schisandrin b and observed that schisandrin b did not exhibit any toxicity toward M146 cells. Moreover, they found that medium and high doses of schisandrin b effectively reduced the production of Aβ42 in a dose-dependent manner. Zeng et al27 conducted experiments on the N2a/APP695swe cell line, which is a specialized N2A cell line capable of producing its own Aβ deposition. The results of their study confirmed that schisandrin b reduces BACE1 activity and APP miscleavage by inhibiting BACE1 transcription and translation. This ultimately leads to a reduction in Aβ production and this study provides a new mechanism for schisandrin b in regulating Aβ metabolism. Chen Qin et al28 established an AD model by injecting cohesive Aβ1-40 combined with gooseberry muscarinic acid into the right basal nucleus region of rats. After 30 days of intervention with senegenin, the results showed that it improved Aβ plaque deposition within the basal nucleus of Meynert and increased the number of neurons around plaques compared to the model group.28 Transmission electron microscopy revealed significant improvements in the structural integrity of the nuclear bilayer, nuclear state, chromatin distribution, endoplasmic reticulum, and mitochondrial swelling. Based on these findings, it can be hypothesized that senegenin has a protective effect on AD.28 The above evidence suggests that natural products of TCM may treat AD by inhibiting Aβ deposition.
TCM Natural Products Treat AD by Regulating ERK in Aβ Deposition
In recent years, studies have found that extracellular regulated protein kinases (ERKs) play a role in the development of AD pathology. Scholars conducted a comparison of the expression of ERK in the brain tissue of AD patients at different stages of the disease. The findings revealed that p-ERK levels were increased in white matter astrocytes in early AD patients, and the severity of the lesion showed a strong correlation with p-ERK levels.29 Moreover, in the brains of late-stage patients, p-ERK was significantly increased in neuronal cell cytosomes and neuronal protrusions around SPs.29 QI et al30 discovered that dysregulation of ERK2 alone can result in abnormal phosphorylation of Tau protein, which ultimately leads to the pathological process of AD. This finding provides strong evidence for the involvement of the ERK transduction pathway in the pathogenesis of AD. Li et al31 induced an AD rat model by injecting Aβ1-40 into the right hippocampus. After intraperitoneal injection of low and high doses of curcumin, they observed that curcumin may influence the production and metabolism of Aβ by reducing the expression of ERK1 and ERK2 proteins, thereby potentially playing a neuroprotective role against neurotoxicity. In another study, curcumin was shown to strongly attract fibrillar Aβ. It also enhances the breakdown of Aβ in its β-folded form and has the ability to directly bind to free Aβ in the brains of AD patients, thereby reducing its accumulation.32 Ma et al33 utilized circular dichroism (CD) chromatography to examine the impact of water extract and its naphtha of acorus tatarinowii on the secondary structure of Aβ 25–35. The findings indicated a significant decrease in the α-helix structure of Aβ25-35 and a notable increase in the β-sheet structure over time.33 After culturing the water extract and its naphtha of acorus tatarinowii with Aβ25-35, the β-fold structure disappeared, and there was a significant increase in α-helix content. When the 100% water extract and 100% naphtha of acorus tatarinowii were incubated together for 7 days, the α-helix content reached 100%.33 These results suggest that a specific concentration of the water extract and its naphtha of acorus tatarinowii can effectively inhibit the transformation of Aβ25-35 from an α-helix to a β-fold structure.33
TCM Natural Products Treat AD by Regulating PI3K/Akt in Aβ Deposition
The phosphoinositide-3 kinase/protein kinase B (PI3K/Akt) signaling pathway is a well-known pathway found in various regions of the cerebral cortex, hypothalamus, and hippocampus. It can be activated or inhibited through different mechanisms.34,35 In the brain, the PI3K/Akt signaling pathway plays a crucial role in various physiological functions, including synapse formation, neuronal cell growth, and survival.36 This pathway can hinder the expression of enzymes necessary for Aβ degradation, resulting in reduced Aβ clearance. Additionally, increased expression of mammalian target of rapamycin (mTOR), a downstream component of the PI3K/Akt signaling pathway, inhibits cellular autophagy and leads to Tau protein phosphorylation. Zhang et al37 conducted a study using APP/BACE transgenic AD model Drosophila and mouse neurons as subjects. They intervened with salidroside, a phenylpropanoid glycoside isolated from Rhodiola rosea L., and assessed changes in lifespan and motor function in Drosophila using a lifespan assay and climbing test, respectively. The degree of Aβ aggregation in each group was measured using an immunofluorescence assay. The results indicated that salidroside could reduce Aβ plaque aggregation by modulating the PI3K/Akt/mTOR signaling pathway, thereby providing protective effects against neurotoxicity. Additionally, Wang et al38 demonstrated that salidroside could reduce Aβ levels and alleviate synaptic dysfunction in APP/PS1 double transgenic AD model mice by activating the PI3K/Akt/mTOR signaling pathway. Glycogen synthase kinase 3β (GSK3β) is a multifunctional serine/threonine kinase that plays a critical role in regulating energy metabolism, cell growth, and apoptosis.39 It acts as a key downstream substrate and effector of PI3K/AKT.40 Another important protein involved in cellular processes is protein phosphodiesterase 2A (PP2A), which is encoded by the PPP2CA gene. PP2A is a heterotrimeric serine/threonine phosphatase that is implicated in cell cycle regulation, growth, metabolism, proliferation, and apoptosis. Notably, dysregulation of PP2A protein expression has been observed in neurodegenerative diseases.41 In their study, Ma et al42 investigated the effects of bilateral ventricular injections of Aβ25-35 to induce AD in rats. After 60 days of gavage treatment with astragalus polysaccharide, they observed that astragalus polysaccharide had the ability to repair hippocampal damage in the model rats. Additionally, it was found to downregulate proteins such as APP, Aβ, p-tau, GSK3β, and BACE1 while upregulating the expression of the protein PP2A. These findings suggest that astragalus polysaccharide has a protective effect against the neurotoxicity caused by Aβ. Schematic representation of the PI3K/AKT/GSK-3β-mediated phosphorylation signaling pathway and its role in autophagy in AD was shown in Figure 2.
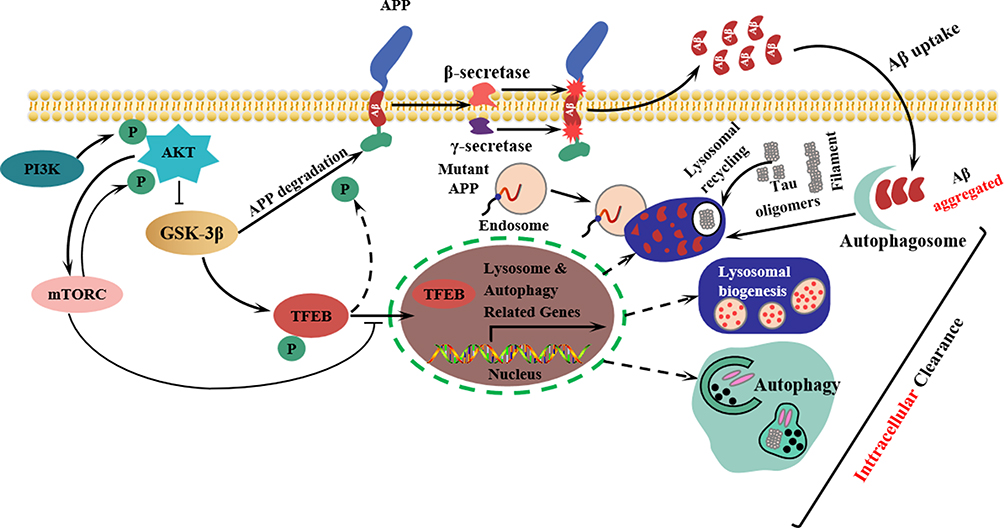 |
Figure 2 Schematic representation of the PI3K/AKT/GSK-3β-mediated phosphorylation signaling pathway and its role in autophagy in AD. Endosomes are membrane-enclosed organelles found in animal cells. Their primary function is to transport substances that are newly ingested through endocytosis to lysosomes, where they are degraded. |
Natural TCM Products Treat AD by Regulating Transthyretin (TTR) in Aβ Deposition
Transthyretin (TTR) is a transport protein that is stably folded and exists as a homotetramer. It is present in the blood and cerebrospinal fluid. According to a study, TTR has the ability to bind to Aβ and inhibit its aggregation through the hydrophobic T4 region.43 In their experiment, Lin et al44 used a combination of 120 mg/kg D-galactose and 10 mg/kg aluminum chloride to create an AD mouse model. They administered resveratrol orally for a period of 90 days, and the results revealed a significant improvement in cognitive function after intervention with resveratrol.44 Additionally, there was a notable reduction in the positive expression of Aβ1-40 and Aβ1-42 in the cortical neuronal cells of the mouse brain.44 In their study, Santos LM et al45 utilized the AβPPswe/PS1A246E/TTR mouse model to investigate the effects of resveratrol intervention. After a two-month intervention, the researchers observed an increase in plasma TTR protein expression levels in the mice treated with resveratrol.45 They also found that resveratrol enhanced TTR tetramer stability, improved its ability to bind free Aβ peptides in the brain, and reduced Aβ aggregation in the brains of AD mice.45 These results suggest that targeting TTR stability could be a potential therapeutic approach for AD.
