")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
The role of MAPT gene in Chinese dementia patients: a P301L pedigree study and brief literature review
Authors He S, Chen S, Xia MR, Sun ZK, Huang Y, Zhang JW
Received 30 October 2017
Accepted for publication 26 March 2018
Published 18 June 2018 Volume 2018:14 Pages 1627—1633
DOI https://doi.org/10.2147/NDT.S155521
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Shuang He,* Shuai Chen,* Ming-Rong Xia,* Zhi-Kun Sun, Yue Huang, Jie-Wen Zhang
Department of Neurology, Zhengzhou University People’s Hospital, Zhengzhou, Henan, 450003, China
*These authors contributed equally to the work
Background and purpose: Frontotemporal dementia (FTD) is the second most common presenile dementia characterized by behavioral changes and language impairment. The diagnosis of FTD relies heavily on neuroimaging, and sometimes on genetic screening. However, the genetic components in Chinese FTD patients remain largely unknown. Only a few FTD cases with established mutations have been reported in China. This study reported the detailed clinical and neuroimaging features in a Chinese behavioral variant FTD family. The role of MAPT gene mutation in Chinese dementia patients was also reviewed.
Methods: By detailed inquiry of all affected individuals in the family, this study summarized the main clinical features of the disease. Four candidate genes (MAPT, PSEN1, PSEN2, and APP) were screened by direct sequencing. Structural magnetic resonance imaging (MRI), functional imaging of cerebral blood flow with arterial spin-labeled MRI (ASL-MRI), and cerebral metabolism with fluorodeoxyglucose positron emission tomography (FDG-PET) were collected in the proband and healthy mutation carriers.
Results: By direct sequencing of candidate genes (MAPT, PSEN1, PSEN2, and APP), this study identified the P301L mutation in the MAPT gene in the proband and three unaffected family members. The phenotype of the affected cases was consistent within the pedigree. In this genetically proven behavioral variant FTD (bvFTD) patient, the maps of hypoperfusion on ASL-MRI look fairly similar to the hypometabolism on FDG-PET. The clinical feature for this bvFTD was in line with the hypoperfusion or hypometabolism pattern on functional neuroimagings. The phenotype of P301L in east Asia seems similar to western countries.
Conclusion: For the inherited FTD patients, ASL-MRI and genetic identification were strongly recommended for the final diagnosis. In case of being underestimated, the role of MAPT gene mutation in Chinese FTD patients warrants further investigation.
Keywords: frontotemporal dementia, MAPT, arterial spin labeling MRI, FDG-PET
Introduction
Frontotemporal dementia (FTD) is the second most common presenile dementia, characterized by prominent behavioral changes and language impairment. The main clinical presentations include behavioral variant frontotemporal dementia (bvFTD), semantic variant primary progressive aphasia (svPPA), and nonfluent/agrammatic variant PPA. Although not uncommon, the diagnosis of bvFTD sometimes was not straightforward due to overlapping symptoms with other dementias, even parkinsonism and motor neuron disease. Currently, structural T1WI imaging on brain magnetic resonance imaging (MRI) serves as an important diagnostic test for FTD. Besides structural MRI, functional imaging of cerebral metabolic rate of glucose by fluorodeoxyglucose positron emission tomography (FDG-PET) was critical when the diagnosis was uncertain. Due to the cost and exposure to ionizing radiation, FDG-PET was not readily available for dementia patients. Recently, arterial spin labeling (ASL) MRI, a novel functional imaging of cerebral perfusion using magnetically labeled arterial blood as the endogenous tracer, showed great promise in the diagnosis of neurodegenerative diseases. Increasing studies have shown that, compared to FDG-PET, ASL-MRI could provide similar information in the diagnosis or differential diagnosis of degenerative dementias.1
Approximately 40% of FTD patients have a positive family history in European populations, with mutations in the microtubule associated protein tau gene (MAPT) being the most common.2 Since cloned in 1998, more than 55 MAPT mutations have been identified, mainly in the FTD pedigrees. Being the first mutation identified in the MAPT gene, the P301L mutation has been associated with clinical and pathologic heterogeneity. The common clinical presentations of P301L mutation included bvFTD and parkinsonism. The explanations for clinicopathologic heterogeneity of P301L mutation carriers were largely unknown. Recently, the largest P301L case series in the same geographical origin from Spain showed relatively homogeneous clinical features. However, the role of MAPT gene in Chinese FTD patients remain largely unknown. So far, only a few cases with established mutations have been reported in China.
