")
Back to Journals » Journal of Inflammation Research » Volume 16
The Role of Inflammation in Cholestatic Liver Injury
Received 16 July 2023
Accepted for publication 6 October 2023
Published 13 October 2023 Volume 2023:16 Pages 4527—4540
DOI https://doi.org/10.2147/JIR.S430730
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Jie Chen, Shujun Zhang
Chongqing Key Laboratory of Infectious Diseases and Parasitic Diseases, Department of Infectious Diseases, the First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China
Correspondence: Shujun Zhang, Tel/Fax +86-23-89012427, Email [email protected]
Abstract: Cholestasis is a common clinical event in which bile formation and excretion are blocked, leading to retention of bile acids or bile salts; whether it occurs intra- or extrahepatically, primary or secondary, its pathogenesis is still unclear and is influenced by a combination of factors. In a variety of inflammatory and immune cells such as neutrophils, macrophages (intrahepatic macrophages are also known as Kupffer cells), mast cells, NK cells, and even T cells in humoral immunity and B cells in cellular immunity, inflammation can be a “second strike” against cholestatic liver injury. These cells, stimulated by a variety of factors such as bile acids, inflammatory chemokines, and complement, can be activated and accumulate in the cholestatic liver, and with the involvement of inflammatory mediators and modulation by cytokines, can lead to destruction of hepatocytes and bile duct epithelial cells and exacerbate (and occasionally retard) the progression of cholestatic liver disease. In this paper, we summarized the new research advances proposed so far regarding the relationship between inflammation and cholestasis, aiming to provide reference for researchers and clinicians in the field of cholestatic liver injury research.
Keywords: inflammation, immune cells, cholestasis, cholestatic liver injury
Introduction
Cholestasis is a pathological condition in which the formation, secretion, and excretion of bile flow are inhibited by various factors, resulting in the inability of bile to drain smoothly into the duodenum and thus return to the blood.1 In the early stage, there is no specific clinical manifestation, only an increase in serum alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT) levels; some patients may experience weakness, nausea, poor appetite, and epigastric discomfort. After gradual progression, jaundice, skin itching, fatigue, steatorrhea, yellow tumor, hepatic osteodystrophy, and fat-soluble vitamin deficiency may occur. In severe cases, it may even lead to liver failure and death.2 Hepatobiliary disorders characterized by cholestasis are called cholestatic liver diseases, including primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), intrahepatic cholestasis in pregnancy syndrome (ICP), IgG4-related sclerosing cholangitis, and progressive familial intrahepatic cholestasis (PFIC). Cholestasis can also occur in alcoholic liver disease, viral hepatitis, drug-induced liver injury, and non-alcoholic fatty liver disease.3
The occurrence of cholestasis is associated with abnormal bile duct cell proliferation, peribiliary fibrosis, myofibroblast activation, toxic bile acid accumulation, and bile duct reactions, all of which can drive disease progression.4 The processes of cholestasis and cholestatic liver injury are extremely complex, and the etiology of cholestatic liver injury varies greatly. The invasion of inflammatory cells, which facilitates bile duct cell death, results in blockage of bile ducts and abnormal bile acid excretion in PBC and PSC, etc. And the accumulating bile acids might act as a second hit, causing inflammation and fibrosis.5 In In cholestatic liver diseases such as ICP and PFIC, which are due to abnormalities of the bile salt transporter pump, the first strike is bile acid accumulation due to inadequate bile acid excretion, which causes a second hit of immunological imbalance, inflammation, and fibrosis, leading to aggravation of cholestatic liver injury.6,7 Inflammation might be the initial cause of cholestasis and the following hit to the liver. As a result, many scientists regard regulation of the inflammatory response as a viable therapeutic target for cholestasis and its accompanying liver harm.8
Therefore, this article provides a review of the mechanisms of inflammation and cholestasis from the aspects of inflammatory cells, humoral immunity, inflammatory factors, and Inflammatory vesicles, etc.
Discussion
Inflammatory Cells in Cholestasis
Neutrophils
Neutrophils are usually the first responders to the inflammatory response. During bacterial infections, neutrophils are able to transform from a resting state to an activated state stimulated by C5a, lipopolysaccharides (LPS), and cytokines, and thus have a direct role in clearing pathogens.9 Usually, neutrophils execute their cytotoxicity through the production of reactive oxygen species (ROS) and hypochlorous acid, both of which have strong toxic effects on liver cells.10 In addition, neutrophils can produce a substance called serine protease, which not only has a direct cytotoxic effect but also participates in the activation of inflammatory cytokines, and thus exacerbate liver damage.11
Neutrophil recruitment in the hepatic microvascular system can be found in bile duct ligation (BDL) or bile-fed mice and has been demonstrated in several previous studies.12,13 This recruitment process begins 6 hours after BDL14 and the level of neutrophils will peak after two to three days, becoming the main inflammatory cell in the cholestasis model.15 This was also found in the livers of ICP patients, accompanied by the decrease in serum neutrophil counts.16 After recruitment, neutrophils exocytose into the surrounding parenchymal tissue and portal bundles in an intercellular adhesion molecule-1 (ICAM-1)-dependent manner on the endothelium, at which point the Na(+)/H(+) exchange factor 1 (NHERF-1) and the cytoskeletal protein ezrin-radixin-moesin (ERM) act as scaffolding proteins for the neutrophils. The process involves a variety of inflammatory mediators, which are involved in the migration of neutrophils across the endothelial and epithelial cells.17,18 The process is mediated by a variety of inflammatory mediators, which are involved in the migration of neutrophils across the endothelial and epithelial cells. In addition, in a mouse model of obstructive cholestasis, the bone bridge protein (OPN) released from the bile duct cells acted as a pro-inflammatory mediator in initiating the early neutrophil-mediated phase of injury after cleavage to a pro-inflammatory form by the matrix metalloproteinases (MMP).19 We are, therefore, expected to reduce liver damage by exploring multiple receptor blockers of inflammatory mediators and blocking neutrophil chemotaxis.20
Earlier studies have found that neutrophils’ bactericidal ability, phagocytosis and intracellular killing ability are significantly reduced in the early and advanced stages of PBC.21 In the rat model of obstructive cholestasis, the decrease in phagocytic function of polymorphonuclear leukocytes can also be found.22 On the contrary, it was found that in the rat model of common bile duct obstruction, neutrophils have enhanced phagocytosis, and can produce superoxide.23 Therefore, we speculate that in the early stages of cholestasis, neutrophils will recruit locally to promote the occurrence and progression of inflammation, and subsequently its function will be weakened, resulting in a series of complications, such as the occurrence of bacterial peritonitis. Therefore, neutrophils may have a contradictory role in the development of cholestatic liver diseases. On the one hand, they may produce an inflammatory protective response, on the other hand, also can promote disease progression.
Macrophages
The liver possesses the largest population of macrophages in the body, including two main cell subpopulations, Kupffer cells (KCs) and infiltrating macrophages (ie monocyte-derived macrophages, MoMFs). KCs express danger recognition receptors and clearance of receptors, and they are the main natural immune cells that eliminate a large number of bacteria, fungi and viruses.24,25 The use of gadolinium chloride (an inhibitor of KCs) attenuated liver injury and fibrosis in a BDL model, suggesting that KCs can promote BDL-induced liver injury.26 Studies have confirmed that KCs can be activated after BDL, leading to the release of reactive oxygen species (ROS)27 and the production of pro-inflammatory cytokines induced by LPS; this further activates the signaling pathways, such as nuclear factor kappa B (NF-kB) and mitogen-activated protein kinase (MAPK) in the hepatocytes and bile duct cells, affecting the function of the bile acid transport proteins and causing bile acid accumulation.28,29 In recent years, it has been found in mouse models of PBC that hepatic macrophages not only secrete cytokines, but also regulate natural killer (NK) cells to jointly participate in the inflammatory process through a signaling pathway mediated by natural killer group 2, member D (NKG2D) and its ligand RAE-1.30 Interestingly, KCs can also reduce cholestatic liver injury by expressing TGR5 (a bile acid-activated G protein-coupled receptor), inhibiting NF-kB and c-Jun N-terminal kinase (JNK) signaling pathways, and inhibiting NLRP3 inflammatory vesicle activation so they may play different roles in the different stages of BDL-induced liver injury.31,32 However, further studies are needed to elucidate whether inhibiting TGR5 activation in Kupffer cells is beneficial for the progression of cholestatic diseases.