TCM Natural Products Treat AD by Regulating Insulin-Degrading Enzyme (IDE) in Aβ Deposition
In organisms, insulin-degrading enzyme (IDE) plays a crucial role in insulin degradation.46 Under normal conditions, microglia and astrocytes in the brain secrete IDE into the extracellular fluid, contributing to the degradation of extracellular Aβ.47 The distribution of IDEs in neurons is more intricate. A study demonstrated that undifferentiated PC12 cells secrete a small amount of IDE extracellularly, while differentiated cells retain most of the IDE in the cell membrane and overexpress IDE intracellularly.48 This leads to a significant decrease in the levels of Aβ40 and Aβ42, further emphasizing the vital role of IDE in Aβ degradation. Iwata et al49 discovered that the expression levels of localized Neprilysin (NEP) were decreased in the aging brain, particularly in areas where symptoms related to disease appear early in patients with AD, such as the hippocampus and cortex. In their study, when NEP expression was increased twofold in the brain of an AD model, the deposition of Aβ was reduced eightfold. This suggests that increasing NEP expression can effectively reduce Aβ levels in the brain, highlighting the significant role of NEP in Aβ degradation.49
Lei et al50 conducted a study using the APP/PS1 double transgenic AD mouse model and administered triptolide treatment for 45 days to investigate the effects of triptolide treatment. Immunohistochemical and Congo red staining combined with unbiased somatology methods were used to stain and quantify Aβ deposition and SP formation in the hippocampal region. The results demonstrated that triptolide dose-dependently inhibited Aβ deposition and SP formation, leading to reduced Aβ protein levels in the hippocampus. In a similar study, Cheng et al51 also utilized APP/PS1 double transgenic AD model mice and administered triptolide for a 60-day intervention. Their findings revealed that triptolide promoted the degradation of Aβ and improved cognitive impairment in AD mice by upregulating IDE and NEP expression. In their study, Wang et al52 focused on the APP/PS1 mouse model. After an 8-week treatment with triptolide, the researchers analyzed the brain tissues of mice in each group. They used enzyme-linked immunosorbent assay (ELISA) to measure the expression levels of Aβ40, Aβ42, glutathione peroxidase (GSH-Px), superoxide dismutase (SOD), catalase (CAT), and the oxidative products of lipids and proteins, such as malondialdehyde (MDA). Additionally, they detected the levels of SPs and Iba1 in the cortex and hippocampus using immunohistochemistry. Furthermore, the researchers examined the expression of BACE1, APP, and NEP through Western blotting. The findings indicated that triptolide exhibited both anti-inflammatory and antioxidant effects. It achieved this by inhibiting the expression of BACE1, which is the rate-limiting enzyme for Aβ production, and increasing the expression of NEP, which is a key enzyme for Aβ degradation. According to Huang et al.53 N-butanol (NB) of radix notoginseng had positive effects on PC12 cell proliferation and protection against Aβ25-35-induced PC12 cytotoxicity in vitro. In their in vivo experiments, they observed that NB improved spatial learning and memory deficits in rapidly aging SAMP8 mice by inhibiting the expression of APP and BACE1 in their brains. Additionally, NB extract reduced SOD and GSH-PX activities, thereby attenuating oxidative stress levels and reducing Aβ production. The researchers also noted that NB exhibited lower toxicity in vivo,54 suggesting its potential as an effective alternative to the current treatment of AD. Liu et al55 conducted a study using AD model mice. They administered D-galactose combined with aluminum trichloride to the mice intraperitoneally for 90 days. Additionally, they administered low and high doses of ligustrazine for 40 days. The results of the study demonstrated that ligustrazine treatment significantly improved the learning and memory ability of the mice. Furthermore, the number of APP-positive cells in the hippocampus was significantly reduced, while the number of NEP-positive cells was significantly increased. This suggests that ligustrazine has the potential to enhance Aβ degradation and protect neurons in brain tissue by upregulating NEP expression. Panax notoginseng saponins (PNS), which have been widely used in the treatment of cardiovascular and cerebrovascular diseases, have also been found to have beneficial effects on nerve cell growth, axon lengthening, and synaptic plasticity. Liu et al56 conducted a study in which they injected Aβ1-42 into the CA1 region of the hippocampus to establish a rat model of AD. They observed that this injection resulted in a decrease in the number of neurons in the CA1 and dentate gyrus regions of the hippocampus.56 However, when panax notoginseng saponin Rg1 was administered, it reduced phosphorylation at the phosphorylation site APP-Thr668, lowered the expression of BACE1 and Psen1, and reduced Aβ production.56 Additionally, the intervention increased the expression of a disintegrin and metalloprotease domain-containing protein 10 (ADAM10) and IDE, which effectively rescued cognitive dysfunction and neuronal loss and promoted learning and memory abilities.56
Natural TCM Products Treat AD by Regulating Neurogenesis in Aβ Deposition
Neurogenesis is crucial in maintaining brain functions such as cognition and emotion regulation in the adult mammalian brain. Numerous studies have indicated that new hippocampal neurons can be generated during adulthood, a process referred to as neurogenesis.57 In response to brain pathology, neural progenitor cells (NPCs) readily proliferate, migrate to areas of neurodegeneration, and contribute to neurogenesis and the repair of neural injuries.58,59 In both Alzheimer’s disease patients and animal models, dysregulation of NPC proliferation and self-renewal leads to abnormal neurogenesis.54,60–62 According to Elena et al57 neurogenesis is still observed in the brains of healthy individuals up to the age of 90. However, in patients with AD, the generation of new neurons is significantly reduced. In ancient China, Ganoderma lucidum (Leyss. Ex Fr.) Karst. was revered as an immortal herb due to its purported effects on immortality and rejuvenation. Several studies have demonstrated that the aqueous extract of Ganoderma lucidum (Leyss. Ex Fr.) Karst. can stimulate neuronal differentiation and promote neurite growth in PC12 cells. Additionally, it has been found to possess hypnotic and antidepressant effects in vivo.63–65 Qin et al66 demonstrated that treatment of APP/PS1 mice with a ganoderma lucidum preparation resulted in a significant increase in the number of naive neuronal cells in the hippocampus and dentate gyrus compared to the AD model group. Additionally, cerebellar neural stem cells appeared early and in high numbers.66 The nucleus membrane remained intact, along with the cytoplasmic mitochondria, endoplasmic reticulum, Golgi apparatus, microtubular structures, and synapses.66 Moreover, no abnormalities were observed in microglia.66 Huang et al67 conducted a study in which they discovered that oral administration of ganoderma lucidum polysaccharides and aqueous extracts had a positive effect on the proliferation of NPCs and hippocampal neurogenesis in APP/PS1 mice. This administration also helped reduce cognitive deficits. Further investigations revealed that ganoderma lucidum polysaccharides enhanced the activation of fibroblast growth factor receptor 1 (FGFR1), along with the downstream ERK and AKT cascades.67 These findings indicate that ganoderma lucidum polysaccharides could potentially be used as therapeutic agents for the regenerative treatment of cognitive decline associated with neurodegenerative diseases.
TCM Natural Products Treat AD by Regulating Apoptosis in Aβ Deposition
Apoptosis, also known as programmed cell death, is a highly regulated process involving enzymes, genes, and specific protein families. The Bcl-2 family and caspase family play crucial roles in apoptosis. The Bcl-2 family includes both pro-apoptotic genes such as Bax and Bak and anti-apoptotic genes such as Bcl-2 and Bcl-xL. Bax can form homodimers (Bcl-2/Bcl-2, Bax/Bax) or heterodimers (Bcl-2/Bax) with Bcl-2, inhibiting the anti-apoptotic function of Bcl-2.68,69 Caspase family enzymes serve as marker enzymes indicating the initiation of apoptosis, with Caspase3 playing a particularly crucial role. When activated, Caspase3 triggers a cascade of reactions that ultimately result in apoptosis.70,71 Furthermore, the Bcl-2 family is capable of inducing apoptosis by modulating the mitochondrial membrane potential, releasing cytochrome c (Cytc), activating downstream factors of Caspase3, and ultimately leading to apoptosis.72 Liu et al73 conducted a study that demonstrated the potential of total saponin from anemarrhena asphodeloides in reducing the rate of Aβ25-35-induced apoptosis in PC12 cells. This effect was achieved by downregulating the expression levels of Bax mRNA and protein. Similarly, Yin Gang et al74 investigated the impact of cistanche polysaccharide on neuronal cells injected with intracerebral Aβ. Their findings revealed that cistanche polysaccharide effectively inhibited the toxicity of Aβ injection, promoted the expression of Bcl-2 protein, suppressed the activation of Caspase-3 factor, and reduced neuronal apoptosis in rats with Aβ25-35-induced AD. In a study by Roland et al75 total cistanche glycosides (GCs), including echolin and vermiculin, were administered to a rat model of AD induced by bilateral hippocampal injection of Aβ25-35 for 14 days. The researchers observed that the neuronal structure of the hippocampal CA1 region in the rats remained largely normal, and no apoptotic or necrotic cells were detected.75 Further analysis revealed that in the neuronal cells of the model group, the nuclear border was disrupted, the cell matrix exhibited unevenness, and the coarse endoplasm showed signs of degranulation.75 The synaptic structure appeared blurred, with the synaptic gap disappearing and the anterior and posterior synaptic membranes becoming unclear. Additionally, the synaptic vesicles exhibited vacuole-like changes.75 The neural progenitor fibers and microtubules were observed to be disorganized.75 In glial cells, the nuclear matrix appeared pale, and the mitochondria exhibited vacuole-like changes.75 GCs could reverse the aforementioned pathological changes, indicating a protective effect of GCs on hippocampal neurons.75 Xiao et al76 utilized the human APP and mutant PS1 gene double-transfected CHO cell line M146L as a model. They observed that intervention with Guangdong Haifengvine polysaccharide did not result in any cytotoxic effects. Moreover, they found that high and medium doses of the polysaccharide were able to significantly inhibit the production of Aβ42. Xue et al77 conducted a study to investigate the effects of wogonoside on antioxidant enzyme activity and the accumulation of ROS in H2O2-induced SH-SY5Y and PC12 cells. The results showed that wogonoside enhanced the activity of the antioxidant enzyme GSH and reduced the accumulation of ROS by regulating the NRF2/HO-1 signaling pathway, which is associated with oxidative stress.77 In addition, wogonoside was found to downregulate Bax, upregulate Bcl-2 protein expression, elevate the Bcl-2/Bax ratio, and inhibit apoptosis in Aβ25-35-induced SH-SY5Y and PC12 cell injury models.77 In addition, wogonoside upregulated the expression of synaptic-related proteins such as synapsin-1, synaptophysin, postsynaptic density 95 (PSD95), and brain-derived neurotrophic factor (BDNF), which improved neuronal cell function and provided a protective effect. Moreover, further in vivo experiments demonstrated that wogonoside significantly reduced the levels of Aβ1-40 and Aβ1-42 in the cerebral cortex and hippocampus of APP/PS1 mice after 60 days of gavage administration.77 This confirmed that the neuroprotective mechanism observed in vitro was consistently effective in vivo.
Hu et al78 conducted a study using 25 mmol/L L-glutamic acid (L-Glu) to induce neurotoxicity in HT22 cells. They found that intervention with different concentrations of phillyrin could prevent apoptosis and mitochondrial membrane potential imbalance induced by L-Glu.78 Further in vivo studies using APP/PS1 double transgenic mice showed that phillyrin inhibited the expression of pro-apoptotic proteins such as Bad, Bax, and Bid while promoting the expression of anti-apoptotic proteins such as Bcl-2 and Bcl-XL.78 This led to a reduction in neuronal cell apoptosis, decreased Aβ deposits in brain tissues, and improved learning and memory ability in AD mice.
Natural TCM Products Treat AD by Regulating the blood‒brain Barrier (BBB) in Aβ Deposition
There are various methods to clear Aβ in the brain, with the most significant one being the blood brain barrier (BBB) pathway.79 When BBB function is impaired, Aβ clearance is reduced, leading to the accumulation of Aβ in the brain parenchyma. This abnormal accumulation can further disrupt BBB permeability and worsen its dysfunction. Diagram of the association between the breakdown of the BBB and AD was shown in Figure 3. The key proteins involved in this pathway are the receptor of advanced glycation end products (RAGE), ATP-binding cassette transporter (ABC), and low-density lipoprotein receptor-related protein-1 (LRP-1). Among these, RAGE is responsible for transporting Aβ from peripheral blood to the brain, and this transportation is achieved through synergy with ABC and LRP-1.80 LRP-1, a signaling receptor produced by neuronal cells, plays a crucial role in the transport of Aβ across the BBB and its subsequent clearance. When Aβ binds to the LRP-1 ligand, it forms a complex that facilitates the transport of Aβ out of the brain and into the circulation. This complex also coactivates inflammatory pathways with insulin in the liver, enhancing the clearance of Aβ.81 Additionally, the LRP-1 endocytic complex translocates to the lysosome for degradation.82 Autopsy findings from a study on AD patients revealed lower levels of LRP-1 and increased accumulation of Aβ in brain tissue compared to normal aging brain tissue.83 According to Hongda et al84 baicalin intervention for 16 weeks resulted in the upregulation of LRP-1 protein expression in the hippocampal region of APP/PS1 double transgenic AD model mice. This intervention also led to a reduction in Aβ1-42 deposition and microvascular injury, as well as an increase in Aβ transport metabolism across the BBB. The study demonstrated that baicalin could enhance LRP-1 expression in neuronal cells and facilitate the outward transport of Aβ through the BBB. As a result, Aβ deposition in the brain was reduced, learning and memory ability improved, and the development of AD was prevented.
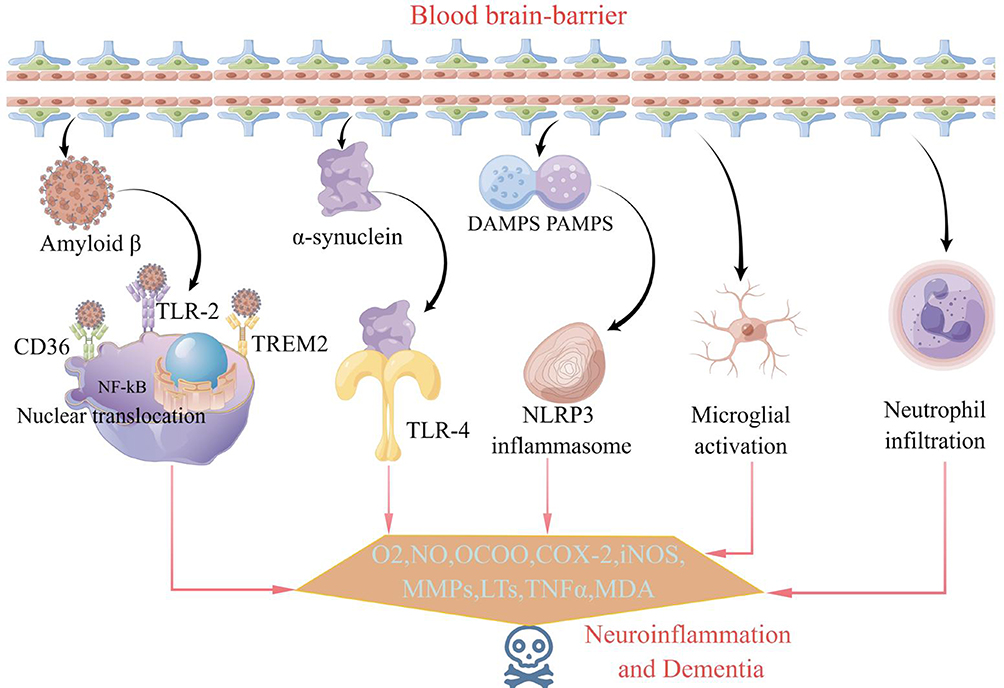 |
Figure 3 Diagram of the association between the breakdown of the BBB and AD. |
TCM Natural Products Treat AD by Regulating microRNAs in Aβ Deposition
MicroRNAs (miRNAs) are small noncoding RNAs that regulate endogenous genes. Their main role is to inhibit the translation or promote the degradation of mRNAs.85 Pharmacological studies have demonstrated that miRNAs can regulate various physiological processes in the body and can serve as diagnostic markers for many diseases.86 Recent research has revealed that the occurrence of AD is linked to abnormal expression of multiple miRNAs.86 Lin Ying et al87 utilized gene microarray technology to screen for differentially expressed miRNAs following osthole intervention in APP/PS1 mice. Their findings revealed an upregulation in the expression of miRNA-9. Furthermore, when the miRNA-9 inhibitor was introduced into the AD cell model with high APP expression (SHSY-5Y), there was an observed elevation in BACE1 protein expression. However, upon administration of osthole, the expression of BACE1 protein was reduced. These results indicate that osthole inhibits BACE-1 activity and decreases Aβ production through the upregulation of miRNA-9.