Here, we report a Han Chinese bvFTD family with the MAPT P301L mutation. Detailed clinical and neuroimaging features were described. The MAPT gene mutations in China and East Asia were also reviewed.
Patients and methods
Clinical examination
We personally examined the proband patient (IV2), her siblings (IV3, IV4) and her children (V5, V6, V7, and V8) (Figure 1). They all underwent clinical and neurological examinations and cognitive assessment. By detailed inquiry of all affected individuals in the family, we summarized the main clinical features of the disease. Routine blood tests mandatory for dementia diagnosis were carried out. The study was approved by the Ethics Committee of Zhengzhou University People’s Hospital. All subjects or their legal representatives gave written informed consent to have the case details published.
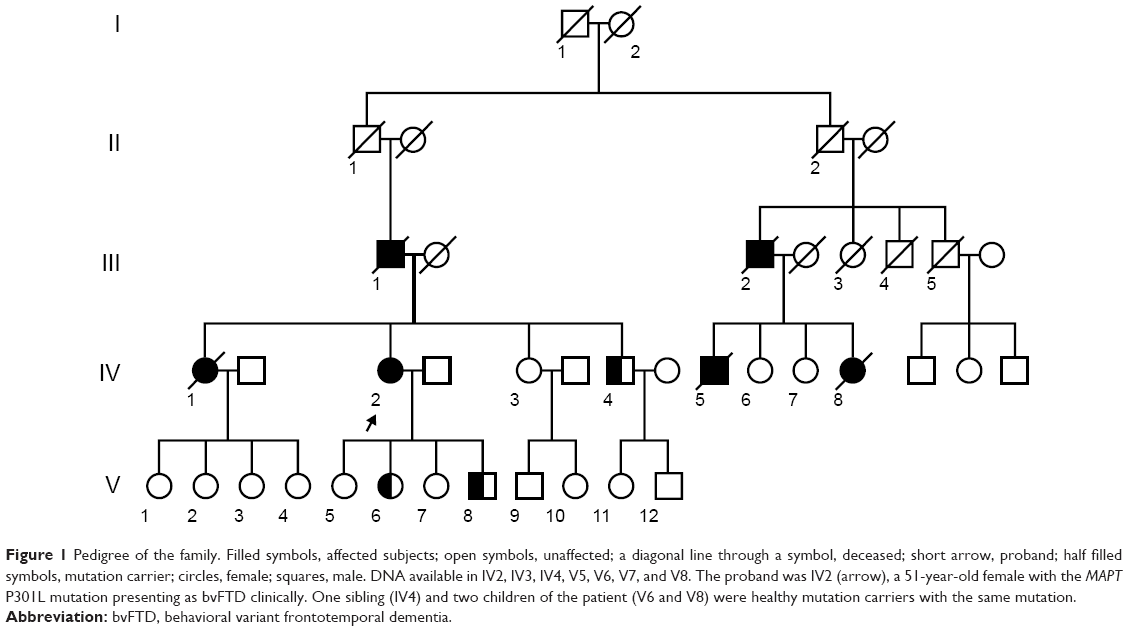 | Figure 1 Pedigree of the family. Filled symbols, affected subjects; open symbols, unaffected; a diagonal line through a symbol, deceased; short arrow, proband; half filled symbols, mutation carrier; circles, female; squares, male. DNA available in IV2, IV3, IV4, V5, V6, V7, and V8. The proband was IV2 (arrow), a 51-year-old female with the MAPT P301L mutation presenting as bvFTD clinically. One sibling (IV4) and two children of the patient (V6 and V8) were healthy mutation carriers with the same mutation. |
Neuroimaging
Conventional MRI, ASL-MRI (3.0T MRI scanner GE Discovery 750, GE Healthcare, Milwaukee, WI, USA), and 18F-FDG-PET scans (Siemens Biograph TruePoint PET-CT, Siemens, Munich, Germany) were performed in the proband (IV2) and her siblings (IV3, IV4). Pseudocontinuous ASL perfusion images were collected using 3D fast spin echo acquisition with background suppression, with a postlabeling delay of 1,525 ms, repetition time of 4,601 ms, and echo time of 10.5 ms. Cerebral blood flow maps were calculated from the perfusion-weighted images using a 2-compartment model with a finite labeling duration.
