")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
The Role of Toll-Like Receptors 2 and 4 in the Pathogenesis of Chronic Obstructive Pulmonary Disease
Authors Sidletskaya K , Vitkina T , Denisenko Y
Received 10 February 2020
Accepted for publication 24 May 2020
Published 23 June 2020 Volume 2020:15 Pages 1481—1493
DOI https://doi.org/10.2147/COPD.S249131
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Karolina Sidletskaya, Tatyana Vitkina, Yulia Denisenko
Vladivostok Branch of Federal State Budgetary Science Institution “Far Eastern Scientific Center of Physiology and Pathology of Respiration” – Institute of Medical Climatology and Rehabilitative Treatment, Vladivostok, Russia
Correspondence: Karolina Sidletskaya
Vladivostok Branch of Federal State Budgetary Science Institution “Far Eastern Scientific Center of Physiology and Pathology of Respiration” – Institute of Medical Climatology and Rehabilitative Treatment, Russian street 73-g, Vladivostok 690105, Russia
Email [email protected]
Abstract: Currently, chronic obstructive pulmonary disease (COPD) is one of the leading causes of morbidity and mortality worldwide. The determination of immune mechanisms of inflammation in the disease presents an important challenge for fundamental medical research. According to modern views, Toll-like receptors (TLRs), among which TLR2 and TLR4 play a key role, are one of the essential components of inflammatory process in COPD. This review focuses on following aspects: the role of TLR2 and TLR4 in the initiation of inflammatory process in COPD; the mechanisms of influence of various exogenous factors (cigarette smoke, suspended particulate matter, and bacteria) on the expression of TLR2 and TLR4; the contribution of these TLRs to the T-helper (Th) immune response development in COPD, in particular to the Th17 immune response, which contributes to the progression of the disease and therapeutic implications of TLR2 and TLR4 in COPD.
Keywords: toll-like receptor 2, toll-like receptor 4, T-helper immune response, chronic obstructive pulmonary disease
Introduction
Chronic obstructive pulmonary disease (COPD) ranks third in terms of total mortality and is one of the most pressing challenges to modern medicine. This disease is characterized by gradually progressing airway obstruction caused by an atypical inflammatory response to inhaled harmful particles and gases, which intensifies during the exacerbation phase.1 The inflammatory process in COPD is both local and systemic.2 A large amount of recent data suggests the importance of Toll-like receptors (TLRs) in the initiation and development of the inflammatory process in COPD. These receptors belong to the family of pathogen-recognizing receptors (PRRs). Their major functions are to recognize the molecular structures of pathogens (pathogen-associated molecular patterns, PAMPs) and the products of cellular damage (damage-associated molecular patterns, DAMPs), and thus ensure a quick response of cells to microbial/viral infection or violation of tissue homeostasis, respectively. So far, 10 human TLRs have been identified. They are localized on organelle (TLR3, TLR7, TLR8, and TLR9) and cellular (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10) membranes.3 A number of studies report the involvement of TLR3, TLR7, and TLR9 in an inflammatory response to viral infection in COPD.4–9 However, numerous studies have shown that TLR2 and TLR4 are the most significant receptors for the development of COPD. They are involved in the pathogenesis of this disease in several ways.10–13
The aim of the review is to analyze the modern views on the role of TLR2 and TLR4 in the pathogenesis of COPD.
Biology of TLR2 and TLR4
Toll-like receptors, including TLR2 and TLR4, are type 1 transmembrane proteins with three domains: 1) extracellular domain with leucine-rich repeats (LRRs) responsible for ligand recognition; 2) transmembrane domain; and 3) intracellular Toll/interleukin-1 receptor (TIR) domain that provides signal transmission from the cell surface to adapter proteins. TLR2 and TLR4 are membrane-associated receptors involved in lipid recognition. TLR2 recognizes glycolipids and lipoproteins/lipopeptides of Gram-positive bacterial cell walls. The receptor was shown to form heterodimers with TLR1 (TLR2/1) and TLR6 (TLR2/6), that are able to recognize triacylated lipopeptides of Gram-negative bacteria and diacylated lipopeptides of Gram-positive bacteria, respectively. The main ligand of TLR4 is lipopolysaccharide (LPS)/endotoxin, the main component of Gram-negative bacterial cell walls; CD14 and MD-2 molecules are also involved in its recognition. In addition to pathogens, both of these TLRs are able to bind endogenous ligands (DAMPs) released as a result of cell death due to oxidative stress caused by environmental factors. It is known that TLR2 and TLR4 are expressed both by innate (neutrophils, macrophages, dendritic cells, endothelial cells, epithelial cells of the skin and mucous membranes) and adaptive immunity cells. Ligand binding to TLR initiates either one of two key signal cascades: MyD88 (myeloid differentiation primary response gene 88) or TRIF-dependent (TIR-domain-containing adapter-inducing interferon-β) pathways mediated by MyD88/TIRAP (TIR-domain-containing adapter protein) and TRIF/TRAM (TRIF-related adapter molecule) complexes of adapter proteins, respectively. TLR2, like most TLRs, implements its functions via MyD88-dependent signaling. The activation of TLR4 can initiate both MyD88-dependent and TRIF-dependent signaling pathways. The MyD88-dependent signal cascade activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) or mitogen-activated protein kinase (MAPK) signaling pathway followed by a synthesis of molecules associated with inflammation. The initiation of TRIF-dependent signaling pathway via TLR4 located on the endosomal membrane leads to a phosphorylation of transcription factor IRF3, which regulates the expression of type 1 interferon genes involved in antiviral immunity (Table 1).3,11