Therefore, natural products of herbal medicine have been found to have several beneficial effects in promoting Aβ degradation. The mechanisms by which natural products affect Aβ deposition are shown in Table 1. These effects include inhibiting the activity of β-secretase and reducing the expression of the Aβ precursor protein APP. Additionally, they can increase the stability of TTR and upregulate the expression of the Aβ-degrading enzymes IDE and NEP. Furthermore, these natural products promote NPC proliferation, enhance hippocampal neurogenesis, regulate cell autophagy and apoptosis, maintain BBB stability, and regulate noncoding small RNA expression of endogenous genes. All of these mechanisms work together to reduce Aβ content and decrease its neurotoxicity, ultimately leading to an improvement in cognitive dysfunction.
 |
Table 1 The Mechanisms of Natural Products in the Deposition of Aβ Deposition |
Inhibition of Tau Protein Hyperphosphorylation by Natural Products of Traditional Chinese Medicine
The pathology of AD is characterized by the loss of synapses and neurons, progressive impairment of cognitive function, and accumulation of Aβ and NFTs in the brain. NFTs are specifically caused by hyperphosphorylation of the intracellular microtubule-stabilizing protein known as Tau protein. The severity of clinical dementia symptoms is directly correlated with the amount of NFTs.88 In recent years, researchers have made efforts to treat AD by focusing on inhibiting Aβ protein aggregation or enhancing its clearance. However, these attempts have not succeeded in reversing AD or slowing down its progression. Notably, the failure of Phase III clinical trials for monoclonal antibodies such as solanezumab and bapineuzumab, which target Aβ clearance, has raised doubts among scholars regarding the effectiveness of Aβ as a therapeutic target.89–91 Currently, there is a growing interest among researchers in investigating the inhibition of Tau protein phosphorylation and aggregation as a possible target for therapeutic interventions. Tau proteins belong to a class of proteins that play a crucial role in stabilizing microtubules within neurons. They are believed to be vital for the assembly and stabilization of the microtubule cytoskeleton, particularly in axons, dendrites and other parts of neurons. In their soluble form, Tau proteins effectively stabilize and bind microtubules, preventing their depolymerization and maintaining normal physiological functions.92 Overphosphorylated Tau proteins weaken their binding to microtubules, which results in the destruction of the cytoskeleton. Additionally, they shed and aggregate to form paired helical filaments and further aggregate to form NFTs.93 These abnormal aggregations impede synaptic transport and cause neuronal degeneration, ultimately leading to the development of AD.94 Therefore, the hyperphosphorylation of tau protein is widely recognized as a crucial early event in the pathogenesis of AD.
Tau is a protein that is prone to phosphorylation and contains 21 abnormal serine/threonine phosphorylation sites (Ser46, Thr123, Ser198, Ser199, Ser202, Thr205, Ser208, Ser210, Thr212, Thr217, Thr231, Ser235, Ser262, Ser396, Ser400, Thr403, Ser404, Ser409, Ser 412, Ser413, and Ser422). The presence of these different phosphorylation sites reflects the progression of the disease. Schematic representation of disease-associated phosphorylation sites in tau was shown in Figure 4. In the early stages of AD, NFTs have not yet formed, and the phosphorylation sites are mainly found at Ser199 and Ser422. As the disease progresses, there is increased phosphorylation of the Ser202 and Thr205 sites. The phosphorylation of the Thr231 site indicates the presence of more mature p-Tau assemblies, leading to the advancement of AD. A recent study discovered a pattern of “dependent regulation” in the phosphorylation of Tau proteins. This means that the phosphorylation of Thr50, Thr69, and Thr181 can regulate the phosphorylation of other sites and consequently affect the overall phosphorylation level of Tau proteins.95 Intracellular phosphorylation and dephosphorylation of Tau protein are balanced and primarily controlled by intracellular protein kinases and protein phosphatases. Protein kinases catalyze the phosphorylation of Tau protein, while protein phosphatases dephosphorylate abnormally phosphorylated Tau proteins. This process restores the biological activity of Tau protein and reduces the production of NFTs.96 The protein kinases responsible for phosphorylation reactions in vivo are serine/threonine kinases. These kinases can be classified into proline-directed kinase (PDPK) and nonproline-directed kinase (NPDPK), depending on whether the substrate catalyzing tau protein phosphorylation requires proline. The three most significant tau protein phosphorylation kinases identified thus far are cyclin-dependent kinase-5 (CDK-5), glycogen synthase kinase-3 (GSK-3), and cyclic-AMP dependent protein kinase A (PKA).97 PKA plays a crucial role in the early stages of tau protein hyperphosphorylation and in the initial stages of AD pathogenesis. It catalyzes the phosphorylation of three phosphorylation sites of tau protein: Ser214, Ser262, and Ser409. Once PKA phosphorylates tau protein, it can impact the phosphorylation of tau protein by other protein kinases.97 A study found that prephosphorylation of tau protein with PKA (NPDPK) enhanced the efficiency of GSK-3/CDK-5 (PDPK) phosphorylation of tau protein and even replicated NFT-like structures.98 In vivo studies demonstrated that coactivation of PKA (NPDPK) and GSK-3/CDK-5 (PDPK) led to more rapid activation of the tau protein phosphorylation sites ser-199/202 and ser-396/404 by GSK-3/CDK-5. This activation is likely facilitated by the activation of the tau protein phosphorylation sites Ser-262 and Ser-214 of PKA (NPDPK), which alters the biological properties of tau protein as a substrate and increases the likelihood of phosphorylation at corresponding sites by GSK-3/CDK-5 (PDPK), etc.99 The above findings suggest that the phosphorylation of all 21 phosphorylation sites of tau protein may require the combined action of multiple phosphorylation kinases, including NPDPK and PDPK.
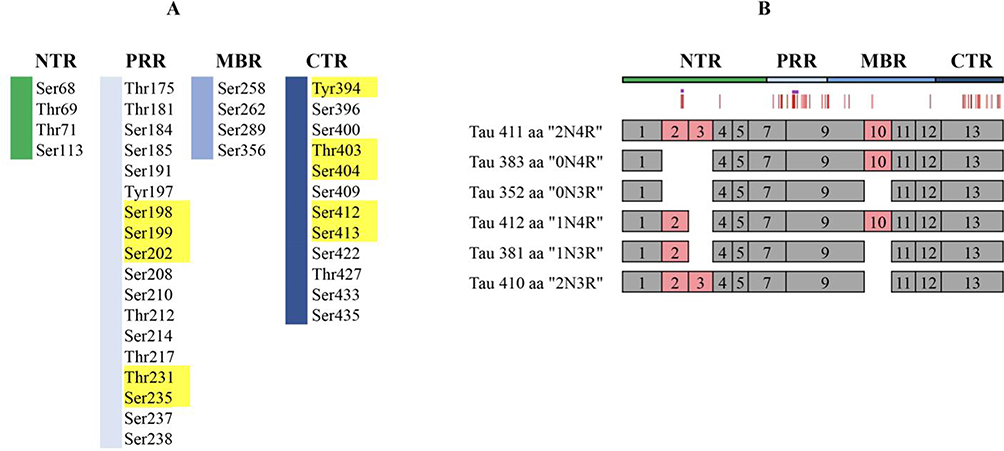 |
Figure 4 Schematic representation of disease-associated phosphorylation sites in tau. (A) The distribution of tau phosphorylation sites in Homo sapiens (441 aa CNS isoform) was determined by mass spectrometry of PHF tau. Different tau regions are indicated in various colors, with major phosphorylation sites shown in yellow. (B) Schematic representation of splice isoforms of tau in the CNS is provided, with alternatively spliced exons indicated as red boxes. The vertical red lines on top represent the phosphorylation sites listed in (A). Reprinted from Trushina NI, Bakota L, Mulkidjanian AY et al. The Evolution of Tau Phosphorylation and Interactions. Front Aging Neurosci. 2019;11:256. Creative Commons.99 |
Li et al100 utilized the okadaic acid (OA)-induced rat culture brain slice method to establish an AD model of Tau protein hyperphosphorylation. The findings revealed a significant decrease in the expression of PKA and P-tau following the administration of ginsenoside Rg1 intervention compared to the model group. This suggests that ginsenoside Rg1 effectively mitigates OA-induced Tau protein hyperphosphorylation, leading to a reduction in the production of NFTs. Li et al101 conducted an experiment in which they injected Aβ25-35 into the left ventricle of SD rats to create an AD model. They then administered panax notoginseng saponin Rg1 as an intervention for 28 days. The results showed that panax notoginseng saponin Rg1 reduced the expression of tau protein phosphorylation at the key site Thr231 in the DG, CAl, and CA3 regions of the hippocampus. Additionally, it decreased the overall level of Tau protein phosphorylation in the hippocampus.
