Back to Journals » Drug Design, Development and Therapy » Volume 19
The Role of Mitochondrial Dysfunction in Sepsis-Associated Acute Kidney Injury: A Narrative Review
Authors Liu J, Zheng J, Xu Y, Jiang Y ![]()
Received 15 July 2025
Accepted for publication 29 September 2025
Published 24 October 2025 Volume 2025:19 Pages 9545—9558
DOI https://doi.org/10.2147/DDDT.S553838
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Tamer Ibrahim
Junfei Liu, Jingmin Zheng, Yinghe Xu, Yongpo Jiang
Department of Critical Care Medicine, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Taizhou, 317000, People’s Republic of China
Correspondence: Yinghe Xu, Email [email protected] Yongpo Jiang, Email [email protected]
Abstract: Sepsis-associated acute kidney injury (SA-AKI) is one of the most frequent complications of sepsis and one of the leading causes of acute kidney injury (AKI), posing a significant threat to patient survival. The pathogenesis of SA-AKI has been linked to mitochondrial dysfunction, according to emerging evidence. Mitochondria serve as the primary energy-producing organelles in cells, and mitochondrial dysfunction can result in insufficient renal energy supply, oxidative stress, and inflammatory responses, all of which can both initiate and exacerbate SA-AKI. Thus, elucidating the role of mitochondrial dysfunction in SA-AKI is of critical importance. Starting from the fundamental mechanisms of mitochondrial dysfunction, this review draws upon more than 130 relevant publications through August 2025, comprehensively summarizes the role of mitochondrial dysfunction in the pathophysiological processes of SA-AKI and the drugs for treating SA-AKI through the improvement of mitochondrial function, while also discussing in detail the potential therapeutic applications of several well-characterized therapeutic targets in SA-AKI, providing important insights into the diagnosis and treatment of this disease.
Keywords: sepsis, SA-AKI, mitochondrial dysfunction, therapeutic
Introduction
Sepsis-associated acute kidney injury (SA-AKI) is characterized as the onset of acute kidney injury (AKI) within a seven-day timeframe following the onset of sepsis.1 This definition is based on the Kidney Disease: Improving Global Outcomes (KDIGO) criteria and the Sepsis-3 Consensus.2,3 SA-AKI represents one of the most prevalent complications arising from sepsis.4 AKI occurs in approximately 60% of sepsis patients, and the mortality rate in sepsis patients with AKI is 2 to 3 times higher than in those without AKI.5,6 Meanwhile, sepsis is the most frequent cause influencing the development of AKI, and existing data indicate that sepsis-associated AKI (SA-AKI) is associated with a higher risk of mortality and a reduced likelihood of renal recovery compared to other forms of AKI.7 At present, the management of SA-AKI patients is largely dependent on early identification and supportive care, due to the absence of effective targeted treatments, which complicates clinical rescue efforts.8 Therefore, research into novel and effective therapeutic targets and approaches to enhance patient prognosis is crucial.
Currently, the pathophysiological mechanisms underlying SA-AKI are recognized to primarily involve inflammation, microcirculatory dysfunction, and metabolic reprogramming.9 There is a growing body of research suggesting mitochondrial dysfunction plays an integral role in several of these processes.10,11 Also, despite constituting less than 1% of total body weight, the kidneys account for approximately 7% of daily resting energy expenditure.12 Their substantial energy demand is further evidenced by their high mitochondrial density and oxygen consumption rate, both of which are second only to those of the heart.13,14 Thus, any impairment in mitochondrial function significantly impacts renal function. However, the degree to which mitochondrial dysfunction contributes to the progression of SA-AKI remains to be elucidated.
Although previous reviews have explored the role of mitochondria in SA-AKI, most have focused on specific mechanistic aspects of mitochondrial dysfunction and lacked an integrated discussion of therapeutic strategies and their clinical translation potential. In this review, we not only provide a comprehensive overview of the multidimensional mechanisms of mitochondrial dysfunction in SA-AKI but also delve into targeted drug and clinical translation strategies based on these mechanisms, with the aim of providing guidance for novel drug development for this disease.
Influence of Mitochondrial Dysfunction on SA-AKI
It is widely known that mitochondria are the powerhouses of the cell, but they perform many other functions besides oxidative phosphorylation. They play an essential role in calcium signaling, production of reactive oxygen species, antiviral defenses, and cell death.15 Mitochondrial dysfunction manifests as a reduced ability to produce ATP and is accompanied by an increased generation of free radicals, notably reactive oxygen species (ROS).16 Mitochondrial dysfunction, encompassing defects in mitochondrial integrity, metabolic and regulatory functions, and bioenergetics, is strongly linked to the development and prognosis of a wide variety of human diseases.17
Abnormal Mitochondrial Membrane Permeability
A mitochondrial membrane is made up of two membranes with distinct structural and functional characteristics, the inner mitochondrial membrane (IMM) and the outer mitochondrial membrane (OMM). The integrity of these membranes is crucial for preserving the unique mitochondrial compartments and preventing the release of mitochondrial components into the cytoplasm, where they can function as apoptotic factors or damage-associated molecular patterns (DAMPs).18
Cell Death and SA-AKI
In diverse cell death mechanisms, mitochondria play an essential role.19 Notably, mitochondrial outer membrane permeabilization (MOMP) and mitochondrial permeability transition (MPT) are two vital processes, acting as key markers of apoptosis and necrosis, respectively.20,21 The BCL-2 family contains pro-apoptotic and anti-apoptotic members that regulate mitochondrial apoptosis.22 Under pro-apoptotic stress conditions, activated BAX and BAK proteins create pores in the outer mitochondrial membrane, a critical event termed MOMP.23 MOMP triggers the release of apoptotic factors such as cytochrome c and SMAC into the cytoplasm.24 Cytochrome c binds to apoptosis protease activating factor 1 (APAF1), leading to the formation of apoptosomes in the cytosol.25 This complex subsequently activates procaspase-9 and caspase-3, propelling the cell into the execution phase of apoptosis.25
Mitochondria, beyond their well-known role in apoptosis, are crucially involved in various cell death pathways.26 When intracellular mitochondrial Ca2+ overload occurs, especially under oxidative stress and/or ATP depletion, it triggers a sudden increase in IMM permeability by opening the mitochondrial permeability transition pore (MPTP).21 This permeability transition allows solutes up to 1.5 kDa to diffuse across the IMM, resulting in mitochondrial depolarization, uncoupling, swelling, ATP depletion, and ultimately, necrotic cell death.27 Furthermore, impaired mitochondrial membrane barrier function has been linked to other cell death pathways, such as pyroptosis and ferroptosis.28,29
Tan et al demonstrated that Pectolinarigenin (PEC), a natural flavonoid, mitigates septic acute kidney injury (SAKI) by upregulating Bcl-2 protein expression and inhibiting the apoptotic protein BAX in a mouse model of SAKI.30 Similarly, Li et al reported that the lipid mediator Maresin 1 (MaR1) confers protection against LPS-induced septic AKI by downregulating BAX and cleaved caspase-3 expression while upregulating Bcl-2 in damaged renal tissues of SAKI mice.31 Overall, these findings suggest that inhibiting cell death via the mitochondrial pathway may serve as a potential therapeutic target for septic AKI.
Inflammatory Responses and SA-AKI
Through the induction of lysogenic forms of cell death, mitochondrial membrane permeability contributes to inflammation by releasing intracellular components that function as DAMPs in the surrounding microenvironment.32 Additionally, disruptions in mitochondrial membrane barrier function can trigger inflammatory signals independent of cell death, including mitochondrial DNA (mtDNA), cardiolipin, ROS, and ATP.18,32 Among these, mtDNA has garnered significant attention as a DAMP due to its similarity to bacterial DNA, serving as a “danger signal” upon release.33 In the following sections, we will focus on mtDNA in both the cytosol and circulation.
Mitochondria possess their own genome, referred to as mtDNA. Unlike nuclear DNA, mtDNA maintains a hypomethylated CpG motif, akin to bacterial and viral DNA.34 This characteristic enables mtDNA to be recognized by traditional “foreign” DNA-sensing pattern recognition receptors (PRRs).35,36
In the context of SAKI, animal studies have revealed that plasma mtDNA levels are elevated in SAKI model mice, correlating with increased severity of kidney injury and poor prognosis.37 Tsuji et al observed that both plasma and urinary mtDNA levels were consistently elevated in SAKI model mice, aligning with the degree of inflammation and renal damage.38 In a clinical investigation, Elisabeth et al examined renal tissue biopsies from fourteen patients with septic renal injury and twelve control subjects, discovering diminished mitochondrial DNA integrity and heightened damage levels, which positively correlated with the expression of renal injury markers.39 These studies underscore the association between mtDNA damage and release with the severity of renal injury and adverse outcomes in SAKI, suggesting that cytosolic and circulating mtDNA could be potential markers and therapeutic targets.
