Back to Journals » Journal of Pain Research » Volume 15
The Role of Neuro-Immune Interactions in Chronic Pain: Implications for Clinical Practice
Authors Su PP, Zhang L, He L, Zhao N, Guan Z ![]()
Received 27 April 2022
Accepted for publication 19 July 2022
Published 4 August 2022 Volume 2022:15 Pages 2223—2248
DOI https://doi.org/10.2147/JPR.S246883
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Überall
Po-Yi Paul Su,1 Lingyi Zhang,1,2 Liangliang He,1,3 Na Zhao,1 Zhonghui Guan1
1Department of Anesthesia and Perioperative Care, University of California San Francisco, San Francisco, CA, USA; 2Department of Anesthesiology, the First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, People’s Republic of China; 3Department of Pain Management, Xuanwu Hospital, Capital Medical University, Beijing, People’s Republic of China
Correspondence: Zhonghui Guan, Department of Anesthesia and Perioperative Care, University of California San Francisco, San Francisco, CA, USA, Tel +415.885.7246, Fax +415.885.7575, Email [email protected]
Abstract: Chronic pain remains a public health problem and contributes to the ongoing opioid epidemic. Current pain management therapies still leave many patients with poorly controlled pain, thus new or improved treatments are desperately needed. One major challenge in pain research is the translation of preclinical findings into effective clinical practice. The local neuroimmune interface plays an important role in the initiation and maintenance of chronic pain and is therefore a promising target for novel therapeutic development. Neurons interface with immune and immunocompetent cells in many distinct microenvironments along the nociceptive circuitry. The local neuroimmune interface can modulate the activity and property of the neurons to affect peripheral and central sensitization. In this review, we highlight a specific subset of many neuroimmune interfaces. In the central nervous system, we examine the interface between neurons and microglia, astrocytes, and T lymphocytes. In the periphery, we profile the interface between neurons in the dorsal root ganglion with T lymphocytes, satellite glial cells, and macrophages. To bridge the gap between preclinical research and clinical practice, we review the preclinical studies of each neuroimmune interface, discuss current clinical treatments in pain medicine that may exert its action at the neuroimmune interface, and highlight opportunities for future clinical research efforts.
Keywords: neuroimmune, chronic pain, glial cells, macrophage, T-cells
Introduction
Chronic pain remains a major public health problem. The most recent Center for Disease Control and Prevention (CDC) survey between 2016 and 2019 reveals an estimated 50 million (20.4%) of US adults affected by chronic pain; and up to 60% of emergency department visits are for pain-related complaints.1–3 This staggering number has many implications: chronic pain results in about $61 billion dollars per year in lost productivity, the treatment of chronic pain costs between $560 to $635 billion dollars annually (more than cancer and heart disease combined), and chronic pain perpetuates health disparities by preferentially impacting females, elderly patients, and those from lower socioeconomic backgrounds.1,2,4,5 Despite these ramifications, the current status quo of chronic pain treatment still leaves many patients with poorly managed pain – and poorly controlled pain contributes to the current opioid epidemic.6,7
The bidirectional interface between neurons and immune or immunocompetent cells is an area of interest in the pathogenesis of chronic pain. Immunogenic inflammation, the response of the immune system to injuries or harmful stimuli, evokes nociception by activating and sensitizing nociceptors. Reciprocally, neurogenic inflammation, which is caused by the release of mediators from activation or injury of nociceptors, can activate innate and adaptive immune systems.8 While acutely, the neuroimmune interface may play a protective role in avoiding harmful stimuli and encouraging tissue healing, current research points to the role of sustained, localized neuroinflammation in the pathophysiology of chronic pain conditions.9,10 For example, in chronic migraine, the level of calcitonin gene-related peptide (CGRP) is elevated in the cranial but not peripheral circulation.11 Translation of preclinical research into successful clinical treatments for patients has historically been challenging due to a multitude of possible factors discussed elsewhere.12 Nevertheless, in this review, we highlight some of the preclinical work on central and peripheral neuroimmune interfaces and emphasize current clinical therapies that target each interface. Where applicable, we highlight clinical trials specific to the treatment of chronic pain conditions. While many neuroimmune interfaces exist along the nociceptive circuitry, the scope of this review is limited to profiling a subset of these interfaces.9,13 Within the central nervous system (CNS), we will examine the interactions between neurons with microglia, astrocytes, and T lymphocytes (Figure 1); and within the peripheral nervous system (PNS), we will highlight the interactions between sensory neurons within the dorsal root ganglion (DRG) with satellite glial cells (SGC), peripheral T lymphocytes and macrophages (Figure 2).
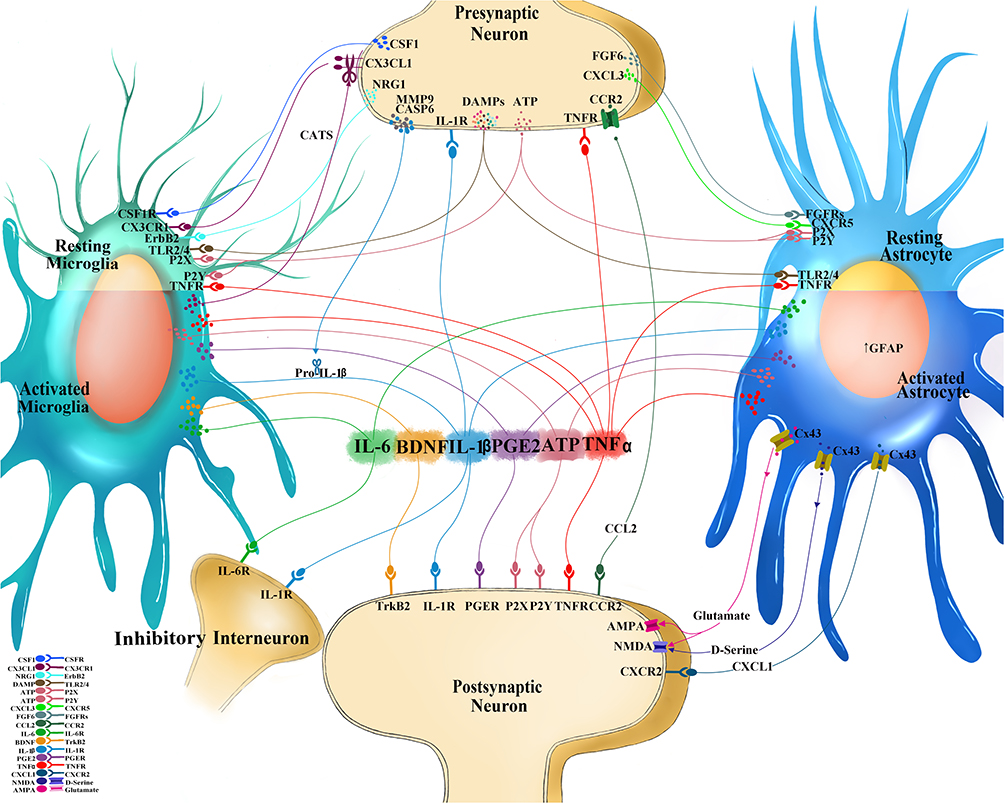 |
Figure 1 Interactions at the central neuroimmune interface. Sensory neurons after injury or high-stimulation activation releases mediators from the presynaptic central terminal in the dorsal horn of the spinal cord to engage cognate receptors on immunocompetent cells in the central nervous system (CNS) such as microglia and astrocytes. Colony-stimulating factor 1 (CSF1) binds to CSF1 receptor (CSF1R), neuregulin 1 (NRG1) binds to tyrosine-protein kinase (ErbB2), CX3-C-chemokine ligand 1(CX3CL1) binds to CX3C receptor 1 (CX3CR1), matrix metalloprotease 9 (MMP9) and caspase 6 (CASP6) both facilitate the cleavage of Pro-interleukin-1 beta (IL-1b) into the mature IL-1b, fibroblast growth factor 6 (FGF6) binds to FGF receptors (FGFRs), CXC chemokine ligand 3 (CXCL3) binds to CXC receptor 5 (CXCR5), and damage associated molecular patterns (DAMPs), which include adenosine triphosphate (ATP; which can bind to purinoceptors (P2X, P2Y)), heat shock proteins (HSP) 70, HSP90, fibronectin, high mobility group box 1 (HMGB1) that all bind to toll-like receptors (TLR) 2 and 4. Unique and shared soluble mediators from neurons, immune, and immunocompetent cells of the spinal cord form feedback and feedforward signaling mechanisms which affect the nociceptive excitability and contribute to pain behavior: IL-1b binds to IL-1 receptor (IL-1R), IL-6 binds to IL-6 receptor (IL-6R), prostaglandin E2 (PGE2) binds to PGE receptor (PGER), tumor necrosis factor alpha (TNFa) binds to TNF receptor (TNFR). Activated microglia release cathepsin S (CatS) which further cleaves CX3CL1. Activate astrocytes are identified by the increased expression of glial fibrillary acidic protein (GFAP). Activated astrocytes increase Connexin 43 (Cx43) hemichannels which allows for extracellular release of CXCL1 (binding to CXCR2 on postsynaptic dorsal horn neuron), C-C chemokine ligand 2 (CCL2; binding to CCR2 on neurons), and glutamate (binding to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartic acid (NMDA) glutamatergic receptors). D-serine, a potent co-agonist of the NMDA receptor is also released from activated astrocytes. |
 |
Figure 2 Interactions at the peripheral neuroimmune interface. Sensory neurons cell bodies and satellite glial cells (SGC) form tight neuron-glial units in the dorsal root ganglion (DRG). Soluble mediators are released from the sensory neuron soma and readily bind to receptors on SGC and also serve as chemoattractant for macrophages and T cells: Glutamate activate N-methyl-D-aspartic acid (NMDA) glutamatergic receptors, substance P (SP) binds to neurokinin 1 receptor (NK1R), C-C motif chemokine ligand 2 (CCL2) binds to C-C motif chemokine receptor 2 (CCR2), calcitonin gene-related peptide (CGRP) binds to CGRP receptors (CGRPR), adenosine triphosphate (ATP) binds to various purinoceptors (P2X, P2Y), damage associated molecular patterns (DAMPs) bind to toll-like receptors (TLR2, TLR4). Injured sensory neurons also release microRNAs (MiR-21 and miR-124 which can have pro-inflammatory and anti-inflammatory effects). Activated SGC, T cells, and macrophages in the DRG respond with shared and unique mediators that in turn bind to receptors on the DRG sensory neurons to affect neuronal excitability. Interleukin-1-beta (IL-1b) binds to IL-1 receptor (IL-1R), IL-6 binds to IL-6 receptor (IL-6R), prostaglandin E2 (PGE2) binds to PGE receptor (PGER), tumor necrosis factor alpha (TNFa) binds to TNF receptor (TNFR). Activated SGC increase Connexin-43 (Cx43) gap junctions between adjacent cells to allow for increased electrical coupling. Activated SGC also increase extracellular channels such as Pannexin-1, which allows for ATP release. Inward rectifying potassium channels (Kir4.1) are downregulated in activated SGC which results in increased extracellular potassium gradient and making neurons hyperexcitable. Activated macrophage produce hydrogen sulfide (H2S) that can activate neuronal transient receptor potential cation channel A1 (TRPA1). Infiltrating T cells depending on the subtype can contribute to pro-excitatory neuronal and is a source of interferon gamma (IFNg) which can bind to IFN receptor (IFNR). Subtypes of macrophage and T cells can contribute to anti-inflammation by releasing IL-4, IL-10, and opioids. |
A Common Repertoire of Proinflammatory Mediators
Immune cells secrete proinflammatory mediators to exert an immune response.14 Except for microglia, which are myeloid cells of the CNS, other glial cells (astrocytes, satellite glial cells) are developmentally different from immune cells but are considered immunocompetent since they too can secrete immune mediators upon activation. In addition to cell-type-specific mediators, activated immune and glial cells release a shared repertoire of proinflammatory mediators that contribute to neuronal plasticity and overall nociceptive hyperexcitability, highlighted among them are tumor necrosis factor alpha (TNFa), interleukin-1-beta (IL-1b), interleukin-6 (IL-6), and prostaglandin E2 (PGE2).
TNFa promotes the excitability of neurons through TNF receptor subtypes 1 and 2 (TNFR1, TNFR2) located on presynaptic and postsynaptic neurons in the spinal cord. Presynaptically, TNFa facilitates glutamate release from presynaptic terminals of primary sensory neurons through a transient receptor potential cation channel subfamily V member 1 (TRPV1)-dependent mechanism and increases the frequency of excitatory postsynaptic current (EPSC). Postsynaptically, TNFa potentiates the function of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartic acid (NMDA) glutamatergic receptors and increases the amplitude of EPSC in spinal dorsal cord (SDH) neurons.17–19 IL-1b overlaps with TNFa to mediate central sensitization of spinal cord neurons through both enhanced excitation and decreased inhibition. IL-1b binds to IL-1 receptor (IL-1R) to cause postsynaptic NMDA facilitation with increased amplitude of EPSC of SDH neurons; and IL-1b also suppresses inhibitory neurotransmission from interneurons.18–22 IL-6 binds to membrane-bound and soluble IL-6 receptors (IL-6R) to mediate its signaling. In the spinal cord, IL-6 can affect spinal cord interneurons to suppress the release of γ-aminobutyric acid (GABA) and glycine. The decrease in frequency and amplitude of inhibitory postsynaptic currents (IPSC) in interneurons results in the overall excitability of the SDH neurons.19 PGE2, generated by the cyclooxygenase-2 (COX-2) enzyme, can act directly on spinal cord neurons by activating non-selective cation channels and via prostanoid receptors to mediate presynaptic neurotransmitter release from presynaptic terminals of primary sensory neurons and depolarize postsynaptic spinal cord neurons.23–25 Nonsteroid anti-inflammatory drugs (NSAIDs) may exert their effects within the neuroimmune interface at the COX-2 control of PGE2 production.26 Each proinflammatory cytokine alone is sufficient to sensitize ascending SDH neurons and each mediator reflects a potential target for therapeutic intervention that will be reviewed in the subsequent clinical section.