Mutation analysis
Blood samples taken from IV2, IV3, IV4, V5, V6, V7, and V8 were stored in the eathylene diamine tetraacetic acid anticoagulant tubes. Genomic DNA were extracted from peripheral blood leukocytes. Owing to the overlapping clinical features of FTD and Alzheimer’s disease (AD), all coding exons and their flanking introns of the MAPT(NM_001123066.3), PSEN1(NM_000021.3), PSEN2(NM_000447.2), and Aβ precursor protein (APP, NM_000484.3) were amplified by polymerase chain reaction (PCR) and sequenced on an ABI 3730 automated sequencer (ABI, Carlsbad, CA, USA). The primer sets (Table S1), gene amplification methods, and electropherogram (Figure S1) are presented in the Supplementary materials. The MAPT haplotype was not genotyped because almost all Chinese harbor the H1 haplotype.
Results
Clinical presentations
Six affected individuals were recorded in the pedigree with one patient still alive (the proband). The proband was a 49-year-old Han Chinese female. She came to our hospital with the speech problem and aberrant behaviors as the chief complaint. Two years earlier, the family members noticed her language expression difficulties. Her active speech reduced, shortened, but preferring repeating others’ words. She often smiled for no reason and purpose. Six months later, the prementioned symptoms got worse. Slowness in reacting was severe. Her speech became stereotyped. When asked questions, she often gave irrelevant or simple answers with limited words, such as “no problem.” She gradually appeared impolite, lacking sympathy, and, thus, had communication difficulties with friends. She seemed timid, nervous, and occasionally emotionally unstable. Unlike before, she seldom did the housework actively. Instead, she loved watching movies, especially war movies in foreign languages that she couldn’t understand at all. Her eating habits changed, preferring sweet foods. Her memory mildly declined. There were no walking difficulties or hallucinations. She could take care of herself independently. No sleep problems were reported.
She had no history of cerebrovascular disease, diabetes, or hypertension. There was no toxic substances exposure. Her medication was unremarkable.
Her father had a similar disease, mainly manifested as repetitive speech and aimless laughing at the age of 63, and died 6 years later. She had three siblings; two sisters and one brother. Her elder sister had similar symptoms at the age of 59 and died at 63. Her father’s cousin and one of her children died with similar symptoms, but no detailed information was obtained (Figure 1).
On neurological examination, she was right-handed, with slight euphoria. Orientation was preserved. She presented fluent speech, but language comprehension was impaired. Eye movement was full. The muscle strength and tone were normal. Tendon reflexes were symmetrical and moderate. Primitive reflexes, but no pathological reflex, were elicited. There was no parkinsonism or ataxia.
Thyroid function, folic acid, and vitamin B12 levels were normal. Syphilis and HIV serological examination were negative.
Neuropsychology
As she was illiterate, she couldn’t finish the detailed neuropsychological tests. The Memory and Executive Screening scale (MES) was utilized, which was acceptable for illiterate and low-educated Chinese.3 The memory and executive function domain was impaired (Table 1). Her siblings (IV3, IV4) and children (V5, V6, V7, and V8) underwent Mini Mental State Examination (MMSE) and Montreal Cognitive Assessment (MoCA) examination. The scores were all within the normal range.
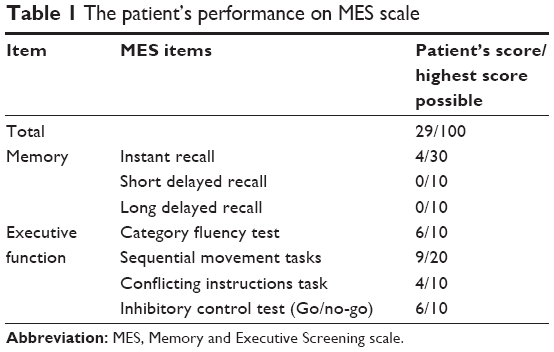 | Table 1 The patient’s performance on MES scale |
Genetic studies
For the index case, sequencing analysis of MAPT identified the c.1907C>T mutation (p.P301L) in exon 10 of MAPT. The same mutation was present in her healthy sibling (IV4) and two of her healthy children (V6, V8). Although P301L mutation proved to be common and pathogenic in western countries, however, rare P301L cases have been reported in China, even in East Asia.