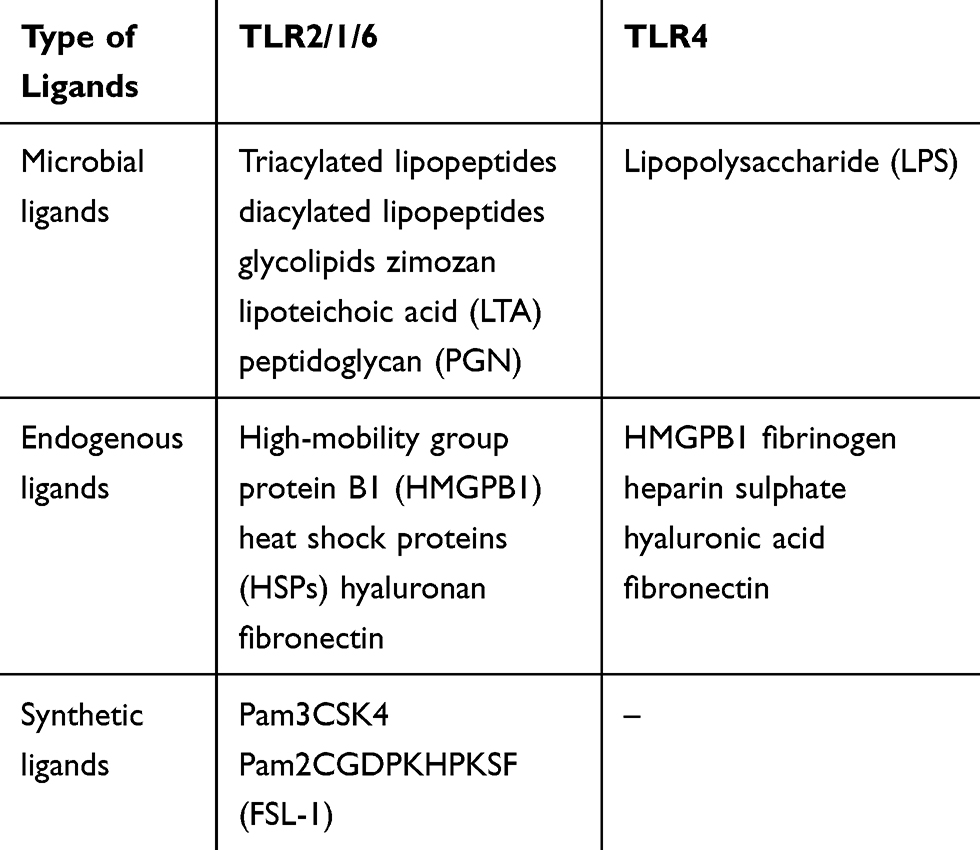 |
Table 1 Ligands of TLR2 and TLR4 |
TLR2 and TLR4 in the Initiation of Inflammation in COPD
Cigarette smoke and components of polluted air (combustion products of diesel and bioorganic fuels, industrial dust, etc.) act as the triggers of inflammatory process in COPD. These substances can activate TLRs both via LPS, which is often found in tobacco smoke and particles of polluted air, and by releasing DAMPs (high-mobility group protein B1 (HMGPB1), heat shock proteins (HSP60, HSP70), β defensin, hyaluronic acid, fibronectin, heparin sulphate, biglycan).14–18
Cigarette smoke has been revealed to cause increased expression and activation of TLR2 and TLR4 on immune and epithelial cells of the respiratory tract both in humans and experimental animals.19–21 These receptors initiate the MyD88-dependent signal cascades that result in the activation of NF-κB followed by the synthesis of cytokines and chemokines. In turn, these mediators are responsible for the recruitment of monocytes and neutrophils in the lung. These inflammatory cells release proteolytic enzymes (matrix metalloproteinases, neutrophil elastase) contributing to the destruction of lung tissue, along with TGF-β1 that is involved in the processes of airway remodeling.13 Using human bronchial cell culture, Pace et al have shown that TLR4/MyD88 signaling plays a key role in the initiation of immune response to exposure to cigarette smoke. The main result of the activation of this signaling pathway is the synthesis of the neutrophil chemoattractant C-X-C Motif Chemokine Ligand 8/interleukin-8 (CXCL8/IL-8).19 The increased number of neutrophils in bronchoalveolar lavage is an important feature of COPD. The study in a mouse model of cigarette smoke-induced emphysema has found that TLR4 is involved in the development of the main clinical manifestations of COPD, such as fibrosis, emphysema, and pulmonary function impairment (Table 2).21
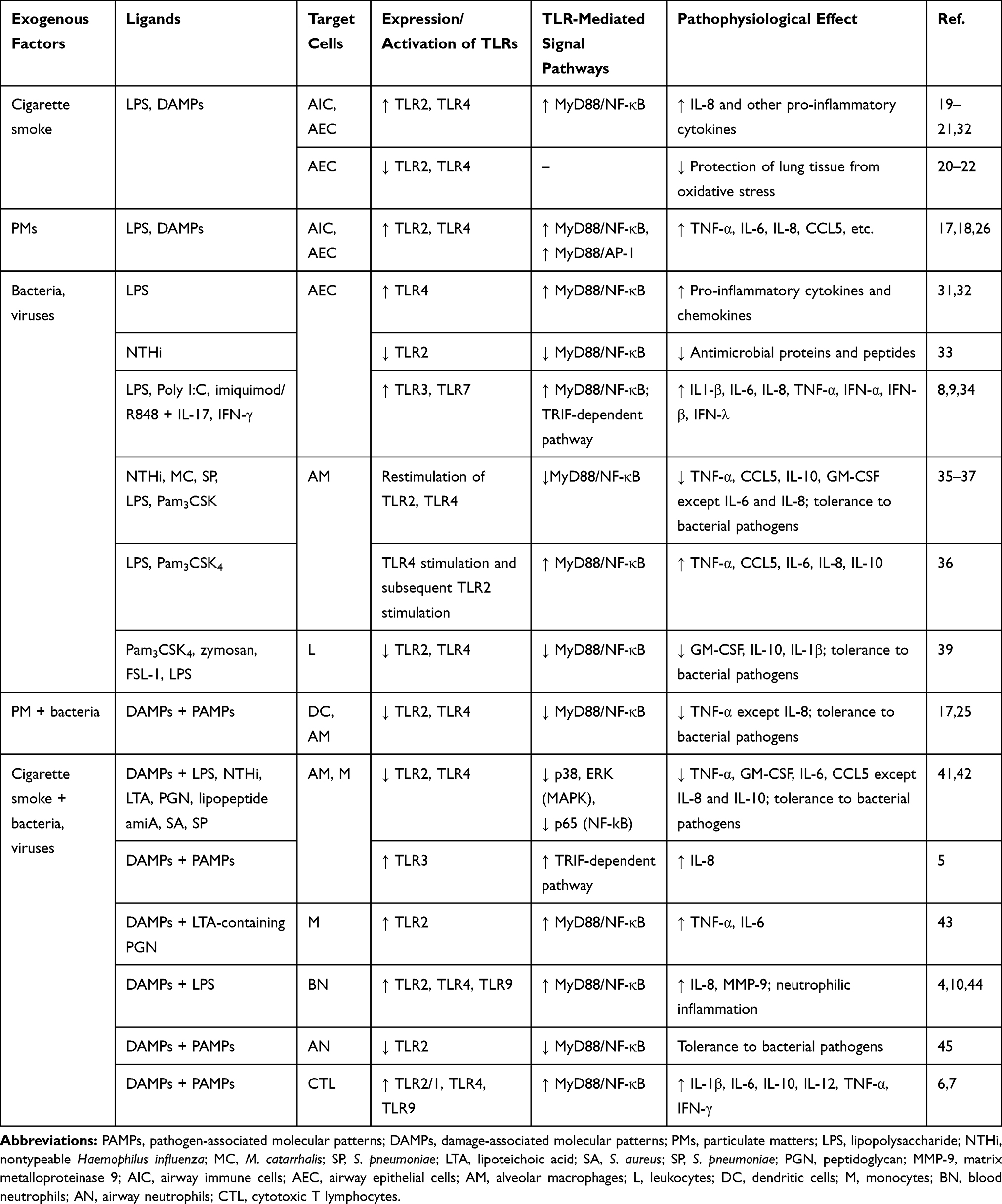 |
Table 2 The Mechanisms of the Influence of Exogenous Factors on TLR Signal Pathways in COPD |
Besides participating in the cigarette smoke-induced pro-inflammatory mechanisms, TLR2 and TLR4 are also involved in protecting the lung tissue against oxidative stress caused by cigarette smoke. This is evidenced by the results of experimental studies conducted on TLR-deficient mice with cigarette smoke-induced emphysema.20,21 The correlation between the development of emphysema, airflow limitation and low expression of TLR4 in the lung tissue of smokers has been found.22 The experimental study has shown that cigarette smoke and Hsp70, which may be released as a result of tobacco smoking, inhibit the expression of TLR2 and TLR4 mRNA in bronchial epithelial cells in COPD. However, this effect was not observed at the time of combined exposure to these factors.23 Thus, there is a need to study the mechanisms of the influence of cigarette smoking on the expression and activity of TLRs in COPD (Table 2).
Indoor and outdoor air pollution is a significant risk factor for COPD along with smoking. The main component of polluted air is suspended particulate matter (PMs). Experimental studies have shown that PMs are also able to increase expression and activation of TLR2 and TLR4 on alveolar macrophages and epithelial cells of the respiratory tract, thereby enhancing the inflammatory response. These particles initiate TLR2/MyD88 and TLR4/MyD88 signaling cascades, which lead to the activation of transcription factors AP-1, NF-κB. The activation is followed by a synthesis of pro-inflammatory mediators (tumor necrosis factor-α (TNF-α), IL-6, IL-8, chemokine C‐C motif receptor 5/regulated on activation, normal T cell expressed and secreted (CCL5/RANTES), etc.).17,18,24 It should be noted that the activation of TLR signaling pathways depends on the particle diameter. PMs with a diameter of ≤2.5 µm (PM2.5) activate TLR2, while PMs with a diameter of ≤10 µm (PM10) mediate their action through TLR4.25,26 The effect of PMs on TLR2 and TLR4 is related to airway obstruction and emphysema (Table 2).17
Budulac et al have shown that polymorphism of the TLR2 and TLR4 genes is associated with a reduced pulmonary function and an increased number of inflammatory cells in the sputum in COPD patients, which indicates that these receptors are involved not only at triggering local inflammatory response at the initial stages of the disease, but also during further development of the disease.27 A number of other genetic studies have also revealed a relationship between TLR2 and TLR4 gene polymorphism and the risk of developing COPD.28,29 Haw et al have found that the mRNA levels of TLR2, TLR4, and their coreceptors (TLR1, TLR6, CD14, and MD2) were increased in the airway epithelium of patients with mild and moderate COPD. However, the expression of these receptors was decreased in lung parenchymal cores from patients with severe COPD. The authors believe that the increased TLR expression at the initial stage of COPD is caused by cigarette smoke exposure, but the reduced levels of receptors observed during the disease progression are due to tissue destruction.21 It should be mentioned that the comparison of TLR mRNA levels between mild-to-moderate and advanced COPD was carried out in samples of various tissues (airway epithelium and lung parenchymal cores), which is a limitation of the study.