Neuroimmune Interfaces Within the Central Nervous System
The Microglia-Neuron Interface
Microglia are canonical immune cells of the CNS.27 In a conserved fashion across species, microglia originate from yolk sac progenitor cells and migrate into the developing brain where they expand to colonize the entire CNS.28–30 The microglia-neuron interface is implicated in the pathogenesis of many brain diseases including chronic pain and is one of the most studied CNS neuroimmune interfaces.16,31–35
Clinical correlation of the microglia-neuronal interface is largely absent despite much work in preclinical models of chronic pain. The inability to identify the microglia-neuron interface clinically is a major limitation, let alone to follow how this interface changes over the course of chronic pain. Postmortem spinal cord section of a patient with complex regional pain syndrome (CRPS) on chronic opioid therapy revealed increased microglia throughout the entire spine.36 Positron emission tomography (PET) scanning using a tracer binding to the translocator protein 18 kDa (TSPO) allows for the visualization of glial activation – however, this is not specific to microglia and includes astrocytes and myeloid cells.37 Nevertheless, positive TSPO accumulation in the nervous system of patients with CRPS, fibromyalgia, and chronic lower back pain point to a link between overall glial activation and chronic pain conditions.38–41 While there are no microglia-specific radioligands, there are radioligands that are more specific for astrocytes (radioligand [11C]-L-deprenyl-D2 targets the astrocyte-specific monoamine oxidase B).42 An interesting PET study examining fibromyalgia patients observed elevated TSPO but not [11C]-L-deprenyl-D2 signals in the frontal and parietal lobes, supporting that microglia, but not astrocytes, contribute to neuroinflammation in these patients.43 The development of more specific PET tracers for microglia (such as targeting other receptors like P2X purinoreceptor 7; P2XR7) and tracers capable of differentiating between microglia phenotypes will help advance our clinical understanding of the dynamics of microglial activation in pain and in other neurologic diseases.37
Neuron to Microglia Interactions
Microglia constantly survey the local microenvironment of the CNS.27 Neurons, particularly the central terminals of DRG neurons, release mediators through injury- and/or stimulation-dependent mechanisms to activate microglia; examples of secreted proinflammatory mediators include damage associated molecular patterns (DAMPs), colony stimulating factor 1 (CSF1), neuregulin 1 (NRG-1), CX3C-chemokine ligand 1 (CX3CL1), matrix metalloprotease 9 (MMP9), caspase 6 (CASP6). Different DAMPs such as adenosine triphosphate (ATP), heat shock proteins (HSP60, HSP90), fibronectin, and high mobility group box 1 (HMGB1) can bind to toll-like receptors 2 and 4 (TLR2, TLR4) expressed on microglia.44–48 Transgenic mice with neuronal knockout of HMGB1, or global knockout of TLR2 or TLR4 receptors all exhibit reduced pain behaviors in inflammatory and neuropathic models of pain. Notably, TLRs are found in immune cells, astrocytes and neurons; thus, the specific contribution of microglial TLRs to global knockout genetic models remains unclear.48–51
ATP binds to numerous purinergic receptors expressed on microglia and the various binding interactions have contributing roles in neuropathic pain.52 In particular, P2X purinoceptor 4 (P2XR4) is exclusively upregulated on spinal cord microglia in response to C-C motif chemokine ligand 21 (CCL21) secreted from injured DRG neurons.53 Blockade or knockout of P2X4R is sufficient to suppress mechanical hypersensitivity in preclinical models of inflammatory and neuropathic pain.33,54
CSF1 receptor (CSF1R) is expressed on microglia. Tonic signaling via CSF1R is critical for the development and maintenance of microglia in the adult nervous system.28,55 Additional release of CSF1 induced by injured DRG sensory neurons leads to local spinal cord microglia activation and proliferation via microglia CSF1R.56–58 Transgenic mice lacking CSF1 in DRG neurons fail to develop pain behavior after nerve injury, while administration of intrathecal CSF1 alone is sufficient to induce pain behavior in uninjured mice.56 CSF1R is also present in peripheral myeloid cells and regulates the differentiation of macrophages. Dysregulation of the CSF1-CSF1R pathway is implicated in certain myeloid cancers.59 Currently, there are small molecules and monoclonal antibodies targeting CSF1R or CSF1 - some already with the United States Food and Drug Administration (FDA) approval (such as pexidartinib, a CSF1R inhibitor) for cancer therapy. Research into whether these medications can provide analgesic benefit in the context of chronic pain will be of great interest.60
CX3CL1 is a neuronal transmembrane protein that can be cleaved to release the soluble chemokine. Cleaved CX3CL1 is a ligand for the CX3CL1 receptor 1 (CX3CR1) found exclusively on microglia in CNS.61 Intrathecal injection of soluble CX3CL1 induces nociceptive behavior in uninjured animals, while global CX3CR1 knockout mice display less severe neuropathic pain behavior in a model-specific manner.62–64 CX3CL1 cleavage from neuronal membrane requires the cysteine protease, cathepsin S (CatS), which is expressed by activated microglia – this highlights the bidirectionality of the neuron-microglia interface.62 KAND567, a small-molecule inhibitor of CX3CR1, is a potential clinical anti-inflammatory agent. Current clinical trials are primarily studying this molecule in the context of myocardial infarction and SARS-CoV-2 (COVID-19) infections, but the manufacturer has advertised future plans to explore this medication for pain treatment.65
MMP9 and CASP6 are neuronal proteases induced under various animal pain models. MMP9 exemplifies an indirect interaction between neurons and microglia. MMP9 is transiently induced in DRG neurons after nerve injury.66 Released MMP9 mediates the cleavage of extracellular pro-interleukin-1b (pro-IL-1b) to the active IL-1b whereby it can act directly on neurons, microglia, astrocytes, and other IL-1R expressing cells.67 The central terminals of injured or activated DRG nociceptors release CASP6 into the spinal cord.68 In vitro culture of microglia with CASP6 elicits a dose-dependent release of microglial TNFa and the pretreatment of rodents with microglial inhibitor minocycline reduces CASP6-evoked pain behavior, but the exact interaction of CASP6 with microglia remains a mystery.68,69
NRG-1 is released from primary afferent terminals after injury. NRG-1 activates microglia through receptor tyrosine-protein kinase ErbB2 to drive the release of IL-1b, microglia proliferation, and pain behavior after injury.70
Microglia to Neuron Interactions
The activation of microglia ultimately results in the secretion of the aforementioned repertoire of proinflammatory cytokines in the spinal cord to mediate central sensitization and pain behavior.15,19,21,71–73 Neurons express tropomyosin receptor kinase B (TrkB), a receptor for brain-derived neurotrophic factor (BDNF), and the binding of BDNF to TrkB on second-order SDH neurons leads to a depolarizing reversal in anion gradient such that SDH neurons become excited upon stimulation by conventionally inhibitory GABA or glycine.74–76 BDNF also disinhibits NMDA receptor (NMDAR) potentiation and facilitates action potential firing in SDH neurons.77 While BDNF signaling via TrkB in SDH neurons is characterized, the source of BDNF is not clear. One potential source of BDNF is from de novo synthesis and release of microglia through activation via ATP-P2X4R signaling, but the detection of BDNF in spinal microglia is inconsistent and thus continues to spark debate.57,78,79 Regardless of the source of BDNF, blocking of TrkB signaling might hold potential therapeutic promise. Two FDA-approved, selective Trk inhibitors are available for the treatment of solid tumors with Trk gene mutations, larotrectinib and entrectinib; however, they have yet to be studied for the treatment of pain.80,81
Microglia can also biosynthesize specialized pro-resolving mediators (SPM) that bind to specific SPM receptors expressed on DRG nociceptors and SDH neurons.82 Direct stimulation of human vagal nerve is sufficient to enhance endogenous SPM production.83 In fact, activation of SPM receptors on nociceptors can block capsaicin-induced EPSC and is a mechanism by which microglia can mediate analgesia at the central microglia-neuron interface.84 Supplementation with marine lipids, which can be metabolized into pro-resolving mediators, may improve clinical quality of life and pain in adults with chronic pain.85,86
The Astrocyte–Neuron Interface
Astrocytes are the most abundant cell type in the CNS and comprise about 40–50% of all glial cells or 20–40% of the total number of CNS cells. Astrocytes tile the entire CNS in a non-overlapping, organized fashion. Each individual astrocyte has innumerous processes that contact multiple neurons and envelop over 100,000 synapses.87 Astrocytes not only maintain the extracellular environment of CNS but also participate in synaptic activity; in fact, the interactions between neurons, microglia, astrocytes, and other glial cells are commonly viewed to form a quad- or even penta-partite synapse.88–90 Networks of astrocytes form a functional syncytium through gap junctions that allows cytoplasmic continuity for exchange of small molecules and electrical coupling.91 Connexin 43 (Cx43) is a specific gap junction protein, which forms the building blocks of these gap junctions. Each adjacent astrocyte contributes a hemichannel to form a full cell–cell channel. Each hemichannel, however, can also be unopposed and open directly to the extracellular space, which has implications for the astrocyte–neuron interface.
Given the abundance of astrocytes in the CNS, it is not surprising that astrocytes play important roles in preclinical models of pain.9,10,92 Activated astrocytes exhibit increased expression of their prototypical glial fibrillary acidic protein (GFAP); and the extent of astrocyte activation correlates with observed pain behavior.93–98 Inhibition of astrocytes using toxins attenuates pain hypersensitivity after injury, while intrathecal injection of TNFa-activated astrocytes or glial-restricted precursor-derived astrocytes in uninjured animals promotes mechanical hypersensitivity.99–102 Elegantly, optogenetic stimulation of spinal astrocytes is sufficient to produce pain behavior in naïve, uninjured rats.103
Like microglia, clinical correlates of the astrocyte–neuron interface in chronic pain are not well established. In a postmortem analysis of matched human patients (1. Without HIV, 2. With HIV, but without chronic pain and 3. With HIV and with chronic pain), HIV-positive patients with chronic pain exhibited greater astrocyte activation in the SDH compared to HIV-positive patients without chronic pain. The expression levels of microglia markers were comparable between the three groups.104 Advances in molecular imaging technology have heralded potential visualization strategies for astrocytes but have been studied primarily in other neurologic diseases and not necessarily for chronic pain.105 The PET tracer targeting TSPO, as discussed previously, cannot distinguish between microglia and astrocytic activity, but the tracer ligand [11C]-L-deprenyl-D2 is generally accepted for the detection of astrocytes due to the selective expression of its monoamine oxidase B target.42,106–108
Neuron to Astrocyte Interactions
Different cell types likely interact with astrocytes and influence the transition to reactive astrogliosis, so it is difficult to distinguish the direct or indirect actions between the various CNS cell types and astrocytes. For example, TNFa signaling through TNFR is a major axis for astrocyte activation, and intrathecal injection of TNFa-activated astrocytes into the spinal cord is sufficient to induce pain behavior in the absence of any injury.109 However, the source of TNFa in vivo is indistinguishable from astrocytes or microglia, though less likely from neurons.68,100,109
Neural activity is sufficient and required to induce astrocyte activation. Electrical stimulation of nerve fibers leads to increased intracellular calcium and GFAP levels in astrocytes, while pretreatment with local anesthetics prevented GFAP increase after inflammatory pain model.110–112 Neural activation also releases neurotransmitters and other mediators that may interface with astrocytes. For example, glutamate is the predominant excitatory amino acid released by nociceptors, and glutamate-evoked membrane currents in astrocytes result in astrocyte activation through p38 mitogen-activated protein kinase (p38 MAPK) signaling.113,114 ATP from synaptic terminals or as a result of nerve injury can also interface with astrocytes through many surface-expressed ionotropic and metabotropic purinergic receptor subtypes.115 Intrathecal injection of ATP is sufficient to produce astrocyte activation and pain behavior even when microglia activation is inhibited by minocycline.116 Similar to microglia, astrocytes also express DAMP-sensing TLR4 receptors and may contribute to inflammatory and neuropathic pain.50,117
An inducible C-X-C motif chemokine ligand 13 (CXCL13), otherwise not expressed in the healthy CNS, is upregulated in the ipsilateral SDH neurons after spinal nerve ligation model of neuropathic pain.118 Interestingly, the expression of C-X-C chemokine receptor 5 (CXCR5), the sole receptor for CXCL13, is also significantly increased in spinal cord astrocytes after nerve injury. Inhibition or genetic deletion of CXCR5 results in less astrocyte GFAP signal after nerve injury and less mechanical hypersensitivity, while intrathecal injection of CXCL13 is sufficient to induce pain hypersensitivity and activation of astrocytes.118 The CXCL13-CXCR5 axis also contributes to certain autoimmune diseases and cancers, and neutralizing antibodies to human CXCL13 are currently under development.119
It has long been recognized that the basic fibroblast growth factor (bFGF) induces mitosis, growth, differentiation and activation of astrocytes by binding to its cognate FGF receptors (FGFR1-4).120,121 bFGF is expressed by astrocytes and DRG sensory neurons; and the level of expression further increases after nerve injury. Neutralizing bFGF reduces astrocyte activation and pain behaviors after nerve injury.122–124 Drugs capable of trapping bFGF are being actively explored for treating endometrial carcinoma.125
Astrocyte to Neuron Interactions
Activation of astrocytes occurs through downstream activation of kinase and protease pathways including c-Jun N-terminal Kinase (JNK) and Extracellular-signal-Regulated Kinase (ERK).126 Astrocyte activation leads to the expression of receptors and mediators to allow astrocytes to interface with neurons and other cells of the nervous system. In immunocompetent cells, activated astrocytes synthesize and release a shared repertoire of proinflammatory mediators (TNFa, IL1b, and IL6, PGE2) and alter the neuronal activity in widespread models of pain as previously described.98,127–129 Astrocytes also upregulate matrix metalloprotease 2 (MMP2) to cleave pro-IL-1b into the active form, although this induction is delayed and contributes more to the maintenance of IL-1b levels.67 Astrocytes also release unique mediators capable of altering neuronal activity. For example, D-serine is synthesized by activated astrocytes and is a potent co-agonist at NMDAR to facilitate long-term potentiation; intrathecal injection of D-serine is sufficient to induce pain behavior in uninjured animals.130,131 In this section, we will further highlight some astrocyte-mediated neuronal interactions.
Extracellular facing Cx43 hemichannels are present on astrocytes and the opening of these hemichannels allows for the astrocytic release of ATP and glutamate, as well as the chemokines CXCL1 and CCL2.132,133 The level and activity of Cx43 hemichannels on astrocytes increase following nerve injury or activation by TNFa.132 Intracellular CXCL1 and CCL2 expressions also increase following astrocyte activation and provide a larger pool of chemokines primed for release.132,134 CXCL1 binds its cognate receptor CXCR2 expressed on SDH neurons, and CXCL1-CXCR2 signaling induces increased spontaneous EPSC frequencies. CXCR2 antagonists suppress spontaneous EPSC and pain behavior after nerve injury in animal models.132 Danirixin, a selective CXCR2 inhibitor, has already been studied clinically for chronic obstructive pulmonary disease (COPD) and could be of interest for investigation in chronic pain.135 Similarly, drugs and gene therapies against Cx43 also exist – in particular, one company has been testing a combination of FDA-approved amitriptyline and mefloquine, a potential Cx43 channel blocker, for neuropathic pain.136,137 The Cx43 channel blocker, tonabersat, is utilized to target cortical spreading of depression observed in acute migraines.137,138 On the other hand, CCL2 signals through the C-C motif chemokine receptor 2 (CCR2) expressed on DRG and spinal cord neurons whose levels increase after nerve injury.139,140 Binding of CCL2 to CCR2 results in overall neuronal hyperexcitability via increased presynaptic glutamate release, enhancement of AMPA- and NMDA-associated glutamatergic synaptic transmission, and suppression of inhibitory synaptic transmission.134,139,141 CCL2 neutralizing antibodies and antagonists or genetic knockout of CCR2 result in reduced pain behaviors after nerve injury and in HIV-associated neuropathy.134,142–145 Of note, the CCL2-CCR2 signaling also exists between peripheral nerves, DRG neurons, and macrophages.145
Astrocytes contribute to extracellular glutamate homeostasis in the synaptic microenvironment by regulating the clearance of glutamate via glutamate transporter 1 (GLT1).146 Nerve injury induces an initial increase then persistent decrease levels of GLT1. The decrease in GLT1 results in increased spinal extracellular glutamate and elicits mechanical and thermal hypersensitivity.147–149 Inhibition of GLT1 is sufficient to induce spontaneous pain behavior without injury, while gene therapy introduction of GLT1 into the spinal cord attenuates inflammatory and neuropathic pain behaviors.150,151 Riluzole, an FDA-approved medication for the treatment of amyotrophic lateral sclerosis, has positive regulatory activities against GLT1 and is capable of attenuating neuropathic pain in preclinical models of nerve injury.147,152 Unfortunately, riluzole was unsuccessful in two separate randomized, cross-over, placebo-controlled trials of patients with peripheral neuropathy in pain or neuropathy outcomes.153
The T Lymphocyte–Neuron Interface
The adaptive immune system consists of B and T lymphocytes. A unique feature of the adaptive immune system is the expression of highly variable antigen recognition receptors generated from random genetic rearrangement during the lymphocyte maturation process. The highly variable repertoire of antigen receptors allows for recognition of specific antigen epitopes presented by antigen presenting cells. T cells can further be distinguished into subsets based on their functions. CD8+ T cells function as cytotoxic T cells, which can kill virus-infected and tumor cells, while CD4+ T cells, which help optimize the immune response, can be further subdivided into additional niches: T-helper cells (Th1 and Th17) produce proinflammatory cytokines, Th2 and regulatory T cells (Treg) produce anti-inflammatory cytokines and suppresses the activity of other immune cells. The dynamic functionality within the family of T cells may explain the conflicting observations regarding the role of T cells across different models of pain. In neuropathic pain models, T cells are mostly detrimental, and depletion or neutralization of T cells reduces pain behavior.154–158 However, in models of chemotherapy-induced peripheral neuropathy (CIPN) and inflammatory pain, depletion of T cells either has no effect or actually worsened pain behavior, suggesting a potential protective or analgesic role.159–163 In clinical studies, while the absolute number of circulating T cells between patients with and without neuropathic pain remains comparable, some studies report an imbalance between the subsets of CD4+ T cells in chronic pain patients.164–166 Unfortunately, the relevance and meaning of this finding in peripheral circulating T cells to the maintenance or development of chronic pain remains unclear.
At baseline, T cells are seldomly found in the peripheral nerve, DRG, or spinal cord. The role of the T cell–neuron interface at the spinal cord level in the pathogenesis of chronic pain is unclear. Lymphocyte attractants are induced in the spinal cord after nerve injury, but there are conflicting reports as to whether T cells infiltrate into the spinal cord after peripheral nerve injury.167 Some studies reported detectable CD4+ T cells, although the number remains low, while other studies did not find evidence to support T cell infiltration after nerve injury.168,169 Additional confounders include localization of T cells in meningeal structures, including leptomeninges and perivascular spaces; while these are technically extra-parenchymal to the spinal cord, these T cells may still functionally participate in the neuroimmune interface as these areas are bathed by the cerebral spinal fluid.170 The development of pain hypersensitivity does not correlate with T cell infiltration into the spinal cord since maximal pain behavior is observed before any detectable infiltration of T cells.154,167,171,172 However, CD4+ T cells infiltrate the peripheral nerve and DRG late (7 days after injury) and persist (lasting beyond 40 days) after nerve injury and may contribute to maintenance or transition from acute to chronic pain.155,157,169
T cells contribute to sexual dimorphism observed in certain preclinical models of chronic pain. First published by Sorge et al, pharmacologic inhibition and depletion of microglia abolished pain behavior in only male rodents but had no effects in females, which still exhibited pain hypersensitivity after nerve injury; this effect was dependent on testosterone. However, depletion of T cells renders female mice sensitive to glial inhibitors comparable to their male counterparts and this observation is reversed with adoptive transfer of T cells into T cell-deficient females.158 Meningeal Tregs in females are capable of interacting with spinal cord microglia to suppress the microglia-dependent pathway for pain hypersensitivity.173 Sexual dimorphism in pain is a fascinating area of research, and current preclinical studies span beyond T cell and microglia function and implicate other neuroimmune players, such as myeloid cells and even neurons themselves.51,174
Neuron and T Lymphocyte Interactions
T cells respond to neuronal outputs as they express receptors including ionotropic and metabotropic glutamate receptors, CGRP receptors, and neurokinin-1 (NK1) receptors.175–177 Depending on the specific subtype of T cells (Th1, Th17), they can release IL-1b, interference gamma (IFNg), TNFa, and IL-17 that can increase neuronal excitability and promote neuroinflammation.178,179 Infiltrating T cells in the DRG release leukocyte elastase (LE), a serine protease, which activates MMP9 to facilitate the cleavage of pro-IL-1b into the active IL-1b.180 IFNg can directly bind AMPA receptors to increase postsynaptic calcium influx.181–183 T cells can also serve as a source of BDNF – although studies of T cell-derived BDNF have been in the setting of neurodevelopment, whether this source of BDNF can mediate anion reverse potential in the setting of neuropathic pain in adults still needs to be addressed.184 Conversely, other subsets of T cells such as Treg and Th2 cells reduce or resolve pain likely through secretion of anti-inflammatory cytokines (IL-4, IL-10, transforming growth factor beta (TGFb)), which most likely interact with glial and immune components. T cells can also produce endogenous opioids (enkephalins and b-endorphins) that act directly on neuronal opioid receptors to provide analgesia.159,185 T-cell-derived opioids also exert anti-inflammatory effects by reducing proinflammatory CD4+ subtypes (Th1, Th17) and increasing anti-inflammatory CD4+ subtypes (Th2).186,187 Modulating specific subsets of T cells not only have the potential to interrupt but also possibly reverse neuroinflammation.