Neuroimaging studies
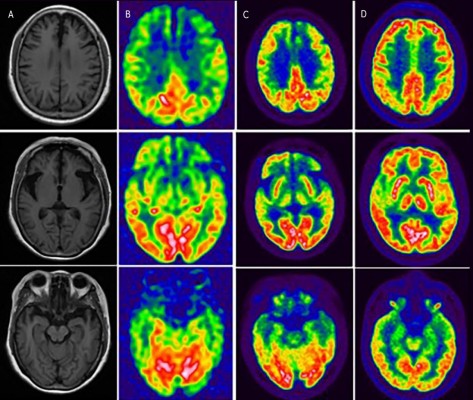
For the proband, structural brain imaging on MRI scan showed bilateral asymmetry of frontotemporal atrophy, more significant on the left. The predominantly frontotemporal hypoperfusion and hypometabolism were observed on ASL-MRI and FDG-PET, respectively. The maps of hypoperfusion look very similar to the maps of hypometabolism. The clinical feature of bvFTD was in line with the hypoperfusion or hypometabolism pattern on functional neuroimaging. Although mild hypometabolism was seen at basal ganglia on FDG-PET, no parkinsonism was found in the proband. The MRI and PET studies in her sibling (IV4, mutation carrier) revealed no significant abnormalities on visual inspection (Figure 2).
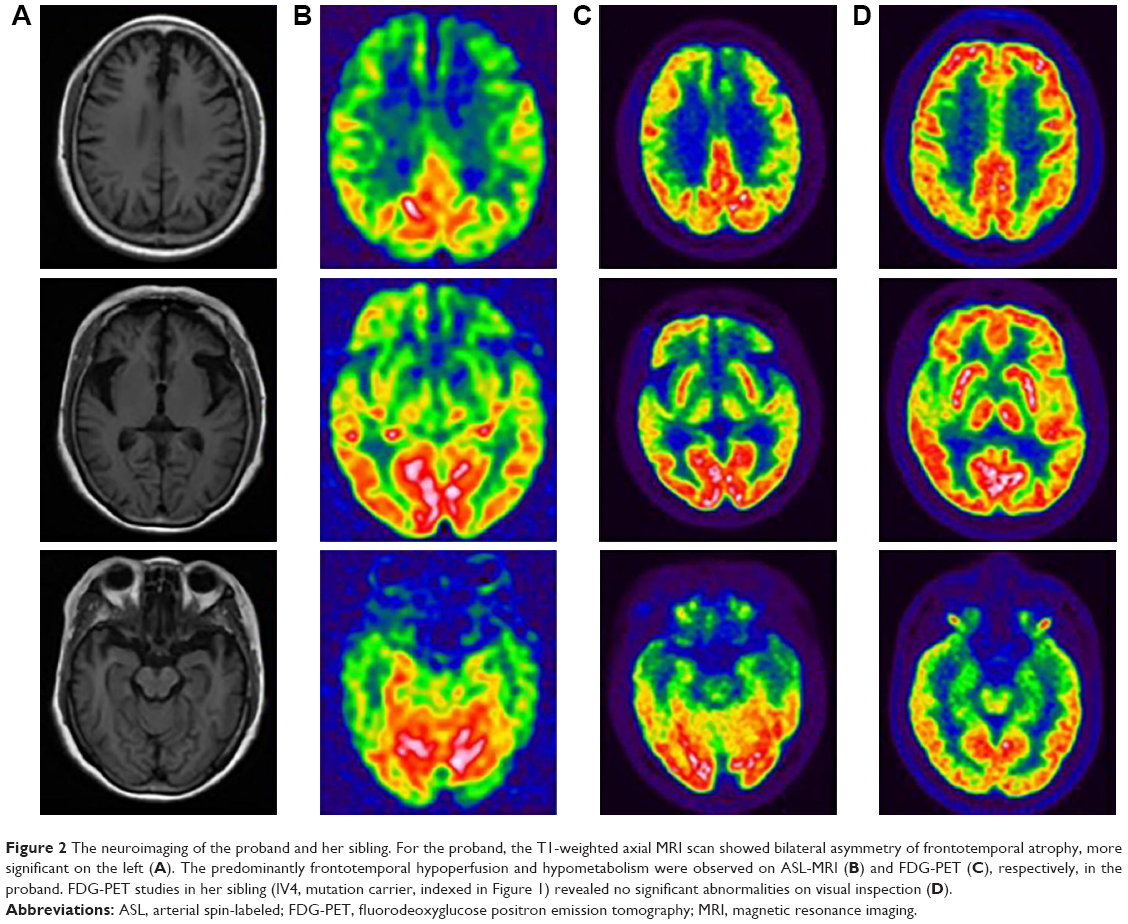 | Figure 2 The neuroimaging of the proband and her sibling. For the proband, the T1-weighted axial MRI scan showed bilateral asymmetry of frontotemporal atrophy, more significant on the left (A). The predominantly frontotemporal hypoperfusion and hypometabolism were observed on ASL-MRI (B) and FDG-PET (C), respectively, in the proband. FDG-PET studies in her sibling (IV4, mutation carrier, indexed in Figure 1) revealed no significant abnormalities on visual inspection (D). |
Discussion
Here, we reported the P301L MAPT mutation in an Han Chinese family. The clinical features and radiological findings support the diagnosis of bvFTD. There was no parkinsonism. The phenotype of the affected cases was consistent within the pedigree.