Thus, the data on the role of TLR2 and TLR4 in local inflammation at COPD are contradictory. On the one hand, the constant exposure of respiratory tract cells to the main COPD risk factors (cigarette smoke and air pollutants) has been established to cause excessive activation of the TLR2 and TLR4 signaling pathways. On the other hand, there are data indicating the protective role of these receptors in COPD. The described mechanisms and their participation in the progression of inflammation in COPD require further studies.
TLR2 and TLR4 in the Development of COPD Exacerbation
Exacerbations of COPD are associated with changes in microbiota and increased inflammation of the respiratory tract. Bacteria and viruses can colonize the lower respiratory tract in COPD, facilitating secondary infections that lead to an acute exacerbation of the disease.30
Bacterial colonization of the lungs is a common feature of COPD. It contributes to the development of exacerbations and the progression of the pathology by maintaining the inflammatory process in the respiratory tract. Streptococcus pneumoniae, nontypeable Haemophilus influenzae (NTHi), Moraxella catarrhalis, and Pseudomonas aeruginosa are the bacteria most often detected in the lower airways of COPD patients.14,30 According to research, TLR2 and TLR4 are the main TLRs that provide an immune response to bacterial invasion of the respiratory tract, thereby participating in the development of COPD exacerbation. There are a number of PAMPs recognized by TLR2, including lipoteichoic acid (LTA), pneumolysin (S. pneumoniae), outer membrane porin protein 2, outer membrane porin protein 6 (NTHi). The ligands for TLR4 are LTA, pneumolysin (S. pneumoniae), LPS (NTHi, M. catarrhalis, P. aeruginosa).14 However, the possibility of LPS contamination in these studies should be considered. The most common viruses that cause acute exacerbations of COPD are rhinovirus, adenovirus, and influenza virus.30
During exacerbation of COPD, the violation of TLR signal transduction is observed in the bronchial epithelial cells in response to bacterial or viral infection. The patients with exacerbated COPD exhibited an elevated expression of TLR4 and MyD88 in the lung.31 An increased airway bacterial load (P. aeruginosa) and high levels of TLR4 expression in bronchial epithelial cells have been observed in severe and very severe COPD. It has been demonstrated that these indicators positively correlate with the degree of airflow obstruction.32 Besides that, TLR2 is also involved in the regulation of an immune response of the airway epithelium against bacteria in COPD. In a recent study, Amatngalim et al have shown that airway epithelial cells from mild-to-moderate COPD patients exhibited attenuated antibacterial activity in response to NTHi infection compared to the cells from smokers/ex-smokers with a normal pulmonary function. The authors suggest that the deterioration in the synthesis of antimicrobial proteins and peptides in COPD is caused by impaired induction of the TLR2 signaling pathway in airway epithelial cells.33 In addition, there is evidence of an increase in TLR3 and TLR7 mRNA levels in airway epithelial cells under high levels of IL-17 and IFN-γ in the microenvironment, that is a characteristic feature of COPD. Enhanced signaling via these receptors can increase the inflammatory response due to the production of IL-1β, IL-6, IL-8, TNF-α, IFNs, and worsen the symptoms of virus-induced COPD exacerbation.9,34 Similar results were obtained in a study of lung whole tissue explants from COPD patients.8 Thus, current literature data indicate that increased TLR4/MyD88, TLR3 and TLR7 signaling and defective TLR2 signaling in airway epithelial cells contribute to COPD exacerbation and progression under viral or bacterial infection (Table 2).
Another proposed mechanism for the development of COPD exacerbation is the formation of TLR-induced tolerance of alveolar macrophages to persistent bacteria. Berenson et al have demonstrated that the tolerance is reflected in a reduced cytokine response (TNF-α, IL-8) to NTHi, M. catarrhalis, and S. pneumoniae, mediated by TLR2, TLR4, MyD88, and NF-κB. A correlation was found between the impaired antibacterial response of alveolar macrophages and the clinical tendency to COPD exacerbation.35 As shown by Lea et al, the tolerance may be associated with repetitive stimulation of alveolar macrophages with the same TLR2 (Pam3CSK4) or TLR4 (LPS) ligand, which leads to a decrease in the production of TNF-α, CCL5, and IL-10, except IL-6 and neutrophilic attractant IL-8.36 Knobloch et al have also revealed a high IL-8 level and a decrease in the production of other pro-inflammatory cytokines by alveolar macrophages in response to a single exposure to LPS in COPD patients.37 This suggests a cytokine-specific effect of bacterial infection on alveolar macrophages. Apparently, IL-8 plays a key role in maintaining the inflammatory response against bacteria in the lungs in COPD patients by recruiting neutrophils. However, TLR4 stimulation and subsequent TLR2 stimulation, alternatively, increased the secretion of pro-inflammatory cytokines. This effect is associated with TLR4 activation that elevates the expression of the TLR2 gene.36 The enhanced production of cytokines, which is induced by various TLR stimuli, can be of great clinical importance for COPD patients simultaneously exposed to several bacterial strains in both stable and exacerbated COPD (Table 2).
Bacterial PAMPs can also enter the bloodstream and participate in the regulation of systemic inflammation in COPD. There is evidence of a decrease in TLR2 expression on monocytes as COPD progresses.38 According to the results of in vitro study, the inhibition of TLR-2/1-, TLR-2/6- and TLR4-dependent secretion of cytokines (granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-10, and IL-1β) by leukocytes has occurred at the last stages of COPD in the presence of chronic bacterial lung infection (Table 2).39
The results of modern studies attest to the fact that components of cigarette smoke and polluted air may influence PAMP-induced inflammatory response in COPD at local and systemic levels. Suspended particles have been reported to suppress the expression of TLR2 and TLR4 on dendritic cells and alveolar macrophages, except for IL-8, thereby resulting in the development of tolerance to bacterial pathogens and more frequent exacerbations of COPD.17,25 In addition, Allard et al have reported that alveolar macrophages, as in the case of bacterial infection, are capable of causing adaptation to the repeated exposure to inhaled oxidative toxicants present in ambient air via the activation of the transcription factor forkhead box P3 (FoxP3) which is involved in the mechanisms of limiting inflammation.40 Metcalfe et al have demonstrated in an experimental study that the exposure to cigarette smoke on alveolar macrophages derived from COPD patients leads to the suppression of TLR4-induced synthesis of TNF-α, GM-CSF, IL-6, CCL5 (with the exception of IL-8) in response to bacterial ligands (LPS, NTHi). This effect is due to a decrease in the expression of MAPK kinases (p38, extracellular signal-regulated kinase (ERK)) and p65 (NF-κB subunit).41 Knobloch et al have also shown that a monocyte-dependent immune response to a bacterial infection is suppressed by cigarette smoke and is impaired in COPD. A decreased cytokine response (GM-CSF, TNF-α) of circulating monocytes to ligands of TLR2, TLR4 and nucleotide-binding oligomerization domain-containing protein (NOD2), such as peptidoglycan (PGN), lipoteichoic acid (LTA), lipopeptide amiA, S. aureus (SA) and S. pneumoniae (SP) extracts, has been detected in smokers and patients with COPD. The authors believe that this effect is associated with down-regulation of these PRRs. Additionally, it was noted that patients with COPD have increased secretion of the pro-inflammatory cytokine IL-10 by monocytes, which also suppresses the synthesis of inflammatory mediators in this respiratory pathology. This study has also shown that the elevated levels of IL-8 were maintained, which indicates the participation of this cytokine not only in local but also in systemic inflammatory process in COPD.42 Thus, according to recent studies, the influence of cigarette smoke and PMs on alveolar macrophages and circulating monocytes causes a decrease in the responsiveness of these cells to bacterial invasion in COPD, while the inflammatory process is maintained by a high concentration of IL-8.
Controversially, earlier data obtained by Pons et al indicate an elevated TLR2 expression on blood monocytes in smokers with stable and exacerbated COPD compared to non-smokers and smokers with normal pulmonary function. Upon stimulation with TLR2 ligand, monocytes isolated from the blood of COPD patients secreted higher levels of IL-6 и TNF-α than monocytes from smokers and non-smokers without COPD.43 It should be noted that the agonist used in this study (LTA-containing PGN) most likely caused simultaneous stimulation of TLR2 and NOD2 signaling pathways that could affect the increased secretion of the mediators.