Neuroimmune Interfaces Within the Peripheral Nervous System
The Satellite Glial Cell–Neuron Interface
Satellite glial cells (SGC) are derivatives of pluripotent neural crest cells and comprise the glial cells of the peripheral nervous system (along with Schwann cells).188 Multiple SGC, connected by adhesive and gap junctions, wrap around the surface of typically one sensory neuron cell body and its proximal axon to form a functionally distinct neuron-glial unit. Each functional neuron-SGC unit is separated by connective tissues, but in 4–9% of DRG neurons, SGC share glial envelopments and connections with one or two other neurons. Distinct neuron-glial functional units are important to mention because nerve injury induces the formation of new intra- and inter-ganglion gap junctions that contribute to pain behavior.189–191 SGCs are often compared to astrocytes of the CNS as they share molecular and functional similarities – expression of GFAP, maintenance of microenvironmental glutamate and potassium via glutamate aspartate transporter and inward rectifying potassium channels (Kir4.1), respectively, and electrical coupling between SGCs through adjoining gap junctions.190 Although several different preclinical models of pain implicate SGC in the initiation and maintenance of pain, the specific role of SGC in humans is not well characterized.192–196
Neuron to SGC Interactions
The soma of sensory neurons in DRG releases excitatory mediators like glutamate, substance P, CGRP, CCL2, and ATP upon depolarization or injury.197–201 Extracellular glutamate is carefully regulated by glutamate transporters expressed by SGC;202 glutamate can also activate SGC by binding NMDAR expressed by SGC.203 Substance P binds to NK1 on SGC and leads to downstream release of IL-1b.204,205 CGRP released from the soma of sensory neurons binds to its cognate receptor on SGC, receptor activity modifying protein 1 (RAMP1), and mediates nitric oxide release to, in turn, sensitize neurons.198 CGRP receptor-blocking antibodies (ie, erenumab) represent the successful stories in targeted drug developments for the treatment of chronic migraines.206 CCL2 induced in DRG neurons upon stimulation or injury can activate SGC through CCR2 receptors.140,200
ATP plays a critical bidirectional role in neuron-SGC signaling because purinergic receptors are expressed on both neurons and SGC. Injury or stimulation elicits robust release of neuron-derived ATP, and the activation of P2Y metabotropic receptors on SGC leads to alteration of the extracellular potassium gradient through potassium channels and ultimately increases downstream neuronal excitability.201,207 Activation of SGC P2X purinoceptors (in particular, P2X7) can also lead to SGC TNFa release.201 Additionally, P2X7 can inhibit neuron excitability via tonic inhibition of P2X3R expression on neurons (P2X3R promotes neuron excitability).201,208,209 Despite the divergent effects of purinergic signaling in preclinical studies, there is a paucity of knowledge on the effects of purinergic signaling of SGCs in humans.
SGC to Neuron Interactions
SGC and neurons interact through secreted mediators, and given their proximity, through rapid ion channels and electrophysiologic signaling. SGCs are immunocompetent cells and release the common repertoire of proinflammatory mediators upon activation.195,210–212 These mediators act not only on neurons but also on neighboring SGCs facilitating paracrine and autocrine positive feedback.213–215 SGCs represent a source of ATP through vesicular- or channel-mediated release. For example, activation of SGC P2X7 channel releases ATP from SGC, which in turn acts on purinergic receptors to exert inhibitory control of P2X3R on neurons.208 SGCs upregulate ATP-permeable pannexin channels after injury, and mice with glial-specific knockout of pannexin channels do not develop pain after injury.216–219
The local extracellular potassium concentration is regulated by Kir4.1 on SGC.190 After injury, Kir4.1 expression on SGC decreases, which increases the extracellular potassium rendering neurons more excitable. Accordingly, silencing Kir4.1 in rat trigeminal ganglion alone is sufficient to induce spontaneous facial pain behavior.220 In naïve animals, SGC couples with adjacent SGC to surround a single DRG neuron via gap junctions (prominently via Cx43, similar to astrocytes). After injury, there is enhanced cell–cell coupling through gap junctions expanding beyond SGCs and neurons of a neuron-glia functional unit to include coupling between adjacent units, to allow for vast propagation of calcium waves.221–227 Blocking gap junctions prevents the cell–cell coupling and reduces the severity of pain behavior in nerve injury, CIPN, and inflammatory pain models.192,221,225 Blockers of gap junctions such as those specific to Cx43 are clinically evaluated and have been discussed previously.
The Macrophage-Sensory Neuron Interface
Tissue-resident and infiltrating macrophages are two types of macrophages found in healthy or inflamed tissues.228 Tissue-resident macrophages integrate into various tissue subtypes during development and co-exist in a homeostatic state in adulthood. Infiltrating macrophages derive from monocytes that originate from the hematopoietic stem cells of the bone marrow and migrate into tissues in response to injury or infection. Infiltrating macrophages can be found in peripheral tissue at the site of injury in inflammatory pain or in the peripheral nerves and DRG in case of neuropathic pain (ie, nerve injury, CIPN, diabetic neuropathy). Both subtypes of macrophages contribute to the regulation of pain.229,230 Macrophages derived from either tissue-resident or infiltrating macrophages can be polarized into pro-inflammatory (M1) or anti-inflammatory (M2) mature macrophages depending on the local milieu.231
The best clinical evidence supporting the role of macrophage in chronic pain is in osteoarthritis and rheumatoid arthritis.232–234 Rheumatoid arthritis patients exhibit increased macrophage numbers in the inflamed synovial membrane, and the radiologic progression of joint destruction correlates with the extent of macrophage infiltration. In fact, macrophage levels serve as a direct biomarker for the responsiveness to disease-modifying antirheumatic drug therapy.234,235 Similarly, synovial macrophages play a role in the onset and progression of osteoarthritis, with a direct correlation between synovial macrophage infiltration and clinical osteoarthritis pain severity.236,237 Many of the clinical anti-inflammatory therapies will be discussed in subsequent sections.
The development of CIPN from specific chemotherapeutics also appears to be dependent on the macrophage–neuron interface, particularly paclitaxel. Infiltrating macrophages are found along the nerve as well as in the DRG after treatment with paclitaxel. Paclitaxel directly stimulates macrophages to release HMGB1 to sensitize neurons.238,239 Clinical support for this pathway comes from a randomized, Phase II clinical trial of recombinant human thrombomodulin. Thrombomodulin degrades HMGB1 and is approved in Japan for the treatment of disseminated intravascular coagulation.240 Patients with colon cancer undergoing chemotherapy that receive thrombomodulin are less likely to develop severe sensory neuropathy.241
Neuron to Macrophage Interactions
Sensory neurons can regulate macrophage states. The release of neuronal ATP attracts infiltrating macrophages (into the DRG for example) and activates P2X and P2Y receptors on macrophages.219 Activation of macrophage P2X4 receptors induces the release of IL-1b and PGE2.242,243 Meanwhile, activation of P2Y7 on macrophage leads to a further wave of IL-1b through inflammasome activation.244 Substance P released by sensory neurons stimulates macrophages NK-1 receptors and leads to macrophage release of IL-1b, TNFa and CCL2.245,246 Similarly, the release of CCL2 and CX3CL1 from injured sensory neurons can also activate macrophages through CCR2 and CX3CR1.247,248
Non-coding micro-RNAs in exosomes are another interesting form of communication between injured sensory neurons and macrophages: MiR-21 enhances pain by promoting M1 macrophage polarization, while miR-124 reduces pain by promoting M2 macrophage polarization.249 Even more surprisingly, macrophages can even transfer mitochondria to sensory neurons; this transfer is hypothesized to help with resolution of oxidative stress in sensory neurons during the resolution phase of inflammation.250
Macrophage to Sensory Neuron Interactions
Most of the mediators secreted by macrophages, especially by the M1 subtype, are pro-inflammatory. As mentioned above, IL-1b, IL-6, PGE2, TNFa and CCL2 released by macrophages can stimulate specific cognate receptors on nociceptive neurons contributing to the development of neuropathic pain.213,251–254 Activated macrophages secrete HMGB1, which binds to TLR4 receptors on sensory neurons to play a role in the pathogenesis of neuropathic pain in animal models.46,239,240 Hydrogen sulfide (H2S) is generated in activated macrophages and contributes to neuronal hyperexcitation by upregulating and activating neuronal calcium channels (transient receptor potential cation channel A1; TRPA1).255–257 Macrophages also secrete anti-inflammatory mediators. More specifically, M1 macrophages can secrete endogenous opioids by IL-4 signaling to downregulate the excitability of neurons;258 meanwhile, M2 macrophages can secrete IL-10 to reverse TNFa-induced upregulation of sodium channels in DRG neurons and alleviate pain behavior.259
Clinical Pain Therapies Targeting the Neuroimmune Interface
In the subsequent sections, we will explore current clinical therapies in pain management and how they mediate analgesia by potentially targeting the neuroimmune interface.
Anti-Inflammatory Mediators and the Neuroimmune Interface
Certain therapeutics targeting specific pro-inflammatory signaling pathways are clinically successful in the management of inflammatory pain, especially in rheumatoid arthritis (RA) or low back pain due to ankylosing spondylitis.260,261 Modulators of the IL-1b/IL-1R signaling include anakinra (a monoclonal IL-1 receptor antagonist), riloncept (IL-1 soluble decoy receptor), and canakinumab (anti-IL-1b monoclonal antibody). All three IL-1b inhibitors are well tolerated and effective in patients with RA, gouty arthritis, ankylosing spondylitis, systemic juvenile idiopathic arthritis, and other auto-inflammatory diseases that involve deregulation of IL-1b.260–266 In a systematic review and meta-analysis of six clinical trials, RA patients treated with anakinra are 42% more likely to have clinical benefits compared to placebo; reduction in clinical markers of inflammation and slowed radiographic evidence of joint damage are also observed in anakinra-treated patients.267–269 In another clinical application, a small pilot trial of a single intraarticular injection of anakinra for acute anterior cruciate ligament knee injury provided a reduction in knee pain and improvement in function over a 2-week interval.270 On the horizon is the promising potential of IL-1 antagonists for treating neuropathic pain. CRPS can be modelled by injecting serum immunoglobulin G from patients with CRPS into mice with hind-paw injuries. Treatment with anakinra prevents the transferred CRPS pain behavior and blocks microglia and astrocyte activation in this translational CRPS mouse model.271
Like IL-1R antagonist, antagonists of TNFa, including etanercept (soluble TNFa decoy receptor), adalimumab and infliximab (both monoclonal TNFa antibodies), are used clinically to alleviate pain and symptoms of RA, psoriatic arthritis, ankylosing spondylitis, and inflammatory bowel diseases.272–277 The combination of TNFa antagonists with methotrexate is the most potent therapy against RA.279–280 TNFa antagonists are also beneficial for the treatment in disc-related pain including radicular and axial back/neck pain. The mechanism is thought to arise partly from the penetration of anti-TNFα into the cerebrospinal fluid and selective delivery to spinal structures, although evidence to support this is weak and the benefits are not seen in all clinical studies; thus, anti-TNFa therapy is not widely adopted in regular pain practice.281–284
IL-6 is another dysregulated proinflammatory cytokine described in chronic pain conditions as well as inflammatory diseases.285–289 Tocilizumab, sarilumab, and satralizumab are currently available monoclonal IL-6 receptor inhibiting antibodies; and siltuximab is an IL-6 sequestering antibody. IL-6 antagonists are effective clinically and offer another FDA-approved biologic therapeutic option for the treatment of RA.290–292 Current research into additional applications of IL-6 antagonists includes the context of lower back pain. In a few small clinical studies of lower back pain, systemic and local application of tocilizumab was effective in reducing pain and disability; in fact, the results suggest that tocilizumab is more effective than the current practice of steroid injections.293–295
The clinical benefit of CGRP antagonists in the preventative treatment of migraine headaches is a model example of drug development born out of preclinical research.206 CGRP dysregulation in the trigeminal system is implicated in the pathophysiology of migraine. Injection of CGRP into migraine patients precipitates headaches.296 This ultimately led to the development of erenumab, an inhibitory monoclonal antibody against the CGRP receptor – approved by the FDA for the preventative treatment of migraines.297–300 Current clinical studies are underway to study the effect of CGRP antagonists on other pain conditions, such as trigeminal neuralgia and fibromyalgia.301–303
Unfortunately, not all drugs targeting neuroimmune signaling translate successfully into clinical practice. A double-blind, randomized, multicenter study of CCR2 antagonist (AZD2423) demonstrated no clinical benefit over placebo in the treatment of posttraumatic neuralgia.304 A TLR4-blocking antibody, NI-0101, was unsuccessful in improving clinical outcomes in RA despite inhibiting in vivo LPS-induced cytokine release in healthy volunteers.305 Antagonist of the P2X7 purinergic receptor (AZD9056) was also ineffective for the treatment of RA-associated joint pain.306 However, the effects of these drugs on other pain conditions have yet to be explored, and many more available drugs targeting the neuroimmune interface in the context of inflammatory, immune, and cancer disorders have yet to be explored in the context of pain too.
Glial Inhibitors/Modulations and the Neuroimmune Interface
Several drugs capable of inhibiting glial action in preclinical studies have not been effective clinically. Minocycline is a semi-synthetic tetracycline, which has been used as an antibiotic for more than 30 years. Minocycline has properties beyond its antibiotic activity, including being anti-inflammatory, anti-apoptotic, anti-angiogenic307,308 – and most relevantly, able to inhibit microglial activation.309 Several preclinical studies supported minocycline’s benefits in attenuating pain behavior in several neuropathic pain models.16,310,311 However, the clinical evidence for minocycline is weak and mixed. Several clinical studies across various pain settings suggested minocycline either did not provide clinically meaningful benefits to pain or actually increased the duration of pain in certain patient populations.312–314 In one small randomized control study with 25 subjects with type 2 diabetes randomized to receive minocycline, the minocycline group recorded improvement in peripheral and autonomic neuropathy outcomes.315 The discrepancy between preclinical and clinical results may be due in part to the small sample sizes of the clinical studies, the assumption that microglial activation is the predominant driver of pathogenesis in the various clinical pain settings, and minocycline likely has non-microglia targets.316 Propentofylline suffered a similar fate to minocycline. Propentofylline is a nonspecific microglia and astrocyte glial inhibitor with promising preclinical results in preventing and reversing neuropathic pain, but it was not effective when administered to patients with post-herpetic neuralgia in a randomized clinical trial.317–319 Ibudilast is a non-selective cyclic nucleotide phosphodiesterase (PDE) inhibitor used as a bronchodilator for asthma but is reported to have glial inhibitory effects via stimulating IL-10 release from activated microglia and suppressing production of TNFa, IL-1β, IL-6.320 While ibudilast is not effective in clinical studies of chronic migraines, diabetic neuropathy or CRPS, there may be promise in its use for the treatment of substance use (stimulant, alcohol, and opioid) disorders.321–326
Steroids and Neuro-Immune Interface
Injection of steroids into or around pain generators is one of the keystones of interventional pain management.327–329 Epidural glucocorticoid injection, for example, is one of the most commonly performed interventional pain procedures for the treatment of chronic spinal pain, with over 2.2 million lumbar epidural glucocorticoid injections performed each year in the Medicare population alone.330–332 Epidural steroids are effective in reducing pain severity, decreasing opioid use, improving function, avoiding the need for surgery, and even treating pain in patients who have not responded to surgical intervention.329,333–337 Steroids may exert anti-inflammatory effects on the neuroimmune interface. Consistently with preclinical models, steroid administration (local, intrathecal, or systemic administration) before or at the time of injury reduces proinflammatory cytokines, neuronal firing rates, glial cell activation in the spinal cord, and correlates with a reduction in pain behavior after nerve injury.338–340 It is interesting to point out that, although steroids reduce proinflammatory cytokines after injury, anti-inflammatory cytokine such as IL-4 and IL-10, which also decrease after injury, are not affected by steroids – supporting a steroid-independent anti-inflammatory process.339
However, there are mixed results regarding the ability of steroids to reverse established pain behavior in animal models.338–340 Similarly, clinical evidence supporting the use of epidural steroids for spinal pain is also not always positive.331,341 Steroids used in clinical practice (dexamethasone, triamcinolone, methylprednisolone, betamethasone) can activate pro-inflammatory mineralocorticoid receptors found in DRG sensory neurons.342–344 Co-administration of mineralocorticoid receptor antagonist with dexamethasone is more effective in reducing SGC activation in the DRG, evoked and spontaneous pain behaviors than dexamethasone alone in rodent models of lower back pain.342,343,345 It is interesting to wonder if the propensity to activate different steroid receptors may account for why steroids are sometimes ineffective or wane in efficacy over time in clinical practice. Ultimately, this could be a promising area of future clinical research as mineralocorticoid receptor antagonists (spironolactone, eplerenone) are widely used in the clinical treatment of hypertension and heart failure.