Although MAPT gene mutations were common for genetic FTD in western populations, the frequency was lower in East Asia. According to the Japanese Familial Alzheimer’s Disease (JFAD) database, 19 MAPT mutations were reported in 32 pedigrees so far in Japan.4 In a Japanese study including 75 patients with FTD, progressive supranuclear palsy (PSP), or corticobasal syndrome, six patients with atypical PSP were found to be harboring the MAPT mutations.5 In the Korean study of 75 FTD patients, no pathogenic MAPT mutations were identified.6 So far, no MAPT mutations have been reported in Korea. A Chinese cohort screened the MAPT, GRN, and C9orf72 genes in 52 FTD patients. They found three MAPT mutations in two progressive nonfluent aphasia (PNFA) patients and in one FTD-parkinsonism family.7 Another study identified two MAPT mutations in 38 FTD Chinese patients, including a novel L48V mutation.8 The latest research, including 82 Chinese sporadic FTD patients, found one novel H299Y mutation in a semantic dementia patient.9 Taken together, MAPT mutations seem rare in FTD patients in East Asia. Currently, there are no precise epidemiological data on the incidence of FTD and the portion of the familial form in all FTD cases in China. The genetic studies on FTD were limited, with relatively small sample sizes. In case of being underestimated, the role of MAPT in Chinese FTD patients needs further studies with larger sample sizes.
By reviewing the publications in PubMed and Chinese medical literature, we identified 10 Chinese patients or families with MAPT mutations (including the present one). The clinical and genetic features are summarized in Table 2.7–13 Seven of the ten MAPT mutations are found in “the hot spot” region of exons 9–13, with three exceptions (L48V, D177V, R5H). It was well documented that the P301L MAPT mutation was associated with mainly FTD and sometimes parkinsonism in the western population.14 The two P301L families (including the present one) in China presented with bvFTD or PNFA.8 Unlike the present one, the phenotypes of the other Chinese P301L family were heterogeneous with both bvFTD and PNFA. In Japan, the P301L patients had phenotypes of bvFTD, semantic dementia, or other language deficits.15–17 All these P301L cases in China and Japan showed no prominent parkinsonism. The N279K mutation presenting with early and predominant parkinsonism in Chinese was similar to Europeans.10,11
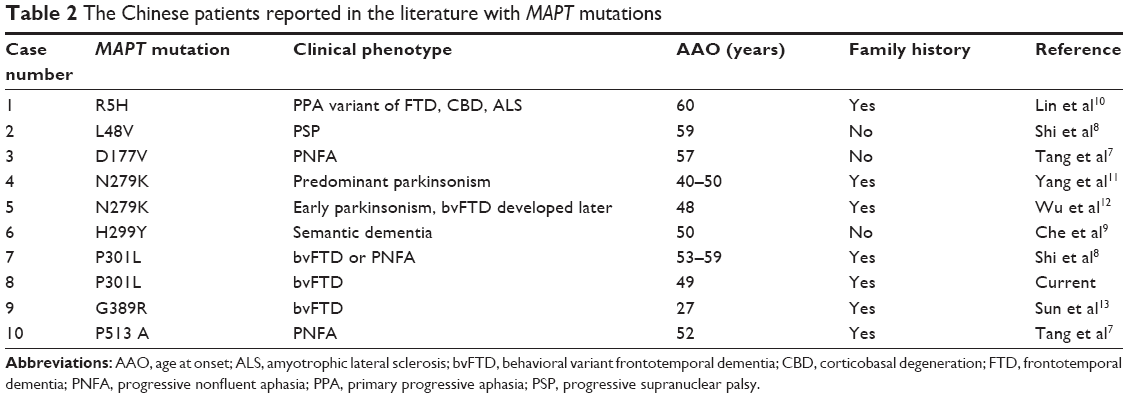 | Table 2 The Chinese patients reported in the literature with MAPT mutations |