In patients with PSC, not only macrophage activation was found, but also bile acids were found to prompt macrophages to produce more histamine through paracrine.33 In patients with biliary atresia and Alagille syndrome, Taylor et al defined three types of macrophages through single-cell RNA sequencing analysis and immunofluorescence isolation: lipid-associated macrophages, monocytoid macrophages and adaptive macrophages, all of which were pathogenic, and can express immunomodulatory gene RORA and TREM2, which may serve as a new therapeutic target for alleviating inflammatory damage in cholestatic liver disease.34 Tian et al also proposed a new potential therapeutic strategy: selectively depletion of long chain non-coding RNA-H19 (lncRNA-H19) of macrophages, which involved in the proliferation of cholangiocytes in cholestatic livers and promotes hepatic fibrosis, helps to inhibit cholestatic liver damage and fibrosis.35 However, macrophages in cholestasis models do not always have significant pathogenicity, such as the reduction in macrophage clearance found in infants with biliary atresia,36 which may contribute to the accumulation of autoimmune complexes in the liver, and indirectly leads to liver damage.
Mast Cells
Mast cells (MC) are immune cells of the bone marrow spectrum with pro/anti-inflammatory, pro-fibrotic, and immunomodulatory effects,37,38 which were found to be increased in PBC, PSC, bile duct obstruction, hepatitis, fatty liver, liver fibrosis, hepatocellular carcinoma, cholangiocarcinoma, and liver failure.39 Mast cells infiltrating the liver can secrete a large number of pro-inflammatory mediators, such as histamine, heparin, trypsin, chymotrypsin, carboxypeptidase A3, granzyme B, and cytokines including TNF-α, IL-1β and IL-3, etc. Among them, histamine (which has vasodilatory and bi-directional inflammatory effects) has been shown to promote cholangiocytes proliferation and hepatic fibrosis through specific pathways such as Notch/Jagged, thereby exacerbating the degree of cholestasis.40–42 It has recently been shown that mast cells can also secrete autologous sorting proteins (ATX), which contribute to itching in the model of bile duct ligation.43 Meanwhile, it has been shown that MC can regulate the enterohepatic circulation of TBA through regulating FXR/FGF15 expression in the biliary and intestinal tracts via MC-FXR.44,45
Patients with PBC and PSC have higher amounts of MC accumulated in the biliary tree and gallbladder, especially on the cholangiocyte membrane, suggesting that senescent cholangiocytes may promote MC activation and proliferation. And histamine levels were also found to be elevated, suggesting increased secretory capacity of MC.46 The reduction of MC in the cholestasis models can reduce liver inflammation47 and the development of liver fibrosis.48,49 Therefore, reducing the degree of inflammation in cholestatic liver by decreasing MC levels may be an effective therapeutic target. This is similar to the drug UDCA, which is commonly used clinically to reduce inflammatory damage by interfering with the degranulation process of MC while decreasing macrophage infiltration and reducing oxidative stress.50
Lymphocytes
T-lymphocytes
T-lymphocytes are major players in specific cellular immunity, killing target cells directly and also releasing lymphokines and regulating the immune process.51 Monoclonal expansion of the CD4+ and CD8+ T cells can be found in the liver of patients with PBC as well as biliary atresia. The CD4+ T cells play a major role in the development of PBC, causing systemic inflammation through the production of pro-inflammatory cytokines, while the CD8+ T cells mainly mediate the process of bile duct injury.52,53 The recruitment of CD8+ T cells has been shown to affect BA metabolism, by reducing the expression of the basolateral bile acid uptake transporters NTCP and OATP, and increasing the expression of the transporter BSEP involved in bile acid excretion, so as to reduce the persistent toxic damage of BA to the liver, thereby playing a positive role in regulating cholestasis.54
Regulatory T cells (Tregs) are T cells capable of negatively regulating the body’s immune response. Earlier studies have suggested that Tregs expression levels are significantly reduced in patients with PBC and BA, suggesting that defects in the Tregs can also promote the development of inflammation in cholestatic liver damage.55,56 It was found that using a combination of immunosuppressive drug vitamin D3 and dexamethasone induced the differentiation of naive CD4+ T cells into Tregs that produce IL-10 in vitro, thus acting to reduce inflammation.57
B-Lymphocytes
B-lymphocytes, including memory B cells, plasma blasts and plasma cells are also involved in specific immune processes, which further produce various immunoglobulins (Ig) and their isoforms, including IgM, IgD, IgG (types 1–4), IgA, and IgE.58 B cells play an immune role by neutralizing antibodies, producing cytokines, participating in complement cascade reactions, and communicating with immune cells, such as macrophages and T cells.59 Overexpression of serum B-cell activating factor (BAFF) is suggestive of autoimmune hyperactivity, it was found to be highly expressed in patients with PBC and positively correlated with aspartate aminotransferase (AST) and total bilirubin levels, suggesting a possible association with persistent necrotizing inflammation of hepatocytes.60 In PBC patients, CD20 and CD19 were also found in lymphoid follicle-like aggregates at some distance from the portal bundle, and CD19 levels were positively correlated with ALP levels, further suggesting the presence of B-cell activation in patients with PBC.61,62 Increased infiltration of B cells is also found in the liver of patients with biliary atresia, with IgM and IgG deposition in the epithelial basement membrane of the bile ducts.63 Further studies have shown that B cells can directly produce pro-inflammatory factors such as IL-6, IL-19 and TNF-α, triggering the activation of CD4+ and CD8+ T cells, as well as inducing apoptosis of Treg cells, and decreases the expression of the anti-inflammatory cytokines IL-10 and TGF-β. And CD38 plasma cells produced by B cells can be directly involved in the bile duct injury process.64,65 However, therapeutic approaches to ameliorate inflammatory damage by depleting B cells are not necessarily effective, and an earlier experimental study showed that anti-CD20/CD79-treated mice had more CD4 and CD8+ T cell infiltration in the portal region and more severe bile duct inflammation, suggesting that simply depleting a kind of inflammatory cell may cannot achieve the desired anti-inflammatory effect.66
Natural Killer (NK) Cells
NK cells are natural lymphocytes that play an important immunomodulatory role in multiple liver diseases, such as PBC, chronic hepatitis B, chronic hepatitis C, alcoholic liver disease, and hepatocellular carcinoma (HCC).67,68 In patients with cholestatic liver injury, the NK cells in the portal region and around the hepatic bile ducts usually show stronger expression activity than the controls, and its recruitment from the blood to the liver is associated with the stimulation of a large number of chemokines, such as CCL3, CCL5, CCL7, CCL8, CXCL11-3, and CX1CL30.69,70 In contrast, a study in bile duct-ligated mice with combined MCMV infection found reduced recruitment of NK cells to the liver, a phenomenon that may be related to cholestasis impairing the expression of pathogen-induced chemokines in the liver.71 After the recruitment process is completed, NK cells are able to rigger non-specific cytotoxicity in the cellular epithelial lining, destroy normal hepatic duct cells and bile duct cells (through perforin/granzyme B-dependent and TRAIL-dependent mechanisms),72,73 and stimulate inflammation by producing both IFN-γ and TNF and granulocyte-macrophage colony-stimulating factors (GM-CSF)74 and bind to Toll-like receptors to induce lysis of biliary epithelial cells, thus aggravating liver damage.75 While, NK cells can also play a negative regulatory role in liver inflammation in PBC, a process that may be achieved by inhibiting the proliferation of the CD4+ T cells.76,77
Humoral Immunity
Immunoglobulin
IgM
IgM is the first immunoglobulin expressed during B-cell development and plays a role in the direct encapsulation and destruction of antigens and immobilization of the complement cells.78 Most cases of autoimmune hepatitis are dominated by elevated IgG levels. Elevated IgM levels are predominant in the serum of PBC patients; however, the mechanism for this elevation is unclear and may be related to methylation of CD40L, the promoter of CD4 + T cells, which affects immunoglobulin class switching.79 High levels of autophagy in the B cells of patients with PBC play an important factor in the synthesis.80 Patients with biliary atresia develop IgM deposits around the basement membrane of the hepatobiliary epithelium and novel IgM autoantibodies against cholangiocyte-associated proteins (CHI3L1, DLL-4, and SFTPD) have been identified, which are involved in the formation of antigen-antibody complexes, complement activation, and cholangiocyte injury, and are positively correlated with serum bilirubin levels and the incidence of liver transplantation.81,82 Thus, IgM may play a major driving role in cholestatic liver injury and its autoantibodies may be a potential target for future therapeutic interventions.
