")
Back to Journals » OncoTargets and Therapy » Volume 11
The role of histone lysine methyltransferase NSD3 in cancer
Authors Han X , Piao L, Zhuang Q, Yuan X , Liu Z, He X
Received 27 February 2018
Accepted for publication 8 April 2018
Published 5 July 2018 Volume 2018:11 Pages 3847—3852
DOI https://doi.org/10.2147/OTT.S166006
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samir Farghaly
Xu Han,1,* Lianhua Piao,2,* Qianfeng Zhuang,1 Xiaofeng Yuan,3 Zhiwei Liu,3 Xiaozhou He1
1Department of Urology, The Third Affiliated Hospital of Soochow University, 2Institute of Bioinformatics and Medical Engineering, Jiangsu University of Technology, 3Department of Orthopaedics, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Abstract: The growing number of findings demonstrate that nuclear receptor suppressor of variegation, enhancer of zeste, and trithorax domain-containing 3 (NSD3) is amplified and overexpressed in multiple cancer types. Nevertheless, the biological roles of NSD3 in carcinogenesis have not been well understood. In this review, we summarize the current knowledge on the mechanisms underlying NSD3 regulation in different cancers. In addition, NSD3 may serve as a potential druggable target for selective cancer therapy in the future.
Keywords: NSD3, cancer, carcinogenesis
Introduction
Nuclear receptor suppressor of variegation, enhancer of zeste, and trithorax (SET) domain-containing 3 (NSD3) is a well-known histone lysine methyltransferase (HMTase), a member of the NSD protein family. This family is composed of three HMTases, such as NSD1, NSD2 (WHSC1/MMSET), and NSD3 (Wolf–Hirschhorn syndrome candidate 1-like 1 [WHSC1L1]), which are primarily known to be involved in chromatin integrity and gene expression through mono-, di-, or tri-methylating lysine 36 of histone H3 (H3K36), respectively.1–3
Accumulating evidence reveals that the amplification of NSDs results in cellular transformation and plays crucial roles in cancer pathogenesis.4 Herein, we review the fundamental characteristics of the third member NSD3, with particular focus on the biological functions of NSD3 in a great variety of cancers (Table 1). Notably, NSD3 contributes to tumorigenesis by interacting with bromodomain-containing protein 4 (BRD4), the bromodomain and extraterminal (BET) protein, which is a potential therapeutic target in acute myeloid leukemia (AML).
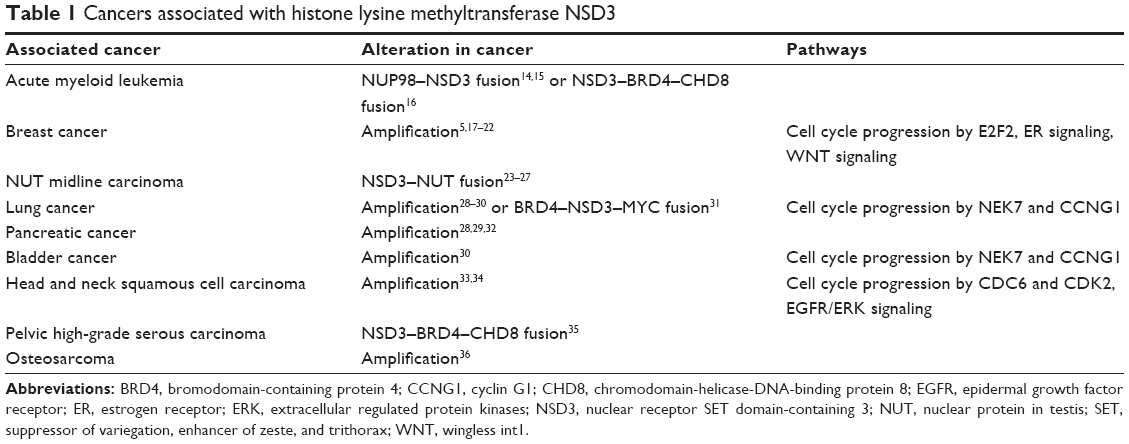 | Table 1 Cancers associated with histone lysine methyltransferase NSD3 |
Identification and characterization of NSD3
NSD3, also known as WHSC1L1, is originally identified in 2001.5,6 Similar to NSD1 and NSD2, the NSD3 gene also possesses a C-terminal block containing ~700 amino acids. Nonetheless, the conserved PHD5–C5HCH module of NSD3 prefers to recognize and bind to the N-terminal peptides of histone H3, such as unmodified K4 and tri-methylated K9, which is greatly different from the other two members.7 Importantly, the NSD3 gene is located on chromosome 8p11.23, along with strong cancer relevance. A total of three protein products of NSD3 deserve to be reported and characterized as long, short, and whistle (Figure 1).8