Neuromodulation and the Neuro-Immune Interface
Neuromodulation is a growing field within pain medicine, which treats chronic pain by using electrical stimulus.346 The types of interventions range from peripheral nerve stimulation, dorsal root ganglion stimulation, spinal cord stimulation (SCS), to brain stimulation. The role of non-neuronal activity including glial cells builds to our understanding of the mechanism of neuromodulation.
The most prevalent therapy in neuromodulation, SCS, may exert benefits in part by modulating the neuroimmune interface, and is an effective treatment for many neuropathic pain conditions, such as post-laminectomy pain syndrome, complex regional pain syndrome, and diabetic neuropathy.347–353 Rodent models of SCS after various nerve injury models demonstrate reductions in pain behavior with SCS treatment, which correlates with reduced glial activation markers and transcriptomic changes in genes implicated in neuroinflammation and immune response.318,353–356 One example of bench-to-bedside translation is a novel SCS waveform designed based on preclinical experiments to engage the transcriptomic signatures of neuronal and glial populations, and the stimulation parameters are designed such that the resulting transcriptomic signatures mimic those of the naïve uninjured profiles.357–359 In a multicenter clinical trial, this novel SCS waveform was able to achieve 80% responder rate in patients with chronic back pain compared to 50% using conventional waveform.360
Another promising avenue for neuromodulation is vagal nerve stimulation. Vagal nerve stimulation has historically been used in the treatment of refractory epilepsy and the treatment of resistant depression.361 The vagal nerve is appreciated as a critical component of an inflammatory reflex in which the vagal nerve signaling suppresses excessive cytokine release and inflammation.362,363 Indeed, epilepsy patients with vagal nerve stimulators have lowered peripheral levels of TNFa, IL-1b, and IL-6.364 A more accessible, noninvasive transcutaneous vagal nerve stimulator is available for the management of acute migraine; this non-invasive device is also capable of decreasing peripheral proinflammatory cytokines.365,366 Since then, vagal nerve stimulation is being adapted to other inflammatory/autoimmune diseases with success, such as rheumatoid arthritis, Crohn’s disease, and COVID-19.364,367–369
Regenerative Medicine and the Neuro-Immune Interface
Regenerative medicine is also another frontier of pain medicine. The initial concept was to bolster the body’s own regenerative capacity to address degenerative pain conditions. This topic was eloquently reviewed recently.370 Clinical data examining physical function and pain severity support the use of blood- and cell-derived pain therapies (such as platelet-rich plasma, autologous conditioned serum, mesenchymal stromal cells) in osteoarthritis of knees and hips, tendinopathy, and degenerative spine disease. Although there is some evidence to support tissue regeneration, the results are not consistent.371–374 The current evidence emphasizes a paradigm shift suggesting that the analgesic effects of regenerative medicine may be largely due to immunomodulatory effects (by providing SPM, IL-4, IL-10, TGFb) and less so from tissue regeneration.375–378
Conclusions
Neurons interact with immune and nonimmune cells through classic immune mediators – this bidirectional neuroimmune interface spans the entire nociceptive circuitry from the peripheral tissue to the CNS. Ultimately, neurons change their firing property and manifests on an organism level as pain behavior. In this review, we highlighted how interfaces of the CNS (microglia, astrocytes, T lymphocytes) and PNS (SGC, macrophages) influence pain. Yet, many other neuroimmune interfaces exist and affect pain that are beyond the scope of our review (such as oligodendrocytes, Schwann cells, endothelial cells, cancer cells, gut microbiome).9 A recurring theme is the paucity of clinical evidence to support neuroimmune interaction in humans despite mounting evidence of the involvement of the neuroimmune interface at a micro- and macrocosm level in preclinical models of chronic pain. Criticism of failed therapeutic trials highlights the assumption that neuroimmune dysfunction drives chronic pain in humans. Future efforts must bridge the gap to safely and non-invasively study pathogenesis of chronic pain in patients. On the positive side, targeted small molecules and antibodies already exist for many ligand–receptor interactions developed mainly in the context of autoimmune diseases and cancers.379–382 There are significant parallels between the neuroimmune interactions in chronic pain and autoimmune diseases and cancers. The opportunity to pivot preexisting targeted therapeutics to clinical studies of pain remains wide open.
Funding
Z.G. is supported by NINDS R01NS121144.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dahlhamer J, Lucas J, Zelaya C, et al. Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults — United States, 2016. MMWR Morb Mortal Wkly Rep. 2018;67(36):1001–1006. doi:10.15585/mmwr.mm6736a2
2. Zelaya CE, Dahlhamer JM, Lucas JW, Connor EM. Chronic Pain and High-impact Chronic Pain Among U.S. Adults, 2019. NCHS Data Brief. 2020;1(390):1–8.
3. Cordell WH, Keene KK, Giles BK, Jones JB, Jones JH, Brizendine EJ. The high prevalence of pain in emergency medical care. Am J Emerg Med. 2002;20(3):165–169. doi:10.1053/ajem.2002.32643
4. Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving Pain in America: a Blueprint for Transforming Prevention, Care, Education, and Research. National Academies Press (US); 2011. Available from: http://www.ncbi.nlm.nih.gov/books/NBK91497/.
5. Stewart WF, Ricci JA, Chee E, Morganstein D, Lipton R. Lost Productive Time and Cost Due to Common Pain Conditions in the US Workforce. JAMA. 2003;290(18):2443–2454. doi:10.1001/jama.290.18.2443
6. Bonnie RJ, Schumacher MA, Clark JD, Kesselheim AS, Management P. Opioid Regulation: continuing Public Health Challenges. Am J Public Health. 2019;109(1):31–34. doi:10.2105/AJPH.2018.304881
7. Volkow ND, McLellan AT. Opioid Abuse in Chronic Pain — misconceptions and Mitigation Strategies. N Eng J Med. 2016;374(13):1253–1263. doi:10.1056/NEJMra1507771
8. Chiu IM, von Hehn CA, Woolf CJ. Neurogenic Inflammation – the Peripheral Nervous System’s Role in Host Defense and Immunopathology. Nat Neurosci. 2012;15(8):1063–1067. doi:10.1038/nn.3144
9. Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354(6312):572–577. doi:10.1126/science.aaf8924
10. Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16(11):1267–1276. doi:10.1038/nm.2234
11. Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28(2):183–187. doi:10.1002/ana.410280213
12. Mouraux A, Bannister K, Becker S, et al. Challenges and opportunities in translational pain research – an opinion paper of the working group on translational pain research of the European pain federation (EFIC). European J Pain. 2021;25(4):731–756. doi:10.1002/ejp.1730
13. Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14(4):217–231. doi:10.1038/nri3621
14. Baral P, Udit S, Chiu IM. Pain and immunity: implications for host defence. Nat Rev Immunol. 2019;19(7):433–447. doi:10.1038/s41577-019-0147-2
15. Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–155. doi:10.1002/glia.10161
16. Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306(2):624–630. doi:10.1124/jpet.103.052407
17. Park CK, Lü N, Xu ZZ, Liu T, Serhan CN, Ji RR. Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci. 2011;31(42):15072–15085. doi:10.1523/JNEUROSCI.2443-11.2011
18. Gruber-Schoffnegger D, Drdla-Schutting R, Hönigsperger C, Wunderbaldinger G, Gassner M, Sandkühler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-α and IL-1β is mediated by glial cells. J Neurosci. 2013;33(15):6540–6551. doi:10.1523/JNEUROSCI.5087-12.2013
19. Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28(20):5189–5194. doi:10.1523/JNEUROSCI.3338-07.2008
20. Chirila AM, Brown TE, Bishop RA, Bellono NW, Pucci FG, Kauer JA. Long-term potentiation of glycinergic synapses triggered by interleukin 1β. Proc Natl Acad Sci U S A. 2014;111(22):8263–8268. doi:10.1073/pnas.1401013111
21. Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkühler J. Selective activation of microglia facilitates synaptic strength. J Neurosci. 2015;35(11):4552–4570. doi:10.1523/JNEUROSCI.2061-14.2015
22. Gardoni F, Boraso M, Zianni E, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1β and NMDA stimulation. J Neuroinflammation. 2011;8(1):14. doi:10.1186/1742-2094-8-14
23. Baba H, Kohno T, Moore KA, Woolf CJ. Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci. 2001;21(5):1750–1756.
24. Meves H. The Action of Prostaglandins on Ion Channels. Curr Neuropharmacol. 2006;4(1):41–57.
25. Nicol GD, Klingberg DK, Vasko MR. Prostaglandin E2 increases calcium conductance and stimulates release of substance P in avian sensory neurons. J Neurosci. 1992;12(5):1917–1927.
26. Samad TA, Moore KA, Sapirstein A, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410(6827):471–475. doi:10.1038/35068566
27. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi:10.1126/science.1110647
28. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi:10.1126/science.1194637
29. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45. doi:10.3389/fncel.2013.00045
30. Rezaie P, Male D. Colonisation of the developing human brain and spinal cord by microglia: a review. Microsc Res Tech. 1999;45(6):359–382. doi:10.1002/(SICI)1097-0029(19990615)45:6<359::AID-JEMT4>3.0.CO;2-D
31. Svensson CI, Marsala M, Westerlund A, et al. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86(6):1534–1544. doi:10.1046/j.1471-4159.2003.01969.x
32. Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T. Bidirectional Microglia-Neuron Communication in Health and Disease. Front Cell Neurosci. 2018;12:323. doi:10.3389/fncel.2018.00323
33. Tsuda M, Shigemoto-Mogami Y, Koizumi S, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424(6950):778–783. doi:10.1038/nature01786
34. Zhou Z, Peng X, Hao S, Fink DJ, Mata M. HSV-mediated transfer of interleukin-10 reduces inflammatory pain through modulation of membrane tumor necrosis factor alpha in spinal cord microglia. Gene Ther. 2008;15(3):183–190. doi:10.1038/sj.gt.3303054
35. Kohno K, Shirasaka R, Yoshihara K, et al. A spinal microglia population involved in remitting and relapsing neuropathic pain. Science. 2022;376(6588):86–90. doi:10.1126/science.abf6805
36. Del Valle L, Schwartzman RJ, Alexander G. Spinal cord histopathological alterations in a patient with longstanding complex regional pain syndrome. Brain Behav Immun. 2009;23(1):85–91. doi:10.1016/j.bbi.2008.08.004
37. Janssen B, Vugts DJ, Windhorst AD, Mach RH. PET Imaging of Microglial Activation-Beyond Targeting TSPO. Molecules. 2018;23(3):E607. doi:10.3390/molecules23030607
38. Jeon SY, Seo S, Lee JS, et al. [11C]-(R)-PK11195 positron emission tomography in patients with complex regional pain syndrome. Medicine. 2017;96(1):e5735. doi:10.1097/MD.0000000000005735
39. Loggia ML, Chonde DB, Akeju O, et al. Evidence for brain glial activation in chronic pain patients. Brain. 2015;138(Pt 3):604–615. doi:10.1093/brain/awu377
40. Seo S, Jung YH, Lee D, et al. Abnormal neuroinflammation in fibromyalgia and CRPS using [11C]-(R)-PK11195 PET. PLoS One. 2021;16(2):e0246152. doi:10.1371/journal.pone.0246152
41. Albrecht DS, Ahmed SU, Kettner NW, et al. Neuroinflammation of the spinal cord and nerve roots in chronic radicular pain patients. Pain. 2018;159(5):968–977. doi:10.1097/j.pain.0000000000001171
42. Meyer JH, Braga J. Development and Clinical Application of Positron Emission Tomography Imaging Agents for Monoamine Oxidase B. Front Neurosci. 2021;15:773404. doi:10.3389/fnins.2021.773404
43. Albrecht DS, Forsberg A, Sandström A, et al. Brain glial activation in fibromyalgia - A multi-site positron emission tomography investigation. Brain Behav Immun. 2019;75:72–83. doi:10.1016/j.bbi.2018.09.018
44. Hutchinson MR, Ramos KM, Loram LC, et al. Evidence for a role of heat shock protein-90 in toll like receptor 4 mediated pain enhancement in rats. Neuroscience. 2009;164(4):1821–1832. doi:10.1016/j.neuroscience.2009.09.046
45. Masuda T, Ozono Y, Mikuriya S, et al. Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat Commun. 2016;7:12529. doi:10.1038/ncomms12529
46. Park JS, Svetkauskaite D, He Q, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279(9):7370–7377. doi:10.1074/jbc.M306793200
47. Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107(26):11942–11947. doi:10.1073/pnas.1003893107
48. Yang H, Zeng Q, Silverman HA, et al. HMGB1 released from nociceptors mediates inflammation. PNAS. 2021;118:33. doi:10.1073/pnas.2102034118
49. Kim D, Kim MA, Cho IH, et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. 2007;282(20):14975–14983. doi:10.1074/jbc.M607277200
50. Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102(16):5856–5861. doi:10.1073/pnas.0501634102
51. Huck NA, Siliezar-Doyle J, Haight ES, et al. Temporal Contribution of Myeloid-Lineage TLR4 to the Transition to Chronic Pain: a Focus on Sex Differences. J Neurosci. 2021;41(19):4349–4365. doi:10.1523/JNEUROSCI.1940-20.2021
52. Kobayashi K, Yamanaka H, Yanamoto F, Okubo M, Noguchi K. Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia. 2012;60(10):1529–1539. doi:10.1002/glia.22373
53. Biber K, Tsuda M, Tozaki-Saitoh H, et al. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO J. 2011;30(9):1864–1873. doi:10.1038/emboj.2011.89
54. Tsuda M, Kuboyama K, Inoue T, Nagata K, Tozaki-Saitoh H, Inoue K. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol Pain. 2009;5:28. doi:10.1186/1744-8069-5-28
55. Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. 2011;6(10):e26317. doi:10.1371/journal.pone.0026317
56. Guan Z, Kuhn JA, Wang X, et al. Injured sensory neuron–derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci. 2016;19(1):94–101. doi:10.1038/nn.4189
57. Zhou LJ, Peng J, Xu YN, et al. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep. 2019;27(13):3844–3859.e6. doi:10.1016/j.celrep.2019.05.087
58. Gu N, Peng J, Murugan M, et al. Spinal Microgliosis Due to Resident Microglial Proliferation Is Required for Pain Hypersensitivity after Peripheral Nerve Injury. Cell Rep. 2016;16(3):605–614. doi:10.1016/j.celrep.2016.06.018
59. Dai XM, Ryan GR, Hapel AJ, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99(1):111–120. doi:10.1182/blood.v99.1.111
60. Benner B, Good L, Quiroga D, et al. Pexidartinib, a Novel Small Molecule CSF-1R Inhibitor in Use for Tenosynovial Giant Cell Tumor: a Systematic Review of Pre-Clinical and Clinical Development. Drug Des Devel Ther. 2020;14:1693–1704. doi:10.2147/DDDT.S253232
61. Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. 2004;20(5):1150–1160. doi:10.1111/j.1460-9568.2004.03593.x
62. Clark AK, Yip PK, Grist J, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. PNAS. 2007;104(25):10655–10660. doi:10.1073/pnas.0610811104
63. Milligan E, Zapata V, Schoeniger D, et al. An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur J Neurosci. 2005;22(11):2775–2782. doi:10.1111/j.1460-9568.2005.04470.x
64. Staniland AA, Clark AK, Wodarski R, et al. Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J Neurochem. 2010;114(4):1143–1157. doi:10.1111/j.1471-4159.2010.06837.x
65. The future | kancera. Available from: https://kancera.com/en/research/the-future/.
66. Ji RR, Xu ZZ, Wang X, Lo EH. Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol Sci. 2009;30(7):336–340. doi:10.1016/j.tips.2009.04.002
67. Kawasaki Y, Xu ZZ, Wang X, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14(3):331–336. doi:10.1038/nm1723
68. Berta T, Park CK, Xu ZZ, et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-α secretion. J Clin Invest. 2014;124(3):1173–1186. doi:10.1172/JCI72230
69. Berta T, Lee JE, Park CK. Unconventional Role of Caspase-6 in Spinal Microglia Activation and Chronic Pain. Mediators Inflamm. 2017;2017:9383184. doi:10.1155/2017/9383184
70. Calvo M, Zhu N, Tsantoulas C, et al. Neuregulin-ErbB signaling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury. J Neurosci. 2010;30(15):5437–5450. doi:10.1523/JNEUROSCI.5169-09.2010
71. Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23(25):8692–8700.
72. Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR. Microglia in Pain: detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron. 2018;100(6):1292–1311. doi:10.1016/j.neuron.2018.11.009
73. Yi MH, Liu YU, Umpierre AD, et al. Optogenetic activation of spinal microglia triggers chronic pain in mice. PLoS Biol. 2021;19(3):e3001154. doi:10.1371/journal.pbio.3001154
74. Coull JAM, Boudreau D, Bachand K, et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424(6951):938–942. doi:10.1038/nature01868
75. Coull JAM, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–1021. doi:10.1038/nature04223
76. Keller AF, Beggs S, Salter MW, De Koninck Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol Pain. 2007;3:27. doi:10.1186/1744-8069-3-27
77. Dedek A, Xu J, Kandegedara CM, et al. Loss of STEP61 couples disinhibition to N-methyl-d-aspartate receptor potentiation in rodent and human spinal pain processing. Brain. 2019;142(6):1535–1546. doi:10.1093/brain/awz105
78. Malcangio M. Spinal mechanisms of neuropathic pain: is there a P2X4-BDNF controversy? Neurobiol Pain. 2017;1:1–5. doi:10.1016/j.ynpai.2017.04.001
79. Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29(11):3518–3528. doi:10.1523/JNEUROSCI.5714-08.2009
80. Drilon A, Siena S, Ou SHI, et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400–409. doi:10.1158/2159-8290.CD-16-1237
81. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med. 2018;378(8):731–739. doi:10.1056/NEJMoa1714448
82. Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem. 2003;278(17):14677–14687. doi:10.1074/jbc.M300218200
83. Serhan CN, de la Rosa X, Jouvene CC. Cutting Edge: human Vagus Produces Specialized Proresolving Mediators of Inflammation with Electrical Stimulation Reducing Proinflammatory Eicosanoids. J Immunol. 2018;201(11):3161–3165. doi:10.4049/jimmunol.1800806
84. Xu ZZ, Zhang L, Liu T, et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat Med. 2010;16(5):592–597. doi:10.1038/nm.2123
85. Callan N, Hanes D, Bradley R. Early evidence of efficacy for orally administered SPM-enriched marine lipid fraction on quality of life and pain in a sample of adults with chronic pain. J Transl Med. 2020;18(1):401. doi:10.1186/s12967-020-02569-5
86. Geusens P, Wouters C, Nijs J, Jiang Y, Dequeker J. Long-term effect of omega-3 fatty acid supplementation in active rheumatoid arthritis. A 12-month, double-blind, controlled study. Arthritis Rheum. 1994;37(6):824–829. doi:10.1002/art.1780370608
87. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35. doi:10.1007/s00401-009-0619-8
88. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22(5):208–215. doi:10.1016/s0166-2236(98)
89. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi:10.1038/nature21029
90. Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia. 2013;61(1):24–36. doi:10.1002/glia.22389
91. Bennett MVL, Contreras JE, Bukauskas FF, Sáez JC. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 2003;26(11):610–617. doi:10.1016/j.tins.2003.09.008
92. Kohro Y, Matsuda T, Yoshihara K, et al. Spinal astrocytes in superficial laminae gate brainstem descending control of mechanosensory hypersensitivity. Nat Neurosci. 2020;23(11):1376–1387. doi:10.1038/s41593-020-00713-4
93. Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol. 1999;157(2):289–304. doi:10.1006/exnr.1999.7065
94. Gao YJ, Cheng JK, Zeng Q, et al. Selective inhibition of JNK with a peptide inhibitor attenuates pain hypersensitivity and tumor growth in a mouse skin cancer pain model. Exp Neurol. 2009;219(1):146–155. doi:10.1016/j.expneurol.2009.05.006
95. Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565(1):1–7. doi:10.1016/0006-8993(91)
96. Mathewson AJ, Berry M. Observations on the astrocyte response to a cerebral stab wound in adult rats. Brain Res. 1985;327(1–2):61–69. doi:10.1016/0006-8993(85)
97. Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20(2):467–473. doi:10.1111/j.1460-9568.2004.03514.x
98. Zhang RX, Liu B, Wang L, et al. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain. 2005;118(1–2):125–136. doi:10.1016/j.pain.2005.08.001
99. Davies JE, Pröschel C, Zhang N, Noble M, Mayer-Pröschel M, Davies SJA. Transplanted astrocytes derived from BMP- or CNTF-treated glial-restricted precursors have opposite effects on recovery and allodynia after spinal cord injury. J Biol. 2008;7(7):24. doi:10.1186/jbiol85
100. Gao Y-J, Zhang L, Ji -R-R. Spinal injection of TNF-α-activated astrocytes produces persistent pain symptom mechanical allodynia by releasing monocyte chemoattractant protein-1. Glia. 2010;58(15):1871–1880. doi:10.1002/glia.21056
101. Okada-Ogawa A, Suzuki I, Sessle BJ, et al. Astroglia in Medullary Dorsal Horn (Trigeminal Spinal Subnucleus Caudalis) Are Involved in Trigeminal Neuropathic Pain Mechanisms. J Neurosci. 2009;29(36):11161–11171. doi:10.1523/JNEUROSCI.3365-09.2009
102. Watkins RL, Martin D, Ulrich P, Tracey JK, Maier FS. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71(3):225–235. doi:10.1016/S0304-3959(97)03369-1
103. Nam Y, Kim J-H, Kim J-H, et al. Reversible Induction of Pain Hypersensitivity following Optogenetic Stimulation of Spinal Astrocytes. Cell Rep. 2016;17(11):3049–3061. doi:10.1016/j.celrep.2016.11.043
104. Shi Y, Gelman BB, Lisinicchia JG, Tang S-J. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci. 2012;32(32):10833–10840. doi:10.1523/JNEUROSCI.5628-11.2012
105. Liu Y, Jiang H, Qin X, Tian M, Zhang H. PET imaging of reactive astrocytes in neurological disorders. Eur J Nucl Med Mol Imaging. 2021. doi:10.1007/s00259-021-05640-5
106. Boche D, Gerhard A, Rodriguez-Vieitez E. Prospects and challenges of imaging neuroinflammation beyond TSPO in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2019;46(13):2831–2847. doi:10.1007/s00259-019-04462-w
107. Guilarte TR. TSPO in diverse CNS pathologies and psychiatric disease: a critical review and a way forward. Pharmacol Ther. 2019;194:44–58. doi:10.1016/j.pharmthera.2018.09.003
108. Tournier BB, Tsartsalis S, Ceyzériat K, et al. Astrocytic TSPO Upregulation Appears Before Microglial TSPO in Alzheimer’s Disease. J Alzheimers Dis. 2020;77(3):1043–1056. doi:10.3233/JAD-200136
109. Lu Y, Jiang B-C, Cao D-L, et al. TRAF6 upregulation in spinal astrocytes maintains neuropathic pain by integrating TNF-α and IL-1β signaling. Pain. 2014;155(12):2618–2629. doi:10.1016/j.pain.2014.09.027
110. Grosche J, Matyash V, Möller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron–glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999;2(2):139–143. doi:10.1038/5692
111. Guo W, Wang H, Watanabe M, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27(22):6006–6018. doi:10.1523/JNEUROSCI.0176-07.2007
112. Xie W, Strong JA, Meij JTA, Zhang JM, Yu L. Neuropathic pain: early spontaneous afferent activity is the trigger. Pain. 2005;116(3):243–256. doi:10.1016/j.pain.2005.04.017
113. Ikeda H, Tsuda M, Inoue K, Murase K. Long-term potentiation of neuronal excitation by neuron-glia interactions in the rat spinal dorsal horn. Eur J Neurosci. 2007;25(5):1297–1306. doi:10.1111/j.1460-9568.2007.05386.x
114. Ziak D, Chvátal A, Syková E. Glutamate-, kainate- and NMDA-evoked membrane currents in identified glial cells in rat spinal cord slice. Physiol Res. 1998;47(5):365–375.
115. Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006;7(6):423–436. doi:10.1038/nrn1928
116. Nakagawa T, Wakamatsu K, Zhang N, et al. Intrathecal administration of ATP produces long-lasting allodynia in rats: differential mechanisms in the phase of the induction and maintenance. Neuroscience. 2007;147(2):445–455. doi:10.1016/j.neuroscience.2007.03.045
117. Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J Pain. 2014;15(7):712–725. doi:10.1016/j.jpain.2014.04.001
118. Jiang BC, Cao DL, Zhang X, et al. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. 2016;126(2):745–761. doi:10.1172/JCI81950
119. Klimatcheva E, Pandina T, Reilly C, et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. 2015;16(1):6. doi:10.1186/s12865-015-0068-1
120. Eclancher F, Perraud F, Faltin J, Labourdette G, Sensenbrenner M. Reactive astrogliosis after basic fibroblast growth factor (bFGF) injection in injured neonatal rat brain. Glia. 1990;3(6):502–509. doi:10.1002/glia.440030609
121. Ferrara N, Ousley F, Gospodarowicz D. Bovine brain astrocytes express basic fibroblast growth factor, a neurotropic and angiogenic mitogen. Brain Res. 1988;462(2):223–232. doi:10.1016/0006-8993(88)
122. Ji RR, Zhang Q, Zhang X, et al. Prominent expression of bFGF in dorsal root ganglia after axotomy. Eur J Neurosci. 1995;7(12):2458–2468. doi:10.1111/j.1460-9568.1995.tb01044.x
123. Madiai F, Hussain SRA, Goettl VM, Burry RW, Stephens RL, Hackshaw KV. Upregulation of FGF-2 in reactive spinal cord astrocytes following unilateral lumbar spinal nerve ligation. Exp Brain Res. 2003;148(3):366–376. doi:10.1007/s00221-002-1286-3
124. Madiai F, Goettl VM, Hussain SR, Clairmont AR, Stephens RL, Hackshaw KV. Anti-fibroblast growth factor-2 antibodies attenuate mechanical allodynia in a rat model of neuropathic pain. J Mol Neurosci. 2005;27(3):315–324. doi:10.1385/JMN:27:3:
125. Hui Q, Jin Z, Li X, Liu C, Wang X, Family: FGF. From Drug Development to Clinical Application. Int J Mol Sci. 2018;19(7):1875. doi:10.3390/ijms19071875
126. Ji RR, Donnelly CR, Nedergaard M. Astrocytes in chronic pain and itch. Nat Rev Neurosci. 2019;20(11):667–685. doi:10.1038/s41583-019-0218-1
127. Guptarak J, Wanchoo S, Durham-Lee J, et al. Inhibition of IL-6 signaling: a novel therapeutic approach to treating spinal cord injury pain. Pain. 2013;154(7):1115–1128. doi:10.1016/j.pain.2013.03.026
128. Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal Glial–Neuronal Interactions Contribute to Descending Pain Facilitation. J Neurosci. 2008;28(42):10482–10495. doi:10.1523/JNEUROSCI.3593-08.2008
129. Zhang RX, Li A, Liu B, et al. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2008;135(3):232–239. doi:10.1016/j.pain.2007.05.023
130. Kolhekar R, Meller ST, Gebhart GF. N-methyl-D-aspartate receptor-mediated changes in thermal nociception: allosteric modulation at glycine and polyamine recognition sites. Neuroscience. 1994;63(4):925–936. doi:10.1016/0306-4522(94)
131. Wolosker H. D-serine regulation of NMDA receptor activity. Sci STKE. 2006;2006(356):pe41. doi:10.1126/stke.3562006pe41
132. Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain. 2014;137(8):2193–2209. doi:10.1093/brain/awu140
133. Kang J, Kang N, Lovatt D, et al. Connexin 43 hemichannels are permeable to ATP. J Neurosci. 2008;28(18):4702–4711. doi:10.1523/JNEUROSCI.5048-07.2008
134. Gao YJ, Zhang L, Samad OA, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29(13):4096–4108. doi:10.1523/JNEUROSCI.3623-08.2009
135. Lazaar AL, Miller BE, Donald AC, et al. CXCR2 antagonist for patients with chronic obstructive pulmonary disease with chronic mucus hypersecretion: a Phase 2b trial. Respir Res. 2020;21(1):149. doi:10.1186/s12931-020-01401-4
136. Jeanson T, Duchêne A, Richard D, et al. Potentiation of Amitriptyline Anti-Hyperalgesic-Like Action By Astroglial Connexin 43 Inhibition in Neuropathic Rats. Sci Rep. 2016;6:38766. doi:10.1038/srep38766
137. Laird DW, Lampe PD. Therapeutic strategies targeting connexins. Nat Rev Drug Discov. 2018;17(12):905–921. doi:10.1038/nrd.2018.138
138. Silberstein SD. Tonabersat, a novel gap-junction modulator for the prevention of migraine. Cephalalgia. 2009;29(Suppl 2):28–35. doi:10.1111/j.1468-2982.2009.01973.x
139. Gosselin RD, Varela C, Banisadr G, et al. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J Neurochem. 2005;95(4):1023–1034. doi:10.1111/j.1471-4159.2005.03431.x
140. White FA, Sun J, Waters SM, et al. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005;102(39):14092–14097. doi:10.1073/pnas.0503496102
141. Ma SB, Xian H, Wu WB, et al. CCL2 facilitates spinal synaptic transmission and pain via interaction with presynaptic CCR2 in spinal nociceptor terminals. Mol Brain. 2020;13(1):161. doi:10.1186/s13041-020-00701-6
142. Abbadie C, Lindia JA, Cumiskey AM, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100(13):7947–7952. doi:10.1073/pnas.1331358100
143. Bhangoo S, Ren D, Miller RJ, et al. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol Pain. 2007;3:38. doi:10.1186/1744-8069-3-38
144. Bhangoo SK, Ripsch MS, Buchanan DJ, Miller RJ, White FA. Increased chemokine signaling in a model of HIV1-associated peripheral neuropathy. Mol Pain. 2009;5:48. doi:10.1186/1744-8069-5-48
145. Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27(45):12396–12406. doi:10.1523/JNEUROSCI.3016-07.2007
146. Huang YH, Bergles DE. Glutamate transporters bring competition to the synapse. Curr Opin Neurobiol. 2004;14(3):346–352. doi:10.1016/j.conb.2004.05.007
147. Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23(7):2899–2910.