TCM Natural Products Treat AD by Regulating Kinases in Tau Protein Hyperphosphorylation
GSK-3 is an important proline-directed protein kinase responsible for phosphorylating multiple sites on the tau protein, including Thr181, Ser199, Ser202, Thr205, Thr212, Thr217, Thr231, Thr235, Ser396, Ser404, and Ser422. The activity of GSK3β is primarily regulated by phosphorylation at two sites: tyrosine (Tyr216) and serine (Ser9). GSK3β is activated when the Tyr216 site is phosphorylated, while its activity is inhibited when the Ser9 site is phosphorylated. In their study, Sun et al102 investigated the effects of curcumin on APP/V717 double transgenic mice. They observed that after 8 weeks of curcumin intervention, the positive expression of Aβ and P-tau protein in the brain tissues of mice was reduced. Additionally, curcumin also decreased the mRNA content of Aβ and P-tau protein. Wang et al103 conducted further research on an animal model of AD that was induced by Tau hyperphosphorylation through OA. This study examined the effects of curcumin-macrophage exosome (Exo-cur) intervention. The results demonstrated that Exo-cur interacts with endothelial intercellular adhesion molecule 1 (ICAM-1) through lymphocyte function-associated antigen 1 (LFA-1). This interaction facilitated Exo-cur’s ability to penetrate the BBB and reduce Tau hyperphosphorylation. The reduction in Tau hyperphosphorylation was achieved by inhibiting the AKT/GSK-3β signaling pathway, leading to neuroprotective effects. The study also revealed that overactivation of GSK-3β can cause hyperphosphorylation of Tau by regulating the Ser-396 site. This, in turn, promotes microtubule depolymerization and the formation of NFTs. These processes disrupt cellular metabolism and affect the transport of substances between axons and dendrites.104,105 Under normal conditions, the intracellular Akt/GSK3β pathway is inhibited to maintain the normal physiological function of the Tau protein. Chronic cerebral hypoperfusion is a frequently observed pathophysiological process in both AD and vascular dementia. Studies have indicated that 40% of AD patients exhibit cerebrovascular pathology, indicating a substantial connection between AD and vascular dementia. Yan et al106 conducted a study using a bilateral common carotid artery ligation (2VO) model with angelica sinensis intervention for 28 days. The study revealed that the levels of hippocampal phosphorylated Tau (Thr231), phosphorylated Tau (Ser396), and phosphorylated Tau/Tau expression were significantly higher in the 2VO group than in the sham-operated group. However, these levels were significantly lower in the low- and high-dose angelica sinensis groups. Further studies have shown that angelica sinensis has the ability to suppress Tau protein hyperphosphorylation and Aβ production in the hippocampus of rats with chronic cerebral hypoperfusion. This leads to a delay in the progression of AD-like changes. The mechanism behind this effect involves the inhibition of the Akt/GSK3β pathway and the promotion of BACE-1 and BDNF expression. BDNF is a crucial peptide hormone in the mammalian central nervous system that plays a significant role in promoting the survival, growth, and maintenance of neural network plasticity. It does so by binding to Trk tyrosine kinase transmembrane receptors. During early development, high levels of BDNF expression are necessary for hippocampal neurons to maintain synaptic integrity and dendritic spine stability.107
Several studies have shown that abnormalities in the Wnt/β-catenin signaling pathway play a significant role in the development of AD. The expression levels of β-catenin, GSK 3β, and transcription factor 7-like 1 (TCF7L1)/T-cell factor 3 (TCF3) in the Wnt/β-catenin signaling pathway differed significantly between the internal olfactory cortical area and the hippocampal area in AD patients compared to non-AD patients.108 In a mouse model of AD, the early activation of the Wnt/β-catenin signaling pathway through the use of lithium has been found to promote the proliferation and differentiation of neural precursor cells and inhibit Aβ deposition in the hippocampal region of adult mice. During the progressive phase, lithium inhibits Aβ deposition in the hippocampal region of adult mice. However, it does not promote neural precursor cell proliferation.109 The product encoded by the Wnt gene is secreted extracellularly and specifically binds to the frizzled protein receptor on neighboring or own cell surfaces. This binding forms a coreceptor with the LRP family, leading to the inhibition of intracellular GSK-3β. Additionally, the scaffolding protein Axin and adenomatous polyposis coli (APC) form a complex that increases the concentration of intracellular β-catenin. This elevated concentration allows β-catenin to enter the nucleus,110 where it binds to TCF/lymphoid enhancer factor (LEF). This binding activates downstream transcription factors and leads to the transcription of target genes such as oncogene c (c-myc), cell cycle protein D (Cyclin D), and paired box gene 6 (Pax 6), among others.111 When the Wnt signaling pathway is downregulated, the levels of GSK-3β, Axin, and APC complexes increase, leading to the degradation of β-catenin through N-terminal ubiquitination. This highlights the central role of β-catenin in the classical Wnt signaling pathway.112 Huo et al113 conducted a study to demonstrate the effects of ligustrazine on the hippocampal tissue of AD rats. The results showed that ligustrazine can downregulate GSK-3β expression, inhibit β-catenin degradation, and consequently inhibit Tau protein hyperphosphorylation. Additionally, ligustrazine was found to activate the Wnt signaling pathway and inhibit Aβ-induced neurotoxicity, thereby exerting neuroprotective effects. Yao et al114 utilized D-galactose in combination with Aβ1~40 to establish an AD model. They observed that the positive staining of tau protein in the CA3 region of the hippocampus was reduced, while the positive staining of p-tau(Ser422) and p-tau(Ser396) proteins was significantly increased in the model group. In comparison to the model group, the Cornus officinalis polysaccharide group exhibited a significant decrease in the positive staining of p-tau(Ser422) and p-tau(Ser396) proteins, along with a notable increase in the positive staining of tau protein. In a study conducted by Su et al115 it was discovered that administering Cornus officinalis polysaccharide to AD model rats for 30 days had a positive impact. The administration resulted in a decrease in the expression of p-tau(Ser422) phosphosites and a reduction in Tau protein hyperphosphorylation. This effect was achieved by inhibiting the expression of GSK-3β in the hippocampal CA1 region of AD rats. Li He et al116 administered schisandra chinensis acidic polysaccharide as a drug through continuous gavage for 14 days. On the 8th day, they induced an AD model in SD rats by injecting Aβ25-35 into the lateral ventricle. The results indicated that in the hippocampal tissue of rats in the model group, there was an increase in the relative expression levels of Tau protein phosphorylation sites (Tau Ser199, Tau Ser396, and Tau Ser404) as well as GSK-3β and phosphorylated protein GSK-3β Tyr216. Additionally, the expression level of GSK-3β Ser9 was found to be decreased. Conversely, the SCP-A group exhibited the opposite trend, and these differences were statistically significant. The findings suggest that schisandra chinensis acidic polysaccharide has an anti-AD effect, which may be attributed to its ability to regulate GSK-3β activity and subsequently decrease the phosphorylation level of Tau protein.116 In a study by Ni et al117 6-month-old APP/PS1 mice were selected and treated with osthole for 42 days. The findings revealed a significant reduction in the expression of p-tau(ser202) protein in the osthole group compared to the model group. Additionally, there were notable increases in the expression levels of PI3K, p-Akt/Akt, and p-Gsk3β/Gsk3β. These results suggest that osthole may regulate the activity of the PI3K/AKT/GSK-3β signaling pathway to reduce tau protein hyperphosphorylation.
Normal CDK-5 is primarily obtained by binding to its neuron-specific activator p35, which is crucial for normal cellular function. Previous studies have demonstrated that neurotoxic substances such as Aβ, glutamate, and oxidative stress can cause calcium inward flow and trigger the cleavage of p35 into p25.118 The half-life of p25 is 5–10 times longer than that of p35. p25 is predominantly found in and around the nucleus, while p35 is concentrated in the cell periphery and plasma membrane. Notably, the binding of p25 to CDK-5 leads to the constitutive activation of CDK-5 and the formation of a p25/CDK-5 complex. This complex hyperphosphorylates tau protein, reducing its ability to bind to microtubules, disrupting the cytoskeleton, and promoting neuronal apoptosis.119,120 The abnormal expression levels of p25 in the brains of AD patients and its overexpression in transgenic mice result in tau protein hyperphosphorylation, neurofibrillary tangles, and the development of cognitive deficits.121 In a study using the Aβ25-35-induced AD injury model in primary cultured cortical neurons, it was demonstrated that tanshinone IIA inhibits Aβ25-35-induced tau protein hyperphosphorylation by suppressing cytoplasmic p25 expression and CDK-5 activation.121 In their study, Li et al122 observed an increase in the expression of the GSK-3β phosphorylation site Tyr216 and a decrease in the expression of the Ser9 site in an Aβ1-42-induced injury model in AD rats. Additionally, the expression of unphosphorylated Tau1 protein was downregulated. Treatment with tanshinone IIA was found to restore the spatial learning and memory deficits induced by Aβ1-42, along with the aforementioned changes. This suggests that tanshinone IIA has the potential to inhibit tau hyperphosphorylation by modulating GSK-3β activation.
TCM Natural Products Treat AD by Regulating Phosphatases in Tau Protein Hyperphosphorylation
PKA plays a crucial role in catalyzing tau protein phosphorylation, either directly or indirectly. It can alter the conformation of tau protein, making it more susceptible to phosphorylation by protein kinases such as GSK-3β and CDK5. PKA achieves this by phosphorylating multiple sites on the tau protein.123 PP2A, the most active phosphatase in neuronal cells, plays a crucial role in promoting the dephosphorylation of abnormally phosphorylated tau proteins. This process leads to the release of free tau proteins and restoration of their biological activity, resulting in the loosening of the NFT tangle structure.124,125 Inhibitors of PP2A have been found to cause synaptic and dendritic loss of hippocampal neurons in AD rats, accompanied by hyperphosphorylation of tau proteins.126 The interaction of PP2A and tau proteins in Alzheimer’s disease is shown in Figure 5. Furthermore, studies have shown that PP2A activity in the brains of AD patients is lower than that in the brains of normal elderly individuals. These findings suggest that defective PKA and PP2A in neuronal cells of AD patients’ brains may be the primary cause of tau protein hyperphosphorylation. Xu et al96 discovered that in the brain neurons of the Aβ1-40-induced AD model group, there was a notable increase in the total tau protein content, phosphorylation level of tau protein at the Ser396 site, and expression level of PKA protein compared to the control group. Conversely, the expression level of PP2A protein was significantly lower. However, after 30 days of senegenin administration, these effects were significantly reversed.
 |
Figure 5 Deregulation of PP2A-tau protein–protein interactions in AD. ((A) The PP2A/Bα holoenzyme has the ability to directly interact with human tau isoforms that have three or four repeats. This interaction occurs through a domain that includes the microtubule-binding repeats, leading to the dephosphorylation of tau. A crucial role in regulating the protein‒protein interactions between PP2A and tau is played by a specific proline-rich motif that contains the Thr231 phosphorylation site. (B) The PP2A-Tau protein‒protein interaction can be inhibited in vitro by these six methods). Adapted from Sontag JM, Sontag E. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci. 2014;7. Creative Commons.127 |
Chen et al128 demonstrated that Banqiao Codonopsis Pilosula (BCP) effectively improves cognitive dysfunction in rats with AD induced by OA. The possible mechanism of action involves inhibiting the expression of PME protein in the hippocampus and increasing the methylation level of PP2Ac. This leads to a reduction in the demethylation and phosphorylation levels of PP2A, resulting in the upregulation of PP2A activity.128 Consequently, the hyperphosphorylation of Tau protein is reduced by decreasing the phosphorylation levels of Tau protein phosphorylation sites pT181, pT231, pS396, and pS404.128 Additionally, BCP enhances the expression levels of Synapsin-1, NMDA-2B, and Synaptophysin in hippocampal tissue by upregulating PP2A activity, promoting neuronal repair.125 It is worth noting that the effectiveness of BCP may vary with the duration and dosage of use. Xie et al129 discovered that Trillium Tschonoskii Maxim has the ability to enhance the methylation level of PP2Ac and decrease the demethylation level. Consequently, the inhibition of PP2A activity in the hippocampus of AD rats caused by OA can be effectively mitigated. This leads to a reduction in the level of Tau hyperphosphorylation induced by OA and an increase in the number of Nissl bodies in the hippocampus in a dose-dependent manner. Additionally, the duration of administration has a certain degree of correlation with these effects. rTg4510 mice are transgenic mice that express the P301L mutant tau protein. They have been developed as an animal model for tauopathy, including AD. In a study by Ma et al130 it was demonstrated that cornel iridoid glycoside increased the activity of PP2A, elevated the levels of PP2Ac at leucine 309, decreased the phosphorylation of PP2Ac at tyrosine 307, and increased the protein expression of leucine carboxyl methyltransferase 1 (LCMT-1), protein tyrosine phosphatase 1B (PTP1B), and protein phosphatase 2A phosphatase activator (PTPA). These effects improved learning and memory abilities, prevented neuronal and synapse loss and reduced tau hyperphosphorylation and aggregation in the brains of rTg4510 mice.130
TCM Natural Products Treat AD by Enhancing Autophagy in Tau Protein Hyperphosphorylation
Autophagy is a highly conserved catabolic process that is mediated by lysosomes. Its main function is to remove abnormally folded proteins and senescent or damaged organelles, which is crucial for maintaining intracellular homeostasis.131–134 Autophagic dysfunction has been implicated in various disorders, including neurodegenerative diseases.135–139 The pathogenesis of AD is associated with multiple stages of autophagic damage, such as autophagosome maturation, autophagosome formation, and autophagosome-lysosome fusion. Certain AD-related genes, such as PS1, PS2, and phosphatidylinositol binding clathrin assembly protein (PICALM), have been found to impact autophagy and/or lysosomal function.133 In AD mouse models, the lack of essential autophagy-related genes such as ATG7 affects the production and secretion of Aβ.140,141 Autophagy has been shown to clear Aβ deposits, soluble tau proteins, and NFT polymers from neurons in animals with AD.142 These findings suggest that autophagy plays a significant role in the pathogenesis of AD. LC3, originally known as microtubule-associated proteins 1A and 1B (MAP1LC3), is commonly used to monitor autophagy. During autophagy initiation, LC3-I is converted to LC3-II, and the amount of LC3-II corresponds to the number of autophagic vesicles, which diminishes as autophagy progresses.143 Another crucial factor in autophagy induction is the Beclin 1 protein, which plays a central role in autophagy initiation.144 Previous studies have demonstrated that the expression of Beclin 1 is decreased in patients with AD as well as in transgenic mice.136,145 Moreover, the deletion of Beclin 1 has been found to enhance the accumulation of intracellular and extracellular Aβ, thereby accelerating the progression of AD.137 Zhou et al146 observed a noteworthy rise in the expression of autophagy-related protein p62 and a significant decline in the expression of Beclin-1 and LC3B in the hippocampus and cortex of Tau transgenic mice (TauP310L). This suggests that autophagy activity was significantly suppressed in Tau transgenic mice. In contrast, baicalin decreased the expression of p62 protein and increased the levels of Beclin-1 and LC3B, suggesting that baicalin enhances autophagy in transgenic mice. Additionally, baicalin was observed to reduce cellular aging in the hippocampus and cortex of Tau transgenic mice, enhance autophagy capability, decrease the formation of tau protein and phosphorylated tau protein, and ultimately improve cognitive dysfunction in Tau transgenic mice. Transcription factor EB (TFEB) is a crucial regulator of the autophagic lysosomal pathway.147–149 In various animal models of AD, TFEB overexpression has been shown to enhance autophagy, leading to the degradation of Tau protein and the improvement of cognitive function.150–152 Yang et al153 demonstrated that celastrol, a new TFEB activator, facilitates the translocation of TFEB from the cytoplasm to the nucleus. This activation enhances autophagy and lysosomal biological activity, specifically reducing insoluble phosphorylated Tau protein aggregates and promoting the degradation of phosphorylated Tau aggregates.