IgG
IgG activates a cascade of complement reactions and has antibody-dependent cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Elevated levels of IgG has been found in various cholestatic liver injury.83 In patients with PBC, elevated levels of the IgG subclasses (IgG3 and IgG5) can be found, and the IgG3 subtype of anti-mitochondrial antibodies in PBC-specific autoantibodies may be associated with the severity of PBC.84 IgG deposit can also be found on the biliary epithelial basement membrane in patients with biliary obstruction, with a positivity rate of up to 56.5%,85 and the biliary epithelial target protein that has been shown to react strongly with IgG is human alpha enolase (αENOL), which is also one of the antigenic targets of the anti-neutrophil cytoplasmic antibodies present in autoimmune liver disease.86 The formation of an immune complex with IgG by αENOL induces endothelial damage via the complement classical pathway and cell death via the apoptotic process, thus suggesting higher liver failure-related mortality in patients with PBC.87 A recent study suggests that IgG can also contribute to the development of inflammation in PBC by binding to FcγRIIB (a negative regulatory receptor for lymphocytes, but which may have a different role in B cells) and thus in turn modulating B cell function, but the exact mechanism of this regulatory role needs to be further explored.88
IgA
Cholangiocytes can actively secrete secretory IgA in the biliary tree and participate in the process of mucosal immunity. At the same time, IgA is actively deposited in the hepatobiliary epithelium of patients with PBC and may enter the cells via recombinant pyruvate dehydrogenase complex (PDC-E2)-specific IgA binding to the human polymeric immunoglobulin receptor (pIgR) to activate cysteine asparaginase (Caspase), which induces hepatocyte apoptosis and biliary tract injury.89,90 Deposition of circulating immune complexes of IgA is found in bile duct ligated mice, which may be associated with reduced hepatic clearance of IgA and increased renal secretory IgA production.91
IgE
IgE is a key effector in the type I allergic immune response, and its activation by B cells leads to the differentiation of IgE-secreting cells.92 Elevated IgE levels were found in approximately 40% of patients with PSC, but there is no relationship between the prognosis of cholangiocarcinoma, liver transplantation, or death.93 Whereas earlier studies have suggested that this is more likely to be related to the presence of hypereosinophilic syndromes and anaphylactic reactions during the pathogenesis of PSC, the question of whether IgE acts as a positive or a negative regulator of the progression of cholestasis needs to be explored further.94 We know that the mast cells play an important role in cholestasis and that the IgE receptor FcεRI is present on the surface of the mast cells; hence, IgE may be involved in the process of cholestatic liver damage by inducing an immune response in the mast cells.95 Furthermore, the liver X receptor (LXR) is a member of the nuclear receptor superfamily in bile acid metabolism, and activation of LXR was found to reduce the levels of IgE secreted by the B cells and activated by the CD68 and IL-11, suggesting that activation of LXR can inhibit IgE and the inflammatory and allergic responses.96
Cytokines
Pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6 and IL-8, produced by the inflammatory cells, such as KCs and bile duct cells stimulated by LPS, can contribute to the development of cholestasis by inducing a sustained decrease in the levels of bile acid transport proteins.97–99 In the early stages of inflammation, TNF-α and IL-1β are involved in the downregulation of NTCP, while in the later stages of inflammation, they are regulated by IL-6.100,101 Higher levels of IL-8 may be associated with the development of PBC and reflect the progression of the disease towards cirrhosis.102 Elevated levels of TNF-α are often thought to reflect a more severe disease in patients with PBC,103 while IL-12 and IFN-γ have also been found to be associated with the severity of PBC, contributing to inflammation by inducing a Th1 immune response during the initial stages of the disease.104 Also, the inflammatory cytokines can slow bile flow by inhibiting chloride and bicarbonate ion transport via the bile duct cells.105
KCs secrete the cytokines with anti-inflammatory functions, such as IL-10, IL-4, and IL-13.106,107 Decreased levels of IL-10 can be found in patients with PBC, which inhibits the production of the pro-inflammatory factors, IFN-γ and TNF-α, by the NK cells and inhibits the proliferation of the Th1 cells, reducing the degree of liver inflammation by blocking the process of antigen presentation.108,109 Recently, a large case-control study found that some of the immune-related genes of IL-10 were associated with Biliary Atresia, further emphasizing its protective role.110 Meanwhile, IL-6 has been found to act as an anti-inflammatory cytokine in ICP patients, inhibiting TNF-α and IL-1 while activating IL-10.111 Thus, in models of cholestatic liver damage, cytokine networks may drive disease progression and may also be protective against inflammation. More cytokines need to be tapped as well as further explored, and there is a need to further define which cytokines play a major role in cholestatic liver disease and how we will slow disease progression by intervening in the cytokine network.
Chemokines
Inflammatory chemokines are able to act specifically on the inflammatory cells during the adaptive immune response and have the ability to induce cell migration. In senescent BEC from PBC patients, upregulated expression of various chemokines and chemotactic activity, including CCL2 and CXCL1, CXCR3, and increased expression of chemokine receptors, such as CX3CR1, can be found,112,113 and the levels of chemokines increase with disease progression.114 CCL11, CCL24, CCL26, and other eosinophils CCL11, CCL24, and CCL26 are also found in PBC as potent chemokines for acidophilic granulocytes, with CCL11 and CCL26 being shown to be associated with fibrosis progression in PBC.115 As mentioned earlier, these chemokines help to activate the innate immune system around the damaged bile ducts and mediate the migration of various inflammatory cells, which in turn leads to persistent inflammation and further exacerbates bile duct injury. However, the role of chemokines and their receptors is not limited to inducing inflammation. In recent years, CXCR2 has been found to be capable of directly damaging hepatocytes in bile duct ligated livers, Thus chemokines may play an important role in inflammatory cell recruitment and subsequent immune-mediated hepatocyte damage.116
Complement
Complement is synthesized predominantly in the liver and mediates immune complex formation, clearance of apoptotic cells and tissue regeneration, and is activated predominantly via the classical, alternative and lectin pathways.117 Activation of the complement system triggers a cascade reaction that leads to the rupture of C3, the core molecule of the complement system, which in turn leads to the rupture of C5 downstream, resulting in the production of the products C3a, C5a.118,119 Complement activation results in the production of membrane attack complexes (MACs), which lead to hepatocyte lysis and induce cellular release of inflammatory cytokines.120 C3 and C4 levels were significantly higher in the congenital biliary atresia model than in the healthy population, and there was a significant positive correlation with GGT levels.121 Deficiency of complement C3 attenuates cholestatic liver injury in mice and reduces neutrophil and macrophage infiltration and activation in the liver by regulating the expression of adhesion factors, further confirming the role of elevated complement levels in driving the progression of cholestatic liver disease.122 In contrast, cholestasis directly induces complement activation, and FXR has been shown to directly encode the C3 gene, increasing C3 mRNA and protein levels and thereby exacerbating liver inflammation.123
Inflammatory Vesicles
Inflammatory vesicles are polymorphic complexes of proteins, including NLR family members NLRP1, NLRP3, NLRC4, as well as AIM2 and pyrin. Activation of inflammatory vesicles can induce or promote inflammation through further activation of the shear cysteine asparaginase-1 that cleaves pro-interleukin-1β (pro-IL-1β) to IL-1β, which then triggers activation of NF-κB signaling via receptors IL-1R and IL-18R.124 This process is seen in a variety of liver diseases: PBC, alcoholic hepatitis, ischemia-reperfusion injury.125–127 Activation of NLRP3 inflammatory vesicles in the cholestatic model can be activated by the production of galactoglucan-3 by macrophages,128,129 whereas bile acids inhibit the activation of NLRP3 inflammatory vesicles via the TGR3-cAMP-PKA axis, thus controlling inflammation and metabolic disturbances. This may therefore contribute to the different roles of NLRP3 in acute and chronic cholestatic liver injury, with a lack of NLRP3 leading to reduced inflammation in chronic cholestatic liver injury, whereas hepatic inflammation is exacerbated in acute patients.130
Bile Acids and Inflammation
Bile acids are closely associated with the inflammatory response and biliary stasis that leads to the accumulation of toxic bile acids, which directly activate inflammatory and pro-fibroblastic cells and stimulate the release of pro-inflammatory and pro-fibrotic mediators from the hepatocytes and bile duct cells. This, in turn, leads to persistent biliary stasis and promotes the development of liver fibrosis. For example, bile acids are known to trigger a neutrophil-mediated inflammatory response and can act synergistically with LPS to promote inflammatory vesicle activation.131,132 In addition, bile acids activate various isoforms of early growth response protein 1 (Egr-1), protein kinase C family, p38, c-Jun N-terminal kinase, and pregnane X receptor, stimulating the upregulation of pro-inflammatory genes by activating one or more of these pathways.133–135 Recently, it has been found that goose deoxycholic acid (CDCA) can trigger the excessive accumulation of mitochondrial reactive oxygen species and promote the activation of inflammatory vesicles by targeting heme oxygenase-1 (HO-1), which in turn promotes the development of inflammation in hepatocellular carcinoma.135 But bile acids also have anti-inflammatory effects and have been shown to inhibit lymphocyte proliferation, immunoglobulin production, and cytokine secretion,136,137 and reduce phagocytosis of KCs.138 Therefore, the mechanisms by which bile acids determine the development of inflammation and their specific role in the injury process still need to be further explored.