It was noted that one sibling and two children of the patient were healthy mutation carriers with the same P301L mutations. There were some possible explanations. One is that the P301L mutation in this pedigree may had an autosomal dominant trait with incomplete penetrance. Other genetic factors may exist which may affect the heritability of the P301L mutation. Besides, a previous case series reported the mean age at disease onset for P301L carriers was 51 years.18 The proband in the current study was 49 years old at onset. Her younger brother, carrying the mutation, was 45 years old. Her two children harboring the mutation were in their 30s, possibly both too young for disease onset currently.
The recognition of bvFTD depends on specialized clinicians, and the diagnosis relies heavily on neuroimaging. Besides structural MRI, functional imaging of cerebral perfusion and metabolism using FDG-PET or single-photon emission computed tomography (SPECT) can improve the accuracy of diagnosis in FTD. Recently, the readily available ASL-MRI perfusion methods have shown great promise in differential diagnosis of AD and FTD.1,19 The cerebral hypoperfusion regions on ASL-MRI matched well with hypometabolism regions on FDG-PET. Here, in this genetically proved FTD patient, the maps of hypoperfusion on ASL-MRI look very similar to the maps of hypometabolism on FDG-PET. For the mutation carriers, ASL-MRI seems more suitable to follow-up and diagnosis.
Conclusion
We reported the detailed clinical and neuroimaging features in a Chinese family with MAPT P301L mutation. The clinical features of this bvFTD patient were in line with the hypoperfusion or hypometabolism pattern on functional neuroimaging. For the inherited FTD patients, ASL-MRI and genetic identification were strongly recommended for the final diagnosis. In case of being underestimated, the role of MAPT gene mutation in Chinese FTD patients warrants further investigation.
Acknowledgments
This study was supported by the National Natural Science Fund (81671068). The authors would like to thank all the family members in this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Tosun D, Schuff N, Rabinovici GD, et al. Diagnostic utility of ASL-MRI and FDG-PET in the behavioral variant of FTD and AD. Ann Clin Transl Neurol. 2016;3(10):740–751. | ||
Olszewska DA, Lonergan R, Fallon EM, Lynch T. Genetics of frontotemporal dementia. Curr Neurol Neurosci Rep. 2016;16(12):107. | ||
Guo QH, Zhou B, Zhao QH, Wang B, Hong Z. Memory and Executive Screening (MES): a brief cognitive test for detecting mild cognitive impairment. BMC Neurol. 2012;12:119. | ||
Kasuga K, Kikuchi M, Tokutake T, et al. Systematic review and meta-analysis of Japanese familial Alzheimer’s disease and FTDP-17. J Hum Genet. 2015;60(5):281–283. | ||
Ogaki K, Li Y, Takanashi M, Ishikawa K, et al. Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord. 2013;19(1):15–20. | ||
Kim EJ, Kwon JC, Park KH, et al. Clinical and genetic analysis of MAPT, GRN, and C9orf72 genes in Korean patients with frontotemporal dementia. Neurobiol Aging. 2014;35(5):1213.e13–17. | ||