Mitochondrial Quality Control Barriers
Mitochondrial quality control (MQC) encompasses many processes, including mitophagy, mitochondrial biogenesis, and mitochondrial dynamics.40 These mechanisms involve the selective elimination of damaged mitochondria, their replacement via biosynthesis, the redistribution of mitochondrial components through fusion, and the segregation of defective mitochondria by fission before mitophagy.40,41 A breakdown in MQC can lead to mitochondrial dysfunction, ultimately resulting in cell death, tissue damage, and organ failure.42
Mitochondrial Biogenesis and SA-AKI
The mitochondrial biosynthesis process involves the synthesis of mitochondrial membranes and mitochondria-encoded proteins, mitochondrial DNA (mtDNA) replication, and the integration of mitochondrial proteins encoded by nuclear genes.43 TFAM-PGC1-NRF1/2 is central to this biosynthetic process. Activation of PGC-1α triggers a cascade of nuclear transcription factors, such as nuclear respiratory factor 1 (NRF-1) and NRF-2, leading to the upregulation of TFAM expression.44 A key regulator of mitochondrial biosynthesis, PGC1, regulates the expression of nuclear genes essential for this process by influencing numerous transcription factors.45 SIRT1 and AMPK are major modulators of PGC1α activity, activating it through acetylation and phosphorylation, respectively.46 TFAM, on the other hand, is indispensable for the transcription and replication of mtDNA, binding to these sequences and playing a key role in new mitochondrial formation.47
Mitochondrial biosynthesis is pivotal in the pathogenesis of SA-AKI. Tran et al revealed that in a mouse model of SAKI caused by lipopolysaccharide (LPS) or cecal ligation and puncture (CLP), the expression of PGC1 in the kidneys was inversely correlated with renal damage and normalized after renal repair. Moreover, the study indicated that systemic or tubule-specific knockout of PGC1α hindered renal recovery in LPS-induced SAKI mice, highlighting the necessity of PGC1α induction for recovery from septic acute kidney injury.48 Similarly, Smith et al reported diminished PGC1α expression correlated with impaired renal function in an LPS-induced SAKI mouse model and demonstrated that neutralizing TNF-α mitigated the suppression of PGC1α expression.49 Pei et al demonstrated that high-concentration hydrogen inhalation alleviates SA-AKI by promoting mitochondrial biogenesis through the reversal of downregulated PGC-1α, Nrf2, and TFAM expression in kidney tissues of CLP-induced mice.50 These findings highlight the essential function of mitochondrial biosynthesis in SAKI and propose possible targets for upcoming treatment strategies. Nevertheless, excessive mitochondrial biosynthesis can result in structural abnormalities, diminished mitochondrial function, and increased production of ROS, which could have detrimental effects.51
Mitophagy and SA-AKI
Mitophagy is a selective autophagic pathway essential for MQC. During this process, damaged mitochondria are engulfed by double-membrane autophagosomes and degraded by lysosomes.52 A ubiquitin-dependent pathway and a ubiquitin-independent pathway are the two principal mechanisms of mitophagy.53 The ubiquitin-dependent mechanisms include PTEN-induced mitophagy mediated by the putative kinase protein 1 (PINK1)-Parkin pathway and other ubiquitin-mediated processes independent of Parkin, with the PINK1/Parkin pathway being the most extensively studied.54 Ubiquitin-independent mechanisms comprise receptor-mediated and lipid-mediated mitophagy.55 Notably, several receptors have been implicated in mitophagy, such as Nip3-like protein X (NIX), FUN14 domain-containing protein 1 (FUNDC1), and BCL2-interacting protein 3 (BNIP3).56 LC3-interacting region (LIR) motifs enable these receptors to interact with LC3 independently of ubiquitin, therefore facilitating phagocytosis of defective mitochondria.57 Mitophagy is crucial for mitochondrial quality control by eliminating dysfunctional mitochondria that not only fail to produce ATP and other biosynthetic products but also generate excessive ROS, which can induce apoptosis.58,59
In SA-AKI, mitophagy has a vital role to play. Sunahara et al revealed that in SAKI model mice, the renal LC3-II/LC3-I ratio increased initially at 6–8 hours and then significantly declined by 24 hours, and that rapamycin-induced autophagy was associated with improved renal function.60 Wang et al demonstrated that in a SAKI mouse model, pink1 or park2 knockout mice showed a higher level of renal injury and apoptosis than wild-type mice, suggesting that the PINK1/PARK2-mediated mitophagy pathway plays a critical role in SAKI.61 Similarly, Gao et al revealed that SIRT1 activation upregulated Parkin-dependent mitophagy, alleviating mitochondrial dysfunction and NLRP3 inflammatory responses in SAKI.62 Recent studies have revealed that modulating the Nrf2/PINK1/Parkin axis can regulate mitophagy levels, offering a promising therapeutic strategy for SA-AKI.63,64 In the realm of ubiquitin-independent mitophagy, Pan et al showed that lacking the mitophagy receptor BNIP3L/Nix accumulated damaged mitochondria and overactivated platelet inflammasomes, exacerbating SAKI in a CLP mouse model. Their findings suggest that targeting TREM-1 and NLRP3/BNIP3L in platelets may provide a novel therapeutic strategy for SAKI.65 In conclusion, the aforementioned studies suggest that the mitophagy pathway mediated by Parkin plays an essential role in SAKI. Inhibition of Parkin suppresses mitophagy, thereby exacerbating SAKI, while activation of Parkin mitigates mitochondrial dysfunction and kidney injury. Concurrently, emerging research highlights the significance of non-ubiquitin-dependent mitophagy receptors in SAKI. These findings suggest that therapeutic strategies aimed at enhancing mitophagy could hold promise for the treatment of SAKI.
Mitochondrial Dynamics and SA-AKI
Mitochondrial dynamics, the balance between the fusion and fission of the mitochondrial network, is essential for maintaining mitochondrial structure and function.66 Fusion involves the merging of multiple mitochondria into a larger organelle, which aids in the distribution of mitochondrial DNA and metabolites; in contrast, fission divides a mitochondrion into smaller organelles, a process involved in apoptosis, mitochondrial biosynthesis, and mitophagy.67,68 The interplay between fusion and fission endows mitochondria with several advantages, including efficient transport, optimized oxidative phosphorylation, and homogenization of the mitochondrial population.69 Mfn1, Mfn2, and Opa1 are involved in fusion, and Drp1 is involved in fission, which is regulated by a conserved family of large GTPases known as dynamin-related proteins (DRPs).70 The dysregulation of these proteins is associated with a variety of pathologies linked to abnormal mitochondrial fusion or fission.71
In a murine model of CLP-induced SAKI, Liu et al observed an aberrant mitochondrial dynamic balance skewed towards fission, accompanied by a concomitant rise in mitochondria-associated apoptosis throughout SAKI progression.72 In response to procyanidin B2 treatment (PBC2), this balance is effectively shifted towards fusion, mitochondrial integrity was enhanced, mitochondrial-mediated apoptosis was reduced, and renal dysfunction, tubular cell vacuolization, and oxidative stress were alleviated in SAKI mice.73 Concurrently, studies employing an endotoxin-induced HK-2 cell model indicated that human trypsin inhibitor (UTI) may preserve mitochondrial function by fostering fusion and curbing fission, thus mitigating apoptosis.74 As a result, these studies indicate that SA-AKI induces mitochondrial fission/fusion imbalances to favor fission, facilitating the removal of damaged mitochondrial components through redistribution and mitophagy. However, excessive fission in the later stages exacerbates oxidative stress and cell death. Consequently, strategies aimed at enhancing mitochondrial fusion may provide protective effects against SA-AKI.