Sepsis and Cholestasis
Sepsis-associated cholestasis is very common in hospitalized patients with jaundice, most especially infants and immunocompromised populations.139 Patients with cirrhosis are more likely to develop bacterial infections, possibly related to translocation of intestinal bacteria.140 In biliary stasis, pathogenic bacteria travel retrograde through the bile ducts to the liver and interact with receptors (especially Toll-like receptors or TLRs) on the immune cells stored in the liver to induce inflammation.141 At the same time, bacterial toxins and bacterial metabolites cause direct damage to hepatocytes, and several studies have found that LPS and LPS-induced cytokines are associated with cholestasis.142,143 LPS stimulates the activation of KCs and neutrophils during cholestasis, leading to the production of large amounts of pro-inflammatory cytokines, thus promoting the development of liver inflammation.144 Notably, this process often occurs in the chronic rather than the acute phase of cholestasis.145
In patients who have developed sepsis, abnormalities in the biochemical parameters of liver function can be detected, suggesting “hypoxic hepatitis” and “cholestatic liver dysfunction”.146 At this time, the enlargement in cell size from Kupffer cells in combination with infiltrating polymorphonuclear cells, eosinocytes, and platelets induces the formation of aggregates, which leads to sinus cavity obstruction. At the same time, the loss of actin and myosin microfilament activity due to inflammation and infection during septicemia directly induces apoptosis and necrosis of hepatocytes, leading to paralysis and dilatation of the bile ducts as well as impaired bile secretion.147,148 However, the liver can rapidly reduce bile formation by adjusting the expression of key molecular components involved in bile flow in response to the infection and inflammatory process, thus elevated biochemical markers of cholestasis do not necessarily persist throughout the course of the disease.149,150
Bile Duct Cell Senescence and Inflammation
Cellular senescence is increasingly recognized as a pathological feature of various inflammatory liver diseases. Bile duct cells in cholestatic liver damage exhibit a senescence-associated secretory phenotype (SASP) and resistance to apoptosis, which contribute to the onset and exacerbation of inflammation by initiating paracrine signaling pathways that enhance the secretion of cytokines (IL-1 and IL-6), chemokines (IL-8, CCR2, MCP-1), growth factors, and pro-fibrotic factors.151–153 This in turn recruits and/or activates immune cells such as macrophages, NK (NK) cells, and T lymphocytes, to promote the removal of senescent cells. A recent study found that senescent liver cells inhibit autophagy by producing BMP9, a subfamily of TGF-β, which downregulate the expression of the ATG3 and ATG7 genes. When autophagic flux is inhibited, macrophages acquire a proinflammatory phenotype and promote tissue damage.154 Hence, bile duct cell senescence plays an important pro-inflammatory role in the late stages of cholestasis.
Conclusions
In summary, the causes of cholestasis are complex, and its pathogenesis remains unclear. Numerous studies have suggested that inflammation plays an important role in cholestasis, including cellular immunity, humoral immunity, and pattern recognition receptors. A large number of inflammatory and immune cells such as neutrophils, macrophages, mast cells, T and B lymphocytes, NK cells, as well as cytokines, inflammatory vesicles, and complement are involved and interact with each other, which not only directly lead to biliary epithelial cell damage, but also regulate disease progression by exacerbating/attenuating biliary inflammation and thereby modulating cholestatic liver damage (see Figure 1). Components of this process with anti-inflammatory effects deserve further exploration to unearth potential therapeutic targets. Cholestasis itself can also have an effect on hepatocyte inflammation; for example, bile acids can act directly as pro-inflammatory agents, inducing inflammatory cells and inflammatory factors causing inflammatory damage. In different models of cholestatic liver damage, cholestasis and inflammation are inextricably linked, and therefore, both should be taken into account in the treatment process; however, the possibility of reducing inflammation to improve the outcome of cholestatic liver damage and delay its progression to liver fibrosis, cirrhosis, or even liver failure remains to be explored.
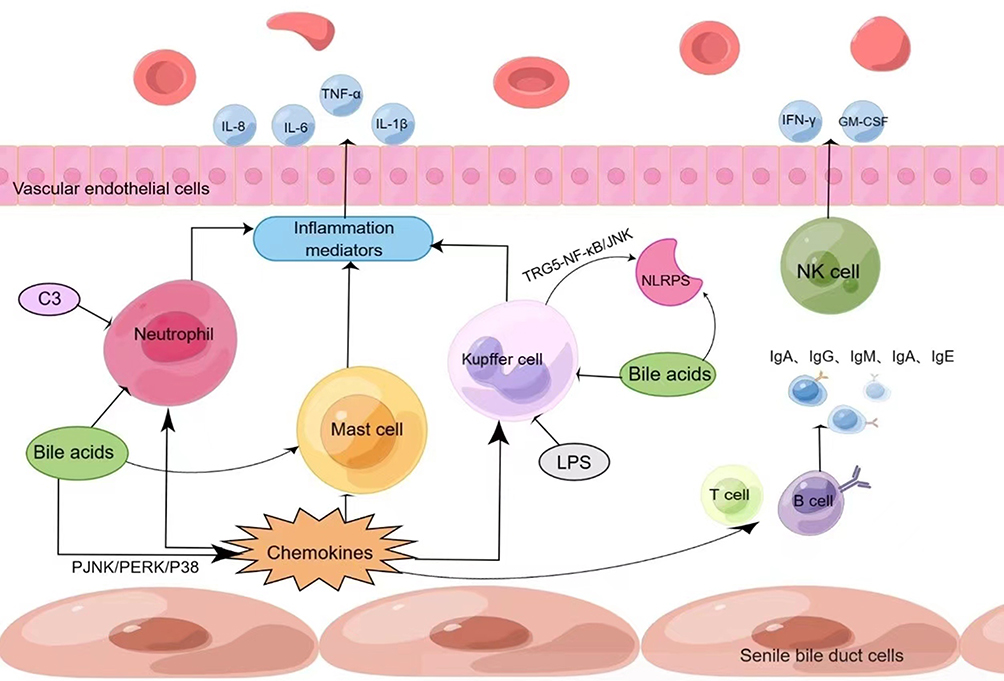 |
Figure 1 Inflammation is inextricably linked to cholestasis. (By Figdraw). Notes: As shown in the figure above, on the surface of senescent bile duct epithelial cells, neutrophils, macrophages, mast cells, T cells, B cells and other inflammatory cells aggregate under the action of chemokines, and release a large number of inflammatory mediators and cytokines, which jointly promote the progression of inflammation in cholestatic liver damage. |
Consent for Publication
All authors gave their consent for publication.
Acknowledgments
The authors would like to express their gratitude to all those who helped during the writing of this thesis.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Sponsors include Chongqing Science and Health Joint Medical Research Project (2020FYYX007), Chongqing young and middle-aged medical high-end talent project (2019-181). The funders had no role in study design, data collection and analysis, publication decisions, or manuscript preparation.
Disclosure
The authors have no relevant financial or non-financial interests to disclose for this work.