Natural products of TCM have been found to reduce Tau protein hyperphosphorylation, increase the dephosphorylation of abnormally phosphorylated Tau protein, restore the biological activity of Tau protein, and reduce the production of NFTs. This is achieved by inhibiting intracellular protein kinases such as PKA and GSK-3β or by promoting the expression of protein phosphatases such as PP2A and PP2B. The mechanisms of natural products in the deposition of Tau protein hyperphosphorylation are shown in Table 2. Additionally, these natural products can promote autophagy mediated by lysosomes, which helps remove abnormally folded proteins or senescent and damaged organelles. They can also facilitate the degradation of phosphorylated Tau aggregates, thereby reducing the formation of NFTs in brain tissue and potentially treating AD.
 |
Table 2 The Mechanisms of Natural Products in the Deposition of Tau Protein Hyperphosphorylation |
Microglia May Be Critical Intermediaries of Aβ–Tau Synergy
Evidence from multiple models and experimental settings presents compelling evidence for the synergistic interaction between Aβ and tau. In in vitro studies, the introduction of Aβ to human cells expressing tau (wild-type) led to the formation of tau aggregates known as paired helical filaments (PHFs), which are the principal components of tangles.154 Moreover, the administration of Aβ (either synthetic or derived from the brain) into P301L-mutant tau mice via cortical and/or hippocampal injections not only expedited tangle development near the injection site but also in interconnected synaptic regions.155,156
The results from animal studies often indicate that tau hyperphosphorylation is a downstream change induced by Aβ. However, this is not the case clinically. It has been observed that in patients with cognitive impairment and negative hyperphosphorylated Tau (p-Tau) in cerebrospinal fluid, there is no correlation between cerebrospinal fluid Aβ content and cerebral cortical atrophy. On the other hand, the degree of cerebral atrophy is correlated with the expression of Aβ in cerebrospinal fluid in patients with positive p-Tau. This suggests that the presence of p-Tau is an essential factor for the effect of Aβ on cortical atrophy.157,158
Investigations revealed that the mere presence of Aβ lacked the ability to induce neuronal degeneration in the hippocampus lacking endogenous Tau protein. A separate examination demonstrated that solely reducing Aβ levels did not alleviate the impairments in learning and memory observed in AD mice. It was only through the reduction of both Aβ and Tau proteins that cognitive function in AD mice improved. The researchers discovered that levels of tau phosphorylation only correlated with cognitive performance when Aβ deposits were identified in the cerebrospinal fluid.159 Several additional studies have demonstrated that alterations in glucose metabolism and brain atrophy in AD brains are attributable to the synergistic combination of Aβ and tau.160 These findings strongly imply the existence of a highly intricate interplay between Aβ and tau, suggesting that the disease process is not solely driven by a causal effect. Combining anti-Aβ deposition with anti-tau-induced neurofibrillary tangles may potentially be the most effective therapeutic approach for AD.157,158
The TREM2 gene, which is expressed by microglia, has emerged as a prominent risk factor in the development of AD. The activation of microglia plays a crucial role as a neuropathological characteristic of AD.161 It is responsible for releasing inflammatory factors such as interleukin-1β and tumor necrosis factor-α (TNF-α), as well as reactive oxygen and nitrogen species.162 Similarly, microglial activation is also induced by the overexpression of tau, and this activation can be observed even before the formation of neurofibrillary tangles.163 These findings suggest that microglia may act as potential mediators in the interaction between Aβ and tau. However, further investigations are required to determine the impact of differing genetic backgrounds of mouse models on microglial activation profiles and tau propagation patterns.
The spreading and bioactivity of tau in the presence of Aβ can be influenced by various factors. For instance, senescent oligodendrocytes near plaques can release cytokines that may activate microglia. This activation of microglia can lead to the uptake, processing, and subsequent release of tau in a more bioactive form. Neurons, possibly through an interaction with LRP1, can take up the released tau and then release it into the neuropil in a manner dependent on their activity levels. Multiple mechanisms enhance neuronal activity, including the inhibition of glutamate reuptake caused by Aβ, impaired synaptic inhibition, and the breakdown of the blood‒brain barrier, which leads to the leakage of neurotoxic substances (eg, albumin) and the activation of astrocytic TGF-β signaling. Microglia can also contribute to tau seeding and propagation through the release of cytokines, chemokines, and nitric oxide, which enhance tau phosphorylation. It is also possible that there is a direct transfer of tau through the somatic junctions between microglia and neurons. It should be noted that Aβ can also directly seed tau. Therefore, it is crucial to understand the complex interactions between Aβ, tau, microglia, and neurons in the context of tau seeding and propagation. The detailed hypothesized mechanism is shown in Figure 6.
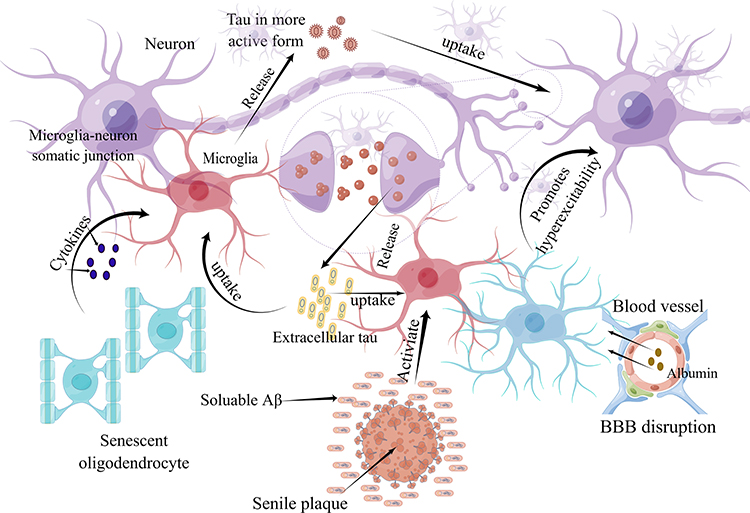 |
Figure 6 Microglia may be critical intermediaries of Aβ–tau synergy. |
Reconceptualizing AD in terms of Aβ-tau synergies could offer a promising treatment avenue for the future. However, our current understanding of Aβ-tau synergy is still in its early stages, and several unanswered questions remain. These include the underlying cause of inconsistencies and other crucial processes, such as the role of glial cell activation. Furthermore, identifying the key mechanism behind the Aβ-tau synergy and conducting additional experiments to validate it are necessary for future research.
Conclusion
According to the World Health Organization (WHO), there are approximately 50 million individuals worldwide suffering from dementia, with an additional 10 million new cases reported annually.164 A meta-analysis conducted between 1990 and 2016 revealed that the prevalence of AD among the elderly population was approximately 4.9%.165 The clinical symptoms of AD are diverse, mainly resulting in cognitive function impairment.164 This condition imposes significant mental pressure and financial burden on patients and their families.164 Currently, there are no drugs available for curing AD. Clinical practice often involves the use of cholinesterase inhibitors and megaline to improve AD symptoms. However, the effectiveness, safety, and cost-effectiveness of these treatments vary. Unfortunately, there is no definitive cure for AD yet, and the available drug treatment options are limited.164 Moreover, there is a lack of uniformity in drug selection, making it difficult to determine the best clinical treatment.164
Currently, the majority of research and development efforts in AD by scholars worldwide are focused on the widely recognized “β-amyloid deposition” and “tau protein hyperphosphorylation”. Aducanumab (BIIB037) is a fully human IgG1 monoclonal antibody that targets Aβ and facilitates the binding to Aβ aggregates, prompting microglia to phagocytose and clear Aβ.4 Lecanemab (BAN2401), another humanized IgG1 antibody, targets soluble Aβ fibrils and has shown good safety and tolerability in Phase 2b clinical trials.166 An 18-month analysis of patients with AD-derived mild cognitive impairment demonstrated a significant reduction of Aβ in the brain and improved cognitive decline in the high-dose group.4 Other phase III studies targeting Aβ, such as gantenerumab, are also currently underway. In terms of new drugs targeting tau protein, there are various categories including small molecule inhibitors, tau monoclonal antibodies, vaccines, etc. LMTM (TRx0237) is a small molecule tau protein aggregation inhibitor that prevents tau protein aggregation,167 but the results of two clinical studies on all-cause dementia and AD were negative.168 Other tau drugs entering the Phase II clinical stage include monoclonal antibodies semorinemab and zagotenemab (LY3303560). The tau protein vaccine, AADvac1, effectively reduced P-tau production in patients with mild AD in Phase II clinical studies.169 Overall, these developments indicate promising progress in the development of drugs targeting the “β-amyloid deposition” and the “tau protein hyperphosphorylation”.
However, a variety of abnormalities in signaling molecules and pathway changes contribute to the pathogenesis of AD. TCM monomer and compound preparations have been shown to effectively regulate multiple signaling pathways associated with AD. They possess characteristics of multitarget and wide-ranging action, which results in distinctive advantages and notable therapeutic effects in the treatment of AD. Chinese medicine scholars, in particular, have made significant efforts in the clearance of β-amyloid deposition and tau protein hyperphosphorylation. They exhibit characteristics of multitarget and wide-ranging action, leading to unique advantages and significant therapeutic effects in the treatment of AD. Nevertheless, the mechanism of drug action in AD has not been fully explored in relevant studies. Most traditional Chinese medicine research has focused on a single signaling pathway experiment, with limited reports on other highly correlated pathways. Furthermore, the identification of signal molecules is unclear, and the indicators used for detection in experiments are often repetitive. To overcome these limitations, future studies can employ biological approaches such as proteomics, transcriptomics, and metabolomics to analyze specific changes and identify key molecules that play a central role in differentiating AD. This will help in the discovery of new therapeutic targets.
The purpose of this review is to provide a comprehensive overview of recent studies on the role of common TCM natural products in β-amyloid deposition and Tau protein hyperphosphorylation in AD. The authors acknowledge that this review does not present quantitative findings but rather offers a consensus of thought among all authors. Although it is not a systematic review, the study includes a thorough examination of the literature and presents the latest research on this topic.
Abbreviations
Aβ, amyloid β-protein; ABC, ATP-binding cassette transporter; AD, alzheimer’s disease; ADAM10, a disintegrin and metalloprotease domain-containing protein 10; APC, adenomatous polyposis coli; APP, amyloid precursor protein; APP/PS1:APPswe/PSEN1dE9; BACE-1, β-APP cleavage enzyme 1; BBB:blood brain barrier; BDNF, brain-derived neurotrophic factor; CAT, catalase; CD, circular dichroism; Cytc:cytochrome c; ELISA, enzyme-linked immunosorbent assay; ERK, extracellular regulated protein kinases; Exo-cur, curcumin-macrophage exosome; GCs:cistanche glycosides; FGFR1, fibroblast growth factor receptor 1; GSH-Px, glutathione peroxidase; ICAM-1, intercellular adhesion molecule 1; IDE, insulin-degrading enzyme; L-Glu, L-glutamic acid; LFA-1, lymphocyte function-associated antigen 1; LRP-1:low-density lipoprotein receptor-related protein-1; MDA, malondialdehyde; miRNAs, MicroRNAs; mTOR, mammalian target of rapamycin; NB, N-butanol; NEP, neprilysin; NFT, neurofibrillary tangles; NPC, neural progenitor cells; NPDPK, nonproline-directed kinase; OA, okadaic acid; PDPK, proline directed kinase; PI3K/Akt, phosphoinositide-3 kinase/protein kinase B; PNS, Panax notoginseng saponins; PSD95, Postsynaptic density 95; RAGE, Receptor of Advanced Glycation Endproducts; SOD, superoxide dismutase; SP, senile plaques; TCF3, T-cell factor 3; TCF7L1, Transcription factor 7-like; TCM, traditional Chinese Medicine; TFEB:Transcription factor EB; TTR, transthyretin; VD, vascular dementia; 2VO, bilateral common carotid artery ligation.
Author Contributions
All authors have made significant contributions to the conception and design, data acquisition, and analysis and interpretation of the data. They have also participated in drafting the article and revising it critically for important intellectual content. Furthermore, all authors have agreed to submit the article to the current journal, given their final approval of the version to be published. Lastly, they have acknowledged their accountability for all aspects of the work.
Funding
Education Department of Jilin Province “13th Five-Year” science and technology project (JJKH20200895KJ); Science and technology ability promotion Plan of Health Commission of Jilin Province (2019J058); Jilin Provincial Science and Technology Department key research and development-medicine and health field (20200404065YY); Natural Science Foundation of Shandong (ZR2023QH159).
Disclosure
The authors report no conflicts of interest associated with this publication.