Synergistic Role of Mitochondrial Quality Control in SA-AKI
Mitochondrial quality control mechanisms play an essential role in maintaining cellular homeostasis during SA-AKI. These processes do not act in isolation but function within an integrated network characterized by functional coupling and signaling crosstalk. Mitochondrial fission facilitates the targeting of damaged mitochondria for mitophagic clearance, while mitochondrial fusion promotes biogenesis through shared regulatory signals such as PGC-1α activation.75,76 The MQC network undergoes dynamic temporal changes across different phases of SA-AKI. During the early acute injury phase, severe stress triggers pathological mitochondrial hyperfission. Although mitophagy is activated to remove fragmented mitochondria, its capacity is often overwhelmed, leading to the accumulation of damaged organelles. In contrast, during the later recovery phase, enhancing mitochondrial fusion and biogenesis becomes crucial. Concurrently, moderate mitophagy helps eliminate impaired mitochondria, creating space for newly generated ones. The coordinated balance among these three mechanisms during this phase supports structural repair and functional recovery.
Mitochondrial Energy and Metabolic Abnormalities
Mitochondria, frequently termed the cell’s powerhouses, play a key role in bioenergetic and biosynthetic processes, serving as the principal locus for energy conversion and ATP synthesis.77 These specialized organelles function as metabolic hubs and signaling platforms, participating in a multitude of essential cellular activities. These include the production of ATP through OXPHOS, the oxidation of fatty acids, calcium homeostasis, the production and regulation of reactive oxygen species, and the modulation of innate immunity.78
Oxidative Stress and SA-AKI
Mitochondria orchestrate critical bioenergetic processes, including the tricarboxylic acid cycle, the β-oxidation of fatty acids, and the electron transport system (ETS), alongside the biosynthesis of amino acids, nucleotides, and lipids, where reactive ROS are generated by products.79 ROS are primarily produced by mitochondria, which account for nearly 90% of cellular ROS.80 Oxidative stress is caused by an imbalance between ROS production and antioxidant defenses, which can impair mitochondrial respiratory chain function, alter membrane permeability, and increase mtDNA heterogeneity, thereby weakening mitochondrial defenses.81 It is well known that ROS may adversely affect cellular biomolecules, especially DNA, and can lead to mitochondrial dysfunction, apoptosis, cellular damage, and various diseases to progress.82
In SA-AKI, Slikke et al reported an up-regulation of mRNA expression for oxidative damage markers and the presence of renal tubular epithelial cells’ cytosolic oxidative DNA damage in patients with SA-AKI compared to controls. Their cellular experiments indicated that LPS-induced HUVEC exposure for 48 hours led to increased manganese superoxide dismutase (mnSOD) expression, decreased mitochondrial transcription factor A (TFAM) expression, and elevated mtDNA damage.39 Oxidative stress, driven by increased ROS release, initiates inflammasome formation and triggers an inflammatory cascade.83,84 Zhao et al reported that SIRT3 inhibits NLRP3 inflammasomes by reducing ROS production, mitigating oxidative stress, and decreasing IL-1 and IL-18 production, thus providing protection against SA-AKI.85 Similarly, although they target different initial pathways, quercetin, irbesartan, and cilostazol have each been shown to attenuate renal histopathological damage and improve renal function in SA-AKI models via their shared anti-inflammatory and antioxidant activities.86–88 Lowes et al suggested that in a rat model of acute sepsis, mitochondria-targeted antioxidants decreased mitochondrial damage, organ dysfunction (liver, kidney, heart), and the inflammatory response.89 Collectively, these studies suggest that SA-AKI leads to oxidative stress and that ROS production is increased, and that reducing ROS or using mitochondria-targeted antioxidants presents a promising therapeutic approach against SA-AKI.
Metabolic Reprogramming and SA-AKI
Metabolic programming rearrangements represent adaptive shifts in cellular energy production and consumption pathways, enabling cells to achieve optimal performance under specific environmental conditions.90 Cells primarily convert nutrients into ATP through two key metabolic pathways: OXPHOS and glycolysis. Most cells use OXPHOS, which occurs in the mitochondrial matrix, as their primary metabolic pathway for producing ATP since it is more efficient than glycolysis at producing energy.91 Mitochondria, being central hubs for metabolic activities such as the tricarboxylic acid (TCA) cycle, play a key role in mammalian cellular bioenergetics.92
The metabolic transition in biphasic metabolic reprogramming from OXPHOS to a glycolytic phenotype is orchestrated by the mammalian target of rapamycin complex 1 (mTORC1) via the activation of HIF-1α and the Akt/mTORC1 pathway.93 This activation can inhibit the conversion of pyruvate to acetyl-coenzyme A and its subsequent entry into mitochondria.93 The ensuing restoration of dominant OXPHOS metabolism is facilitated by the synergistic activation of AMPK, PGC-1α, Sirt1, and Sirt6, which potentially mitigates the influence of HIF-1α on glycolytic enzyme expression.91
In SA-AKI, early metabolic reprogramming of cellular pathways plays a critical role in safeguarding the kidney from further damage and influencing tissue repair outcomes, thereby potentially preventing progression to fibrosis and chronic organ dysfunction.94 During SA-AKI, renal tubular epithelial cells exhibit biphasic metabolic reprogramming, initially shifting from OXPHOS to a glycolytic phenotype, followed by a subsequent return to predominantly OXPHOS metabolism. This metabolic transition mitigates excessive reactive ROS production while ensuring adequate energy supply to avert cell death.11,95 Waltz et al, through animal experiments involving CLP in septic mice, demonstrated that gas chromatography/mass spectrometry analysis of renal biopsies taken 8 hours post-CLP revealed increased glycolytic intermediates and decreased TCA intermediates, indicative of a metabolic shift towards glycolysis.96 Similarly, an early LPS-induced mouse model of AKI reported suppressed gene expression of OXPHOS mediators.48 Furthermore, Yang et al found that treatment of sepsis model mice with pergolide, a potent PKM2 inhibitor, improved host survival when administered 12 or 24 hours before, 24 hours after LPS administration, or 24–48 hours after CLP.97 Collectively, these studies suggest that an early shift towards glycolysis, followed by a late restoration of OXPHOS, is beneficial during SA-AKI.
However, this adaptive hypothesis has been significantly challenged by a series of interventional studies. Accumulating evidence demonstrates that directly inhibiting aerobic glycolysis during the early phase of sepsis improves survival and attenuates renal injury in rodents.91 This indicates that excessive activation of early glycolysis may drive pathological processes rather than merely serving as an adaptive response. The notable controversy over the net effect of early glycolytic shifts in SA-AKI may originate from the following factors: (1) Cell Type Specificity: In immune cells, aerobic glycolysis is crucial for initiating appropriate inflammatory responses and for establishing trained immunity to combat infection.93 However, in renal tubular epithelial cells, the development of trained immunity may lead to excessive responses to subsequent stimuli, thereby augmenting cellular and organ damage.98 (2) Duration: An early and transient burst of glycolysis may represent an adaptive survival strategy by rapidly generating ATP and buying time for cellular repair.99 However, a prolonged shift toward glycolytic metabolism promotes tubular atrophy, fibrosis, and subsequent progression to chronic kidney disease.100 This notion is corroborated in kidney tissues from patients with chronic kidney disease, which exhibit significantly reduced expression of OXPHOS and FAO enzymes alongside increased expression of glycolytic enzymes.101 (3) Model Variability: Animal models used across studies (eg, LPS or CLP) differ inherently in injury patterns, severity, and immune responses, leading to inconsistent manifestations of metabolic reprogramming. This implies that translating such preclinical findings to the complex setting of human septic acute kidney injury is likely to face significant challenges. Furthermore, clear guidelines are still lacking for identifying the metabolic reprogramming status in patients.
Advances in Mitochondrial Dysfunction Targeting Therapies for SA-AKI
Drugs/compounds targeting mitochondrial dysfunction in SA-AKI are summarized in Table 1. The following sections will focus on representative candidates, while others are not discussed in detail. However, before delving into specific drug treatment strategies, it is worth considering that renal mitochondria exhibit significant nephron segments heterogeneity.13 Among these segments, the proximal tubule is responsible for approximately 70% of solute reabsorption, resulting in exceptionally high energy demands. This, in turn, significantly increases its susceptibility to mitochondrial dysfunction, making it the primary site of injury in SA-AKI.102 Conversely, the medullary region inherently operates under hypoxic conditions, and its cells rely more heavily on anaerobic glycolysis for energy production. Consequently, this region demonstrates higher susceptibility to metabolic alterations.103 Therefore, prioritizing therapeutic strategies that target mitochondrial dysfunction in the proximal tubule is crucial for maximizing treatment efficacy, with precise drug delivery to this segment representing a highly promising future direction.