References
1. Zollner G, Trauner M. Mechanisms of cholestasis. Clin Liver Dis. 2008;12(1):1–26, vii. doi:10.1016/j.cld.2007.11.010
2. Younossi ZM, Bernstein D, Shiffman ML, et al. Diagnosis and management of primary biliary cholangitis. Am J Gastroenterol. 2019;114(1):48–63. doi:10.1038/s41395-018-0390-3
3. Jüngst C, Berg T, Cheng J, et al. Intrahepatic cholestasis in common chronic liver diseases. Eur J Clin Invest. 2013;43(10):1069–1083. doi:10.1111/eci.12128
4. You H, Ma X, Efe C, et al. APASL clinical practice guidance: the diagnosis and management of patients with primary biliary cholangitis. Hepatol Int. 2022;16(1):1–23. doi:10.1007/s12072-021-10276-6
5. Zhuang Y, Ortega-Ribera M, Thevkar Nagesh P, et al. Bile acid-induced IRF3 phosphorylation mediates cell death, inflammatory responses and fibrosis in cholestasis-induced liver and kidney injury via regulation of ZBP1. Hepatology. 2023. doi:10.1097/HEP.0000000000000611
6. Xiao J, Li Z, Song Y, et al. Molecular Pathogenesis of Intrahepatic Cholestasis of Pregnancy. Can J Gastroenterol Hepatol. 2021;2021:6679322. doi:10.1155/2021/6679322
7. Hassan S, Hertel P. Overview of progressive familial intrahepatic cholestasis. Clin Liver Dis. 2022;26(3):371–390. doi:10.1016/j.cld.2022.03.003
8. Zhang Y, Lu Y, Ji H, Li Y. Anti-inflammatory, anti-oxidative stress and novel therapeutic targets for cholestatic liver injury. Biosci Trends. 2019;13(1):23–31. doi:10.5582/bst.2018.01247
9. Guo RF, Ward PA. Mediators and regulation of neutrophil accumulation in inflammatory responses in lung: insights from the IgG immune complex model. Free Radic Biol Med. 2002;33(3):303–310. doi:10.1016/S0891-5849(02)00823-7
10. Dold S, Laschke MW, Zhau Y, et al. P-selectin glycoprotein ligand-1-mediated leukocyte recruitment regulates hepatocellular damage in acute obstructive cholestasis in mice. Inflamm Res. 2010;59(4):291–298. doi:10.1007/s00011-009-0099-2
11. Ho JS, Buchweitz JP, Roth RA, Ganey PE. Identification of factors from rat neutrophils responsible for cytotoxicity to isolated hepatocytes. J Leukoc Biol. 1996;59(5):716–724. doi:10.1002/jlb.59.5.716
12. Gujral JS, Hinson JA, Jaeschke H. Chlorotyrosine protein adducts are reliable biomarkers of neutrophil-induced cytotoxicity in vivo. Comp Hepatol. 2004;3(Suppl 1):S48. doi:10.1186/1476-5926-2-S1-S48
13. Gujral JS, Farhood A, Bajt ML, Jaeschke H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology. 2003;38(2):355–363. doi:10.1053/jhep.2003.50341
14. Woolbright BL, Antoine DJ, Jenkins RE, Bajt ML, Park BK, Jaeschke H. Plasma biomarkers of liver injury and inflammation demonstrate a lack of apoptosis during obstructive cholestasis in mice. Toxicol Appl Pharmacol. 2013;273(3):524–531. doi:10.1016/j.taap.2013.09.023
15. Georgiev P, Jochum W, Heinrich S, et al. Characterization of time-related changes after experimental bile duct ligation. Br J Surg. 2008;95(5):646–656. doi:10.1002/bjs.6050
16. Silva J, Magenta M, Sisti G, Serventi L, Gaither K. Association between complete blood count components and intrahepatic cholestasis of pregnancy. Cureus. 2020;12(12):e12381. doi:10.7759/cureus.12381
17. Barreiro O, Yanez-Mo M, Serrador JM, et al. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. 2002;157(7):1233–1245. doi:10.1083/jcb.200112126
18. Carman CV, Jun CD, Salas A, Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol. 2003;171(11):6135–6144. doi:10.4049/jimmunol.171.11.6135
19. Yang M, Ramachandran A, Yan HM, et al. Osteopontin is an initial mediator of inflammation and liver injury during obstructive cholestasis after bile duct ligation in mice. Toxicol Lett. 2014;224(2):186–195. doi:10.1016/j.toxlet.2013.10.030
20. Cai SY, Ouyang X, Chen Y, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight. 2017;2(5):e90780. doi:10.1172/jci.insight.90780
21. De Fernandez MA, Clark A, Triger DR. Neutrophil phagocytic and bactericidal function in primary biliary cirrhosis and other chronic liver diseases. Clin Exp Immunol. 1987;67(3):655–661.
22. Roughneen PT, Drath DB, Kulkarni AD, Kumar SC, Andrassy RJ, Rowlands BJ. Inflammatory cell function in young rodents with experimental cholestasis: investigations of functional deficits, their etiology, and their reversibility. J Pediatr Surg. 1989;24(7):668–673. doi:10.1016/S0022-3468(89)80716-X
23. Levy R, Schlaeffer F, Keynan A, Nagauker O, Yaari A, Sikuler E. Increased neutrophil function induced by bile duct ligation in a rat model. Hepatology. 1993;17(5):908–914. doi:10.1002/hep.1840170522
24. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60(5):1090–1096. doi:10.1016/j.jhep.2013.12.025
25. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14(10):996–1006. doi:10.1038/ni.2691
26. Zandieh A, Payabvash S, Pasalar P, et al. Gadolinium chloride, a Kupffer cell inhibitor, attenuates hepatic injury in a rat model of chronic cholestasis. Hum Exp Toxicol. 2011;30(11):1804–1810. doi:10.1177/0960327111400106
27. Kim SY, Jeong JM, Kim SJ, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun. 2017;8(1):2247. doi:10.1038/s41467-017-02325-2
28. Mulder J, Karpen SJ, Tietge UJ, Kuipers F. Nuclear receptors: mediators and modifiers of inflammation-induced cholestasis. Front Biosci. 2009;14(7):2599–2630. doi:10.2741/3400
29. Lou G, Ma X, Fu X, et al. GPBAR1/TGR5 mediates bile acid-induced cytokine expression in murine Kupffer cells. PLoS One. 2014;9(4):e93567. doi:10.1371/journal.pone.0093567
30. Fu HY, Bao WM, Yang CX, et al. Kupffer cells regulate natural killer cells via the NK group 2, Member D (NKG2D)/Retinoic Acid Early Inducible-1 (RAE-1) interaction and cytokines in a primary biliary cholangitis mouse model. Med Sci Monit. 2020;26:e923726. doi:10.12659/MSM.923726
31. Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology. 2011;54(4):1421–1432. doi:10.1002/hep.24525
32. Li WT, Luo QQ, Wang B, et al. Bile acids induce visceral hypersensitivity via mucosal mast cell-to-nociceptor signaling that involves the farnesoid X receptor/nerve growth factor/transient receptor potential vanilloid 1 axis. FASEB j. 2019;33(2):2435–2450. doi:10.1096/fj.201800935RR
33. Jones H, Hargrove L, Kennedy L, et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(-/-) mice. Hepatology. 2016;64(4):1202–1216. doi:10.1002/hep.28704
34. Taylor SA, Chen SY, Gadhvi G, et al. Transcriptional profiling of pediatric cholestatic livers identifies three distinct macrophage populations. PLoS One. 2021;16(1):e0244743. doi:10.1371/journal.pone.0244743
35. Tian X, Wang Y, Lu Y, et al. Conditional depletion of macrophages ameliorates cholestatic liver injury and fibrosis via lncRNA-H19. Cell Death Dis. 2021;12(7):646. doi:10.1038/s41419-021-03931-1
36. Wang J, Xu Y, Chen Z, et al. Liver immune profiling reveals pathogenesis and therapeutics for biliary atresia. Cell. 2020;183(7):1867–1883.e1826. doi:10.1016/j.cell.2020.10.048
37. Metz M, Maurer M. Mast cells--key effector cells in immune responses. Trends Immunol. 2007;28(5):234–241. doi:10.1016/j.it.2007.03.003
38. Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol. 2007;7(2):93–104. doi:10.1038/nri2018
39. Jarido V, Kennedy L, Hargrove L, et al. The emerging role of mast cells in liver disease. Am J Physiol Gastrointest Liver Physiol. 2017;313(2):G89–G101. doi:10.1152/ajpgi.00333.2016