References
1. Academy of Cognitive Disorder of China, Writing Group of Expert Consensus on Long-term Healthcare of Cognitive Disorders in China. Chinese expert consensus on activity, behavior and cognition comprehensive management of Alzheimer’s disease. Chin J Geriatr. 2020;39(01):1–8. doi:10.3760/cma.j.issn.0254-9026.2020.01.001
2. Lynch C. World Alzheimer Report 2019: attitudes to dementia, a global survey. Alzheimer’s & Dementia. 2020;16(S10):8255. doi:10.1002/alz.038255
3. Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021;17(3):327–406. doi:10.1002/alz.12328
4. Chinese Society of Dementia and Cognitive Impairment. Chinese expert consensus on the diagnosis and treatment of mild cognitive impairment due to Alzheimer′s disease 2021. Chin J Neurol. 2022;55(5):421–440. doi:10.3760/cma.j.cn113694-20211004-00679
5. Jia L, Du Y, Chu L, et al. Prevalence, risk factors, and management of dementia and mild cognitive impairment in adults aged 60 years or older in China: a cross-sectional study. Lancet Public Health. 2020;5(12):e661–e671. doi:10.1016/S2468-2667(20)30185-7
6. Wang WL, Song CS. Recent Advances in the Pathogenesis of Alzheimer′s Disease and Clinical Medication. Chin J Drug Eval. 2019;36(03):204–209. doi:10.3969/j.issn.2095-3593.2019.03.011
7. Tian JZ, Xie HG, Wang LN, et al. the Guideline Panel of the Alzheimer’s Disease Chinese(ADC).Chinese guideline for the diagnosis and treatment of Alzheimer’s disease dementia(2020). Chin J Geriatr. 2021;40(3):269–283. doi:10.3760/cma.j.issn.0254-9026.2021.03.001
8. Liu J, Wang LN, Tian JZ. Recognition of dementia in ancient China. Neurobiol Aging. 2012;33(12):2911–2948. doi:10.1016/j.neurobiolaging.2012.06.019
9. Zhang YG, Liang YQ, Li YL, et al. Talking about Alzheimer’s disease from ‘kidney deficiency phlegm stasis’ Evolution of TCM pathogenesis. Modernization Traditional Chin Med Mater Medica-World Sci Technol. 2021;23(01):159–164. doi:10.11842/wst.20200724001
10. Tian JZ, Shi J, Ni JN, et al. Sequential Therapy Based on Evolvement of Patterns: a New Model for Treatment of Alzheimer’s Disease. Chin J Integr Med. 2019;25(8):565–573. doi:10.1007/s11655-019-3066-y
11. Tian J, Shi J, Wei M, et al. Chinese herbal medicine Qinggongshoutao for the treatment of amnestic mild cognitive impairment: a 52-week randomized controlled trial. Alzheimers Dement. 2019;5:441–449. doi:10.1016/j.trci.2019.03.001.eCollection2019
12. Savaskan E, Mueller H, Hoerr R, et al. Treatment effects of Ginkgo biloba extract EGb 761 on the spectrum of behavioral and psychological symptoms of dementia: meta-analysis of randomized controlled trials. Int Psychogeriatr. 2018;30(3):285–293. doi:10.1017/S1041610217001892
13. Shi J, Ni J, Lu T, et al. Adding Chinese herbal medicine to conventional therapy brings cognitive benefits to patients with Alzheimer’s disease: a retrospective analysis. BMC Complement Altern Med. 2017;17(1):533. doi:10.1186/s12906-017-2040-5
14. Zhou XL. Research Progress of the Prevention and Treatment of Alzheimer’s Disease with Chinese Herbal Monomers and their Active Ingredients. World Latest Medi Inf. 2019;19(46):99–100.
15. Cheng XR, Zhou WX, Zhang YX. Research on the pathogenesis of Alzheimer’s disease and prevention and treatment drugs. Chin J Pharmacol Toxicol. 2017;31(12):1129–1141. doi:10.3867/j.issn.1000-3002.2017.12.001
16. Zhang YH, Yang MH, Wu DH. Research Progress of the Intervention of Monomer and Active Ingredients of Chinese Medicinal in Treatment of AD. Info Tradit Chin Med. 2019;36(03):118–122. doi:10.19656/j.cnki.1002-2406.190092
17. Ryu JC, Zimmer ER, Rosa-Neto P, et al. Consequences of Metabolic Disruption in Alzheimer’s Disease Pathology. Neurotherapeutics. 2019;16(3):600–610. doi:10.1007/s13311-019-00755-y
18. Li C, Zhao L, Zang WD. Amyloid β-protein and neural network abnormality in Alzheimer’s disease. Chin Bull Life Sci. 2021;33(07):801–809. doi:10.13376/j.cbls/2021086
19. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi:10.15252/emmm.201606210
20. Dunys J, Valverde A, Checler F. Are N- and C-terminally truncated Abeta species key pathological triggers in Alzheimer’s disease? J Biol Chem. 2018;293(40):15419–15428. doi:10.1074/jbc.R118.003999
21. Volloch V, Rits S. Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease. Med Sci. 2018;6(2). doi:10.3390/medsci6020045
22. Li L, Xia BL, Ru LQ. Curative Effectiveness of Tanshinone in the Treatment of ModeI Rats with Two Kinds of Learning and Memory Impairment. Acta Med Univ Sei Technol Huazhong. 2008;37(06):819–822. doi:10.3870/j.issn.1672-0741.2008.06.031
23. Zhuang Y, Shi B, Tian X, et al. Effects of ginsenoside Rg2 on learning and memory ability and senile plaque formation in rats with Alzheimer’s disease model. Chin J Gerontol. 2010;30(02):202–204. doi:10.3969/j.issn.1005-9202.2010.02.025
24. Lu TT, Wang YY, Huang ZT, et al. The Influence of Berberine on Learning and Memory Ability and Amyloid Expression in APP/PS1 Transgenic Mice. Chin J Integr Med Cerebrovasc Dis. 2019;17(08):1155–1161.
25. Niu K, Chen Y, Xiao YY, et al. Effect of β-asarone on the deposition of β-amyloid in the brain of AD mice. J Hefei Univ Technol, Nat Sci. 2022;45(04):561–564. doi:10.3969/j.issn.1003-5060.2022.04.022
26. Xiao F, Luo HM, Li XG, et al. Inhibition of γ-schisandrin on the production of amyloid β-protein from M146L cells. Chin J New Drugs. 2005;1(3):290–292. doi:10.3321/j.issn:1003-3734.2005.03.010
27. Li Z, Liu XC, Li R, et al. Reduction of Aβ Generation by Schisandrin B through Restraining Beta-Secretase 1 Transcription and Translation. Med Sci Monit. 2018;24:1219–1224. doi:10.12659/msm.905127
28. Chen Q, Gao CX, Ge LH. The Effect of Senegenin on Neuromorphopathological Changes of AD Rats with β-Amyloid1-40 Injection into Right. Nucleus Basalis Acta Laser Biol Sin. 2006;2006(3):294–298. doi:10.3389/fphar.2022.937333
29. Webster B, Hansen L, Adame A, et al. Astroglial activation of extracellular-regulated kinase in early stages of Alzheimer disease. J Neuropathol Exp Neurol. 2006;65(2):142–151. doi:10.1097/01.jnen.0000199599.63204.6f
30. Qi H, Prabakaran S, Cantrelle FX, et al. Characterization of Neuronal Tau Protein as a Target of Extracellular Signal-regulated Kinase. J Biol Chem. 2016;291(14):7742–7753. doi:10.1074/jbc.M115.700914
31. Li SX, Han YS, Ji XY, et al. The Influence of Curcumin on the Expression of ERK Protein in Hippocampal Neurons Induced by Aβ in Rats with Alzheimer’s. Disease Acta Chin Med Pharmacol. 2014;42(02):21–23. doi:10.19664/j.cnki.1002-2392.2014.02.008
32. Sun T, Zhang DS, Jing YS. Therapeutic effect of curcumin on Alzheimer’s disease and its mechanism. Chin J Pharmacol Toxicol. 2021;35(09):641.
33. Ma YX, Jiao JL, Liu CY, et al. Effect of rhizoma acori graminei on the secondary structure of amyloid beta-protein 25-35. Chin J Pathophysiol. 2007;2:352–355. doi:10.3321/j.issn:1000-4718.2007.02.033
34. Huang X, Liu G, Guo J, et al. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14(11):1483–1496. doi:10.7150/ijbs.27173
35. Xie Y, Shi X, Sheng K, et al. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia(Review). Mol Med Rep. 2019;19(2):783–791. doi:10.3892/mmr.2018.9713
36. Gabbouj S, Ryhanen S, Marttinen M, et al. Altered Insulin Signaling in Alzheimer’s Disease Brain - Special Emphasis on PI3K-Akt Pathway. Front Neurosci. 2019;13:629. doi:10.3389/fnins.2019.00629
37. Zhang B, Wang Y, Li H, et al. Neuroprotective effects of salidroside through PI3K/Akt pathway activation in Alzheimer’s disease models. Drug Des Devel Ther. 2016;10:1335–1343. doi:10.2147/DDDT.S99958
38. Wang H, Li Q, Sun S, et al. Neuroprotective Effects of Salidroside in a Mouse Model of Alzheimer’s Disease. Cell Mol Neurobiol. 2020;40(7):1133–1142. doi:10.1007/s10571-020-00801-w
39. Guo B, Zhang W, Xu S, et al. GSK-3beta mediates dexamethasone-induced pancreatic beta cell apoptosis. Life Sci. 2016;144:1–7. doi:10.1016/j.lfs.2015.11.017
40. Luo CC, Yuan CY, Chen QF. GOLPH3 regulates proliferation and apoptosis of endometrial carcinoma cells through PI3K/AKT/GSK3βsignal. J Int Oncol. 2020;47(02):65–69. doi:10.3760/cma.j.issn.1673-422X.2020.02.001
41. Sun XY, J LL, Dong QX, et al. Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2021;18(1):131. doi:10.1186/s12974-021-02182-3
42. Ma GX, Ding QP, Deng HH, et al. Effect of astragalus polysaccharide and mechanism on Alzheimer’s disease rats. Stroke nerv dis. 2017;24(04):323–327. doi:10.3969/j.issn.1007-0478.2017.04.011
43. Du J, Murphy RM. Characterization of the interaction of beta-amyloid with transthyretin monomers and tetramers. Biochemistry. 2010;49(38):8276–8289. doi:10.1021/bi101280t
44. Lin M, Wang M, Gao SQ, et al. Effect of resveratrol on cognitive function and the expression of Aβ1-40 and Aβ1-42 in cerebral cortex of model mice with Alzheimer’s disease. J Mod Med Health. 2017;33(16):2425–2427. doi:10.3969/j.issn.1009-5519.2017.16.006
45. Santos LM, Rodrigues D, Alemi M, et al. Resveratrol administration increases Transthyretin protein levels ameliorating AD features- importance of transthyretin tetrameric stability. Mol Med. 2016;22:597–607. doi:10.2119/molmed.2016.00124
46. Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev. 1998;19(5):608–624. doi:10.1210/edrv.19.5.0349
47. Son SM, Cha MY, Choi H, et al. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy. 2016;12(5):784–800. doi:10.1080/15548627.2016.1159375
48. Vekrellis K, Ye Z, Qiu WQ, et al. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci. 2000;20(5):1657–1665. doi:10.1523/JNEUROSCI.20-05-01657.2000
49. Jha NK, Jha SK, Kumar D, et al. Impact of Insulin Degrading Enzyme and Neprilysin in Alzheimer’s Disease Biology: characterization of Putative Cognates for Therapeutic Applications. J Alzheimers Dis. 2015;48(4):891–917. doi:10.3233/JAD-150379
50. Lei DL, Li MB, Xiong K, et al. Triptolide inhibits the Aβ deposition and senile plaques formation in the hippocampus of APP/PS1 double transgenic mice. Acta Anat Sin. 2009;40(03):369–373. doi:10.3969/j.issn.0529-1356.2009.03.005
51. Cheng S, Leblanc KJ, Li L. Triptolide preserves cognitive function and reduces neuropathology in a mouse model of Alzheimer’s disease. PLoS One. 2014;9(9):e108845. doi:10.1371/journal.pone.0108845
52. Wang Q, Xiao B, Cui S, et al. Triptolide treatment reduces Alzheimer’s disease (AD)-like pathology through inhibition of BACE1 in a transgenic mouse model of AD. Dis Model Mech. 2014;7(12):1385–1395. doi:10.1242/dmm.018218
53. Huang JL, Feng YQ, Bai LR, et al. Fraction n-Butanol of Radix Notoginseng Protects PC12 Cells from Abeta(25-35)-Induced Cytotoxicity and Alleviates Cognitive Deficits in SAMP8 Mice by Attenuating Oxidative Stress and Abeta Accumulation. Evid Based Complement Alternat Med. 2017;2017:8469754. doi:10.1155/2017/8469754
54. Jin K, Peel AL, Mao XO, et al. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101(1):343–347. doi:10.1073/pnas.2634794100
55. Liu CY, Zhang PX, Rong GY, et al. Effects of tetramethylpyrazine on the expression of APP and NEP in the brain of rats with Alzheimer’s disease (AD) model. Heiilongjiang Medi Pharm. 2016;39(03):8–9. doi:10.3969/j.issn.1008-0104.2016.03.004
56. Liu SZ, Cheng W, Shao JW, et al. Notoginseng Saponin Rg1 Prevents Cognitive Impairment through Modulating APP Processing in Abeta(1-42)-injected Rats. Curr Med Sci. 2019;39(2):196–203. doi:10.1007/s11596-019-2019-1
57. Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019;25(4):554–560. doi:10.1038/s41591-019-0375-9