Cigarette smoke has been shown to cause up-regulation of TLR3 expression in alveolar macrophages and TLR2/1, TLR4 and TLR9 expression in cytotoxic T lymphocytes (CD8+ cells) during viral infection, which leads to increased secretion of pro-inflammatory mediators, such as IL-1β, IL-6, IL-8, IL-10, IL-12p70, TNF-α, and IFN-γ.5–7 Smoking appears to enhance the inflammatory response to viral infection, as opposed to bacterial infection.
It is known that the neutrophilic inflammation is a characteristic feature of COPD. A number of researchers have demonstrated that the effect of LPS on neutrophils from smokers with COPD results in an increase in TLR2/TLR4 expression and a subsequent enhancement of the synthesis of IL-8 and matrix metalloproteinase-9 (MMP-9).10,44 It has been revealed that TLR9 can also contribute to the cigarette smoke-mediated neutrophilic response by providing IL-8 secretion from circulating neutrophils.4 Apparently, an enhanced blood neutrophilic response in COPD can be triggered by both bacterial or viral infection and cigarette smoke. However, there is evidence of decrease in TLR2 expression on neutrophils during their migration from the bloodstream into the airways in smokers with COPD. It may be the cause of decreased effectiveness of recognition and elimination of pathogens as well as bacterial colonization of the lungs in COPD.10,45 Some researchers have reported no changes in TLR4 expression on blood monocytes and neutrophils in patients with COPD compared to non-smokers and smokers with normal pulmonary function (Table 2).10,43,45,46
In general, the results of studies presented in the literature suggest that there are differences between local and systemic effects of PAMP-mediated activation of TLR2/TLR4 in COPD. In addition, it is known that adverse environmental factors, such as cigarette smoking and air pollution, have a great impact on these processes. Nevertheless, the mechanisms through which TLRs participate in the development of COPD exacerbation remain to be investigated due to the contradictory findings and varying methodological approaches.
TLR2 and TLR4 in the Formation of T-Helper Immune Response in COPD
According to modern concepts, T-helper (Th) lymphocytes play an important role in the regulation of the inflammatory process. Each of Th subpopulations (type 1 T-helpers (Th1), Th2, Th17, T-regulatory cells (Treg), etc.) forms its own immunoregulatory pathway, characterized by a specific cytokine profile and a set of effector cells. Whereas Th1, Th2, Th17 cells are responsible for the enhancement of inflammatory response, the main function of Tregs is its inhibition. In COPD, a predominance of Th1 and Th17 types of immune response has been detected.47,48 Moreover, Th17-associated inflammation is characterized by severe airway obstruction and a high risk of infectious complications.48 A lot of researchers argue that an imbalance between subpopulations of Th17 cells and Tregs promotes COPD progression.49–51
There is more evidence of the contribution of TLRs in the formation of Th immune response, including the regulation of Th17/Treg balance. The ligands of these receptors have been shown to have both indirect and direct effects on Th cells.52
An indirect effect is manifested through the activation of antigen-presenting cells (APCs), which leads to the expression of a number of cytokines, chemokines and costimulatory molecules necessary for naive CD4+ cells priming in the lymph nodes. The interaction between APCs and naive Th cells determines the further formation of T cell response.53,54 The activation of dendritic cells via the TLR2-MyD88-mediated signaling pathway induces the differentiation of Th17 cells (Figure 1).52
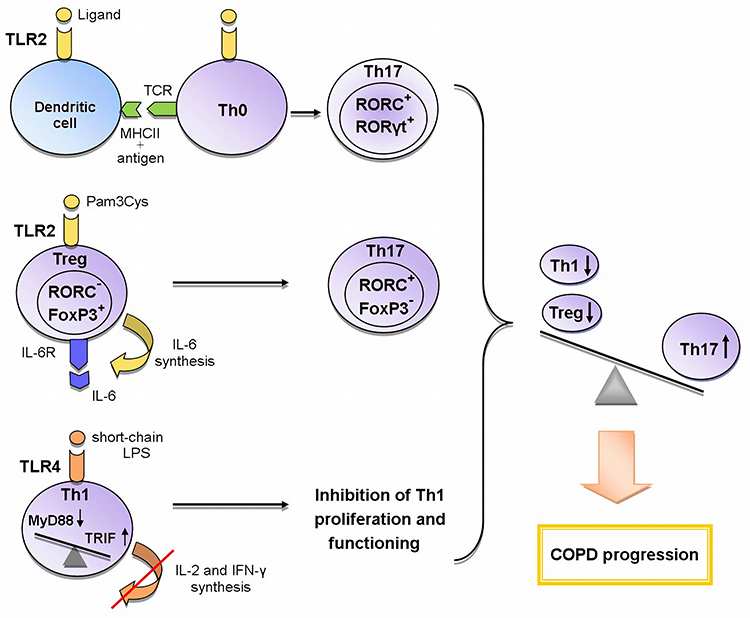 |
Figure 1 The role of TLR2 and TLR4 in the formation of Th17 immune response in COPD.Abbreviations: FoxP3 – forkhead box P3, IL-6 – interleukin-6, IL-6R – IL-6 receptor, MHCII – histocompatibility complex class II, MyD88 – myeloid differentiation primary response gene 88, RORα – retineic-acid-receptor-related orphan nuclear receptor α, RORC – retineic-acid-receptor-related orphan nuclear receptor C, RORγt – retineic-acid-receptor-related orphan nuclear receptor γ, TRIF – toll/interleukin-1 receptor-domain-containing adapter-inducing interferon-β, TCR – T cell receptor, Th0 – naive T helper cell, Th1 – T helper cell type 1, Th17 – T helper cell type 17, Treg – T regulatory cell, TLR – toll-like receptorLegend: Recently three mechanisms of the influence of TLRs on the formation of T-helper immune response in COPD are proposed: 1) the activation of TLR2 signaling in dendritic cells during the presentation of antigen to Th0 cells enhances the expression of RORγt, RORC (Th17-associated transcription factors) and thus contributes to the differentiation of Th17 cells; 2) TLR2 signaling in Tregs initiates the synthesis of IL-6; IL-6, acting in an autocrine manner, inhibits the expression of FoxP3 responsible for the suppressive activity and initiates the expression of RORC in these cells; as a result, the polarization of Tregs towards Th17 cells occurs; 3) the switching from the TLR4/MyD88-dependent pathway to the TLR4/TRIF-dependent pathway in Th1 cells leads to a decrease in synthesis of IL-2 and IFN-γ and the inhibition of IL-2-mediated proliferation of these cells. Thus, the activation of these mechanisms causes an imbalance between T-helper subpopulations towards Th17 cells, which leads to COPD progression. |
Toll-like receptors may directly regulate T-cell function, as evidenced by the fact that T-helpers express most of the TLRs. In vitro studies have shown that TLR2 and TLR4 ligands act as costimulatory molecules during T-cell receptor (TCR) activation, enhancing the proliferation and survival of T-lymphocytes.52–56 In addition, it has been shown that stimulation of TLR2 with endogenous ligand HSP60 induces the adhesion and suppresses the chemotaxis of T cells by up-regulating suppressor of cytokine signaling 3 (SOCS3) expression.57
Another important function of TLR signaling in the development of Th immune response is the participation in CD4+ cell polarization. Recent studies strongly suggest the importance of the TLR2-mediated signaling for Th17 immune response. Several scientific groups have found that during TCR activation TLR2 signal mediated MyD88-dependent signaling pathway enhances the expression of key Th17 transcription factors activating IL-17 synthesis: retineic-acid-receptor-related orphan nuclear receptor γt (RORγt), RORα, and RORC.55,58–60 Thus, TLR2 signaling promotes TCR-dependent development of Th17 cells. Besides that, Nyirenda et al have discovered that TLR2 activation (Pam3Cys) in Tregs increases the proliferation, but inhibits the suppressive activity of these cells. The mechanism of this regulation was also described. The TLR2 signaling in Treg cells increases the synthesis of IL-6 that plays an important role in the differentiation of Th cells. In turn, IL-6, acting on Tregs in an autocrine manner, inhibits the expression of transcription factor forkhead box P3 (FoxP3) mediating the suppressive properties of these cells. The Jak/STAT5 signaling pathway is also involved in the process. If TGF-β is present in the microenvironment, IL-6 enhances the expression of Th17-specific transcription factor RORC via Jak/STAT3 signaling cascade. The sequential activation of TLR2 and IL-6 signaling pathways in Tregs results in the formation of Th17-like cells producing the pro-inflammatory cytokine IL-17 (Figure 1).60,61 Nevertheless, there is evidence of the activation of Tregs suppressor functions via TLR2 signaling initiated by HSP60, Pam3Cys, and Bacteroides fragilis capsular polysaccharide A (PSA). These TLR2 ligands have been shown to modulate Th1/Th2 balance by decreasing the expression of Th1 transcription factor T-bet and increasing the synthesis of Th2 transcription factor GATA-3 in T cells.57 However, in other studies, the effect of TLR2 ligands (Pam3Cys, Pam3Csk4, FSL-1) on these transcription factors has not been detected.55,58 These differences in results may be associated with the use of varying ligands and research methods. It can be concluded that the TLR2 signaling pathway contributes to the control of Treg/Th17 and Th1/Th2 balance. However, the exact mechanisms of this process require further investigation.