40. Grizzi F, Di Caro G, Laghi L, et al. Mast cells and the liver aging process. Immun Ageing. 2013;10(1):9. doi:10.1186/1742-4933-10-9
41. Yamashiro M, Kouda W, Kono N, Tsuneyama K, Matsui O, Nakanuma Y. Distribution of intrahepatic mast cells in various hepatobiliary disorders. An immunohistochemical study. Virchows Arch. 1998;433(5):471–479. doi:10.1007/s004280050276
42. Huntzicker EG, Hötzel K, Choy L, et al. Differential effects of targeting Notch receptors in a mouse model of liver cancer. Hepatology. 2015;61(3):942–952. doi:10.1002/hep.27566
43. Yurdakul Ertan H, Özbakış Dengiz G, Barut F, et al. The Investigation of Therapeutic Implications of Mast Cell Stabilizer Cromolyn Sodium on Cholestasis and Cholestatic Pruritus in Experimental Cholestasis. Turk J Gastroenterol. 2023;34(1):62–72. doi:10.5152/tjg.2022.21744
44. Meadows V, Kennedy L, Ekser B, et al. Mast cells regulate ductular reaction and intestinal inflammation in cholestasis through farnesoid X receptor signaling. Hepatology. 2021;74(5):2684–2698. doi:10.1002/hep.32028
45. Meadows V, Kennedy L, Kundu D, Alpini G, Francis H. Bile acid receptor therapeutics effects on chronic liver diseases. Front Med. 2020;7:15. doi:10.3389/fmed.2020.00015
46. Gonzalez MI, Vannan DT, Eksteen B, Flores-Sotelo I, Reyes JL. Mast cells in immune-mediated cholangitis and cholangiocarcinoma. Cells. 2022;11(3):375. doi:10.3390/cells11030375
47. Aller M, Martínez V, Arias A, et al. Mast cell-mediated splanchnic cholestatic inflammation. Clin Res Hepatol Gastroenterol. 2019;43(5):561–574. doi:10.1016/j.clinre.2019.02.001
48. Komi DEA, Rambasek T, Wöhrl S. Mastocytosis: from a Molecular Point of View. Clin Rev Allergy Immunol. 2018;54(3):397–411. doi:10.1007/s12016-017-8619-2
49. Zhou K, Xie G, Wen J, et al. Histamine is correlated with liver fibrosis in biliary atresia. Dig Liver Dis. 2016;48(8):921–926. doi:10.1016/j.dld.2016.05.001
50. Carotti S, Guarino MP, Cicala M, et al. Effect of ursodeoxycholic acid on inflammatory infiltrate in gallbladder muscle of cholesterol gallstone patients. Neurogastroenterol Motil. 2010;22(8):866–873, e232. doi:10.1111/j.1365-2982.2010.01510.x
51. Shimoda S, Ishikawa F, Kamihira T, et al. Autoreactive T-cell responses in primary biliary cirrhosis are proinflammatory whereas those of controls are regulatory. Gastroenterology. 2006;131(2):606–618. doi:10.1053/j.gastro.2006.05.056
52. Mack CL, Falta MT, Sullivan AK, et al. Oligoclonal expansions of CD4+ and CD8+ T-cells in the target organ of patients with biliary atresia. Gastroenterology. 2007;133(1):278–287. doi:10.1053/j.gastro.2007.04.032
53. Kita H, Lian ZX, Van de Water J, et al. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: t cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med. 2002;195(1):113–123. doi:10.1084/jem.20010956
54. Glaser F, John C, Engel B, et al. Liver infiltrating T cells regulate bile acid metabolism in experimental cholangitis. J Hepatol. 2019;71(4):783–792. doi:10.1016/j.jhep.2019.05.030
55. Lan RY, Cheng C, Lian ZX, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology. 2006;43(4):729–737. doi:10.1002/hep.21123
56. Tucker RM, Feldman AG, Fenner EK, Mack CL. Regulatory T cells inhibit Th1 cell-mediated bile duct injury in murine biliary atresia. J Hepatol. 2013;59(4):790–796. doi:10.1016/j.jhep.2013.05.010
57. Barrat FJ, Cua DJ, Boonstra A, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195(5):603–616. doi:10.1084/jem.20011629
58. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol. 2020;20(4):229–238. doi:10.1038/s41577-019-0244-2
59. Cyster JG, Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. 2019;177(3):524–540. doi:10.1016/j.cell.2019.03.016
60. Migita K, Ilyassova B, Kovzel EF, et al. Serum BAFF and April levels in patients with PBC. Clin Immunol. 2010;134(2):217–225. doi:10.1016/j.clim.2009.09.007
61. Jin Q, Moritoki Y, Lleo A, et al. Comparative analysis of portal cell infiltrates in antimitochondrial autoantibody-positive versus antimitochondrial autoantibody-negative primary biliary cirrhosis. Hepatology. 2012;55(5):1495–1506. doi:10.1002/hep.25511
62. Li Y, Wang W, Tang L, et al. Chemokine (C-X-C motif) ligand 13 promotes intrahepatic chemokine (C-X-C motif) receptor 5+ lymphocyte homing and aberrant B-cell immune responses in primary biliary cirrhosis. Hepatology. 2015;61(6):1998–2007. doi:10.1002/hep.27725
63. Bednarek J, Traxinger B, Brigham D, et al. Cytokine-producing B cells promote immune-mediated bile duct injury in murine biliary atresia. Hepatology. 2018;68(5):1890–1904. doi:10.1002/hep.30051
64. Zhang B, Hu M, Zhang P, et al. BAFF promotes regulatory T-cell apoptosis and blocks cytokine production by activating B cells in primary biliary cirrhosis. Braz J Med Biol Res. 2013;46(5):433–439.
65. Takahashi T, Miura T, Nakamura J, et al. Plasma cells and the chronic nonsuppurative destructive cholangitis of primary biliary cirrhosis. Hepatology. 2012;55(3):846–855.
66. Dhirapong A, Lleo A, Yang GX, et al. B cell depletion therapy exacerbates murine primary biliary cirrhosis. Hepatology. 2011;53(2):527–535. doi:10.1002/hep.24044
67. Bonorino P, Ramzan M, Camous X, et al. Fine characterization of intrahepatic NK cells expressing natural killer receptors in chronic hepatitis B and C. J Hepatol. 2009;51(3):458–467. doi:10.1016/j.jhep.2009.05.030
68. Wu Y, Kuang DM, Pan WD, et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57(3):1107–1116. doi:10.1002/hep.26192
69. Wehr A, Baeck C, Ulmer F, et al. Pharmacological inhibition of the chemokine CXCL16 diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. PLoS One. 2014;9(11):e112327. doi:10.1371/journal.pone.0112327
70. Grégoire C, Chasson L, Luci C, et al. The trafficking of natural killer cells. Immunol Rev. 2007;220(1):169–182. doi:10.1111/j.1600-065X.2007.00563.x
71. Rattay S, Graf D, Kislat A, et al. Anti-inflammatory consequences of bile acid accumulation in virus-infected bile duct ligated mice. PLoS One. 2018;13(6):e0199863. doi:10.1371/journal.pone.0199863
72. Guo C, Zhu J, Pu CL, Deng YH, Zhang MM. Combinatory effects of hepatic CD8+ and NK lymphocytes in bile duct injury from biliary atresia. Pediatr Res. 2012;71(6):638–644. doi:10.1038/pr.2012.17
73. van Dommelen SL, Sumaria N, Schreiber RD, Scalzo AA, Smyth MJ, Degli-Esposti MA. Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity. 2006;25(5):835–848. doi:10.1016/j.immuni.2006.09.010
74. Qian C, Jiang T, Zhang W, et al. Increased IL-23 and IL-17 expression by peripheral blood cells of patients with primary biliary cirrhosis. Cytokine. 2013;64(1):172–180. doi:10.1016/j.cyto.2013.07.005
75. Shimoda S, Harada K, Niiro H, et al. Interaction between Toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology. 2011;53(4):1270–1281. doi:10.1002/hep.24194
76. Chuang YH, Lian ZX, Tsuneyama K, et al. Increased killing activity and decreased cytokine production in NK cells in patients with primary biliary cirrhosis. J Autoimmun. 2006;26(4):232–240. doi:10.1016/j.jaut.2006.04.001
77. Zhao ZB, Lu FT, Ma HD, et al. Liver-resident NK cells suppress autoimmune cholangitis and limit the proliferation of CD4(+) T cells. Cell Mol Immunol. 2020;17(2):178–189. doi:10.1038/s41423-019-0199-z
78. Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J Exp Med. 1998;188(12):2381–2386. doi:10.1084/jem.188.12.2381
79. Lleo A, Liao J, Invernizzi P, et al. Immunoglobulin M levels inversely correlate with CD40 ligand promoter methylation in patients with primary biliary cirrhosis. Hepatology. 2012;55(1):153–160. doi:10.1002/hep.24630
80. Lian C, Zhao Y, Sun J, Zhao L, Zhang F. Role of cell autophagy in the generation of IgM and hepatic fibrosis in primary biliary cholangitis. Clin Rheumatol. 2020;39(11):3499–3506. doi:10.1007/s10067-020-05111-6
81. Luo Y, Brigham D, Bednarek J, et al. Unique cholangiocyte-targeted IgM autoantibodies correlate with poor outcome in biliary atresia. Hepatology. 2021;73(5):1855–1867. doi:10.1002/hep.31504
82. Marshall K, Jin J, Atkinson C, et al. Natural immunoglobulin M initiates an inflammatory response important for both hepatic ischemia reperfusion injury and regeneration in mice. Hepatology. 2018;67(2):721–735. doi:10.1002/hep.29512
83. Abe K, Takahashi A, Nozawa Y, et al. The utility of IgG, IgM, and CD138 immunohistochemistry in the evaluation of autoimmune liver diseases. Med Mol Morphol. 2014;47(3):162–168. doi:10.1007/s00795-014-0082-z
84. Surh CD, Cooper AE, Coppel RL, et al. The predominance of IgG3 and IgM isotype antimitochondrial autoantibodies against recombinant fused mitochondrial polypeptide in patients with primary biliary cirrhosis. Hepatology. 1988;8(2):290–295. doi:10.1002/hep.1840080217
85. Pang SY, Dai YM, Zhang RZ, et al. Autoimmune liver disease-related autoantibodies in patients with biliary atresia. World J Gastroenterol. 2018;24(3):387–396. doi:10.3748/wjg.v24.i3.387
86. Lu BR, Brindley SM, Tucker RM, Lambert CL, Mack CL. α-enolase autoantibodies cross-reactive to viral proteins in a mouse model of biliary atresia. Gastroenterology. 2010;139(5):1753–1761. doi:10.1053/j.gastro.2010.07.042
87. Terrier B, Degand N, Guilpain P, Servettaz A, Guillevin L, Mouthon L. Alpha-enolase: a target of antibodies in infectious and autoimmune diseases. Autoimmun Rev. 2007;6(3):176–182. doi:10.1016/j.autrev.2006.10.004
88. Gao X, Ma H, Niu J, Li D. FcγRIIB expression increases during primary biliary cholangitis. Mol Immunol. 2023;162:30–37. doi:10.1016/j.molimm.2023.08.001
89. Tanaka A, Nezu S, Uegaki S, et al. The clinical significance of IgA antimitochondrial antibodies in sera and saliva in primary biliary cirrhosis. Ann N Y Acad Sci. 2007;1107:259–270. doi:10.1196/annals.1381.028
90. Tanaka A, Nalbandian G, Leung PS, et al. Mucosal immunity and primary biliary cirrhosis: presence of antimitochondrial antibodies in urine. Hepatology. 2000;32(5):910–915. doi:10.1053/jhep.2000.19254
91. Emancipator SN, Gallo GR, Razaboni R, Lamm ME. Experimental cholestasis promotes the deposition of glomerular IgA immune complexes. Am J Pathol. 1983;113(1):19–26.
92. Geha RS, Jabara HH, Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol. 2003;3(9):721–732. doi:10.1038/nri1181
93. Tabibian JH, Enders F, Imam MH, Kolar G, Lindor KD, Talwalkar JA. Association between serum IgE level and adverse clinical endpoints in primary sclerosing cholangitis. Ann Hepatol. 2014;13(3):384–389. doi:10.1016/S1665-2681(19)30869-5
94. Shimomura I, Takase Y, Matsumoto S, et al. Primary sclerosing cholangitis associated with increased peripheral eosinophils and serum IgE. J Gastroenterol. 1996;31(5):737–741. doi:10.1007/BF02347627
95. Shen DZ. A target role for mast cell in the prevention and therapy of hepatic fibrosis. Med Hypotheses. 2008;70(4):760–764. doi:10.1016/j.mehy.2007.07.042
96. Heine G, Dahten A, Hilt K, et al. Liver X receptors control IgE expression in B cells. J Immunol. 2009;182(9):5276–5282. doi:10.4049/jimmunol.0801804
97. Geier A, Dietrich CG, Voigt S, et al. Effects of proinflammatory cytokines on rat organic anion transporters during toxic liver injury and cholestasis. Hepatology. 2003;38(2):345–354. doi:10.1053/jhep.2003.50317
98. Hartmann G, Cheung AK, Piquette-Miller M. Inflammatory cytokines, but not bile acids, regulate expression of murine hepatic anion transporters in endotoxemia. J Pharmacol Exp Ther. 2002;303(1):273–281. doi:10.1124/jpet.102.039404
99. Sharanek A, Burban A, Ciriaci N, Guillouzo A. Pro-inflammatory cytokines enhance dilatation of bile canaliculi caused by cholestatic antibiotics. Toxicol In Vitro. 2019;58:51–59. doi:10.1016/j.tiv.2019.03.015
100. Bruccoleri A, Gallucci R, Germolec DR, et al. Induction of early-immediate genes by tumor necrosis factor alpha contribute to liver repair following chemical-induced hepatotoxicity. Hepatology. 1997;25(1):133–141. doi:10.1002/hep.510250125
101. Siewert E, Dietrich CG, Lammert F, et al. Interleukin-6 regulates hepatic transporters during acute-phase response. Biochem Biophys Res Commun. 2004;322(1):232–238. doi:10.1016/j.bbrc.2004.07.102
102. Liu G, Wang X, Yang T, et al. High interleukin-8 levels associated with decreased survival in patients with cirrhosis following transjugular intrahepatic portosystemic shunt. Front Med. 2022;9:829245. doi:10.3389/fmed.2022.829245
103. Neuman M, Angulo P, Malkiewicz I, et al. Tumor necrosis factor-alpha and transforming growth factor-beta reflect severity of liver damage in primary biliary cirrhosis. J Gastroenterol Hepatol. 2002;17(2):196–202. doi:10.1046/j.1440-1746.2002.02672.x
104. Yang CY, Ma X, Tsuneyama K, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology. 2014;59(5):1944–1953. doi:10.1002/hep.26979
105. Spirlì C, Nathanson MH, Fiorotto R, et al. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology. 2001;121(1):156–169. doi:10.1053/gast.2001.25516
106. Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer Zum Büschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol. 1995;22(2):226–229. doi:10.1016/0168-8278(95)80433-1
107. Milner JD, Orekov T, Ward JM, et al. Sustained IL-4 exposure leads to a novel pathway for hemophagocytosis, inflammation, and tissue macrophage accumulation. Blood. 2010;116(14):2476–2483. doi:10.1182/blood-2009-11-255174
108. Nagano T, Yamamoto K, Matsumoto S, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. 1999;19(6):422–427. doi:10.1023/A:1020511002025
109. Bedke T, Muscate F, Soukou S, Gagliani N, Huber S. Title: IL-10-producing T cells and their dual functions. Semin Immunol. 2019;44:101335. doi:10.1016/j.smim.2019.101335
110. Lin Z, Tian Y, Chai C, et al. The association of immune-related genes and the potential role of IL10 with biliary atresia. Pediatr Res. 2023. doi:10.1038/s41390-023-02626-x
111. Huang S, Liu Y, Guo N, Liu X, Li G, Du Q. Serum profiles of inflammatory cytokines associated with intrahepatic cholestasis of pregnancy. J Matern Fetal Neonatal Med. 2022;35(25):10072–10081. doi:10.1080/14767058.2022.2089551
112. Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J Hepatol. 2010;53(2):318–325. doi:10.1016/j.jhep.2010.03.008
113. Isse K, Harada K, Zen Y, et al. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. 2005;41(3):506–516. doi:10.1002/hep.20582
114. Limongi F. Th1 cytokines and chemokines in primary biliary cirrhosis. Clin Ter. 2015;166(2):e122–e125. doi:10.7417/CT.2015.1834