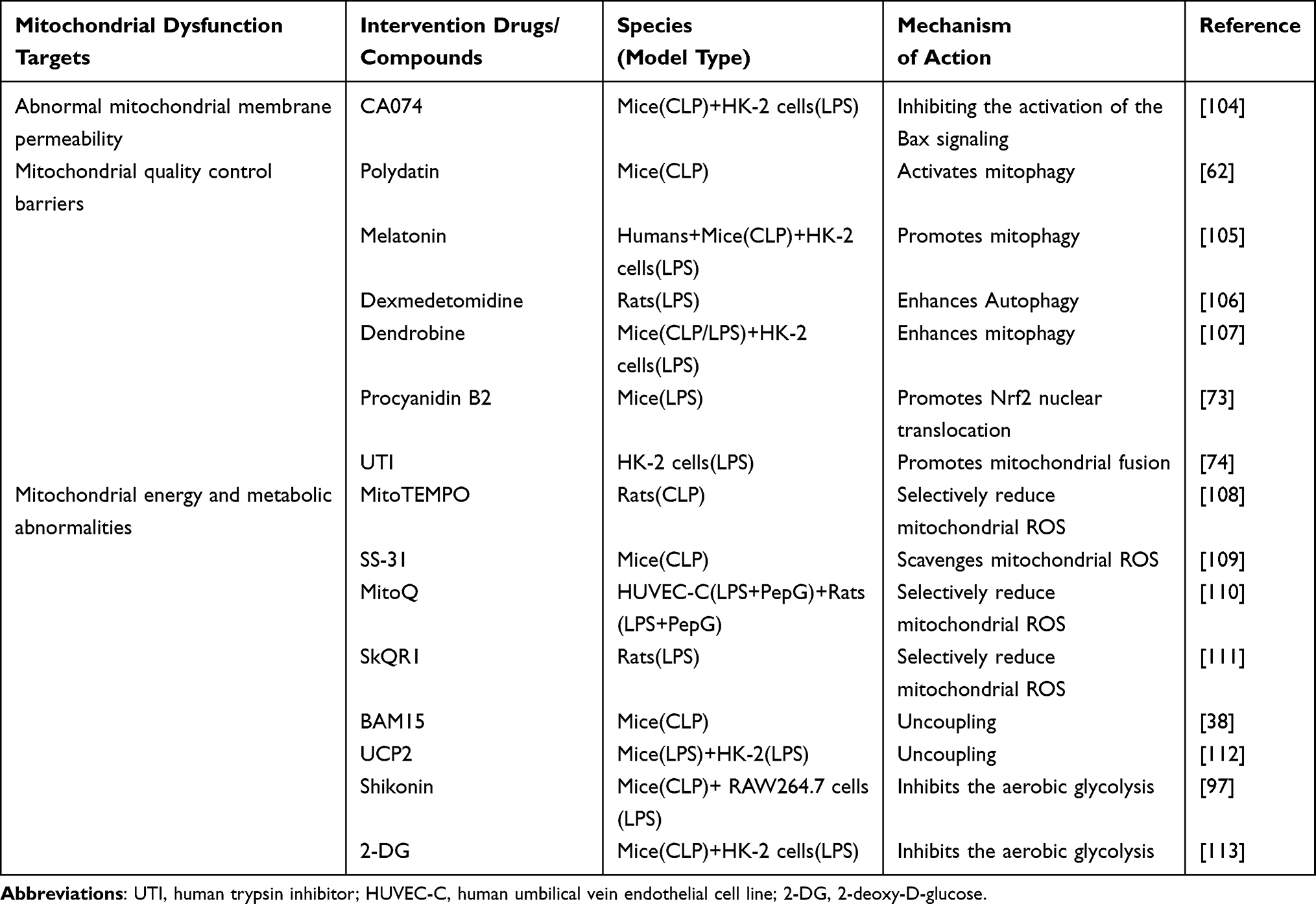 |
Table 1 The Intervention Drugs/Compounds for Mitochondrial Dysfunction in SA-AKI |
The Therapeutic Effects of Mitochondria-Targeted Antioxidants on SA-AKI
The ability of mitochondria-targeted antioxidants to penetrate the mitochondria-phospholipid bilayer and neutralize ROS at their source has attracted significant attention.114 A substantial body of research indicates that these antioxidants can mitigate mitochondrial damage and multi-organ dysfunction in sepsis.115–117 The mitochondrial antioxidant SS-31 ameliorated kidney injury in SA-AKI mice, as reported by Li et al.109 Similarly, Mito-TEMPO, a mitochondria-targeted antioxidant, was also found to decrease mitochondrial superoxide levels and alleviate SA-AKI.118 Moreover, the selective mitochondrial antioxidant MitoQ inhibited mitochondrial oxidative stress in both endothelial cell models of sepsis and in vivo models of sepsis rats, and improved renal function.110 The results of these studies highlight the promising therapeutic potential of mitochondria-targeted antioxidants for the treatment of sepsis-induced organ dysfunction. Additionally, it is worth noting that although the mitochondrial-targeted antioxidants involved in the aforementioned studies have not yet entered clinical trials for SA-AKI, most have advanced to clinical research stages in other disease areas. For example, MitoQ has completed Phase II trials in patients with Parkinson’s disease and hepatitis;119 SkQ1 has demonstrated both safety and efficacy in a Phase II clinical trial for dry eye syndrome;120 and SS-31 is currently undergoing Phase II clinical studies for conditions such as acute kidney injury and heart failure.121
However, despite encouraging laboratory and preclinical results, their clinical translation faces considerable challenges. First, although conventional triphenylphosphonium (TPP)-based mitochondrial targeted antioxidants such as MitoQ, SkQ1, and Mito-TEMPO can be selectively and efficiently accumulated in the mitochondrial matrix by leveraging the high membrane potential across the inner membrane, the buildup of high concentrations of TPP+ cations has been shown to induce mitochondrial membrane depolarization, suppress ATP synthesis, and compromise cell survival.122 Secondly, pharmacokinetic challenges remain significant for novel mitochondrial-targeted antioxidants. For instance, SS-31 circumvents cytotoxicity limitations by targeting cardiolipin on the inner mitochondrial membrane instead of relying on membrane potential, and exerts cytoprotective effects by stabilizing the electron transport chain through inhibition of cytochrome c peroxidase activity.121 However, as a small peptide, it suffers from a short half-life, susceptibility to proteolytic degradation, and a limited volume of distribution. These issues markedly reduce its bioavailability after systemic administration, making it difficult to sustain therapeutically effective concentrations in tissues over prolonged periods.123,124 Finally, additional evidence indicates that dysfunctional mitochondria exhibit altered membrane permeability and generally demonstrate a diminished capacity to actively uptake exogenous antioxidants compared to their healthy counterparts, which further obstructs effective targeting.125 Therefore, future research should focus on developing novel targeted molecules with reduced dependence on membrane potential, actively exploring novel delivery strategies based on nanotechnology or biomaterials such as hydrogels to improve their pharmacokinetic properties, and advancing tubular-specific targeting technologies to ensure efficient drug accumulation at the site of action, thereby overcoming existing challenges.
The Therapeutic Effects of Mitophagy Activators on SA-AKI
Mitophagy, essential for eliminating dysfunctional mitochondria, promotes the turnover of damaged organelles and reduces oxidative stress caused by excessive ROS.72,126 According to numerous studies, mitophagy is suppressed in AKI induced by CLP, whereas its activation offers protective benefits to proximal tubules and improves renal function.60,127,128 In this regard, Cui et al found that enhancing mitophagy with rapamycin markedly attenuated gentamicin-induced AKI in minipigs.129 Moreover, mitophagy-deficient mice experienced more severe renal dysfunction after CLP compared to their wild-type counterparts in a sepsis model.62 The current consensus holds that the activation of mitochondrial autophagy promotes the clearance of damaged mitochondria, thereby reducing the activation of mitochondrial-dependent cell death pathways caused by mitochondrial damage, as well as oxidative stress (by reducing ROS sources) and inflammatory responses.