Knobloch et al have described the contribution of TLR4 signaling pathway to the functioning of Th1 cells in COPD. They have found a reduction in the Th1-mediated immune response to Gram-negative bacterial infections in smokers with and without COPD. This down-regulation was caused by a smoking-induced violation of TLR4/MyD88/IRAK (interleukin-1 receptor-associated kinase)/MAPK signaling that led to the decrease in LPS-activated TLR4 expression in circulating Th1 cells. In turn, the lowered expression of TLR4 gene caused the suppression of the synthesis of IFN-γ, a key cytokine for differentiation and functioning of Th1 cells. It has been shown that TLR4/TRIF/IKKε (IkB kinase ε)/TBK1 (TANK-binding kinase 1) signaling is essential for IFN-γ secretion from Th1 cells. It should be noted that short-chain LPS was used as the TLR4 ligand in this study because this LPS form activates TLR4 independent of co-receptor CD14 which is not expressed by Th1 cells. In addition, the bacteria most commonly detected in the airways of COPD patients synthesize various forms of LPS, including the short-chain form. This mechanism may explain the increased susceptibility to bacterial infections in COPD.62 This research team has revealed yet another disturbance of TLR4 signal transmission on CD4+ cells in COPD. It was manifested in switching from MyD88- to TRIF-dependent signaling pathway and led to the suppression of IL-2-dependent proliferation of CD4+ cells and the inhibition of Th1 immune response (Figure 1).63 Thus, TLR4 signaling pathway plays an important role in the functioning of Th1 cells in COPD.
The activation of the above-described mechanisms causes the imbalance of T-helper subpopulations in COPD towards the increase in the number of Th17 cells and the decrease in the number of Th1 cells and Tregs that eventually leads to a worsening of the disease. Thus, fundamental studies in recent years have made major contributions to the understanding of mechanisms through which TLR signaling pathways influence the polarization of Th immune response. However, their role in the progression and the development of COPD exacerbations is not completely understood.
Therapeutic Implications of TLR2 and TLR4 Signaling Pathways in COPD
Given the important role of TLR2 and TLR4 signaling pathways in COPD pathogenesis, these receptors and their adapter molecules are recently considered as promising molecular targets for therapeutic action in COPD. These receptors include both membrane receptors (MD-2, TLR2, TLR4) and the molecules of intracellular cascades (TIR domain, MyD88, IκB kinases, NF-κB, etc.) (Table 3).64–69
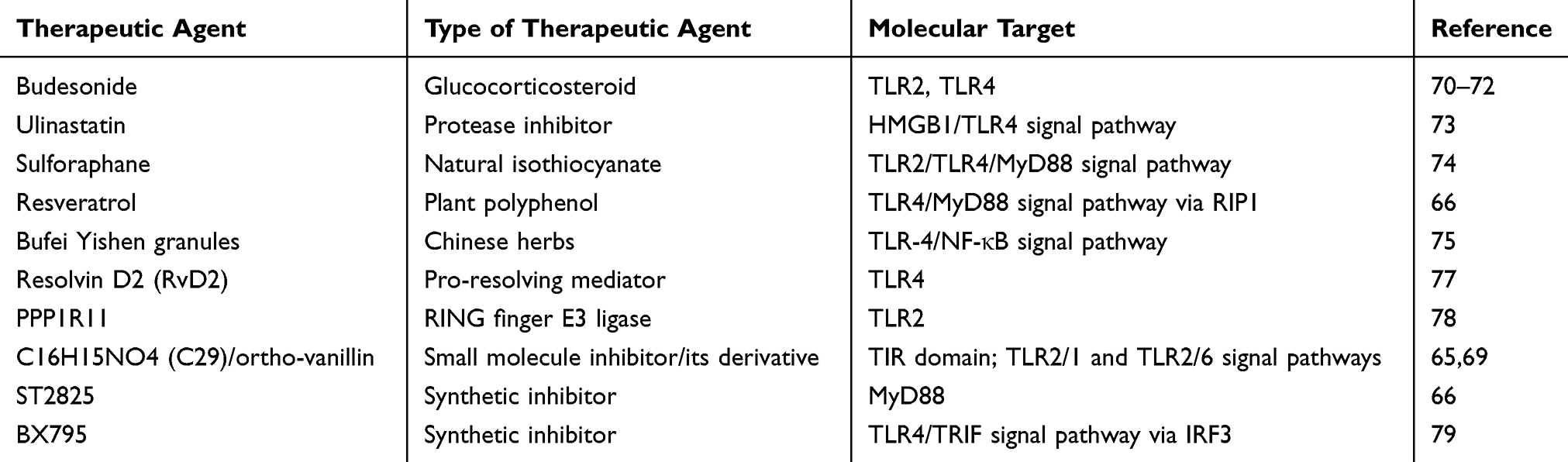 |
Table 3 Proposed Therapeutic Agents for the Treatment of COPD |
According to experimental studies, budesonide, a glucocorticosteroid widely used to treat COPD, may be one of the possible therapeutic agents modulating TLR2 and TLR4 signaling pathways. It has been found that budesonide in combination with pro-inflammatory stimuli (PGN, LPS, TNF, organic dust) synergistically increases TLR2 expression on alveolar macrophages from smokers with COPD and on primary bronchial epithelial cells.70,71 Thus, this glucocorticosteroid has a beneficial effect in acute exacerbations of COPD caused by microorganisms and air pollutants through TLR2-mediated enhancement of the immune response. At the same time, Pace et al have shown that this drug increases the suppressive activity of Tregs due to elevated expression of TLR4 and TLR2 and thereby inhibits the inflammatory process in asthma.72 This effect may also occur in COPD pathogenesis.
According to current evidence, the inhibition of TLR2 and TLR4 activation may be an effective strategy for suppressing an excessive inflammatory response in COPD. In this regard, several candidates for the role of the therapeutic agent in COPD have been investigated.
Ulinastatin, a broad-spectrum protease inhibitor, may be one of the possible TLR inhibitors. Its protective effect on the lungs of COPD rats was shown to be associated with the inhibition of molecules involved in HMGB1/TLR4 signaling. In this way, ulinastatin inhibits the excessive release of inflammatory mediators (TNF-α, IL-6, and IL-8) and reduces lung tissue damage.73
Zeng et al have demonstrated that sulforaphane, an isothiocyanate naturally occurring in cruciferous vegetables, exhibits an anti-inflammatory effect by inhibiting cytokine expression via the TLR2/TLR4/MyD88 signaling pathway in macrophages derived from monocytes from patients with COPD.74 Ma et al have shown in the experimental study that Chinese herbal granules “Bufei Yishen” are effective in reducing inflammation in COPD. The potential mechanism of their action appears to be the suppression of TLR-4/NF-κB signaling.75 There is evidence of an inhibitory effect of plant polyphenols (resveratrol, epigallocatechin gallate, kaempferol-3-O-sophoroside, naringenin, catechin) on TLR4 and TLR2 signal transduction.76 Resveratrol is a particularly promising candidate for the treatment of chronic inflammatory diseases. This drug suppresses the TLR4/MyD88 signaling pathway through exposure to RIP1 which interacts with TRIF and mediates NF-κB activation.66
In accordance with Croasdell et al, resolvin D2 (RvD2) that belongs to specialized pro-resolving mediators (SPMs) and promotes the resolution of inflammation is able to reduce TLR4 expression in human monocytes and weaken LPS-caused inflammation. These data provide a new perspective for using SPMs in target COPD therapy.77
McKelvey et al have described a new mechanism of the regulation of TLR2 expression in murine lung epithelial cells in response to ligand stimulation (Pam3CSK4) via receptor ubiquitination with a RING finger E3 ligase, PPP1R11. Although TLR2 degradation inhibits an excessive inflammatory response, it also impairs bacterial clearance in S. aureus infection.78 These data emphasize the dual role of TLRs in pulmonary inflammation that greatly complicates the development of therapeutic agents that affect these receptors in COPD.