58. Erlandsson A, Lin CH, Yu F, et al. Immunosuppression promotes endogenous neural stem and progenitor cell migration and tissue regeneration after ischemic injury. Exp Neurol. 2011;230(1):48–57. doi:10.1016/j.expneurol.2010.05.018
59. Kolb B, Morshead C, Gonzalez C, et al. Growth factor-stimulated generation of new cortical tissue and functional recovery after stroke damage to the motor cortex of rats. J Cereb Blood Flow Metab. 2007;27(5):983–997. doi:10.1038/sj.jcbfm.9600402
60. Jin K, Galvan V, Xie L, et al. Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw, Ind) mice. Proc Natl Acad Sci U S A. 2004;101(36):13363–13367. doi:10.1073/pnas.0403678101
61. Niidome T, Taniuchi N, Akaike A, et al. Differential regulation of neurogenesis in two neurogenic regions of APPswe/PS1dE9 transgenic mice. Neuroreport. 2008;19(14):1361–1364. doi:10.1097/WNR.0b013e32830e6dd6
62. Rodriguez JJ, Jones VC, Tabuchi M, et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS One. 2008;3(8):e2935. doi:10.1371/journal.pone.0002935
63. Cheung WM, Hui WS, Chu PW, et al. Ganoderma extract activates MAP kinases and induces the neuronal differentiation of rat pheochromocytoma PC12 cells. FEBS Lett. 2000;486(3):291–296. doi:10.1016/s0014-5793(00)02317-6
64. Chu QP, Wang LE, Cui XY, et al. Extract of Ganoderma lucidum potentiates pentobarbital-induced sleep via a GABAergic mechanism. Pharmacol Biochem Behav. 2007;86(4):693–698. doi:10.1016/j.pbb.2007.02.015
65. Matsuzaki H, Shimizu Y, Iwata N, et al. Antidepressant-like effects of a water-soluble extract from the culture medium of Ganoderma lucidum mycelia in rats. BMC Complement Altern Med. 2013;13:370. doi:10.1186/1472-6882-13-370
66. Qin C, Wu SQ, Chen BS, et al. Pathological Changes in APP/PS-1 Transgenic Mouse Models of Alzheimer’s Disease Treated with Ganoderma lucidum Preparation. Acta Acad Med Sin. 2017;39(4):552–561. doi:10.3881/j.issn.1000-503X.2017.04.015
67. Huang S, Mao J, Ding K, et al. Polysaccharides from Ganoderma lucidum Promote Cognitive Function and Neural Progenitor Proliferation in Mouse Model of Alzheimer’s Disease. Stem Cell Reports. 2017;8(1):84–94. doi:10.1016/j.stemcr.2016.12.007
68. Wang Q, Zhang L, Yuan X, et al. The Relationship between the Bcl-2/Bax Proteins and the Mitochondria-Mediated Apoptosis Pathway in the Differentiation of Adipose-Derived Stromal Cells into Neurons. PLoS One. 2016;11(10):e163327. doi:10.1371/journal.pone.0163327
69. Wang C, Yao B, Xu M, et al. RIP1 upregulation promoted tumor progression by activating AKT/Bcl-2/BAX signaling and predicted poor postsurgical prognosis in HCC. Tumour Biol. 2016;37(11):15305–15313. doi:10.1007/s13277-016-5342-1
70. Saito A, Suga K, Ono-Nakagawa R, et al. Time lapse imaging analysis of the effect of ER stress modulators on apoptotic cell assessed by caspase3/7 activation in NG108-15 cells. Data Brief. 2016;6:20–27. doi:10.1016/j.dib.2015.11.030
71. Liu ZZ, Fu X, Chen G, et al. Combined effects and mechanism of lactate dehydrogenase A gene knockout and mitomycin C on prostate cancer cell apoptosis. Chin J Biomed Eng. 2016;22(3):206–209. doi:10.3760/cma.j.issn.1674-1927.2016.03.006
72. Mitupatum T, Aree K, Kittisenachai S, et al. mRNA Expression of Bax, Bcl-2, p53, Cathepsin B, Caspase-3 and Caspase-9 in the HepG2 Cell Line Following Induction by a Novel Monoclonal Ab Hep88 mAb: cross-Talk for Paraptosis and Apoptosis. Asian Pac J Cancer Prev. 2016;17(2):703–712. doi:10.7314/apjcp.2016.17.2.703
73. Liu C, Wang Y, Wang JY, et al. Expression of Bax on beta-amyloid (Aβ) -induced PC12 cell by Anemarrhena total saponins. Chin J Gerontol. 2014;34(18):5163–5165. doi:10.3969/j.issn.1005-9202.2014.18.68
74. Yin G, Gong DK, Liu BH, et al. Effects of Cistanche polysaccharide on learning and memory ability and expression of Bcl-2 and caspase-3 in hippocampal neurons of rats with Alzheimer’s disease. Lishizhen Med Mater Med Res. 2013;24(05):1091–1092. doi:10.3969/j.issn.1008-0805.2013.05.027
75. Luo L, Wang XW, Shi KR, et al. Preliminary study on the pathomorphology of the Glycosides of Cistanche Herba on the hippocampal CA1 area of experimental AD rats induced by β-amyloid. J North Sichuan Med Coll. 2020;35(06):947–951. doi:10.3969/j.issn.1005-3697.2020.06.02
76. Xiao F, Li XG, Gao Q, et al. The inhibition of amyloid β-protein production in M146L cell by Kadsura heteroclita polysaccharide. Chin J Gerontol. 2009;29(19):2458–2460. doi:10.3969/j.issn.1005-9202.2009.19.017
77. Xue JC. Effects of wogonoside on the nerves of APP/PS1 double transgenic mice neuroprotection and its mechanism. Guang zhou: Guangzhou University of Chinese Medicine; 2021. doi:10.27044/d.cnki.ggzzu.2021.001040
78. Hu XY, Sun XQ, Li ZP. Protective effect of phillyrin on HT22 cells and APP/PS1 mice induced by L-glutamate. Chin J Gerontol. 2022;42(07):1710–1714. doi:10.3969/j.issn.1005-9202.2022.07.051
79. Tayler H, Miners JS, Guzel O, et al. Mediators of cerebral hypoperfusion and blood-brain barrier leakiness in Alzheimer’s disease, vascular dementia and mixed dementia. Brain Pathol. 2021;31(4):e12935. doi:10.1111/bpa.12935
80. He Y, Ruganzu JB, Zheng Q, et al. Silencing of LRP1 Exacerbates Inflammatory Response Via TLR4/NF-kappaB/MAPKs Signaling Pathways in APP/PS1 Transgenic Mice. Mol Neurobiol. 2020;57(9):3727–3743. doi:10.1007/s12035-020-01982-7
81. Aurelie N. LRP1 plays a major role in the amyloid-β clearance in microglia. Mol Neurodegener. 2013;8:S1. doi:10.1186/1750-1326-8-S1-P33
82. Pascale CL, Miller MC, Chiu C, et al. Amyloid-beta transporter expression at the blood-CSF barrier is age-dependent. Fluids Barriers CNS. 2011;8:21. doi:10.1186/2045-8118-8-21
83. Zhao XS. Effects and Mechanism of Ox-LDL on Neural Cells Apoptosis Mediated by PCSK9/LRP1. Hu Nan: University of South China; 2016.
84. Chen HD. The Mechanism of the Aβ Clearance by Baicailin Through Blood Brain Barrier Pathwayin Alzheimer’s Disease Mouse. Guang zhou: Guang zhou university of chinese medicine; 2021. doi:10.27044/d.cnki.ggzzu.2021.001038
85. Tang G, Tang X, Mendu V, et al. The art of microRNA: various strategies leading to gene silencing via an ancient pathway. Biochim Biophys Acta. 2008;1779(11):655–662. doi:10.1016/j.bbagrm.2008.06.006
86. Mcgeer PL, Mcgeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2007;28(5):639–647. doi:10.1016/j.neurobiolaging.2006.03.013
87. Lin Y, Yao YJ, Liang XC, et al. Osthole suppresses BACE-1 expression by up-regulating miR-9 in Alzheimer’s disease. Chin Pharmacol Bull. 2019;35(04):524–529. doi:10.3969/j.issn.1001-1978.2019.04.016
88. Canet G, Chevallier N, Zussy C, et al. Central Role of Glucocorticoid Receptors in Alzheimer’s Disease and Depression. Front Neurosci. 2018;12:739. doi:10.3389/fnins.2018.00739
89. Doody RS, Raman R, Farlow M, et al. A Phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341–350. doi:10.1056/NEJMoa1210951
90. Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322–333. doi:10.1056/NEJMoa1304839
91. Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–321. doi:10.1056/NEJMoa1312889
92. Xia ZY, Zhao H, Pan LX, et al. Research progress on the role of Tau protein in Alzheimer’s disease. Neural Inj Functional Reconstruction. 2019;14(09):450–453. doi:10.16780/j.cnki.sjssgncj.2019.09.006
93. Dai XY, Zhang LY, Ren J, et al. Effect of glutamate receptor antagonist on learning-memory impairment and protein Tau hyperphosphorylation in neonatal rats after fetal distress. Chin Med. 2016;11(11):1718–1723. doi:10.3760/cma.j.issn.1673-4777.2016.11.032
94. Zhu ZP, Zhu H. Research progress in imaging agents targeting Aβ and Tau protein in Alzheimer′s disease. Chin J Nucl Med Mol Imaging. 2018;38(4):291–294. doi:10.3760/cma.j.issn.2095-2848.2018.04.018
95. Stefanoska K, Gajwani M, Tan A, et al. Alzheimer’s disease: ablating single master site abolishes tau hyperphosphorylation. Sci Adv. 2022;8(27):l8809. doi:10.1126/sciadv.abl8809
96. Xu KL, Chen Q, Liu W, et al. Effect of senegenin on tau protein phosphorylation at Ser396 site in neurons of AD rats induced by Aβ1-40. Chin J Pathophysiol. 2012;28(09):1605–1609. doi:10.3969/j.issn.1000-4718.2012.09.012
97. Ye J, Yin Y, Liu H, et al. Tau inhibits PKA by nuclear proteasome-dependent PKAR2alpha elevation with suppressed CREB/GluA1 phosphorylation. Aging Cell. 2020;19(1):e13055. doi:10.1111/acel.13055
98. Hu H, Liao ZD. Phosphorylation and dephosphorylation of tau protein in Alzheimer’s disease. Guang X Med J. 2010;32(05):602–605. doi:10.3969/j.issn.0253-4304.2010.05.043
99. Trushina NI, Bakota L, Mulkidjanian AY, et al. The Evolution of Tau Phosphorylation and Interactions. Front Aging Neurosci. 2019;11:256. doi:10.3389/fnagi.2019.00256
100. Li X, Zhang X, Zhang ZY, et al. The effects of Gensenoside Rg1 on the expressions of P-Tau and PKA on the brain slices of AD model rats. Chin J Gerontol. 2009;29(19):2468–2470. doi:10.3969/j.issn.1005-9202.2009.19.021
101. Li MZ, Wu WH, Wu ZP, et al. Panax Notoginseng Saponins Rg1 Improves Spatial Cognitive Ability and Decreases Phosphorylation Level of Tau Protein in Alzheimer’s Disease Rat Model. Acta Med Univ Sci Technol Huazhong. 2017;46(03):248–252. doi:10.3870/j.issn.1672-0741.2017.03.002
102. Sun DM. Therapeutic effects of curcumin in mice with Alzheimer’s disease. Chin J Gerontol. 2019;39(18):4577–4580. doi:10.3969/j.issn.1005-9202.2019.18.063
103. Wang H, Sui H, Zheng Y, et al. Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the Tau protein through the AKT/GSK-3beta pathway. Nanoscale. 2019;11(15):7481–7496. doi:10.1039/c9nr01255a
104. Metaxas A, Kempf SJ. Neurofibrillary tangles in Alzheimer’s disease: elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural Regen Res. 2016;11(10):1579–1581. doi:10.4103/1673-5374.193234
105. Iqbal K, Gong CX, Liu F. Microtubule-associated protein tau as a therapeutic target in Alzheimer’s disease. Expert Opin Ther Targets. 2014;18(3):307–318. doi:10.1517/14728222.2014.870156
106. Yan HM, Du XB, Zhang JJ. Effect of Radix Angelica Sinensis on Alzheimer’s disease-like lesions and cognitive function in rats with chronic cerebral hypoperfusion. Chin J Geriatr Heart, Brain Vessel Dis. 2017;19(07):699–703. doi:10.3969/j.issn.1009-0126.2017.07.007
107. Chen H, Lombes M, Le Menuet D. Glucocorticoid receptor represses brain-derived neurotrophic factor expression in neuron-like cells. Mol Brain. 2017;10(1):12. doi:10.1186/s13041-017-0295-x
108. Riise J, Plath N, Pakkenberg B, et al. Aberrant Wnt signaling pathway in medial temporal lobe structures of Alzheimer’s disease. J Neural Transm. 2015;122(9):1303–1318. doi:10.1007/s00702-015-1375-7
109. Fiorentini A, Rosi MC, Grossi C, et al. Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. PLoS One. 2010;5(12):e14382. doi:10.1371/journal.pone.0014382
110. Kim SE, Huang H, Zhao M, et al. Wnt stabilization of beta-catenin reveals principles for morphogen receptor-scaffold assemblies. Science. 2013;340(6134):867–870. doi:10.1126/science.1232389
111. Willert K, Jones KA. Wnt signaling: is the party in the nucleus? Genes Dev. 2006;20(11):1394–1404. doi:10.1101/gad.1424006
112. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi:10.1016/j.cell.2012.05.012
113. Huo JT, Zhang XQ, Yan J, et al. Activation of Wnt signaling pathway by tetramethylpyrazine improves brain tissue inflammation in rats with Alzheimer’s disease. Zhejiang Clin Med J. 2015;2015(8):1262–1264.