However, despite numerous studies confirming the protective role of mitophagy activation in SA-AKI, some investigations have reported conflicting results. For instance, Wu et al revealed that mitophagy activation exacerbated renal injury in mice, implying a detrimental effect on SA-AKI.130 We speculate that this contradiction may stem from the following factors: 1) Cell type specificity. Excessive activation of mitochondrial autophagy in immune cells may disrupt their function. For example, inhibiting mitochondrial autophagy in macrophages eliminates pro-inflammatory pathway activators such as ROS and mtDNA, thereby blocking their anti-infective processes.131 Similarly, Patoli et al reported that promoting mitochondrial autophagy suppresses macrophage activation, leading to immune paralysis, impaired bacterial clearance, and reduced survival in CLP mice.13 2) Excessive activation of mitochondrial autophagy. Overactivated mitochondrial autophagy may cause severe depletion of mitochondria, resulting in ATP exhaustion and cell death, thereby exacerbating tissue injury.132 3) Timing of mitochondrial autophagy activation. As previously noted, early activation of mitochondrial autophagy in macrophages suppresses their activation. In contrast, early activation of mitochondrial autophagy in renal tubular epithelial cells during SA-AKI progression is often beneficial.107
In addition to the aforementioned controversies, the clinical translation of mitophagy activators faces several bottlenecks. First, existing activators lack renal selectivity, and systemic administration may suppress immune cell function, thereby increasing the risk of uncontrolled infection. Second, it remains challenging to quantitatively assess the extent of mitophagy activation in vivo, which hinders dose optimization and efficacy evaluation in clinical trials. The development of tubule-specific delivery systems and the identification of novel biomarkers capable of dynamically monitoring mitophagic flux are key to overcoming these translational barriers.
The Therapeutic Effects of Improved Mitochondrial Dynamics on SA-AKI
Therapeutic strategies targeting mitochondrial fusion/fission imbalance represent an active area of investigation in SA-AKI research. Current approaches are broadly divided into two categories: inhibiting pathological fission and promoting protective fusion. Inhibiting fission, targeting the key protein Drp1 is a core strategy.133 Drp1 translocates to the mitochondrial outer membrane and contracts mitochondria by forming ring-like oligomers to complete mitochondrial fission.134 Evidence suggests that mdivi-1, a novel pharmacological inhibitor of dynamin-related protein 1 (Drp1), mitigates AKI and reduces renal tubular cell apoptosis.135 In promoting integration, Liu et al reported that procyanidin B2 improves mitochondrial dynamics and diminishes SAKI-induced cell injury by promoting Nrf2 translocation.73 Furthermore, in vitro studies have shown that Sirt3 overexpression attenuates endotoxin-induced mitochondrial damage and apoptosis in renal tubular epithelial cells by enhancing OPA1-mediated mitochondrial fusion.136 Similarly, in HK-2 cells, Human trypsin inhibitors (UTIs) reduce endotoxin-induced apoptosis by promoting mitochondrial fusion and limiting division.74 To summarize, preclinical studies have confirmed that targeting key effector proteins involved in mitochondrial fusion/fission shows promising therapeutic potential. However, research in this area remains at an early stage, and its clinical translation faces multiple challenges. First, indiscriminate inhibition of fission or promotion of fusion may expand dysfunctional mitochondrial networks, thereby exacerbating oxidative stress. Second, regulatory proteins such as Drp1 are widely expressed throughout the body, making renal tubular-specific targeting an urgent delivery challenge. Finally, developing reliable biomarkers for monitoring mitochondrial dynamics in vivo will be essential for patient stratification and efficacy evaluation in clinical practice.
Conclusion and Perspective
The pathophysiology of SA-AKI is highly complex, with mitochondrial dysfunction recognized as a central mechanism. In summary, mitochondrial injury in SA-AKI presents as a cascade of interconnected events, including abnormal opening of the MOMP and the MPTP, dysregulation of mitochondrial quality control (encompassing mitophagy, biogenesis, and dynamics), ROS burst, and energy metabolism reprogramming. Therapeutic strategies targeting these mechanisms—such as mitochondria-targeted antioxidants, mitophagy activators, or dynamics modulators—have shown considerable therapeutic promise.
However, translating these promising therapeutic strategies into clinically effective treatments remains a formidable challenge. Future research must achieve breakthroughs in several key areas: (1) Model optimization: Current studies rely predominantly on rodent models, which exhibit limited translatability to clinical contexts. Future work should prioritize the validation of these mechanisms using human patient tissue samples and clinical data to bridge the gap between basic research and human pathophysiology. The adoption of advanced models that better recapitulate human disease—such as organoids and humanized mouse models—will be essential to advance translational research. (2) Development of precision-targeted therapeutic strategies: Future efforts should focus on creating clinically feasible, highly specific delivery systems capable of renal tubular-targeted intervention. At the same time, drug design should actively incorporate emerging modalities, such as small molecules, peptides, and mRNA-based therapies, to enable more precise regulation of complex mitochondrial processes. (3) Developing Patient-Stratification Biomarkers: The heterogeneity of mitochondrial dysfunction necessitates precise patient stratification in future clinical trials. There is an urgent need to develop and validate novel biomarkers capable of dynamically and non-invasively reflecting mitochondrial functional status. Such biomarkers should be employed in patient screening, dose optimization, and treatment response monitoring to provide a foundation for personalized therapeutic strategies. (4) Integrating Multi-Omics Technologies: Advanced multi-omics approaches such as single-cell sequencing, spatial transcriptomics, and metabolomics should be leveraged to deeply investigate mitochondrial functional states and interaction networks across different renal cell types in SA-AKI. Furthermore, integrating multi-omics-driven machine learning models into clinical workflows could facilitate early diagnosis and enable precision treatment for SA-AKI patients.
Abbreviations
SA-AKI, Sepsis-associated acute kidney injury; KDIGO, Kidney Disease, Improving Global Outcomes; ROS, reactive oxygen species; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; DAMPs, damage-associated molecular patterns; MOMP, mitochondrial outer membrane permeabilization; MPT, mitochondrial permeability transition; APAF1, apoptosis protease activating factor 1; MPTP, mitochondrial permeability transition pore; PEC, Pectolinarigenin; MaR1, Maresin 1; mtDNA, mitochondrial DNA; PRRs, pattern recognition receptors; NRF-1, nuclear respiratory factor 1; MQC, Mitochondrial Quality Control; LPS, lipopolysaccharide; CLP, cecal ligation and puncture; PBC2, procyanidin B2; DRPs, dynamin-related proteins; mnSOD, manganese superoxide dismutase; mRos, mitochondrial ROS; TCA, tricarboxylic acid; mTORC1, mammalian target of rapamycin complex 1; TWNDs, tungsten-based nano-dots; UCPs, uncoupling proteins; ANT, adenine nucleotide translocase; CCCP, carbonyl cyanide m-chlorophenyl hydrazone; UCP2, uncoupling protein 2; Drp1, dynamin-related protein 1; OXPHOS, oxidative phosphorylation; TPP, triphenylphosphonium.
Funding
This work was supported by The Science and Technology Project of Taizhou (23ywa47,25ywa23), The Key R&D Project of Taizhou Science and Technology Program(25ywzd01), The Medicines Health Research Fund of Zhejiang, China (2024ky1784), The National Key Research and Development Program of Zhejiang Province (2023C03083), The Joint Fund of Zhejiang Provincial Natural Science Foundation of China under Grant No. LKLY25H200001.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Graphical abstract was created with BioRender software (https://app.biorender.com/).