Despite the number of preclinical studies, there are no clinical studies of TLR antagonists for COPD treatment. However, therapeutic agents aimed at suppressing the inflammatory process in various diseases are of interest for future studies within this field. These include: OPN-305 (TLR2 antagonizing IgG4 monoclonal antibody) that is in Phase II clinical trials primarily for oncogenic therapy; NI-0101 (anti-TLR4 antibody) that is in Phase II clinical trials for the treatment of rheumatoid arthritis; VB-201 (small molecule inhibitor of TLR2/4) undergoing Phase II clinical trials regarding psoriasis and ulcerative colitis; JKB-122 (small-molecule inhibitor of TLR4) that is in Phase II clinical trials for the treatment of chronic hepatitis C.79,80 As of today, none of the TLR antagonists investigated in various inflammatory pathologies have been approved for therapeutic use. This is due to the fact that these drugs have a negative effect, weakening the immune response and increasing the risk of infection.
A promising new approach to increasing the effectiveness of TLR inhibitors is the use of delivery vehicles for targeted drug delivery,80 including liposomes (spherical vesicles with one or more lipid bilayers),81 hydrogels (hydrophilic network of polymer chains),82 nanoparticles (particles sized 1–100 nm),83 dendrimers (highly branched macromolecular structures),84 and cyclodextrins (cyclic oligosaccharides with a hydrophilic outer surface and a lipophilic central cavity).85 In addition, the possibility of using prodrugs that are activated after a certain enzymatic or chemical reaction is being considered.86 Preclinical studies of these delivery vehicles are currently underway.
In addition to direct targeting of TLRs, a promising strategy for reducing the inflammatory response in COPD may be to influence the adapter molecules of TLR signaling pathways.
One of these approaches is the inhibition of the TIR domain to suppress the TLR-mediated inflammatory process. Mistry et al have found that a small molecule inhibitor C16H15NO4 (C29) and its derivate (ortho-vanillin) inhibit TLR2/1 and TLR2/6 signaling pathways in HEK-TLR2 and THP-1 human cell lines and TLR2/1 signaling in mouse macrophages. A decrease in TLR2-induced inflammation has been observed in mice treated with ortho-vanillin.65 Cell-permeable decoy peptides (segments of the primary sequence of TIR domain) are also proposed as the inhibitors of TIR domain to suppress the cytokine response elicited by TLR agonists.69
The possibility of using selective MyD88 inhibitors in the therapy of COPD is being discussed.68 The inhibitor of MyD88 dimerization, ST2825, is considered as a potential therapeutic agent in the treatment of chronic inflammatory diseases.66 Despite the fact that MyD88 inhibitors can have a significant impact on COPD progression, it should be noted that the complete loss of MyD88 signal transmission can blunt the immune response and increase the risk of infection.
Southworth et al have demonstrated that the inhibition of signal transducer and activator of transcription 1 (STAT1) represses IFN-γ-mediated TLR2 and TLR4 expression in alveolar macrophages in patients with COPD. The scientific group considers the modulation of these signaling pathways by JAK (Janus kinase) and STAT1 inhibitors as a novel anti-inflammatory strategy for COPD.87 The inhibitor of interferon regulatory factor 3 (IRF3), BX795, an important participant in the TRIF-dependent TLR4 signal cascade, can be used as a therapeutic agent for COPD to suppress acute exacerbation caused by a viral infection.66
The pathogenetic mechanisms of TLR2 and TLR4 signaling pathways in COPD should be studied in more detail to develop new therapeutic agents that will modulate the immune response and lead to persistent remission.
Conclusion
TLR2 and TLR4 play an important role in the immunoregulation of the inflammatory process in COPD. They are involved in the mechanisms of local and systemic inflammation, the development of exacerbation and the regulation of T-helper immune response in this pathology. Both the excessive activation of TLR2 and TLR4 signaling pathways and the suppression of signal transmission from these receptors may contribute to COPD progression. Many factors affect the expression and functioning of these TLRs, including smoking, air pollution, and bacterial load. Further studies are needed to detail the mechanisms of TLR2 and TLR4 participation in COPD pathogenesis. Available data on the role of these TLRs in COPD suggest that they might be promising targets for the treatment of the disease. However, further pharmacological studies of their antagonists are required.
Acknowledgments
The reported study was funded by RFBR, project number 19-315-90029/19.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Barnes PJ. Cellular and molecular mechanisms of asthma and COPD. Clin Sci. 2017;131(13):1541–1558. doi:10.1042/CS20160487
2. Vitkina TI, Denisenko YK, Davydova KA. The changes in the profile of cytokines in progressing chronic obstructive pulmonary disease. Int Res J. 2016;49(7–3):6–8. doi:10.18454/IRJ.2016.49.024
3. De Nardo D. Toll-like receptors: activation, signalling and transcriptional modulation. Cytokine. 2015;74(2):181–189. doi:10.1016/j.cyto.2015.02.025
4. Mortaz E, Adcock IM, Ito K, Kraneveld AD, Nijkamp FP, Folkerts G. Cigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptor. Eur Respir J. 2010;36(5):1143–1154. doi:10.1183/09031936.00062209
5. Koarai A, Yanagisawa S, Sugiura H, et al. Cigarette smoking augments toll-like receptor 3 expression and responses in macrophages. Eur Respir J. 2011;38(Suppl 55):218.
6. Nadigel J, Prefontaine D, Baglole CJ, et al. Cigarette smoke increases TLR4 and TLR9 expression and induces cytokine production from CD8+T cells in chronic obstructive pulmonary disease. Respir Res. 2011;12(1):149. doi:10.1186/1465-9921-12-149
7. Freeman CM, Martinez FJ, Han MK, et al. Lung CD8+ T cells in COPD have increased expression of bacterial TLRs. Respir Res. 2013;14(1):13. doi:10.1186/1465-9921-14-13
8. Pomerenke A, Lea SR, Herrick S, Lindsay MA, Singh D. Characterization of TLR-induced inflammatory responses in COPD and control lung tissue explants. Int J Chron Obstruct Pulmon Dis. 2016;11:2409–2417. doi:10.2147/COPD.S105156
9. Gimenes-Junior J, Owuar N, Vari HR, et al. FOXO3a regulates rhinovirus-induced innate immune responses in airway epithelial cells. Sci Rep. 2019;9:18180. doi:10.1038/s41598-019-54567-3
10. Baines KJ, Simpson JL, Gibson PG, Hartl D. Innate immune responses are increased in chronic obstructive pulmonary disease. PLoS One. 2011;6(3):e18426. doi:10.1371/journal.pone.0018426
11. Zuo L, Lucas K, Fortuna CA, Chuang -C-C, Best TM. Molecular regulation of toll-like receptors in asthma and COPD. Front Physiol. 2015;6:312. doi:10.3389/fphys.2015.00312
12. Hansbro PM, Haw TJ, Starkey MR, Miyake K. Toll-like receptors in COPD. Eur Respir J. 2017;49(5):1700739. doi:10.1183/13993003.00739-2017
13. McGrath JJC, Stampfli MR. The immune system as a victim and aggressor in chronic obstructive pulmonary disease. J Thorac Dis. 2018;10(Suppl 17):S2011–S2017. doi:10.21037/jtd.2018.05.63
14. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026. doi:10.1016/S0140-6736(11)60988-4
15. Bauer RN, Diaz-Sanchez D, Jaspers I. Effects of air pollutants on innate immunity: the role of toll-like receptors and nucleotide-binding oligomerization domain–like receptors. J Allergy Clin Immunol. 2012;129(1):14–24. doi:10.1016/j.jaci.2011.11.004
16. Bezemer GFG, Sagar S, van Bergenhenegouwen J, et al. Dual role of toll-like receptors in asthma and chronic obstructive pulmonary disease. Pharmacol Rev. 2012;64(2):337–358. doi:10.1124/pr.111.004622
17. Olloquequi J, Silva OR. Biomass smoke as a risk factor for chronic obstructive pulmonary disease: effects on innate immunity. Innate Immun. 2016;22(5):373–381. doi:10.1177/1753425916650272
18. Silva R, Oyarzún M, Olloquequi J. Pathogenic mechanisms in chronic obstructive pulmonary disease due to biomass smoke exposure. Arch Bronconeumol (English Ed). 2015;51(6):285–292. doi:10.1016/j.arbr.2015.04.013