148. Wang W, Wang W, Wang Y, Huang J, Wu S, Li YQ. Temporal changes of astrocyte activation and glutamate transporter-1 expression in the spinal cord after spinal nerve ligation-induced neuropathic pain. Anat Rec. 2008;291(5):513–518. doi:10.1002/ar.20673
149. Xin WJ, Weng HR, Dougherty PM. Plasticity in expression of the glutamate transporters GLT-1 and GLAST in spinal dorsal horn glial cells following partial sciatic nerve ligation. Mol Pain. 2009;5:15. doi:10.1186/1744-8069-5-15
150. Maeda S, Kawamoto A, Yatani Y, Shirakawa H, Nakagawa T, Kaneko S. Gene transfer of GLT-1, a glial glutamate transporter, into the spinal cord by recombinant adenovirus attenuates inflammatory and neuropathic pain in rats. Mol Pain. 2008;4:65. doi:10.1186/1744-8069-4-65
151. Weng HR, Chen JH, Cata JP. Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience. 2006;138(4):1351–1360. doi:10.1016/j.neuroscience.2005.11.061
152. Carbone M, Duty S, Rattray M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem Int. 2012;60(1):31–38. doi:10.1016/j.neuint.2011.10.017
153. Galer BS, Twilling LL, Harle J, Cluff RS, Friedman E, Rowbotham MC. Lack of efficacy of riluzole in the treatment of peripheral neuropathic pain conditions. Neurology. 2000;55(7):971–975. doi:10.1212/wnl.55.7.971
154. Cao L, DeLeo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol. 2008;38(2):448–458. doi:10.1002/eji.200737485
155. Du B, Ding YQ, Xiao X, Ren HY, Su BY, Qi JG. CD4+ αβ T cell infiltration into the leptomeninges of lumbar dorsal roots contributes to the transition from acute to chronic mechanical allodynia after adult rat tibial nerve injuries. J Neuroinflammation. 2018;15(1):81. doi:10.1186/s12974-018-1115-7
156. Kobayashi Y, Kiguchi N, Fukazawa Y, Saika F, Maeda T, Kishioka S. Macrophage-T cell interactions mediate neuropathic pain through the glucocorticoid-induced tumor necrosis factor ligand system. J Biol Chem. 2015;290(20):12603–12613. doi:10.1074/jbc.M115.636506
157. Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. 2004;129(3):767–777. doi:10.1016/j.neuroscience.2004.08.035
158. Sorge RE, Mapplebeck JCS, Rosen S, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18(8):1081–1083. doi:10.1038/nn.4053
159. Basso L, Boué J, Mahiddine K, et al. Endogenous analgesia mediated by CD4(+) T lymphocytes is dependent on enkephalins in mice. J Neuroinflammation. 2016;13(1):132. doi:10.1186/s12974-016-0591-x
160. Krukowski K, Eijkelkamp N, Laumet G, et al. CD8+ T Cells and Endogenous IL-10 Are Required for Resolution of Chemotherapy-Induced Neuropathic Pain. J Neurosci. 2016;36(43):11074–11083. doi:10.1523/JNEUROSCI.3708-15.2016
161. Laumet G, Edralin JD, Dantzer R, Heijnen CJ, Kavelaars A. Cisplatin educates CD8+ T cells to prevent and resolve chemotherapy-induced peripheral neuropathy in mice. Pain. 2019;160(6):1459–1468. doi:10.1097/j.pain.0000000000001512
162. Liu XJ, Zhang Y, Liu T, et al. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res. 2014;24(11):1374–1377. doi:10.1038/cr.2014.106
163. Petrović J, Silva JR, Bannerman CA, et al. γδ T Cells Modulate Myeloid Cell Recruitment but Not Pain During Peripheral Inflammation. Front Immunol. 2019;10:473. doi:10.3389/fimmu.2019.00473
164. Luchting B, Rachinger-Adam B, Zeitler J, et al. Disrupted TH17/Treg balance in patients with chronic low back pain. PLoS One. 2014;9(8):e104883. doi:10.1371/journal.pone.0104883
165. Luchting B, Rachinger-Adam B, Heyn J, Hinske LC, Kreth S, Azad SC. Anti-inflammatory T-cell shift in neuropathic pain. J Neuroinflammation. 2015;12:12. doi:10.1186/s12974-014-0225-0
166. Mangiacavalli S, Corso A, De Amici M, et al. Emergent T-helper 2 profile with high interleukin-6 levels correlates with the appearance of bortezomib-induced neuropathic pain. Br J Haematol. 2010;149(6):916–918. doi:10.1111/j.1365-2141.2010.08138.x
167. Gattlen C, Clarke CB, Piller N, et al. Spinal Cord T-Cell Infiltration in the Rat Spared Nerve Injury Model: a Time Course Study. Int J Mol Sci. 2016;17(3):352. doi:10.3390/ijms17030352
168. Kipnis J, Gadani S, Derecki NC. Pro-cognitive properties of T cells. Nat Rev Immunol. 2012;12(9):663–669. doi:10.1038/nri3280
169. Hu P, McLachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience. 2002;112(1):23–38. doi:10.1016/s0306-4522(02)
170. Kivisäkk P, Imitola J, Rasmussen S, et al. Localizing central nervous system immune surveillance: meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol. 2009;65(4):457–469. doi:10.1002/ana.21379
171. Costigan M, Moss A, Latremoliere A, et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. 2009;29(46):14415–14422. doi:10.1523/JNEUROSCI.4569-09.2009
172. Leger T, Grist J, D’Acquisto F, Clark AK, Malcangio M. Glatiramer acetate attenuates neuropathic allodynia through modulation of adaptive immune cells. J Neuroimmunol. 2011;234(1–2):19–26. doi:10.1016/j.jneuroim.2011.01.005
173. Kuhn JA, Vainchtein ID, Braz J, et al. Regulatory T-cells inhibit microglia-induced pain hypersensitivity in female mice. Elife. 2021;10:e69056. doi:10.7554/eLife.69056
174. Luo X, Chen O, Wang Z, et al. IL-23/IL-17A/TRPV1 axis produces mechanical pain via macrophage-sensory neuron crosstalk in female mice. Neuron. 2021;109(17):2691–2706.e5. doi:10.1016/j.neuron.2021.06.015
175. Ganor Y, Besser M, Ben-Zakay N, Unger T, Levite M. Human T cells express a functional ionotropic glutamate receptor GluR3, and glutamate by itself triggers integrin-mediated adhesion to laminin and fibronectin and chemotactic migration. J Immunol. 2003;170(8):4362–4372. doi:10.4049/jimmunol.170.8.4362
176. Mikami N, Matsushita H, Kato T, et al. Calcitonin gene-related peptide is an important regulator of cutaneous immunity: effect on dendritic cell and T cell functions. J Immunol. 2011;186(12):6886–6893. doi:10.4049/jimmunol.1100028
177. Rameshwar P, Gascon P, Ganea D. Immunoregulatory effects of neuropeptides. Stimulation of interleukin-2 production by substance p. J Neuroimmunol. 1992;37(1–2):65–74. doi:10.1016/0165-5728(92)90156-f
178. Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp Neurol. 2006;200(2):480–485. doi:10.1016/j.expneurol.2006.03.014
179. Laumet G, Ma J, Robison AJ, Kumari S, Heijnen CJ, Kavelaars AT. Cells as an Emerging Target for Chronic Pain Therapy. Front Mol Neurosci. 2019;12:216. doi:10.3389/fnmol.2019.00216
180. Vicuña L, Strochlic DE, Latremoliere A, et al. The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell-derived leukocyte elastase. Nat Med. 2015;21(5):518–523. doi:10.1038/nm.3852
181. Mizuno T, Zhang G, Takeuchi H, et al. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J. 2008;22(6):1797–1806. doi:10.1096/fj.07-099499
182. Tan PH, Ji J, Yeh CC, Ji RR. Interferons in Pain and Infections: emerging Roles in Neuro-Immune and Neuro-Glial Interactions. Front Immunol. 2021;12:783725. doi:10.3389/fimmu.2021.783725
183. Donnelly CR, Jiang C, Andriessen AS, et al. STING controls nociception via type I interferon signalling in sensory neurons. Nature. 2021;591(7849):275–280. doi:10.1038/s41586-020-03151-1
184. Fargali S, Sadahiro M, Jiang C, et al. Role of neurotrophins in the development and function of neural circuits that regulate energy homeostasis. J Mol Neurosci. 2012;48(3):654–659. doi:10.1007/s12031-012-9790-9
185. Labuz D, Schreiter A, Schmidt Y, Brack A, Machelska H. T lymphocytes containing β-endorphin ameliorate mechanical hypersensitivity following nerve injury. Brain Behav Immun. 2010;24(7):1045–1053. doi:10.1016/j.bbi.2010.04.001
186. Basso L, Garnier L, Bessac A, et al. T-lymphocyte-derived enkephalins reduce Th1/Th17 colitis and associated pain in mice. J Gastroenterol. 2018;53(2):215–226. doi:10.1007/s00535-017-1341-2
187. Boué J, Basso L, Cenac N, et al. Endogenous regulation of visceral pain via production of opioids by colitogenic CD4(+) T cells in mice. Gastroenterology. 2014;146(1):166–175. doi:10.1053/j.gastro.2013.09.020
188. Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6(9):671–682. doi:10.1038/nrn1746
189. Haberberger RV, Barry C, Dominguez N, Matusica D. Human Dorsal Root Ganglia. Front Cell Neurosci. 2019;13:271. doi:10.3389/fncel.2019.00271
190. Hanani M, Spray DC. Emerging importance of satellite glia in nervous system function and dysfunction. Nat Rev Neurosci. 2020;21(9):485–498. doi:10.1038/s41583-020-0333-z
191. Pannese E, Ledda M, Cherkas PS, Huang TY, Hanani M. Satellite cell reactions to axon injury of sensory ganglion neurons: increase in number of gap junctions and formation of bridges connecting previously separate perineuronal sheaths. Anat Embryol (Berl). 2003;206(5):337–347. doi:10.1007/s00429-002-0301-6
192. Dublin P, Hanani M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav Immun. 2007;21(5):592–598. doi:10.1016/j.bbi.2006.11.011
193. Hanani M, Blum E, Liu S, Peng L, Liang S. Satellite glial cells in dorsal root ganglia are activated in streptozotocin-treated rodents. J Cell Mol Med. 2014;18(12):2367–2371. doi:10.1111/jcmm.12406
194. Song J, Ying Y, Wang W, et al. The role of P2X7R/ERK signaling in dorsal root ganglia satellite glial cells in the development of chronic postsurgical pain induced by skin/muscle incision and retraction (SMIR). Brain Behav Immun. 2018;69:180–189. doi:10.1016/j.bbi.2017.11.011
195. Takeda M, Tanimoto T, Kadoi J, et al. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain. 2007;129(1–2):155–166. doi:10.1016/j.pain.2006.10.007
196. Takeda M, Takahashi M, Matsumoto S. Contribution of the activation of satellite glia in sensory ganglia to pathological pain. Neurosci Biobehav Rev. 2009;33(6):784–792. doi:10.1016/j.neubiorev.2008.12.005
197. Huang LY, Neher E. Ca(2+)-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. 1996;17(1):135–145. doi:10.1016/s0896-6273(00)80287-1
198. Li J, Vause CV, Durham PL. Calcitonin gene-related peptide stimulation of nitric oxide synthesis and release from trigeminal ganglion glial cells. Brain Res. 2008;1196:22–32. doi:10.1016/j.brainres.2007.12.028
199. Matsuka Y, Neubert JK, Maidment NT, Spigelman I. Concurrent release of ATP and substance P within Guinea pig trigeminal ganglia in vivo. Brain Res. 2001;915(2):248–255. doi:10.1016/s0006-8993(01)
200. Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48(4):463–469. doi:10.1016/j.neures.2004.01.004
201. Zhang X, Chen Y, Wang C, Huang LYM. Neuronal somatic ATP release triggers neuron-satellite glial cell communication in dorsal root ganglia. Proc Natl Acad Sci U S A. 2007;104(23):9864–9869. doi:10.1073/pnas.0611048104
202. Berger UV, Hediger MA. Distribution of the glutamate transporters GLAST and GLT-1 in rat circumventricular organs, meninges, and dorsal root ganglia. J Comparative Neurol. 2000;421(3):385–399. doi:10.1002/(SICI)1096-9861(20000605)421:3<385::
203. Castillo C, Norcini M, Martin Hernandez LA, Correa G, Blanck TJJ, Recio-Pinto E. Satellite glia cells in dorsal root ganglia express functional NMDA receptors. Neuroscience. 2013;240:135–146. doi:10.1016/j.neuroscience.2013.02.031
204. Marriott I. The role of tachykinins in central nervous system inflammatory responses. Front Biosci. 2004;9:2153–2165. doi:10.2741/1377
205. Neubert JK, Maidment NT, Matsuka Y, Adelson DW, Kruger L, Spigelman I. Inflammation-induced changes in primary afferent-evoked release of substance P within trigeminal ganglia in vivo. Brain Res. 2000;871(2):181–191. doi:10.1016/s0006-8993(00)
206. Urits I, Jones MR, Gress K, et al. CGRP Antagonists for the Treatment of Chronic Migraines: a Comprehensive Review. Curr Pain Headache Rep. 2019;23(5):29. doi:10.1007/s11916-019-0768-y
207. Filippov AK, Fernández-Fernández JM, Marsh SJ, Simon J, Barnard EA, Brown DA. Activation and inhibition of neuronal G protein-gated inwardly rectifying K(+) channels by P2Y nucleotide receptors. Mol Pharmacol. 2004;66(3):468–477. doi:10.1124/mol.66.3
208. Chen Y, Zhang X, Wang C, Li G, Gu Y, Huang LYM. Activation of P2X7 receptors in glial satellite cells reduces pain through downregulation of P2X3 receptors in nociceptive neurons. Proc Natl Acad Sci U S A. 2008;105(43):16773–16778. doi:10.1073/pnas.0801793105
209. Xiang Z, Bo X, Burnstock G. Localization of ATP-gated P2X receptor immunoreactivity in rat sensory and sympathetic ganglia. Neurosci Lett. 1998;256(2):105–108. doi:10.1016/s0304-3940(98)00774-5
210. Dubový P, Jancálek R, Klusáková I, Svízenská I, Pejchalová K. Intra- and extraneuronal changes of immunofluorescence staining for TNF-alpha and TNFR1 in the dorsal root ganglia of rat peripheral neuropathic pain models. Cell Mol Neurobiol. 2006;26(7–8):1205–1217. doi:10.1007/s10571-006-9006-3
211. Jang Y, Kim M, Hwang SW. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J Neuroinflammation. 2020;17:30. doi:10.1186/s12974-020-1703-1
212. Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine. 2004;29(10):1082–1088. doi:10.1097/00007632-200405150-00006
213. Jin X, Gereau RW. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006;26(1):246–255. doi:10.1523/JNEUROSCI.3858-05.2006
214. Li M, Shi J, Tang JR, et al. Effects of complete Freund’s adjuvant on immunohistochemical distribution of IL-1beta and IL-1R I in neurons and glia cells of dorsal root ganglion. Acta Pharmacol Sin. 2005;26(2):192–198. doi:10.1111/j.1745-7254.2005.00522.x
215. Liu B, Li H, Brull SJ, Zhang JM. Increased sensitivity of sensory neurons to tumor necrosis factor alpha in rats with chronic compression of the lumbar ganglia. J Neurophysiol. 2002;88(3):1393–1399. doi:10.1152/jn.2002.88.3.1393
216. Dahl G, Qiu F, Wang J. The bizarre pharmacology of the ATP release channel pannexin1. Neuropharmacology. 2013;75:583–593. doi:10.1016/j.neuropharm.2013.02.019
217. Feldman-Goriachnik R, Belzer V, Hanani M. Systemic inflammation activates satellite glial cells in the mouse nodose ganglion and alters their functions. Glia. 2015;63(11):2121–2132. doi:10.1002/glia.22881
218. Hanstein R, Hanani M, Scemes E, Spray DC. Glial pannexin1 contributes to tactile hypersensitivity in a mouse model of orofacial pain. Sci Rep. 2016;6:38266. doi:10.1038/srep38266
219. Zhang Y, Laumet G, Chen SR, Hittelman WN, Pan HL. Pannexin-1 Up-regulation in the Dorsal Root Ganglion Contributes to Neuropathic Pain Development. J Biol Chem. 2015;290(23):14647–14655. doi:10.1074/jbc.M115.650218
220. Vit JP, Ohara PT, Bhargava A, Kelley K, Jasmin L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J Neurosci. 2008;28(16):4161–4171. doi:10.1523/JNEUROSCI.5053-07.2008
221. Cherkas PS, Huang TY, Pannicke T, Tal M, Reichenbach A, Hanani M. The effects of axotomy on neurons and satellite glial cells in mouse trigeminal ganglion. Pain. 2004;110(1–2):290–298. doi:10.1016/j.pain.2004.04.007
222. Huang TY, Belzer V, Hanani M. Gap junctions in dorsal root ganglia: possible contribution to visceral pain. Eur J Pain. 2010;14(1):
223. Ledda M, Blum E, De Palo S, Hanani M. Augmentation in gap junction-mediated cell coupling in dorsal root ganglia following sciatic nerve neuritis in the mouse. Neuroscience. 2009;164(4):1538–1545. doi:10.1016/j.neuroscience.2009.09.038
224. Suadicani SO, Cherkas PS, Zuckerman J, Smith DN, Spray DC, Hanani M. Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron Glia Biol. 2010;6(1):43–51. doi:10.1017/S1740925X09990408
225. Warwick RA, Hanani M. The contribution of satellite glial cells to chemotherapy-induced neuropathic pain. Eur J Pain. 2013;17(4):571–580. doi:10.1002/j.1532-2149.2012.00219.x
226. Warwick RA, Hanani M. Involvement of aberrant calcium signalling in herpetic neuralgia. Exp Neurol. 2016;277:10–18. doi:10.1016/j.expneurol.2015.12.002