References
1. Zarbock A, Nadim MK, Pickkers P, et al. Sepsis-associated acute kidney injury: consensus report of the 28th acute disease quality initiative workgroup. Nat Rev Nephrol. 2023;19(6):401–417. doi:10.1038/s41581-023-00683-3
2. Kellum JA, Lameire N. Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Crit Care. 2013;17(1):204. doi:10.1186/cc11454
3. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
4. Barbar SD, Clere-Jehl R, Bourredjem A, et al. Timing of renal-replacement therapy in patients with acute kidney injury and sepsis. N Engl J Med. 2018;379(15):1431–1442. doi:10.1056/NEJMoa1803213
5. Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294(7):813–818. doi:10.1001/jama.294.7.813
6. Bagshaw SM, Uchino S, Bellomo R, et al. Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol. 2007;2(3):431–439. doi:10.2215/CJN.03681106
7. Legrand M, Bagshaw SM, Bhatraju PK, et al. Sepsis-associated acute kidney injury: recent advances in enrichment strategies, sub-phenotyping and clinical trials. Crit Care. 2024;28(1):92. doi:10.1186/s13054-024-04877-4
8. Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ. 2019;364:k4891. doi:10.1136/bmj.k4891
9. Qiao J, Cui L. Multi-omics techniques make it possible to analyze sepsis-associated acute kidney injury comprehensively. Front Immunol. 2022;13:905601. doi:10.3389/fimmu.2022.905601
10. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. 2023;23(3):159–173. doi:10.1038/s41577-022-00760-x
11. Gómez H, Kellum JA, Ronco C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat Rev Nephrol. 2017;13(3):143–151. doi:10.1038/nrneph.2016.186
12. Wang Z, Ying Z, Bosy-Westphal A, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. 2010;92(6):1369–1377. doi:10.3945/ajcn.2010.29885
13. Scholz H, Boivin FJ, Schmidt-Ott KM, et al. Kidney physiology and susceptibility to acute kidney injury: implications for renoprotection. Nat Rev Nephrol. 2021;17(5):335–349. doi:10.1038/s41581-021-00394-7
14. O’Connor PM. Renal oxygen delivery: matching delivery to metabolic demand. Clin Exp Pharmacol Physiol. 2006;33(10):961–967. doi:10.1111/j.1440-1681.2006.04475.x
15. Montes de Oca Balderas P. Mitochondria-plasma membrane interactions and communication. J Biol Chem. 2021;297(4):101164. doi:10.1016/j.jbc.2021.101164
16. Reitsema VA, Star BS, de Jager VD, van Meurs M, Henning RH, Bouma HR. Metabolic resuscitation strategies to prevent organ dysfunction in sepsis. Antioxid Redox Signal. 2019;31(2):134–152. doi:10.1089/ars.2018.7537
17. Harrington JS, Ryter SW, Plataki M, Price DR, Choi AMK. Mitochondria in health, disease, and aging. Physiol Rev. 2023;103(4):2349–2422. doi:10.1152/physrev.00058.2021
18. Vringer E, Tait SWG. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023;30(2):304–312. doi:10.1038/s41418-022-01094-w
19. Glover HL, Schreiner A, Dewson G, Tait SWG. Mitochondria and cell death. Nat Cell Biol. 2024;26(9):1434–1446. doi:10.1038/s41556-024-01429-4
20. Popgeorgiev N, Gil C, Berthenet K, Bertolin G, Ichim G. Shedding light on mitochondrial outer-membrane permeabilization and membrane potential: state of the art methods and biosensors. Semin Cell Dev Biol. 2024;156:58–65. doi:10.1016/j.semcdb.2023.07.003
21. Briston T, Selwood DL, Szabadkai G, Duchen MR. Mitochondrial permeability transition: a molecular lesion with multiple drug targets. Trends Pharmacol Sci. 2019;40(1):50–70. doi:10.1016/j.tips.2018.11.004
22. Ader NR, Hoffmann PC, Ganeva I, et al. Molecular and topological reorganizations in mitochondrial architecture interplay during Bax-mediated steps of apoptosis. Elife. 2019;8.
23. Bock FJ, Riley JS. When cell death goes wrong: inflammatory outcomes of failed apoptosis and mitotic cell death. Cell Death Differ. 2023;30(2):293–303. doi:10.1038/s41418-022-01082-0
24. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. doi:10.1016/S0092-8674(00)00008-8
25. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25(1):65–80. doi:10.1038/cdd.2017.186
26. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. doi:10.1038/s41580-019-0173-8
27. Panel M, Ruiz I, Brillet R, et al. Small-molecule inhibitors of cyclophilins block opening of the mitochondrial permeability transition pore and protect mice from hepatic ischemia/reperfusion injury. Gastroenterology. 2019;157(5):1368–1382. doi:10.1053/j.gastro.2019.07.026
28. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689. doi:10.1038/s41467-019-09397-2
29. Pedrera L, Espiritu RA, Ros U, et al. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021;28(5):1644–1657. doi:10.1038/s41418-020-00691-x
30. Tan Z, Liu Q, Chen H, et al. Pectolinarigenin alleviated septic acute kidney injury via inhibiting Jak2/Stat3 signaling and mitochondria dysfunction. Biomed Pharmacother. 2023;159:114286. doi:10.1016/j.biopha.2023.114286
31. Li J, Zhang Z, Wang L, et al. Maresin 1 attenuates lipopolysaccharide-induced acute kidney injury via inhibiting NOX4/ROS/NF-κB pathway. Front Pharmacol. 2021;12:782660. doi:10.3389/fphar.2021.782660
32. Flores-Romero H, Dadsena S, García-Sáez AJ. Mitochondrial pores at the crossroad between cell death and inflammatory signaling. Mol Cell. 2023;83(6):843–856. doi:10.1016/j.molcel.2023.02.021
33. Itagaki K, Riça I, Konecna B, et al. Role of mitochondria-derived danger signals released after injury in systemic inflammation and sepsis. Antioxid Redox Signal. 2021;35(15):1273–1290. doi:10.1089/ars.2021.0052
34. Fang C, Wei X, Wei Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell. 2016;7(1):11–16. doi:10.1007/s13238-015-0222-9
35. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi:10.1016/j.tibs.2016.09.002
36. Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. doi:10.1038/35047123
37. Tsuji N, Tsuji T, Ohashi N, Kato A, Fujigaki Y, Yasuda H. Role of mitochondrial DNA in septic AKI via toll-like receptor 9. J Am Soc Nephrol. 2016;27(7):2009–2020. doi:10.1681/ASN.2015040376
38. Tsuji N, Tsuji T, Yamashita T, et al. BAM15 treats mouse sepsis and kidney injury, linking mortality, mitochondrial DNA, tubule damage, and neutrophils. J Clin Invest. 2023;133(7). doi:10.1172/JCI152401
39. van der Slikke EC, Star BS, van Meurs M, Henning RH, Moser J, Bouma HR. Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-AKI. Crit Care. 2021;25(1):36. doi:10.1186/s13054-020-03424-1
40. Wang J, Toan S, Zhou H. Mitochondrial quality control in cardiac microvascular ischemia-reperfusion injury: new insights into the mechanisms and therapeutic potentials. Pharmacol Res. 2020;156:104771. doi:10.1016/j.phrs.2020.104771
41. Gottlieb RA, Piplani H, Sin J, et al. At the heart of mitochondrial quality control: many roads to the top. Cell Mol Life Sci. 2021;78(8):3791–3801. doi:10.1007/s00018-021-03772-3
42. Tang C, Cai J, Yin XM, Weinberg JM, Venkatachalam MA, Dong Z. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol. 2021;17(5):299–318. doi:10.1038/s41581-020-00369-0
43. Fontecha-Barriuso M, Martin-Sanchez D, Martinez-Moreno JM, et al. The role of PGC-1α and mitochondrial biogenesis in kidney diseases. Biomolecules. 2020;10(2):347. doi:10.3390/biom10020347
44. Popov LD. Mitochondrial biogenesis: an update. J Cell Mol Med. 2020;24(9):4892–4899. doi:10.1111/jcmm.15194
45. Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol. 2013;5(7):a015008–a015008. doi:10.1101/cshperspect.a015008
46. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. 2011;1813(7):1269–1278. doi:10.1016/j.bbamcr.2010.09.019
47. Picca A, Lezza AM. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion. 2015;25:67–75. doi:10.1016/j.mito.2015.10.001
48. Tran M, Tam D, Bardia A, et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121(10):4003–4014. doi:10.1172/JCI58662
49. Smith JA, Stallons LJ, Collier JB, Chavin KD, Schnellmann RG. Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J Pharmacol Exp Ther. 2015;352(2):346–357. doi:10.1124/jpet.114.221085
50. Pei S, Zheng L, Tian Z, et al. High concentration hydrogen attenuates sepsis-induced acute kidney injury by promoting mitochondrial biogenesis and fusion. Int Immunopharmacol. 2024;143(Pt 2):113410. doi:10.1016/j.intimp.2024.113410
51. Li SY, Park J, Qiu C, et al. Increasing the level of peroxisome proliferator-activated receptor γ coactivator-1α in podocytes results in collapsing glomerulopathy. JCI Insight. 2017;2(14). doi:10.1172/jci.insight.92930