19. Pace E, Ferraro M, Siena L, et al. Cigarette smoke increases toll‐like receptor 4 and modifies lipopolysaccharide‐mediated responses in airway epithelial cells. Immunology. 2008;124(3):401–411. doi:10.1111/j.1365-2567.2007.02788.x
20. An CH, Wang XM, Lam HC, et al. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol. 2012;303(9):L748–L757. doi:10.1152/ajplung.00102.2012
21. Haw TJ, Starkey MR, Pavlidis S, et al. Toll-like receptor 2 and 4 have opposing roles in the pathogenesis of cigarette smoke-induced chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2018;314:L298–L317. doi:10.1152/ajplung.00154.2017
22. Lee SW, Kim DR, Kim TJ, et al. The association of downregulated toll-like receptor 4 expression with airflow limitation and emphysema in smokers. Respir Res. 2012;13:106. doi:10.1186/1465-9921-13-106
23. Hulina-Tomaskovic A, Heijin IH, Jonker M, Somborac-Bacura A, Rajkovic MG, Rumora L. Pro-inflammatory effects of extracellular Hsp70 and cigarette smoke in primary airway epithelial cells from COPD patients. Biochimie. 2019;156:47–58. doi:10.1016/j.biochi.2018.09.010
24. Sussan TE, Ingole V, Kim JH, et al. Source of biomass cooking fuel determines pulmonary response to household air pollution. Am J Respir Cell Mol Biol. 2014;50(3):538–548. doi:10.1165/rcmb.2013-0201OC
25. Becker S, Mundandhara S, Devlin RB, Madden M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol Appl Pharmacol. 2005;207(2):269–275. doi:10.1016/j.taap.2005.01.023
26. Shoenfelt J, Mitkus RJ, Zeisler R, et al. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J Leukoc Biol. 2009;86(2):303–312. doi:10.1189/jlb.1008587
27. Budulac SE, Boezen HM, Hiemstra PS, et al. Toll-like receptor (TLR2 and TLR4) polymorphisms and chronic obstructive pulmonary disease. PLoS One. 2012;7(8):e43124. doi:10.1371/journal.pone.0043124
28. Apostolou A, Kerenidi T, Michopoulos A, et al. Association between TLR2/TLR4 gene polymorphisms and COPD phenotype in a Greek cohort. Herz. 2017;42:752–757. doi:10.1007/s00059-016-4510-9
29. Li Z, Mao X, Liu Q, et al. Functional variations of the TLR4 gene in association with chronic obstructive pulmonary disease and pulmonary tuberculosis. BMC Pulm Med. 2019;19(1):184. doi:10.1186/s12890-019-0939-y
30. D’Anna SE, Balbi B, Cappello F, Carone M, Di Stefano A. Bacterial–viral load and the immune response in stable and exacerbated COPD: significance and therapeutic prospects. Int J Chron Obstruct Pulmon Dis. 2016;11:445–453. doi:10.2147/COPD.S93398
31. Pace E, Ferraro M, Giarratano A, et al. TLR4 up-regulation and reduced Foxp3 expression in mechanically ventilated smokers with obstructive chronic bronchitis. COPD: J Chron Obstruct Pulmon Dis. 2013;10(2):147–155. doi:10.3109/15412555.2012.730565
32. Di Stefano A, Ricciardolo FLM, Caramori G, et al. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur Respir J. 2017;49(5):1602006. doi:10.1183/13993003.02006-2016
33. Amatngalim GD, Schrumpf JA, Henic A, et al. Antibacterial defense of human airway epithelial cells from chronic obstructive pulmonary disease patients induced by acute exposure to nontypeable Haemophilus influenzae: modulation by cigarette smoke. J Innate Immun. 2017;9:359–374. doi:10.1159/000455193
34. Chen Y, Kumar RK, Thomas PS, Herbert C. Th1/17-biased inflammatory environment associated with COPD alters the response of airway epithelial cells to viral and bacterial stimuli. Mediators Inflamm. 2019;2019:1–12. doi:10.1155/2019/7281462
35. Berenson CS, Kruzel RL, Eberhardt E, et al. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax. 2014;69(9):811–818. doi:10.1136/thoraxjnl-2013-203669
36. Lea SR, Reynolds SL, Kaur M, et al. The effects of repeated toll-like receptors 2 and 4 stimulation in COPD alveolar macrophages. Int J Chron Obstruct Pulmon Dis. 2018;13:771–780. doi:10.2147/COPD.S97071
37. Knobloch J, Hag H, Jungck D, Urban K, Koch A. Resveratrol impairs the release of steroid resistant cytokines from bacterial endotoxin exposed alveolar macrophages in chronic obstructive pulmonary disease. Basic Clin Pharmacol Toxicol. 2011;109(2):138–143. doi:10.1111/j.1742-7843.2011.00707.x
38. Vitkina TI, Sidletskaya KA, Denisenko YK. Сytokine response and CD14+CD282+ cells in non-smokers with chronic obstructive pulmonary disease. Respirology. 2019;24(Suppl2):110. doi:10.1111/resp.13700_39
39. Fan VS, Gharib SA, Martin TR, Wurfel MM. COPD disease severity and innate immune response to pathogen-associated molecular patterns. Int J Chron Obstruct Pulmon Dis. 2016;11:467–477. doi:10.2147/COPD.S94410
40. Allard B, Panariti A, Pernet E, et al. Tolerogenic signaling of alveolar macrophages induces lung adaptation to oxidative injury. J Allergy Clin Immunol. 2019;144(4):945–961. doi:10.1016/j.jaci.2019.07.015
41. Metcalfe HJ, Lea S, Hughes D, Khalaf R, Abbott‐Banner K, Singh D. Effects of cigarette smoke on Toll‐like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin Expl Immunol. 2014;176(3):461–472. doi:10.1111/cei.12289
42. Knobloch J, Panek S, Yanik SD, et al. The monocyte-dependent immune response to bacteria is suppressed in smoking-induced COPD. J Mol Med. 2019;97(6):817–828. doi:10.1007/s00109-019-01778-w
43. Pons J, Sauleda J, Regueiro V, et al. Expression of toll-like receptor 2 is up-regulated in monocytes from patients with chronic obstructive pulmonary disease. Respir Res. 2006;7(1):64. doi:10.1186/1465-9921-7-64
44. Tripathi PM, Kant S, Yadav RS, et al. Expression of toll-like receptor 2 and 4 in peripheral blood neutrophil cells from patients with chronic obstructive pulmonary disease. Oman Med J. 2017;32(6):477–485. doi:10.5001/omj.2017.92
45. von Scheele I, Larsson K, Dahlen B, et al. Toll-like receptor expression in smokers with and without COPD. Respir Med. 2011;105(8):1222–1230. doi:10.1016/j.rmed.2011.02.012
46. Ulutas GS, Alpaydın AO, Taneli F, et al. Toll-like receptor 2 expression and peripheral blood CD4+/CD8+ T cell ratio in COPD. Turk Thorac J. 2014;15(3):106–111. doi:10.5152/ttd.2014.3842
47. Cosmi L, Liotta F, Annunziato F. Th17 regulating lower airway disease. Curr Opin Allergy Clin Immunol. 2016;16(1):1–6. doi:10.1097/ACI.0000000000000227
48. Kalinina EP, Vitkina TI, Knyshov VV, Fedoseyev EA, Novgorodtseva TP, Gvozdenko TA. Clinical and immunological comparisons in Th-dependent immune response mechanisms among patients with chronic obstructive pulmonary disease. Med Immunol. 2018;20(6):855–864. doi:10.15789/1563-0625-2018-6-855-864
49. Wang H, Ying H, Wang S, et al. Imbalance of peripheral blood Th17 and treg responses in patients with chronic obstructive pulmonary disease. Clin Respir J. 2015;9:330–341. doi:10.1111/crj.12147
50. Li H, Liu Q, Jiang Y, Zhang Y, Xiao W, Zhang Y. Disruption of th17/treg balance in the sputum of patients with chronic obstructive pulmonary disease. Am J Med Sci. 2015;349(5):392–397. doi:10.1097/MAJ.0000000000000447
51. Ito JT, Cervilha DAB, Lourenço JD, et al. Th17/treg imbalance in COPD progression: a temporal analysis using a CS-induced model. PLoS One. 2019;14(1):e0209351. doi:10.1371/journal.pone.0209351
52. Jin B, Sun T, Yu XH, Yang YX, Yeo AET. The effects of TLR activation on T-cell development and differentiation. Clin Dev Immunol. 2012;2012. doi:10.1155/2012/836485
53. Kabelitz D. Expression and function of toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19(1):39–45. doi:10.1016/j.coi.2006.11.007
54. Rahman AH, Taylor DK, Turka LA. The contribution of direct TLR signaling to T cell responses. Immunol Res. 2009;45:25–36. doi:10.1007/s12026-009-8113-x
55. Reynolds JM, Pappu BP, Peng J, et al. Toll-like receptor 2 signaling in CD4+ T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32(5):692–702. doi:10.1016/j.immuni.2010.04.010
56. Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci. 2012;109(32):13064–13069. doi:10.1073/pnas.1120585109
57. Cohen IR, Zanin-Zhorov A. Signaling via TLR2 and TLR4 directly down-regulates T cell effector functions: the regulatory face of danger signals. Front Immunol. 2013;4:211. doi:10.3389/fimmu.2013.00211
58. Zhao RR, Yang XF, Dong J, et al. Toll-like receptor 2 promotes T helper 17 cells response in hepatitis B virus infection. Int J Clin Exp Med. 2015;8(5):7315–7323.