227. Zhang H, Mei X, Zhang P, et al. Altered functional properties of satellite glial cells in compressed spinal ganglia. Glia. 2009;57(15):1588–1599. doi:10.1002/glia.20872
228. Ydens E, Amann L, Asselbergh B, et al. Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat Neurosci. 2020;23(5):676–689. doi:10.1038/s41593-020-0618-6
229. Zigmond RE, Echevarria FD. Macrophage biology in the peripheral nervous system after injury. Prog Neurobiol. 2019;173:102–121. doi:10.1016/j.pneurobio.2018.12.001
230. Peng J, Gu N, Zhou L, et al. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun. 2016;7:12029. doi:10.1038/ncomms12029
231. Domoto R, Sekiguchi F, Tsubota M, Kawabata A. Macrophage as a Peripheral Pain Regulator. Cells. 2021;10(8):1881. doi:10.3390/cells10081881
232. Geraghty T, Winter DR, Miller RJ, Miller RE, Malfait AM. Neuroimmune interactions and osteoarthritis pain: focus on macrophages. PR9. 2021;6(1):e892. doi:10.1097/PR9.0000000000000892
233. Siouti E, Andreakos E. The many facets of macrophages in rheumatoid arthritis. Biochem Pharmacol. 2019;165:152–169. doi:10.1016/j.bcp.2019.03.029
234. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol. 2016;12(8):472–485. doi:10.1038/nrrheum.2016.91
235. Gerlag DM, Tak PP. Novel approaches for the treatment of rheumatoid arthritis: lessons from the evaluation of synovial biomarkers in clinical trials. Best Pract Res Clin Rheumatol. 2008;22(2):311–323. doi:10.1016/j.berh.2008.02.002
236. Wu CL, Harasymowicz NS, Klimak MA, Collins KH, Guilak F. The role of macrophages in osteoarthritis and cartilage repair. Osteoarthritis Cartilage. 2020;28(5):544–554. doi:10.1016/j.joca.2019.12.007
237. Zhang H, Cai D, Bai X. Macrophages regulate the progression of osteoarthritis. Osteoarthritis Cartilage. 2020;28(5):555–561. doi:10.1016/j.joca.2020.01.007
238. Sekiguchi F, Domoto R, Nakashima K, et al. Paclitaxel-induced HMGB1 release from macrophages and its implication for peripheral neuropathy in mice: evidence for a neuroimmune crosstalk. Neuropharmacology. 2018;141:201–213. doi:10.1016/j.neuropharm.2018.08.040
239. Sekiguchi F, Kawabata A. Role of HMGB1 in Chemotherapy-Induced Peripheral Neuropathy. Int J Mol Sci. 2020;22(1):E367. doi:10.3390/ijms22010367
240. Tsujita R, Tsubota M, Sekiguchi F, Kawabata A. Role of high-mobility group box 1 and its modulation by thrombomodulin/thrombin axis in neuropathic and inflammatory pain. Br J Pharmacol. 2021;178(4):798–812. doi:10.1111/bph.15091
241. Kotaka M, Saito Y, Kato T, et al. A placebo-controlled, double-blind, randomized study of recombinant thrombomodulin (ART-123) to prevent oxaliplatin-induced peripheral neuropathy. Cancer Chemother Pharmacol. 2020;86(5):607–618. doi:10.1007/s00280-020-04135-8
242. Oliveira-Fusaro MC, Gregory NS, Kolker SJ, Rasmussen L, Allen LAH, Sluka KA. P2X4 Receptors on Muscle Macrophages Are Required for Development of Hyperalgesia in an Animal Model of Activity-Induced Muscle Pain. Mol Neurobiol. 2020;57(4):1917–1929. doi:10.1007/s12035-019-01852-x
243. Ulmann L, Hirbec H, Rassendren F. P2X4 receptors mediate PGE2 release by tissue-resident macrophages and initiate inflammatory pain. EMBO J. 2010;29(14):2290–2300. doi:10.1038/emboj.2010.126
244. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11(3):201–212. doi:10.1038/nri2938
245. Lucrezi JD, Burns TJ, Matesic DF, Oldham CD, May SW. Inhibition of JNK and p38 MAPK phosphorylation by 5-(acetylamino)-4-oxo-6-phenyl-2-hexenoic acid methyl ester and 4-phenyl-butenoic acid decreases substance P-induced TNF-α upregulation in macrophages. Int Immunopharmacol. 2014;21(1):44–50. doi:10.1016/j.intimp.2014.04.007
246. Sun J, Ramnath RD, Zhi L, Tamizhselvi R, Bhatia M. Substance P enhances NF-κB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am J Physiol Cell Physiol. 2008;294(6):C1586–C1596. doi:10.1152/ajpcell.00129.2008
247. Kwon MJ, Shin HY, Cui Y, et al. CCL2 Mediates Neuron-Macrophage Interactions to Drive Proregenerative Macrophage Activation Following Preconditioning Injury. J Neurosci. 2015;35(48):15934–15947. doi:10.1523/JNEUROSCI.1924-15.2015
248. Huang ZZ, Li D, Liu CC, et al. CX3CL1-mediated macrophage activation contributed to paclitaxel-induced DRG neuronal apoptosis and painful peripheral neuropathy. Brain Behav Immun. 2014;40:155–165. doi:10.1016/j.bbi.2014.03.014
249. Simeoli R, Montague K, Jones HR, et al. Exosomal cargo including microRNA regulates sensory neuron to macrophage communication after nerve trauma. Nat Commun. 2017;8(1):1778. doi:10.1038/s41467-017-01841-5
250. van der Vlist M, Raoof R, Willemen HLDM, et al. Macrophages transfer mitochondria to sensory neurons to resolve inflammatory pain. Neuron. 2022;110(4):613–626.e9. doi:10.1016/j.neuron.2021.11.020
251. Chen O, Donnelly CR, Ji RR. Regulation of pain by neuro-immune interactions between macrophages and nociceptor sensory neurons. Curr Opin Neurobiol. 2020;62:17–25. doi:10.1016/j.conb.2019.11.006
252. Ebbinghaus M, Uhlig B, Richter F, et al. The role of interleukin-1β in arthritic pain: main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum. 2012;64(12):3897–3907. doi:10.1002/art.34675
253. Jeevakumar V, Al Sardar AK, Mohamed F, Smithhart CM, Price T, Dussor G. IL-6 induced upregulation of T-type Ca2+ currents and sensitization of DRG nociceptors is attenuated by MNK inhibition. J Neurophysiol. 2020;124(1):274–283. doi:10.1152/jn.00188.2020
254. Ma W, St-Jacques B, Duarte PC. Targeting pain mediators induced by injured nerve-derived COX2 and PGE2 to treat neuropathic pain. Expert Opin Ther Targets. 2012;16(6):527–540. doi:10.1517/14728222.2012.680955
255. Dilek N, Papapetropoulos A, Toliver-Kinsky T, Szabo C. Hydrogen sulfide: an endogenous regulator of the immune system. Pharmacol Res. 2020;161:105119. doi:10.1016/j.phrs.2020.105119
256. Kawabata A, Ishiki T, Nagasawa K, et al. Hydrogen sulfide as a novel nociceptive messenger. Pain. 2007;132(1–2):74–81. doi:10.1016/j.pain.2007.01.026
257. Terada Y, Fujimura M, Nishimura S, Tsubota M, Sekiguchi F, Kawabata A. Roles of Cav3.2 and TRPA1 channels targeted by hydrogen sulfide in pancreatic nociceptive processing in mice with or without acute pancreatitis. J Neurosci Res. 2015;93(2):361–369. doi:10.1002/jnr.23490
258. Labuz D, Celik MÖ, Seitz V, Machelska H. Interleukin-4 Induces the Release of Opioid Peptides from M1 Macrophages in Pathological Pain. J Neurosci. 2021;41(13):2870–2882. doi:10.1523/JNEUROSCI.3040-20.2021
259. Shen KF, Zhu HQ, Wei XH, et al. Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp Neurol. 2013;247:466–475. doi:10.1016/j.expneurol.2013.01.018
260. Rossi-Semerano L, Fautrel B, Wendling D, et al. Tolerance and efficacy of off-label anti-interleukin-1 treatments in France: a nationwide survey. Orphanet J Rare Dis. 2015;10(1):19. doi:10.1186/s13023-015-0228-7
261. Tan AL, Marzo-Ortega H, O’Connor P, Fraser A, Emery P, McGonagle D. Efficacy of anakinra in active ankylosing spondylitis: a clinical and magnetic resonance imaging study. Ann Rheum Dis. 2004;63(9):1041–1045. doi:10.1136/ard.2004.020800
262. Federici S, Martini A, Gattorno M. The Central Role of Anti-IL-1 Blockade in the Treatment of Monogenic and Multi-Factorial Autoinflammatory Diseases. Front Immunol. 2013;4:351. doi:10.3389/fimmu.2013.00351
263. Kalliolias GD, Liossis C. The future of the IL-1 receptor antagonist anakinra: from rheumatoid arthritis to adult-onset Still’s disease and systemic-onset juvenile idiopathic arthritis. Expert Opin Investig Drugs. 2008;17(3):349–359. doi:10.1517/13543784.17.3.349
264. McGonagle D, Tan AL, Shankaranarayana S, Madden J, Emery P, McDermott MF. Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann Rheum Dis. 2007;66(12):1683–1684. doi:10.1136/ard.2007.073759
265. Saag KG, Khanna PP, Keenan RT, et al. A Randomized, Phase II Study Evaluating the Efficacy and Safety of Anakinra in the Treatment of Gout Flares. Arthritis Rheumatol. 2021;73(8):1533–1542. doi:10.1002/art.41699
266. Terkeltaub R, Sundy JS, Schumacher HR, et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis. 2009;68(10):1613–1617. doi:10.1136/ard.2009.108936
267. Bresnihan B, Cobby M. Clinical and radiological effects of anakinra in patients with rheumatoid arthritis. Rheumatology. 2003;42(Suppl 2):ii22–28. doi:10.1093/rheumatology/keg329
268. Furst DE. Anakinra: review of recombinant human interleukin-I receptor antagonist in the treatment of rheumatoid arthritis. Clin Ther. 2004;26(12):1960–1975. doi:10.1016/j.clinthera.2004.12.019
269. Nikfar S, Saiyarsarai P, Tigabu BM, Abdollahi M. Efficacy and safety of interleukin-1 antagonists in rheumatoid arthritis: a systematic review and meta-analysis. Rheumatol Int. 2018;38(8):1363–1383. doi:10.1007/s00296-018-4041-1
270. Kraus VB, Birmingham J, Stabler TV, et al. Effects of intraarticular IL1-Ra for acute anterior cruciate ligament knee injury: a randomized controlled pilot trial (NCT00332254). Osteoarthritis Cartilage. 2012;20(4):271–278. doi:10.1016/j.joca.2011.12.009
271. Helyes Z, Tékus V, Szentes N, et al. Transfer of complex regional pain syndrome to mice via human autoantibodies is mediated by interleukin-1–induced mechanisms. PNAS. 2019;116(26):13067–13076. doi:10.1073/pnas.1820168116
272. Callhoff J, Sieper J, Weiß A, Zink A, Listing J. Efficacy of TNFα blockers in patients with ankylosing spondylitis and non-radiographic axial spondyloarthritis: a meta-analysis. Ann Rheum Dis. 2015;74(6):1241–1248. doi:10.1136/annrheumdis-2014-205322
273. Czókolyová M, Pusztai A, Végh E, et al. Changes of Metabolic Biomarker Levels upon One-Year Anti-TNF-α Therapy in Rheumatoid Arthritis and Ankylosing Spondylitis: associations with Vascular Pathophysiology. Biomolecules. 2021;11(10):1535. doi:10.3390/biom11101535
274. Ferrari M, Onuoha SC, Fossati-Jimack L, et al. Novel Bispecific Antibody for Synovial-Specific Target Delivery of Anti-TNF Therapy in Rheumatoid Arthritis. Front Immunol. 2021;12:640070. doi:10.3389/fimmu.2021.640070
275. Mantravadi S, Ogdie A, Kraft WK. Tumor necrosis factor inhibitors in psoriatic arthritis. Expert Rev Clin Pharmacol. 2017;10(8):899–910. doi:10.1080/17512433.2017.1329009
276. Papamichael K, Lin S, Moore M, Papaioannou G, Sattler L, Cheifetz AS. Infliximab in inflammatory bowel disease. Ther Adv Chronic Dis. 2019;10:2040622319838443. doi:10.1177/2040622319838443
277. Shim MR. Efficacy of TNF inhibitors in advanced ankylosing spondylitis with total spinal fusion: case report and review of literature. OARRR. 2019;11:173–177. doi:10.2147/OARRR.S212456
278. Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with Adalimumab plus methotrexate versus methotrexate alone or Adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54(1):26–37. doi:10.1002/art.21519
279. Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363(9410):675–681. doi:10.1016/S0140-6736(04)
280. Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun. 2010;11:180–210. doi:10.1159/000289205
281. Freeman BJC, Ludbrook GL, Hall S, et al. Randomized, double-blind, placebo-controlled, trial of transforaminal epidural etanercept for the treatment of symptomatic lumbar disc herniation. Spine. 2013;38(23):1986–1994. doi:10.1097/01.brs.0000435140.61593.4c
282. Okoro T, Tafazal SI, Longworth S, Sell PJ. Tumor necrosis alpha-blocking agent (etanercept): a triple blind randomized controlled trial of its use in treatment of sciatica. J Spinal Disord Tech. 2010;23(1):74–77. doi:10.1097/BSD.0b013e31819afdc4
283. Pimentel DC, El Abd O, Benyamin RM, et al. Anti-tumor necrosis factor antagonists in the treatment of low back pain and radiculopathy: a systematic review and meta-analysis. Pain Physician. 2014;17(1):E27–44.
284. Tobinick E. Perispinal etanercept: a new therapeutic paradigm in neurology. Expert Rev Neurother. 2010;10(6):985–1002. doi:10.1586/ern.10.52
285. Ahmed R, Soliman N. Serum interleukin-6 in primary fibromyalgia syndrome patients: impact on disease burden, severity, quality of life and sleep. Egyptian Rheumatologist. 2022;44(1):15–18. doi:10.1016/j.ejr.2021.07.004
286. Karshikoff B, Martucci KT, Mackey S. Relationship Between Blood Cytokine Levels, Psychological Comorbidity, and Widespreadness of Pain in Chronic Pelvic Pain. Front Psychiatry. 2021;12:548.
287. Sebba A. Pain: a Review of Interleukin-6 and Its Roles in the Pain of Rheumatoid Arthritis. OARRR. 2021;13:31–43. doi:10.2147/OARRR.S291388
288. Teodorczyk-Injeyan JA, Triano JJ, Injeyan HS. Nonspecific Low Back Pain: inflammatory Profiles of Patients With Acute and Chronic Pain. Clin J Pain. 2019;35(10):818–825. doi:10.1097/AJP.0000000000000745
289. Zhou YQ, Liu Z, Liu ZH, et al. Interleukin-6: an emerging regulator of pathological pain. J Neuroinflammation. 2016;13:141. doi:10.1186/s12974-016-0607-6
290. Emery P, Rondon J, Parrino J, et al. Safety and tolerability of subcutaneous sarilumab and intravenous tocilizumab in patients with rheumatoid arthritis. Rheumatology. 2019;58(5):849–858. doi:10.1093/rheumatology/key361
291. Fleischmann R, Genovese MC, Lin Y, et al. Long-term safety of sarilumab in rheumatoid arthritis: an integrated analysis with up to 7 years’ follow-up. Rheumatology. 2020;59(2):292–302. doi:10.1093/rheumatology/kez265
292. Hoffman E, Rahat MA, Feld J, et al. Effects of Tocilizumab, an Anti-Interleukin-6 Receptor Antibody, on Serum Lipid and Adipokine Levels in Patients with Rheumatoid Arthritis. IJMS. 2019;20(18):4633. doi:10.3390/ijms20184633
293. Ohtori S, Miyagi M, Eguchi Y, et al. Efficacy of epidural administration of anti-interleukin-6 receptor antibody onto spinal nerve for treatment of sciatica. Eur Spine J. 2012;21(10):2079–2084. doi:10.1007/s00586-012-2183-5
294. Sainoh T, Orita S, Miyagi M, et al. Single intradiscal injection of the interleukin-6 receptor antibody tocilizumab provides short-term relief of discogenic low back pain; prospective comparative cohort study. J Orthopaedic Sci. 2016;21(1):2–6. doi:10.1016/j.jos.2015.10.005
295. Sainoh T, Orita S, Miyagi M, et al. Improvements in Intractable Lumbar and LowerExtremity Symptoms after Systemic Administration of Tocilizumab, an Anti-interleukin-6 Receptor Antibody. Asian Spine J. 2021. doi:10.31616/asj.2020.0283
296. Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia. 2002;22(1):54–61. doi:10.1046/j.1468-2982.2002.00310.x
297. Dodick DW, Ashina M, Brandes JL, et al. ARISE: a Phase 3 randomized trial of erenumab for episodic migraine. Cephalalgia. 2018;38(6):1026–1037. doi:10.1177/0333102418759786
298. Goadsby PJ, Reuter U, Hallström Y, et al. A Controlled Trial of Erenumab for Episodic Migraine. N Engl J Med. 2017;377(22):2123–2132. doi:10.1056/NEJMoa1705848
299. Tepper S, Ashina M, Reuter U, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16(6):425–434. doi:10.1016/S1474-4422(17)
300. Traynor K. FDA approves licensing of erenumab-aooe to prevent migraine. Am J Health Syst Pharm. 2018;75(13):929–930. doi:10.2146/news180044
301. Biohaven Pharmaceuticals, Inc. BHV3000-202: phase 2: a Double-Blind, Placebo Controlled, Crossover Trial of BHV-3000 (Rimegepant) for Treatment Refractory Trigeminal Neuraligia. 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT03941834.
302. Jonzon MD. A Placebo-Controlled, Double-Blind, Randomized, Proof-of-Concept Study to Evaluate the Efficacy and Tolerability of Erenumab in Patients With Trigeminal Neuralgia. 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT04054024.
303. Teva Branded Pharmaceutical Products R&D, Inc. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Proof of Concept Study of the Efficacy and Safety of Fremanezumab for Treatment of Patients With Fibromyalgia; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT03965091.