52. Cen X, Chen Y, Xu X, et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model. Nat Commun. 2020;11(1):5731. doi:10.1038/s41467-020-19547-6
53. Yao RQ, Ren C, Xia ZF, Yao YM. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 2021;17(2):385–401. doi:10.1080/15548627.2020.1725377
54. Li X, Huang L, Lan J, et al. Molecular mechanisms of mitophagy and its roles in neurodegenerative diseases. Pharmacol Res. 2021;163:105240. doi:10.1016/j.phrs.2020.105240
55. Xian H, Liou YC. Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 2021;28(3):827–842. doi:10.1038/s41418-020-00657-z
56. Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol Cell Biol. 2015;93(1):3–10. doi:10.1038/icb.2014.75
57. Gkikas I, Palikaras K, Tavernarakis N. The Role Of Mitophagy In Innate Immunity. Front Immunol. 2018;9:1283. doi:10.3389/fimmu.2018.01283
58. Shao D, Kolwicz SC Jr, Wang P, et al. Increasing fatty acid oxidation prevents high-fat diet-induced cardiomyopathy through regulating parkin-mediated mitophagy. Circulation. 2020;142(10):983–997. doi:10.1161/CIRCULATIONAHA.119.043319
59. Wang S, Long H, Hou L, et al. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther. 2023;8(1):304. doi:10.1038/s41392-023-01503-7
60. Sunahara S, Watanabe E, Hatano M, et al. Influence of autophagy on acute kidney injury in a murine cecal ligation and puncture sepsis model. Sci Rep. 2018;8(1):1050. doi:10.1038/s41598-018-19350-w
61. Wang Y, Zhu J, Liu Z, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2021;38:101767. doi:10.1016/j.redox.2020.101767
62. Gao Y, Dai X, Li Y, et al. Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J Transl Med. 2020;18(1):114. doi:10.1186/s12967-020-02283-2
63. Sun X, Wang H, Liu Y, et al. 5-methoxytryptophan alleviates lipopolysaccharide-induced acute kidney injury by regulating nrf2-mediated mitophagy. J Inflamm Res. 2024;17:9857–9873. doi:10.2147/JIR.S474040
64. Lei X, Wang J, Zhang F, et al. Micheliolide ameliorates lipopolysaccharide-induced acute kidney injury through suppression of NLRP3 activation by promoting mitophagy via Nrf2/PINK1/Parkin axis. Int Immunopharmacol. 2024;138:112527. doi:10.1016/j.intimp.2024.112527
65. Pan P, Chen J, Xie F, et al. Enhancing Nix-dependent mitophagy relieves AKI by restricting TREM-1-mediated hyperactivation of inflammasome in platelets. FASEB j. 2023;37(11):e23239. doi:10.1096/fj.202202144RRR
66. Sidarala V, Zhu J, Levi-D’Ancona E, et al. Mitofusin 1 and 2 regulation of mitochondrial DNA content is a critical determinant of glucose homeostasis. Nat Commun. 2022;13(1):2340. doi:10.1038/s41467-022-29945-7
67. Quintana-Cabrera R, Scorrano L. Determinants and outcomes of mitochondrial dynamics. Mol Cell. 2023;83(6):857–876. doi:10.1016/j.molcel.2023.02.012
68. Rahmani S, Roohbakhsh A, Karimi G. Inhibition of Drp1-dependent mitochondrial fission by natural compounds as a therapeutic strategy for organ injuries. Pharmacol Res. 2023;188:106672. doi:10.1016/j.phrs.2023.106672
69. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. 2020;15:235–259. doi:10.1146/annurev-pathmechdis-012419-032711
70. Green A, Hossain T, Eckmann DM. Mitochondrial dynamics involves molecular and mechanical events in motility, fusion and fission. Front Cell Dev Biol. 2022;10:1010232. doi:10.3389/fcell.2022.1010232
71. Zacharioudakis E, Gavathiotis E. Mitochondrial dynamics proteins as emerging drug targets. Trends Pharmacol Sci. 2023;44(2):112–127. doi:10.1016/j.tips.2022.11.004
72. Liu JX, Yang C, Zhang WH, et al. Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci. 2019;235:116828. doi:10.1016/j.lfs.2019.116828
73. Liu JX, Yang C, Liu ZJ, et al. Protection of procyanidin B2 on mitochondrial dynamics in sepsis associated acute kidney injury via promoting Nrf2 nuclear translocation. Aging. 2020;12(15):15638–15655. doi:10.18632/aging.103726
74. Liu N, Jiang Z, Liu Y, et al. Human trypsin inhibitor reduces the apoptosis of lipopolysaccharide‑induced human kidney‑2 cells by promoting mitochondrial fusion. Mol Med Rep. 2017;16(3):2899–2906. doi:10.3892/mmr.2017.6927
75. Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011;14(10):1939–1951. doi:10.1089/ars.2010.3779
76. Picca A, Mankowski RT, Burman JL, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. 2018;15(9):543–554. doi:10.1038/s41569-018-0059-z
77. Capece D, Verzella D, Di Francesco B, Alesse E, Franzoso G, Zazzeroni F. NF-κB and mitochondria cross paths in cancer: mitochondrial metabolism and beyond. Semin Cell Dev Biol. 2020;98:118–128. doi:10.1016/j.semcdb.2019.05.021
78. Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20(7):745–754. doi:10.1038/s41556-018-0124-1
79. Srivastava A, Tomar B, Sharma D, Rath SK. Mitochondrial dysfunction and oxidative stress: role in chronic kidney disease. Life Sci. 2023;319:121432. doi:10.1016/j.lfs.2023.121432
80. Avolio R, Matassa DS, Criscuolo D, Landriscina M, Esposito F. Modulation of mitochondrial metabolic reprogramming and oxidative stress to overcome chemoresistance in cancer. Biomolecules. 2020;10(1):135. doi:10.3390/biom10010135
81. Elfawy HA, Das B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: etiologies and therapeutic strategies. Life Sci. 2019;218:165–184. doi:10.1016/j.lfs.2018.12.029
82. Kowalczyk P, Sulejczak D, Kleczkowska P, et al. Mitochondrial oxidative stress-A causative factor and therapeutic target in many diseases. Int J Mol Sci. 2021;22(24):13384. doi:10.3390/ijms222413384
83. Jin L, Batra S, Jeyaseelan S. Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol. 2017;198(3):1253–1262. doi:10.4049/jimmunol.1601745
84. Danielski LG, Giustina AD, Bonfante S, Barichello T, Petronilho F. The NLRP3 inflammasome and its role in sepsis development. Inflammation. 2020;43(1):24–31. doi:10.1007/s10753-019-01124-9
85. Zhao WY, Zhang L, Sui MX, Zhu YH, Zeng L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci Rep. 2016;6:33201. doi:10.1038/srep33201
86. Raheem AK, Abu-Raghif AR, Abbas AH, Ridha-Salman H, Oubaid EN. Quercetin mitigates sepsis-induced renal injury via inhibiting inflammatory and oxidative pathways in mice. J Mol Histol. 2025;56(3):184. doi:10.1007/s10735-025-10442-2
87. Raheem AK, Abu-Raghif AR, Abd-alakhwa SZ. Irbesartan attenuates sepsis-induced renal injury in mice models. J Pharm Negat Results. 2022;13:662–669.
88. Raheem AKK, Abu-Raghif AR, Zigam QA. Cilostazol protects against sepsis-induced kidney impairment in a mice model. J Med Chem Sci. 2023;6(5):1193–1203.
89. Lowes DA, Webster NR, Murphy MP, Galley HF. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth. 2013;110(3):472–480. doi:10.1093/bja/aes577
90. Smith RL, Soeters MR, Wüst RCI, Houtkooper RH. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr Rev. 2018;39(4):489–517. doi:10.1210/er.2017-00211
91. Toro J, Manrique-Caballero CL, Gómez H. Metabolic reprogramming and host tolerance: a novel concept to understand sepsis-associated AKI. J Clin Med. 2021;10(18). doi:10.3390/jcm10184184
92. Podrini C, Cassina L, Boletta A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell Signal. 2020;67:109495. doi:10.1016/j.cellsig.2019.109495
93. Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684. doi:10.1126/science.1250684
94. Zager RA. ‘Biologic memory’ in response to acute kidney injury: cytoresistance, toll-like receptor hyper-responsiveness and the onset of progressive renal disease. Nephrol Dial Transplant. 2013;28(8):1985–1993. doi:10.1093/ndt/gft101
95. Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019;96(5):1083–1099. doi:10.1016/j.kint.2019.05.026
96. Waltz P, Carchman E, Gomez H, Zuckerbraun B. Sepsis results in an altered renal metabolic and osmolyte profile. J Surg Res. 2016;202(1):8–12. doi:10.1016/j.jss.2015.12.011
97. Yang L, Xie M, Yang M, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436. doi:10.1038/ncomms5436
98. Gomez H, Ince C, De Backer D, et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41(1):3–11. doi:10.1097/SHK.0000000000000052
99. Liu C, Wei W, Huang Y, Fu P, Zhang L, Zhao Y. Metabolic reprogramming in septic acute kidney injury: pathogenesis and therapeutic implications. Metabolism. 2024;158:155974.