59. Xu C, Lu Y, Zheng X, et al. TLR2 expression in peripheral CD4+ T cells promotes Th17 response and is associated with disease aggravation of hepatitis B virus-related acute-on-chronic liver failure. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.01609
60. Nyirenda MH, Sanvito L, Darlington PJ, et al. TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J Immunol. 2011;187(5):2278–2290. doi:10.4049/jimmunol.1003715
61. Nyirenda MH, Morandi E, Vinkemeier U, et al. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: a novel mechanism of reduced regulatory T cell function in multiple sclerosis. J Immunol. 2015;194(12):5761–5774. doi:10.4049/jimmunol.1400472
62. Knobloch J, Schild K, Jungck D, et al. The T-helper cell type 1 immune response to gram-negative bacterial infections is impaired in COPD. Am J Respir Crit Care Med. 2011;183(2):204–214. doi:10.1164/rccm.201002-0199OC
63. Knobloch J, Chikosi SJ, Yanik S, Rupp J, Jungck D, Koch A. A systemic defect in toll-like receptor 4 signaling increases lipopolysaccharide-induced suppression of IL-2-dependent T-cell proliferation in COPD. Am J Physiol Lung Cell Mol Physiol. 2015;310:L24–L39. doi:10.1152/ajplung.00367.2014
64. Lucas K, Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol. 2013;48(1):190–204. doi:10.1007/s12035-013-8425-7
65. Mistry P, Laird MHW, Schwarz RS, et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc Natl Acad Sci. 2015;112(7):5455–5460. doi:10.1073/pnas.1422576112
66. Roy A, Srivastava M, Saqib U, et al. Potential therapeutic targets for inflammation in toll-like receptor 4 (TLR4)-mediated signaling pathways. Int Immunopharmacol. 2016;40:79–89. doi:10.1016/j.intimp.2016.08.026
67. Gao W, Xiong Y, Li Q, Yang H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front Physiol. 2017;8:508. doi:10.3389/fphys.2017.00508
68. Di Padova F, Quesniaux VFJ, Ryffel B. MyD88 as a therapeutic target for inflammatory lung diseases. Expert Opin Ther Targets. 2018;22(5):401–408. doi:10.1080/14728222.2018.1464139
69. Toshchakov VY, Javmen A. Targeting the TLR signalosome with TIR domain-derived cell-permeable decoy peptides: the current state and perspectives. Innate Immun. 2020;1:13. doi:10.1177/1753425919844310
70. Ji J, von Scheele I, Billing B, et al. Effects of budesonide on toll-like receptor expression in alveolar macrophages from smokers with and without COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:1035–1043. doi:10.2147/COPD.S102668
71. von Scheele I, Larsson K, Palmberg L. Budesonide enhances toll-like receptor 2 expression in activated bronchial epithelial cells. Inhal Toxicol. 2010;22(6):493–499. doi:10.3109/08958370903521216
72. Pace E, Di Sano C, Ferraro M, et al. Budesonide increases TLR4 and TLR2 expression in treg lymphocytes of allergic asthmatics. Pulm Pharmacol Ther. 2015;32:93–100. doi:10.1016/j.pupt.2015.02.003
73. Liu W, Liu Z, Zhang W, Cai S. Ulinastatin protects the lungs of COPD rats through the HMGB1/TLR4 signaling pathway. Oncol Lett. 2018;16(3):4057–4063. doi:10.3892/ol.2018.9123
74. Zeng X, Med M, Liu X, Bao H, Zhang Y, Tan E. Sulforaphane suppressed LPS and Pam3CSK4 mediated inflammation in COPD through MyD88-dependent toll-like receptors pathway. Chest. 2016;149(4):A351. doi:10.1016/j.chest.2016.02.366
75. Ma J, Tian Y, Li J, et al. Effect of bufei yishen granules combined with electroacupuncture in rats with chronic obstructive pulmonary disease via the regulation of TLR-4/NF-κB signaling. Evid Based Complement Alternat. 2019;2019. doi:10.1155/2019/6708645
76. Azam S, Jakaria M, Kim IS, Kim J, Haque M, Choi DK. Regulation of Toll-Like Receptor (TLR) signaling pathway by polyphenols in the treatment of age-linked neurodegenerative diseases: focus on TLR4 signaling. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.01000
77. Croasdell A, Sime PJ, Phipps RP. Resolvin D2 decreases TLR4 expression to mediate resolution in human monocytes. FASEB J. 2016;30(9):3181–3193. doi:10.1096/fj.201600375R
78. McKelvey AC, Lear TB, Dunn SR, et al. RING finger E3 ligase PPP1R11 regulates TLR2 signaling and innate immunity. Elife. 2016;5. doi:10.7554/eLife.18496.001
79. Anwar MA, Shah M, Kim J, Choi S. Recent clinical trends in toll-like receptor targeting therapeutics. Med Res Rev. 2019;39(3):1053–1090. doi:10.1002/med.21553
80. Javaid N, Yasmeen F, Choi S. Toll-like receptors and relevant emerging therapeutics with reference to delivery methods. Pharmaceutics. 2019;11(9):441. doi:10.3390/pharmaceutics11090441
81. Henriksen-Lacey M, Korsholm KS, Andersen P, Perrie Y, Christensen D. Liposomal vaccine delivery systems. Expert Opin Drug Deliv. 2011;8(4):505–519. doi:10.1517/17425247.2011.558081
82. Li Y, Huang G, Zhang X, et al. Magnetic hydrogels and their potential biomedical applications. Adv Funct Mater. 2013;23:660–672. doi:10.1002/adfm.201201708
83. Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic nanoparticles for vaccines and immunotherapy. Chem Rev. 2015;115(19):11109–11146. doi:10.1021/acs.chemrev.5b00109
84. Kesharwani P, Jain K, Jain NK. Dendrimer as nanocarrier for drug delivery. Prog Polym Sci. 2014;39:268–307. doi:10.1016/j.progpolymsci.2013.07.005
85. Jansook P, Ogawa N, Loftsson T. Cyclodextrins: structure, physicochemical properties and pharmaceutical applications. Int J Pharm. 2018;535(1–2):272–284.
86. Ettmayer P, Amidon GL, Clement B, Testa B. Lessons learned from marketed and investigational prodrugs. J Med Chem. 2004;47:2393–2404. doi:10.1021/jm0303812
87. Southworth T, Metryka A, Lea S, Farrow S, Plumb J, Singh D. IFN-γ synergistically enhances LPS signalling in alveolar macrophages from COPD patients and controls by corticosteroid-resistant STAT1 activation. Br J Pharmacol. 2012;166:2070–2083. doi:10.1111/j.1476-5381.2012.01907.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.