304. Kalliomäki J, Attal N, Jonzon B, et al. A randomized, double-blind, placebo-controlled trial of a chemokine receptor 2 (CCR2) antagonist in posttraumatic neuralgia. Pain. 2013;154(5):761–767. doi:10.1016/j.pain.2013.02.003
305. Monnet E, Choy EH, McInnes I, et al. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: a phase II study. Ann Rheum Dis. 2020;79(3):316–323. doi:10.1136/annrheumdis-2019-216487
306. Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB. D1520C00001 Study Team. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann Rheum Dis. 2012;71(10):1630–1635. doi:10.1136/annrheumdis-2011-143578
307. Garrido-Mesa N, Zarzuelo A, Gálvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169(2):337–352. doi:10.1111/bph.12139
308. Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54(2):258–265. doi:10.1016/j.jaad.2005.10.004
309. Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166(12):7527–7533. doi:10.4049/jimmunol.166.12.7527
310. Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26(16):4308–4317. doi:10.1523/JNEUROSCI.0003-06.2006
311. Owolabi SA, Saab CY. Fractalkine and minocycline alter neuronal activity in the spinal cord dorsal horn. FEBS Lett. 2006;580(18):4306–4310. doi:10.1016/j.febslet.2006.06.087
312. Martinez V, Szekely B, Lemarié J, et al. The efficacy of a glial inhibitor, minocycline, for preventing persistent pain after lumbar discectomy: a randomized, double-blind, controlled study. Pain. 2013;154(8):1197–1203. doi:10.1016/j.pain.2013.03.028
313. Vanelderen P, Van Zundert J, Kozicz T, et al. Effect of minocycline on lumbar radicular neuropathic pain: a randomized, placebo-controlled, double-blind clinical trial with amitriptyline as a comparator. Anesthesiology. 2015;122(2):399–406. doi:10.1097/ALN.0000000000000508
314. Curtin CM, Kenney D, Suarez P, et al. A Double-Blind Placebo Randomized Controlled Trial of Minocycline to Reduce Pain After Carpal Tunnel and Trigger Finger Release. J Hand Surg Am. 2017;42(3):166–174. doi:10.1016/j.jhsa.2016.12.011
315. Syngle A, Verma I, Krishan P, Garg N, Syngle V. Minocycline improves peripheral and autonomic neuropathy in type 2 diabetes: MIND study. Neurol Sci. 2014;35(7):1067–1073. doi:10.1007/s10072-014-1647-2
316. Möller T, Bard F, Bhattacharya A, et al. Critical data-based re-evaluation of minocycline as a putative specific microglia inhibitor. Glia. 2016;64(10):1788–1794. doi:10.1002/glia.23007
317. DeLeo J, Toth L, Schubert P, Rudolphi K, Kreutzberg GW. Ischemia-induced neuronal cell death, calcium accumulation, and glial response in the hippocampus of the Mongolian gerbil and protection by propentofylline (HWA 285). J Cereb Blood Flow Metab. 1987;7(6):745–751. doi:10.1038/jcbfm.1987.129
318. Tawfik VL, Nutile-McMenemy N, LaCroix-Fralish ML, DeLeo JA. Efficacy of propentofylline, a glial modulating agent, on existing mechanical allodynia following peripheral nerve injury. Brain Behav Immun. 2007;21(2):238–246. doi:10.1016/j.bbi.2006.07.001
319. Landry RP, Jacobs VL, Romero-Sandoval EA, DeLeo JA. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Exp Neurol. 2012;234(2):340–350. doi:10.1016/j.expneurol.2011.11.006
320. Mizuno T, Kurotani T, Komatsu Y, et al. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004;46(3):404–411. doi:10.1016/j.neuropharm.2003.09.009
321. Kwok YH, Swift JE, Gazerani P, Rolan P. A double-blind, randomized, placebo-controlled pilot trial to determine the efficacy and safety of ibudilast, a potential glial attenuator, in chronic migraine. J Pain Res. 2016;9:899–907. doi:10.2147/JPR.S116968
322. Johnson JL, Kwok YH, Sumracki NM, et al. Glial Attenuation With Ibudilast in the Treatment of Medication Overuse Headache: a Double-Blind, Randomized, Placebo-Controlled Pilot Trial of Efficacy and Safety. Headache. 2015;55(9):1192–1208. doi:10.1111/head.12655
323. Rolan P, Hutchinson M, Johnson K. Ibudilast: a review of its pharmacology, efficacy and safety in respiratory and neurological disease. Expert Opin Pharmacother. 2009;10(17):2897–2904. doi:10.1517/14656560903426189
324. Metz VE, Jones JD, Manubay J, et al. Effects of Ibudilast on the Subjective, Reinforcing, and Analgesic Effects of Oxycodone in Recently Detoxified Adults with Opioid Dependence. Neuropsychopharmacol. 2017;42(9):1825–1832. doi:10.1038/npp.2017.70
325. Burnette EM, Baskerville WA, Grodin EN, Ray LA. Ibudilast for alcohol use disorder: study protocol for a phase II randomized clinical trial. Trials. 2020;21:779. doi:10.1186/s13063-020-04670-y
326. Grodin EN, Bujarski S, Towns B, et al. Ibudilast, a neuroimmune modulator, reduces heavy drinking and alcohol cue-elicited neural activation: a randomized trial. Transl Psychiatry. 2021;11(1):355. doi:10.1038/s41398-021-01478-5
327. Eker HE, Cok OY, Aribogan A, Arslan G. Management of neuropathic pain with methylprednisolone at the site of nerve injury. Pain Med. 2012;13(3):443–451. doi:10.1111/j.1526-4637.2011.01323.x
328. Kotani N, Kushikata T, Hashimoto H, et al. Intrathecal methylprednisolone for intractable postherpetic neuralgia. N Engl J Med. 2000;343(21):1514–1519. doi:10.1056/NEJM200011233432102
329. Riew KD, Yin Y, Gilula L, et al. The effect of nerve-root injections on the need for operative treatment of lumbar radicular pain. A prospective, randomized, controlled, double-blind study. J Bone Joint Surg Am. 2000;82(11):1589–1593. doi:10.2106/00004623-200011000-00012
330. Friedly J, Chan L, Deyo R. Increases in lumbosacral injections in the Medicare population: 1994 to 2001. Spine. 2007;32(16):1754–1760. doi:10.1097/BRS.0b013e3180b9f96e
331. Friedly JL, Comstock BA, Turner JA, et al. A Randomized Trial of Epidural Glucocorticoid Injections for Spinal Stenosis. N Eng J Med. 2014;371(1):11–21. doi:10.1056/NEJMoa1313265
332. Manchikanti L, Singh V, Pampati V, Smith HS, Hirsch JA. Analysis of growth of interventional techniques in managing chronic pain in the Medicare population: a 10-year evaluation from 1997 to 2006. Pain Physician. 2009;12(1):9–34.
333. Botwin KP, Gruber RD, Bouchlas CG, et al. Fluoroscopically guided lumbar transformational epidural steroid injections in degenerative lumbar stenosis: an outcome study. Am J Phys Med Rehabil. 2002;81(12):898–905. doi:10.1097/00002060-200212000-00003
334. Ciocon JO, Galindo-Ciocon D, Amaranath L, Galindo D. Caudal epidural blocks for elderly patients with lumbar canal stenosis. J Am Geriatr Soc. 1994;42(6):593–596. doi:10.1111/j.1532-5415.1994.tb06855.x
335. Hashemi M, Dadkhah P, Taheri M, Ghasemi M, Hosseinpour A. Lumbar Transforaminal Epidural Steroid Injection in Patients with Lumbar Radicular Pain; Outcome Results of 2-Year Follow-Up. Bull Emerg Trauma. 2019;7(2):144–149. doi:10.29252/beat-070209
336. Klessinger S. Diagnostic Value of Transforaminal Injections of Steroids in Recurrent Disc Herniations. Spine Neurosurgery. 2014;2013:548.
337. Smith CC, McCormick ZL, Mattie R, MacVicar J, Duszynski B, Stojanovic MP. The Effectiveness of Lumbar Transforaminal Injection of Steroid for the Treatment of Radicular Pain: a Comprehensive Review of the Published Data. Pain Med. 2020;21(3):472–487. doi:10.1093/pm/pnz160
338. Johansson A, Bennett GJ. Effect of local methylprednisolone on pain in a nerve injury model. A pilot study. Reg Anesth. 1997;22(1):59–65. doi:10.1016/s1098-7339(06)80057-x
339. Li H, Xie W, Strong JA, Zhang JM. Systemic antiinflammatory corticosteroid reduces mechanical pain behavior, sympathetic sprouting, and elevation of proinflammatory cytokines in a rat model of neuropathic pain. Anesthesiology. 2007;107(3):469–477. doi:10.1097/01.anes.0000278907.37774.8d
340. Takeda K, Sawamura S, Sekiyama H, Tamai H, Hanaoka K. Effect of methylprednisolone on neuropathic pain and spinal glial activation in rats. Anesthesiology. 2004;100(5):1249–1257. doi:10.1097/00000542-200405000-00029
341. Ammendolia C, Stuber K, de Bruin LK, et al. Nonoperative treatment of lumbar spinal stenosis with neurogenic claudication: a systematic review. Spine. 2012;37(10):E609–616. doi:10.1097/BRS.0b013e318240d57d
342. Dong F, Xie W, Strong JA, Zhang JM. Mineralocorticoid receptor blocker eplerenone reduces pain behaviors in vivo and decreases excitability in small-diameter sensory neurons from local inflamed dorsal root ganglia in vitro. Anesthesiology. 2012;117(5):1102–1112. doi:10.1097/ALN.0b013e3182700383
343. Ibrahim SIA, Xie W, Strong JA, Tonello R, Berta T, Zhang JM. Mineralocorticoid Antagonist Improves Glucocorticoid Receptor Signaling and Dexamethasone Analgesia in an Animal Model of Low Back Pain. Front Cell Neurosci. 2018;12:453. doi:10.3389/fncel.2018.00453
344. Joëls M, Karst H, DeRijk R, de Kloet ER. The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 2008;31(1):1–7. doi:10.1016/j.tins.2007.10.005
345. Ye L, Xie W, Strong JA, Zhang JM. Blocking the mineralocorticoid receptor improves effectiveness of steroid treatment for low back pain in rats. Anesthesiology. 2014;121(3):632–643. doi:10.1097/ALN.0000000000000277
346. Knotkova H, Hamani C, Sivanesan E, et al. Neuromodulation for chronic pain. Lancet. 2021;397(10289):2111–2124. doi:10.1016/S0140-6736(21)
347. Caylor J, Reddy R, Yin S, et al. Spinal cord stimulation in chronic pain: evidence and theory for mechanisms of action. Bioelectronic Med. 2019;5(1):12. doi:10.1186/s42234-019-0023-1
348. Deer T, Slavin KV, Amirdelfan K, et al. Success Using Neuromodulation With BURST (SUNBURST) Study: results From a Prospective, Randomized Controlled Trial Using a Novel Burst Waveform. Neuromodulation. 2018;21(1):56–66. doi:10.1111/ner.12698
349. Kapural L, Yu C, Doust MW, et al. Novel 10-kHz High-frequency Therapy (HF10 Therapy) Is Superior to Traditional Low-frequency Spinal Cord Stimulation for the Treatment of Chronic Back and Leg Pain: the SENZA-RCT Randomized Controlled Trial. Anesthesiology. 2015;123(4):851–860. doi:10.1097/ALN.0000000000000774
350. Kemler MA, De Vet HCW, Barendse GAM, Van Den Wildenberg FAJM, Van Kleef M. The Effect of Spinal Cord Stimulation in Patients with Chronic Reflex Sympathetic Dystrophy: two Years’ Follow-up of the Randomized Controlled Trial. Ann Neurol. 2004;55(1):13–18. doi:10.1002/ana.10996
351. Kumar K, Taylor RS, Jacques L, et al. Spinal cord stimulation versus conventional medical management for neuropathic pain: a multicentre randomised controlled trial in patients with failed back surgery syndrome. Pain. 2007;132(1):179–188. doi:10.1016/j.pain.2007.07.028
352. Petersen EA, Stauss TG, Scowcroft JA, et al. Effect of High-frequency (10-kHz) Spinal Cord Stimulation in Patients With Painful Diabetic Neuropathy: a Randomized Clinical Trial. JAMA Neurol. 2021;78(6):687–698. doi:10.1001/jamaneurol.2021.0538
353. Vallejo R, Tilley DM, Cedeño DL, Kelley CA, DeMaegd M, Benyamin R. Genomics of the Effect of Spinal Cord Stimulation on an Animal Model of Neuropathic Pain. Neuromodulation. 2016;19(6):576–586. doi:10.1111/ner.12465
354. Sato KL, Johanek LM, Sanada LS, Sluka KA. Spinal cord stimulation reduces mechanical hyperalgesia and glial cell activation in animals with neuropathic pain. Anesth Analg. 2014;118(2):464–472. doi:10.1213/ANE.0000000000000047
355. Sivanesan E, Stephens KE, Huang Q, et al. Spinal cord stimulation prevents paclitaxel-induced mechanical and cold hypersensitivity and modulates spinal gene expression in rats. Pain Rep. 2019;4(5):e785. doi:10.1097/PR9.0000000000000785
356. Stephens KE, Chen Z, Sivanesan E, et al. RNA-seq of spinal cord from nerve-injured rats after spinal cord stimulation. Mol Pain. 2018;14:1744806918817429. doi:10.1177/1744806918817429
357. Cedeño DL, Smith WJ, Kelley CA, Vallejo R. Spinal cord stimulation using differential target multiplexed programming modulates neural cell-specific transcriptomes in an animal model of neuropathic pain. Mol Pain. 2020;16:1744806920964360. doi:10.1177/1744806920964360
358. Smith WJ, Cedeño DL, Thomas SM, Kelley CA, Vetri F, Vallejo R. Modulation of microglial activation states by spinal cord stimulation in an animal model of neuropathic pain: comparing high rate, low rate, and differential target multiplexed programming. Mol Pain. 2021;17:1744806921999013. doi:10.1177/1744806921999013
359. Vallejo R, Kelley CA, Gupta A, Smith WJ, Vallejo A, Cedeño DL. Modulation of neuroglial interactions using differential target multiplexed spinal cord stimulation in an animal model of neuropathic pain. Mol Pain. 2020;16:1744806920918057. doi:10.1177/1744806920918057
360. Fishman MA, Calodney A, Kim P, et al. Prospective, Multicenter Feasibility Study to Evaluate Differential Target Multiplexed Spinal Cord Stimulation Programming in Subjects With Chronic Intractable Back Pain With or Without Leg Pain. Pain Pract. 2020;20(7):761–768. doi:10.1111/papr.12908
361. Ohemeng KK, Parham K. Vagal Nerve Stimulation: indications, Implantation, and Outcomes. Otolaryngol Clin North Am. 2020;53(1):127–143. doi:10.1016/j.otc.2019.09.008
362. Falvey A, Metz CN, Tracey KJ, Pavlov VA. Peripheral nerve stimulation and immunity: the expanding opportunities for providing mechanistic insight and therapeutic intervention. Int Immunol. 2022;34(2):107–118. doi:10.1093/intimm/dxab068
363. Tracey KJ. The inflammatory reflex. Nature. 2002;420(6917):853–859. doi:10.1038/nature01321
364. Koopman FA, Chavan SS, Miljko S, et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2016;113(29):8284–8289. doi:10.1073/pnas.1605635113
365. Lerman I, Hauger R, Sorkin L, et al. Noninvasive Transcutaneous Vagus Nerve Stimulation Decreases Whole Blood Culture-Derived Cytokines and Chemokines: a Randomized, Blinded, Healthy Control Pilot Trial. Neuromodulation. 2016;19(3):283–290. doi:10.1111/ner.12398
366. Yuan H, Silberstein SD. Vagus Nerve Stimulation and Headache. Headache. 2017;57(Suppl 1):29–33. doi:10.1111/head.12721
367. Covid 19 - gammacore. Available from: https://www.gammacore.com/covid-19/.
368. Genovese MC, Gaylis NB, Sikes D, et al. Safety and efficacy of neurostimulation with a miniaturised vagus nerve stimulation device in patients with multidrug-refractory rheumatoid arthritis: a two-stage multicentre, randomised pilot study. Lancet Rheumatol. 2020;2(9):e527–e538. doi:10.1016/S2665-9913(20)
369. Sinniger V, Pellissier S, Fauvelle F, et al. A 12-month pilot study outcomes of vagus nerve stimulation in Crohn’s disease. Neurogastroenterol Motil. 2020;32(10):e13911. doi:10.1111/nmo.13911
370. Buchheit T, Huh Y, Maixner W, Cheng J, Ji RR. Neuroimmune modulation of pain and regenerative pain medicine. J Clin Invest. 2020;130(5):2164–2176. doi:10.1172/JCI134439
371. Ali M, Mohamed A, Ahmed HE, Malviya A, Atchia I. The use of ultrasound-guided platelet-rich plasma injections in the treatment of Hip osteoarthritis: a systematic review of the literature. J Ultrason. 2018;18(75):332–337. doi:10.15557/JoU.2018.0048
372. Anitua E, Padilla S. Biologic therapies to enhance intervertebral disc repair. Regen Med. 2018;13(1):55–72. doi:10.2217/rme-2017-0111
373. Di Martino A, Di Matteo B, Papio T, et al. Platelet-Rich Plasma Versus Hyaluronic Acid Injections for the Treatment of Knee Osteoarthritis: results at 5 Years of a Double-Blind, Randomized Controlled Trial. Am J Sports Med. 2019;47(2):347–354. doi:10.1177/0363546518814532
374. Mishra AK, Skrepnik NV, Edwards SG, et al. Efficacy of platelet-rich plasma for chronic tennis elbow: a double-blind, prospective, multicenter, randomized controlled trial of 230 patients. Am J Sports Med. 2014;42(2):463–471. doi:10.1177/0363546513494359
375. Chen G, Park CK, Xie RG, Ji RR. Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-β secretion. J Clin Invest. 2015;125(8):3226–3240. doi:10.1172/JCI80883
376. de Witte SFH, Luk F, Sierra Parraga JM, et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells. 2018;36(4):602–615. doi:10.1002/stem.2779
377. Hart R, Safi A, Komzák M, Jajtner P, Puskeiler M, Hartová P. Platelet-rich plasma in patients with tibiofemoral cartilage degeneration. Arch Orthop Trauma Surg. 2013;133(9):1295–1301. doi:10.1007/s00402-013-1782-x
378. Pujol JP, Chadjichristos C, Legendre F, et al. Interleukin-1 and transforming growth factor-beta 1 as crucial factors in osteoarthritic cartilage metabolism. Connect Tissue Res. 2008;49(3):293–297. doi:10.1080/03008200802148355
379. Anwar MA, Shah M, Kim J, Choi S. Recent clinical trends in Toll‐like receptor targeting therapeutics. Med Res Rev. 2019;39(3):1053–1090. doi:10.1002/med.21553
380. Brown PD. Ongoing trials with matrix metalloproteinase inhibitors. Expert Opin Investig Drugs. 2000;9(9):2167–2177. doi:10.1517/13543784.9.9.2167
381. Tap WD, Gelderblom H, Palmerini E, et al. Pexidartinib for advanced tenosynovial giant cell tumor: results of the randomized phase 3 ENLIVEN study. Lancet. 2019;394(10197):478–487. doi:10.1016/S0140-6736(19)30764-0
382. Zhang W, Luo H, Zhu Z. The role of P2X4 receptors in chronic pain: a potential pharmacological target. Biomed Pharmacotherapy. 2020;129:110447. doi:10.1016/j.biopha.2020.110447
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.