100. Wang T, Huang Y, Zhang X, Zhang Y, Zhang X. Advances in metabolic reprogramming of renal tubular epithelial cells in sepsis-associated acute kidney injury. Front Physiol. 2024;15:1329644. doi:10.3389/fphys.2024.1329644
101. Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21(1):37–46. doi:10.1038/nm.3762
102. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13(10):629–646. doi:10.1038/nrneph.2017.107
103. Chen Y, Fry BC, Layton AT. Modeling glucose metabolism and lactate production in the kidney. Math Biosci. 2017;289:116–129. doi:10.1016/j.mbs.2017.04.008
104. Wang Y, Xi W, Zhang X, et al. CTSB promotes sepsis-induced acute kidney injury through activating mitochondrial apoptosis pathway. Front Immunol. 2022;13:1053754. doi:10.3389/fimmu.2022.1053754
105. Deng Z, He M, Hu H, et al. Melatonin attenuates sepsis-induced acute kidney injury by promoting mitophagy through SIRT3-mediated TFAM deacetylation. Autophagy. 2024;20(1):151–165. doi:10.1080/15548627.2023.2252265
106. Zhao Y, Feng X, Li B, et al. Dexmedetomidine protects against lipopolysaccharide-induced acute kidney injury by enhancing autophagy through inhibition of the PI3K/AKT/mTOR pathway. Front Pharmacol. 2020;11:128. doi:10.3389/fphar.2020.00128
107. Hu C, Wu Z, Li T, et al. Dendrobine attenuates sepsis-associated acute kidney injury by promoting PINK1/PARKIN-mediated mitophagy. Int Immunopharmacol. 2025;157:114741. doi:10.1016/j.intimp.2025.114741
108. Arulkumaran N, Pollen SJ, Tidswell R, et al. Selective mitochondrial antioxidant MitoTEMPO reduces renal dysfunction and systemic inflammation in experimental sepsis in rats. Br J Anaesth. 2021;127(4):577–586. doi:10.1016/j.bja.2021.05.036
109. Li G, Wu J, Li R, et al. Protective effects of antioxidant peptide SS-31 against multiple organ dysfunctions during endotoxemia. Inflammation. 2016;39(1):54–64. doi:10.1007/s10753-015-0222-1
110. Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45(11):1559–1565. doi:10.1016/j.freeradbiomed.2008.09.003
111. Plotnikov EY, Pevzner IB, Zorova LD, et al. Mitochondrial damage and mitochondria-targeted antioxidant protection in LPS-induced acute kidney injury. Antioxidants. 2019;8(6):176. doi:10.3390/antiox8060176
112. Ding Y, Zheng Y, Huang J, et al. UCP2 ameliorates mitochondrial dysfunction, inflammation, and oxidative stress in lipopolysaccharide-induced acute kidney injury. Int Immunopharmacol. 2019;71:336–349. doi:10.1016/j.intimp.2019.03.043
113. Tan C, Gu J, Li T, et al. Inhibition of aerobic glycolysis alleviates sepsis‑induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK‑regulated autophagy. Int J Mol Med. 2021;47(3). doi:10.3892/ijmm.2021.4852
114. Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB j. 2015;29(12):4766–4771. doi:10.1096/fj.15-275404
115. Ramsey H, Wu MX. Mitochondrial anti-oxidant protects IEX-1 deficient mice from organ damage during endotoxemia. Int Immunopharmacol. 2014;23(2):658–663. doi:10.1016/j.intimp.2014.10.019
116. Wu J, Zhang M, Hao S, et al. Mitochondria-targeted peptide reverses mitochondrial dysfunction and cognitive deficits in sepsis-associated encephalopathy. Mol Neurobiol. 2015;52(1):783–791. doi:10.1007/s12035-014-8918-z
117. Ji MH, Li GM, Jia M, et al. Valproic acid attenuates lipopolysaccharide-induced acute lung injury in mice. Inflammation. 2013;36(6):1453–1459. doi:10.1007/s10753-013-9686-z
118. Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol. 2014;306(7):F734–743. doi:10.1152/ajprenal.00643.2013
119. Wang JY, Li JQ, Xiao YM, Fu B, Qin ZH. Triphenylphosphonium (TPP)-based antioxidants: a new perspective on antioxidant design. ChemMedChem. 2020;15(5):404–410. doi:10.1002/cmdc.201900695
120. Petrov A, Perekhvatova N, Skulachev M, Stein L, Ousler G. SkQ1 ophthalmic solution for dry eye treatment: results of a phase 2 safety and efficacy clinical study in the environment and during challenge in the controlled adverse environment model. Adv Ther. 2016;33(1):96–115. doi:10.1007/s12325-015-0274-5
121. Du X, Zeng Q, Luo Y, et al. Application research of novel peptide mitochondrial-targeted antioxidant SS-31 in mitigating mitochondrial dysfunction. Mitochondrion. 2024;75:101846. doi:10.1016/j.mito.2024.101846
122. Ross MF, Kelso GF, Blaikie FH, et al. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry. 2005;70(2):222–230. doi:10.1007/s10541-005-0104-5
123. Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81(1):136–147. doi:10.1111/cbdd.12055
124. Szeto HH, Schiller PW. Novel therapies targeting inner mitochondrial membrane--from discovery to clinical development. Pharm Res. 2011;28(11):2669–2679. doi:10.1007/s11095-011-0476-8
125. Plotnikov EY, Zorov DB. Pros and cons of use of mitochondria-targeted antioxidants. Antioxidants. 2019;8(8).
126. Baechler BL, Bloemberg D, Quadrilatero J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy. 2019;15(9):1606–1619. doi:10.1080/15548627.2019.1591672
127. Howell GM, Gomez H, Collage RD, et al. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS One. 2013;8(7):e69520. doi:10.1371/journal.pone.0069520
128. Guo J, Wang R, Liu D. Bone marrow-derived mesenchymal stem cells ameliorate sepsis-induced acute kidney injury by promoting mitophagy of renal tubular epithelial cells via the SIRT1/Parkin axis. Front Endocrinol. 2021;12:639165. doi:10.3389/fendo.2021.639165
129. Cui J, Bai XY, Sun X, et al. Rapamycin protects against gentamicin-induced acute kidney injury via autophagy in mini-pig models. Sci Rep. 2015;5:11256. doi:10.1038/srep11256
130. Wu Y, Wang L, Meng L, Cao GK, Zhao YL, Zhang Y. Biological effects of autophagy in mice with sepsis-induced acute kidney injury. Exp Ther Med. 2019;17(1):316–322. doi:10.3892/etm.2018.6899
131. Choudhuri S, Chowdhury IH, Garg NJ. Mitochondrial regulation of macrophage response against pathogens. Front Immunol. 2020;11:622602. doi:10.3389/fimmu.2020.622602
132. Liu Z, Qin G, Yang J, et al. Targeting mitochondrial degradation by chimeric autophagy-tethering compounds. Chem Sci. 2023;14(40):11192–11202. doi:10.1039/D3SC03600F
133. Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12(8):2245–2256. doi:10.1091/mbc.12.8.2245
134. Kalia R, Wang RY, Yusuf A, et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature. 2018;558(7710):401–405. doi:10.1038/s41586-018-0211-2
135. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119(5):1275–1285. doi:10.1172/JCI37829
136. Jian Y, Yang Y, Cheng L, et al. Sirt3 mitigates LPS-induced mitochondrial damage in renal tubular epithelial cells by deacetylating YME1L1. Cell Prolif. 2023;56(2):e13362. doi:10.1111/cpr.13362
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Deciphering Cuproptosis in Sepsis: Mechanisms, Consequences, and Therapeutic Opportunities
Tan R, Wen K, Zhao T, Guo H, Han X, Wang J, Ge C, Du Q
Journal of Inflammation Research 2025, 18:9879-9890
Published Date: 25 July 2025