Tang M, Gu X, Wei J, et al. Analyses MAPT, GRN, and C9orf72 mutations in Chinese patients with frontotemporal dementia. Neurobiol Aging. 2016;46:235.e11–15. | ||
Shi Z, Liu S, Xiang L, et al. Frontotemporal dementia-related gene mutations in clinical dementia patients from a Chinese population. J Hum Genet. 2016;61(12):1003–1008. | ||
Che XQ, Zhao QH, Huang Y, et al. Genetic features of MAPT, GRN, C9orf72 and CHCHD10 gene mutations in Chinese patients with frontotemporal dementia. Curr Alzheimer Res. 2017;14(10):1102–1108. | ||
Lin HC, Lin CH, Chen PL, Cheng SJ, Chen PH. Intrafamilial phenotypic heterogeneity in a Taiwanese family with a MAPT p.R5H mutation: a case report and literature review. BMC Neurol. 2017;17(1):186. | ||
Yang Y, Tang BS, Weng L, et al. Genetic identification is critical for the diagnosis of parkinsonism: a Chinese pedigree with early onset of parkinsonism. PLoS One. 2015;10(8):e0136245. | ||
Wu LY, Feng XY, Li HZ, et al. Clinical and neuroimaging features of frontotemporal dementia with parkinsonism linked to chromosome 17. China J Neurol. 2017;50(1):11–16. | ||
Sun L, Chen K, Li X, Xiao S. Rapidly progressive frontotemporal dementia associated with MAPT mutation G389R. J Alzheimers Dis. 2017;55(2):777–785. | ||
Siuda J, Fujioka S, Wszolek ZK. Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat Disord. 2014;20(9):957–964. | ||
Sitek EJ, Narozanska E, Barczak A, et al. Agraphia in patients with frontotemporal dementia and parkinsonism linked to chromosome 17 with P301L MAPT mutation: dysexecutive, aphasic, apraxic or spatial phenomenon? Neurocase. 2014;20(1):69–86. | ||
Ishizuka T, Nakamura M, Ichiba M, Sano A. Familial semantic dementia with P301L mutation in the Tau gene. Dement Geriatr Cogn Disord. 2011;31(5):334–340. | ||
Kodama K, Okada S, Iseki E, et al. Familial frontotemporal dementia with a P301L tau mutation in Japan. J Neurol Sci. 2000;176(1):57–64. | ||
Borrego-Écija S, Morgado J, Palencia-Madrid L, et al. Frontotemporal Dementia Caused by the P301L Mutation in the MAPT Gene: Clinicopathological Features of 13 Cases from the Same Geographical Origin in Barcelona, Spain. Dement Geriatr Cogn Disord. 2017;44(3–4):213–221. | ||
Verfaillie SC, Adriaanse SM, Binnewijzend MA, et al. Cerebral perfusion and glucose metabolism in Alzheimer’s disease and frontotemporal dementia: two sides of the same coin? Eur Radiol. 2015;25(10):3050–3059. |
Supplementary materials
Polymerase chain reaction (PCR) protocol
PCR was performed on 10 μL of reaction mixture at 95°C for 5 min, followed by 35 cycles of 95°C for 30 s, an annealing step for 30 s, and 72°C for 30 s, with a final extension at 72°C for 10 min.
 | Table S1 MAPT primers |
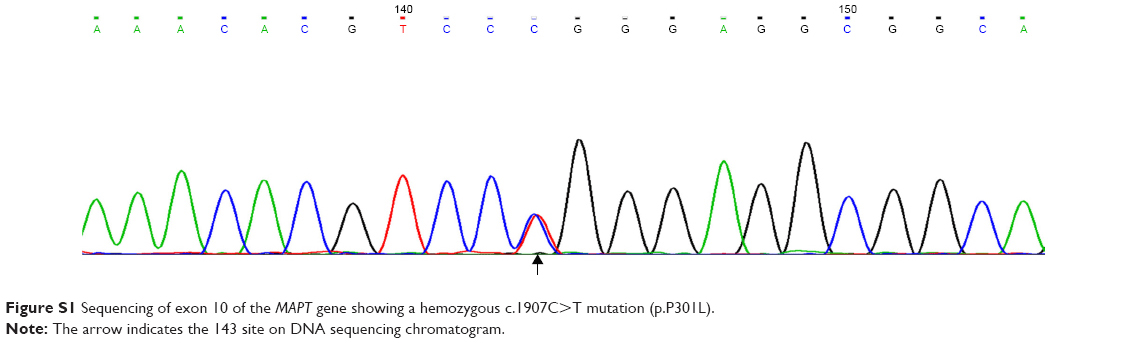 | Figure S1 Sequencing of exon 10 of the MAPT gene showing a hemozygous c.1907C>T mutation (p.P301L). |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.