115. Lin F, Shi H, Liu D, et al. Association of CCL11, CCL24 and CCL26 with primary biliary cholangitis. Int Immunopharmacol. 2019;67:372–377. doi:10.1016/j.intimp.2018.12.026
116. Konishi T, Schuster RM, Goetzman HS, Caldwell CC, Lentsch AB. Cell-specific regulatory effects of CXCR2 on cholestatic liver injury. Am J Physiol Gastrointest Liver Physiol. 2019;317(6):G773–G783. doi:10.1152/ajpgi.00080.2019
117. Köhl J. The role of complement in danger sensing and transmission. Immunol Res. 2006;34(2):157–176. doi:10.1385/IR:34:2:157
118. Bengtson A, Heideman M. Anaphylatoxin formation in sepsis. Arch Surg. 1988;123(5):645–649. doi:10.1001/archsurg.1988.01400290131023
119. Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30(2):83–90. doi:10.1016/j.it.2008.11.003
120. Lin CJ, Hu ZG, Yuan GD, Lei B, He SQ. Complements are involved in alcoholic fatty liver disease, hepatitis and fibrosis. World J Hepatol. 2018;10(10):662–669. doi:10.4254/wjh.v10.i10.662
121. Liang J, Li H, Fu J, et al. A model incorporating serum C3 complement levels may be useful for diagnosing biliary atresia in infants. Gastroenterol Hepatol. 2022;45(1):47–58. doi:10.1016/j.gastrohep.2021.02.020
122. Guo Z, Chen J, Zeng Y, et al. Complement inhibition alleviates cholestatic liver injury through mediating macrophage infiltration and function in mice. Front Immunol. 2021;12:785287. doi:10.3389/fimmu.2021.785287
123. Li J, Pircher PC, Schulman IG, Westin SK. Regulation of complement C3 expression by the bile acid receptor FXR. J Biol Chem. 2005;280(9):7427–7434. doi:10.1074/jbc.M411473200
124. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/S1097-2765(02)00599-3
125. Wan X, Xu C, Yu C, Li Y. Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can J Gastroenterol Hepatol. 2016;2016:6489012. doi:10.1155/2016/6489012
126. Wu Y, Qiu G, Zhang H, et al. Dexmedetomidine alleviates hepatic ischaemia-reperfusion injury via the PI3K/AKT/Nrf2-NLRP3 pathway. J Cell Mol Med. 2021;25(21):9983–9994. doi:10.1111/jcmm.16871
127. Matsushita H, Miyake Y, Takaki A, et al. TLR4, TLR9, and NLRP3 in biliary epithelial cells of primary sclerosing cholangitis: relationship with clinical characteristics. J Gastroenterol Hepatol. 2015;30(3):600–608. doi:10.1111/jgh.12711
128. Tian J, Yang G, Chen HY, et al. Galectin-3 regulates inflammasome activation in cholestatic liver injury. FASEB j. 2016;30(12):4202–4213. doi:10.1096/fj.201600392RR
129. Gaul S, Leszczynska A, Alegre F, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021;74(1):156–167. doi:10.1016/j.jhep.2020.07.041
130. Frissen M, Liao L, Schneider KM, et al. Bidirectional role of NLRP3 during acute and chronic cholestatic liver injury. Hepatology. 2021;73(5):1836–1854. doi:10.1002/hep.31494
131. Hao H, Cao L, Jiang C, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 2017;25(4):856–867.e855. doi:10.1016/j.cmet.2017.03.007
132. Balazs I, Horvath A, Leber B, et al. Serum bile acids in liver cirrhosis promote neutrophil dysfunction. Clin Transl Med. 2022;12(2):e735. doi:10.1002/ctm2.735
133. Cariello M, Piccinin E, Garcia-Irigoyen O, Sabbà C, Moschetta A. Nuclear receptor FXR, bile acids and liver damage: introducing the progressive familial intrahepatic cholestasis with FXR mutations. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4 Pt B):1308–1318. doi:10.1016/j.bbadis.2017.09.019
134. Allen K, Kim ND, Moon JO, Copple BL. Upregulation of early growth response factor-1 by bile acids requires mitogen-activated protein kinase signaling. Toxicol Appl Pharmacol. 2010;243(1):63–67. doi:10.1016/j.taap.2009.11.013
135. Chen W, Ding M, Ji L, et al. Bile acids promote the development of HCC by activating inflammasome. Hepatol Commun. 2023;7(9). doi:10.1097/HC9.0000000000000217
136. Yamada K, Lim BO, Sugano M. Suppression of immunoglobulin production of rat lymphocytes by bile acids. Vitro Cell Dev Biol Anim. 1993;29(11):840–841. doi:10.1007/BF02631360
137. Keane RM, Gadacz TR, Munster AM, Birmingham W, Winchurch RA. Impairment of human lymphocyte function by bile salts. Surgery. 1984;95(4):439–443.
138. Sung JJ, Go MY. Reversible Kupffer cell suppression in biliary obstruction is caused by hydrophobic bile acids. J Hepatol. 1999;30(3):413–418. doi:10.1002/hep.24757
139. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. 2017;14(1):55–66. doi:10.1038/nrgastro.2016.168
140. Jalan R, Fernandez J, Wiest R, et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J Hepatol. 2014;60(6):1310–1324. doi:10.1016/j.jhep.2014.01.024
141. Recknagel P, Gonnert FA, Westermann M, et al. Liver dysfunction and phosphatidylinositol-3-kinase signalling in early sepsis: experimental studies in rodent models of peritonitis. PLoS Med. 2012;9(11):e1001338. doi:10.1371/journal.pmed.1001338
142. Bahador M, Cross AS. From therapy to experimental model: a hundred years of endotoxin administration to human subjects. J Endotoxin Res. 2007;13(5):251–279. doi:10.1177/0968051907085986
143. Remetic J, Ghallab A, Hobloss Z, et al. Loss of bile salt export pump aggravates lipopolysaccharide-induced liver injury in mice due to impaired hepatic endotoxin clearance. Hepatology. 2022;75(5):1095–1109. doi:10.1002/hep.32289
144. Trussoni CE, Tabibian JH, Splinter PL, O’Hara SP. Lipopolysaccharide (LPS)-induced biliary epithelial cell nras activation requires Epidermal Growth Factor Receptor (EGFR). PLoS One. 2015;10(4):e0125793. doi:10.1371/journal.pone.0125793
145. Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13(11):1324–1332. doi:10.1038/nm1663
146. Horvatits T, Trauner M, Fuhrmann V. Hypoxic liver injury and cholestasis in critically ill patients. Curr Opin Crit Care. 2013;19(2):128–132. doi:10.1097/MCC.0b013e32835ec9e6
147. Koskinas J, Gomatos IP, Tiniakos DG, et al. Liver histology in ICU patients dying from sepsis: a clinico-pathological study. World J Gastroenterol. 2008;14(9):1389–1393. doi:10.3748/wjg.14.1389
148. Jenniskens M, Langouche L, Vanwijngaerden YM, Mesotten D, Van den Berghe G. Cholestatic liver (dys) function during sepsis and other critical illnesses. Intensive Care Med. 2016;42(1):16–27. doi:10.1007/s00134-015-4054-0
149. Ramadori G, Christ B. Cytokines and the hepatic acute-phase response. Semin Liver Dis. 1999;19(2):141–155. doi:10.1055/s-2007-1007106
150. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340(6):448–454. doi:10.1056/NEJM199902113400607
151. Sasaki M, Ikeda H, Yamaguchi J, Miyakoshi M, Sato Y, Nakanuma Y. Bile ductular cells undergoing cellular senescence increase in chronic liver diseases along with fibrous progression. Am J Clin Pathol. 2010;133(2):212–223. doi:10.1309/AJCPWMX47TREYWZG
152. Ferreira-Gonzalez S, Lu WY, Raven A, et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat Commun. 2018;9(1):1020. doi:10.1038/s41467-018-03299-5
153. Sasaki M, Ikeda H, Haga H, Manabe T, Nakanuma Y. Frequent cellular senescence in small bile ducts in primary biliary cirrhosis: a possible role in bile duct loss. J Pathol. 2005;205(4):451–459. doi:10.1002/path.1729
154. Liu R, Xu W, Zhu H, Dong Z, Dong H, Yin S. Aging aggravates Acetaminophen-induced acute liver injury and inflammation through inordinate C/EBPα-BMP9 crosstalk. Cell Biosci. 2023;13(1):61. doi:10.1186/s13578-023-01014-6
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.