114. Yao HT, Ma JL, Zhang XB, et al. Effect of Cornus officinalis polysaccharide on tau protein in Alzheimer’s disease model rats. Chin J Gerontol. 2013;33(12):2838–2841. doi:10.3969/j.issn.1005-9202.2013.12.047
115. Su YN, Yao HT, Zhang XB, et al. Effects of Cornus officinale polysaccharide on glycogen synthase kinase-3β and phosphorylated-tau protein in the hippocampus of rats with an Alzheimer’s disease model. Chin J Gerontol. 2017;37(12):2885–2887. doi:10.3969/j.issn.1005-9202.2017.12.010
116. Li H, Liu C, Li N, et al. Improvement of acidic polysaccharose of Schisandra Chinensis on learning and memory functions of Alzheimer’s disease model mice. J Jilin Univ, Med Ed. 2017;43(06):1115–1120. doi:10.13481/j.1671-587x.20170609
117. Ni YN, Wang MY, Kong L, et al. Effect of osthole on tau hyperphosphorylation and PI3K/Akt/Gsk3β signaling pathway in the brain of AD mice. Chin New Drugs J. 2019;28(23):2865–2871. doi:10.3969/j.issn.1003-3734.2019.23.013
118. Lee MS, Kwon YT, Li M, et al. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405(6784):360–364. doi:10.1038/35012636
119. Patrick GN, Zukerberg L, Nikolic M, et al. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615–622. doi:10.1038/45159
120. Alvarez A, Toro R, Caceres A, et al. Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 1999;459(3):421–426. doi:10.1016/s0014-5793(99)01279-x
121. Shi LL, Yang WN, Chen XL, et al. The protective effects of tanshinone IIA on neurotoxicity induced by beta-amyloid protein through calpain and the p35/Cdk5 pathway in primary cortical neurons. Neurochem Int. 2012;61(2):227–235. doi:10.1016/j.neuint.2012.04.019
122. Lin L, Jadoon SS, Liu SZ, et al. Tanshinone IIA Ameliorates Spatial Learning and Memory Deficits by Inhibiting the Activity of ERK and GSK-3beta. J Geriatr Psychiatry Neurol. 2019;32(3):152–163. doi:10.1177/0891988719837373
123. Gong CX, Liu F, Grundke-Iqbal I, et al. Post-translational modifications of tau protein in Alzheimer’s disease. J Neural Transm. 2005;112(6):813–838. doi:10.1007/s00702-004-0221-0
124. Sanchez-Mut JV, Aso E, Heyn H, et al. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus. 2014;24(4):363–368. doi:10.1002/hipo.22245
125. Szegeczki V, Horvath G, Perenyi H, et al. Alzheimer’s Disease Mouse as a Model of Testis Degeneration. Int J Mol Sci. 2020;21(16). doi:10.3390/ijms21165726
126. Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, et al. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol. 2001;168(2):402–412. doi:10.1006/exnr.2001.7630
127. Sontag JM, Sontag E. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci. 2014;7:548.
128. Chen Q, et al. Banqiao Codonopsis pilosula improves cognitive dysfunction in AD model rats by PP2A signaling pathway. Chin Pharmacol Bull. 2019;35:1232–1239. doi:10.3969/j.issn.1001-1978.2019.09.010
129. Xie WZ. Trillium tschonoskii maxim improves cognitive dysfunction in Alzheimer’s disease induced by okadaic acid in rats and its possible mechanism. Chin Pharmacol Bull. 2018;34:1268–1275. doi:10.3969/j.issn.1001-1978.2018.09.017
130. Ma D, et al. Cornel iridoid glycoside suppresses tau hyperphosphorylation and aggregation in a mouse model of tauopathy through increasing activity of PP2A. Curr Alzheimer Res. 2019;16:1316–1331. doi:10.2174/1567205017666200103113158
131. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. doi:10.1016/j.ceb.2009.11.014
132. Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. doi:10.1038/nm.3232
133. Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16(6):345–357. doi:10.1038/nrn3961
134. Menzies FM, Fleming A, Caricasole A, et al. Autophagy and Neurodegeneration: pathogenic Mechanisms and Therapeutic Opportunities. Neuron. 2017;93(5):1015–1034. doi:10.1016/j.neuron.2017.01.022
135. Kumar H, Kim IS, More SV, et al. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat Prod Rep. 2014;31(1):109–139. doi:10.1039/c3np70065h
136. Mbaveng AT, Kuete V, Efferth T. Potential of Central, Eastern and Western Africa Medicinal Plants for Cancer Therapy: spotlight on Resistant Cells and Molecular Targets. Front Pharmacol. 2017;8:343. doi:10.3389/fphar.2017.00343
137. Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118(6):2190–2199. doi:10.1172/JCI33585
138. Ni HM, Chao X, Yang H, et al. Dual Roles of Mammalian Target of Rapamycin in Regulating Liver Injury and Tumorigenesis in Autophagy-Defective Mouse Liver. Hepatology. 2019;70(6):2142–2155. doi:10.1002/hep.30770
139. Ni HM, Mcgill MR, Chao X, et al. Removal of Acetaminophen protein adducts by autophagy protects against Acetaminophen-induced liver injury in mice. J Hepatol. 2016;65(2):354–362. doi:10.1016/j.jhep.2016.04.025
140. Nilsson P, Loganathan K, Sekiguchi M, et al. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013;5(1):61–69. doi:10.1016/j.celrep.2013.08.042
141. Nilsson P, Sekiguchi M, Akagi T, et al. Autophagy-related protein 7 deficiency in amyloid beta (Abeta) precursor protein transgenic mice decreases Abeta in the multivesicular bodies and induces Abeta accumulation in the Golgi. Am J Pathol. 2015;185(2):305–313. doi:10.1016/j.ajpath.2014.10.011
142. Uddin MS, Stachowiak A, Mamun AA, et al. Autophagy and Alzheimer’s Disease: from Molecular Mechanisms to Therapeutic Implications. Front Aging Neurosci. 2018;10:4. doi:10.3389/fnagi.2018.00004
143. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545. doi:10.4161/auto.4600
144. Ohashi Y, Tremel S, Williams RL. VPS34 complexes from a structural perspective. J Lipid Res. 2019;60(2):229–241. doi:10.1194/jlr.R089490
145. Jaeger PA, Wyss-Coray T. Beclin 1 complex in autophagy and Alzheimer disease. Arch Neurol. 2010;67(10):1181–1184. doi:10.1001/archneurol.2010.258
146. Zhou KY, Hu Y, Yang M, et al. Baicalin improves cognitive function by delaying senescence and regulating autophagy activity in Tau transgenic mice. Chin New Drugs J. 2019;28(01):65–72.
147. Settembre C, Di malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi:10.1126/science.1204592
148. Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. doi:10.1126/science.1174447
149. Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–914. doi:10.4161/auto.19653
150. Liu J, Zhang Y, Liu A, et al. Distinct Dasatinib-Induced Mechanisms of Apoptotic Response and Exosome Release in Imatinib-Resistant Human Chronic Myeloid Leukemia Cells. Int J Mol Sci. 2016;17(4):531. doi:10.3390/ijms17040531
151. Polito VA, Li H, Martini-Stoica H, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 2014;6(9):1142–1160. doi:10.15252/emmm.201303671
152. Martini-Stoica H, Xu Y, Ballabio A, et al. The Autophagy-Lysosomal Pathway in Neurodegeneration: a TFEB Perspective. Trends Neurosci. 2016;39(4):221–234. doi:10.1016/j.tins.2016.02.002
153. Yang C, Su C, Iyaswamy A, et al. Celastrol enhances transcription factor EB (TFEB)-mediated autophagy and mitigates Tau pathology: implications for Alzheimer’s disease therapy. Acta Pharm Sin B. 2022;12(4):1707–1722. doi:10.1016/j.apsb.2022.01.017
154. Ferrari A, Hoerndli F, Baechi T, et al. beta-Amyloid induces paired helical filament-like tau filaments in tissue culture. J Biol Chem. 2003;278(41):40162–40168. doi:10.1074/jbc.M308243200
155. Götz J, Chen F, van Dorpe J, et al. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–1495. doi:10.1126/science.1062097
156. Bolmont T, Clavaguera F, Meyer-Luehmann M, et al. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am J Pathol. 2007;171(6):2012–2020. doi:10.2353/ajpath.2007.070403
157. Desikan RS, McEvoy LK, Thompson WK, et al. Amyloid-beta associated volume loss occurs only in the presence of phospho-tau. Ann Neurol. 2011;70(4):657–661. doi:10.1002/ana.22509
158. Fortea J, Vilaplana E, Alcolea D, et al. Cerebrospinal fluid beta-amyloid and phospho-tau biomarker interactions affecting brain structure in preclinical Alzheimer disease. Ann Neurol. 2014;76(2):223–230. doi:10.1002/ana.24186
159. Busche MA, Hyman BT. Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat Neurosci. 2020;23(10):1183–1193. doi:10.1038/s41593-020-0687-6
160. Hanseeuw BJ, Betensky RA, Schultz AP, et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann Neurol. 2017;81(4):583–596. doi:10.1002/ana.24910
161. Perez-Nievas BG, Stein TD, Tai HC, et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain. 2013;136(Pt 8):2510–2526. doi:10.1093/brain/awt171
162. Prinz M, Jung S, Priller J. Microglia Biology: one Century of Evolving Concepts. Cell. 2019;179(2):292–311. doi:10.1016/j.cell.2019.08.053
163. Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351. doi:10.1016/j.neuron.2007.01.010
164. Zhang XL, Zeng JY, Chen X, et al. Comparison and Interpretation of Chinese and British Guidelines about Therapeutic Drugs for Alzheimer’s Disease. Chin Gen Pract. 2021;24(12):1454–1458. doi:10.12114/j.issn.1007-9572.2021.00.121
165. Zhang HB, Xu Y, Chen B, et al. A meta-analysis of the prevalence of Alzheimer’s disease. Chin J Gerontol. 2018;38(9):2157–2162. doi:10.3969/j.issn.1005-9202.2018.09.045
166. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther. 2021;13(1):80. doi:10.1186/s13195-021-00813-8
167. Wischik CM, Edwards PC, Lai RY, et al. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proceed Natl Acad Sci U S A. 1996;93(20):11213–11218. doi:10.1073/pnas.93.20.11213
168. Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer′s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016;388(10062):2873–2884. doi:10.1016/S0140-6736(16)31275-2
169. Novak P, Zilka N, Zilkova M, et al. AADvac1, an active immunotherapy for Alzheimer′s disease and non Alzheimer tauopathies: an overview of preclinical and clinical development. J Prev Alzheimers Dis. 2018;6(1):1–7. doi:10.14283/jpad.2018.45
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.