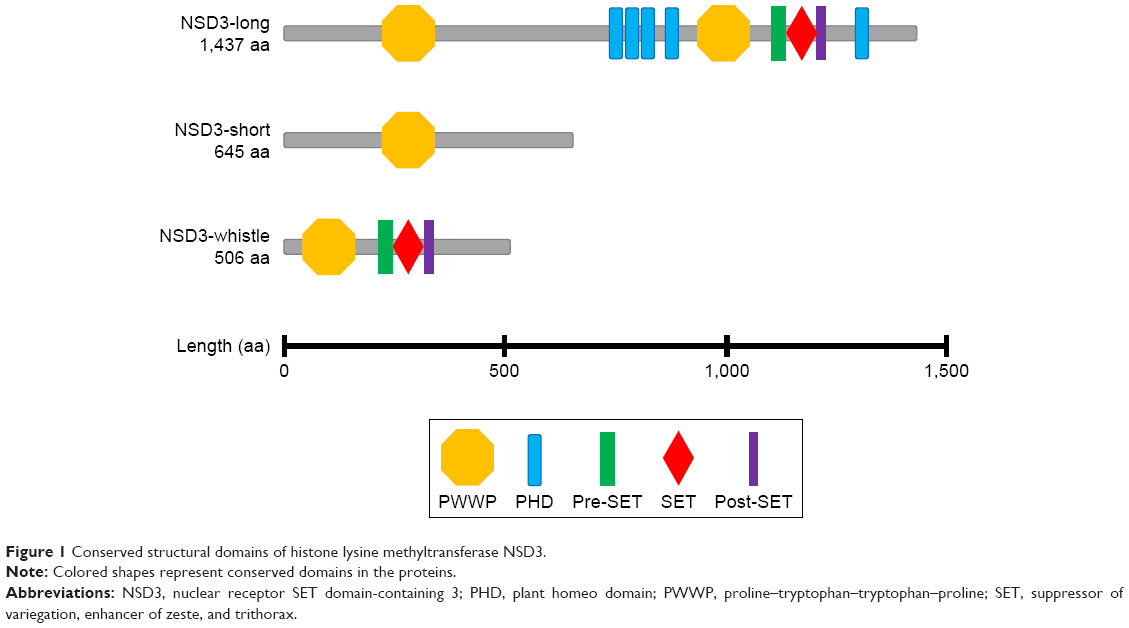 | Figure 1 Conserved structural domains of histone lysine methyltransferase NSD3. |
The NSD3-long isoform encodes a protein of 1,437 amino acids, which contains two proline-tryptophan-tryptophan-proline (PWWP, the conserved motif Pro–Trp–Trp–Pro) domains, five plant homeo domain (PHD)-type zinc finger motifs, one SET-associated Cys (SAC)-rich domain, and one SET domain.5 Notably, the C-terminal of NSD3-long isoform, which contains the pre-SET, SET, and post-SET domains within the catalytic core, is critical for its recognizing and methylating molecular targets of histones H3 and H4 in vitro.9,10
Compared with NSD3-long isoform, the NSD3-short isoform encodes a 645-amino acid protein that lacks the catalytic SET domain, but it reserves one N-terminal PWWP domain of 620 amino acids, which can bind to histone H3 at lysine 36.11,12 Interestingly, both transcripts of NSD3-short and NSD3-long isoforms are coexpressed in tumorous tissues, which mean that they have to compete for protein interaction via the analogous PWWP domain.
Whistle (WHSC1-like 1 isoform 9 with methyltransferase activity to lysine) is the shortest isoform of NSD3 and merely consists of 506 amino acids, which retains the following three distinct domains: PWWP, SET, and post-SET. NSD3-whistle is capable of repressing gene transcription by facilitating specific di-methylation activities on histones H3K4 and H3K27.13
NSD3 in AML
The NSD3 gene has been observed as a transcriptional coactivator in prior analyses of some cases with AML. Rosati et al14 first reported that there is a fusion between the NUP98 and NSD3 genes in a patient accompanied with t(8;11)(p11.2;p15). Afterward, Taketani et al15 presented an atomic bomb survivor in Nagasaki with radiation-associated myelodysplastic syndrome (r-MDS), carrying chromosome abnormalities, including t(8;11)(p11;p15) and del(1)(p22p32). Furthermore, they found that NSD3-long and NSD3-short isoforms are prevalently expressed not only in common leukemic cell lines but also in EBV-B cell lines derived from normal adults. Altogether, these data demonstrate that the NUP98–NSD3 fusion transcript can lead to hematological malignancies. Meanwhile, there is another mechanism for NSD3 in AML. Shen et al16 discovered that NSD3-short isoform may serve as an essential adaptor protein that induces leukemia by linking the BET protein BRD4 to the chromatin remodeling enzyme chromodomain-helicase-DNA-binding protein 8 (CHD8). Remarkably, AML cells merely require the NSD3-short isoform, which lacks the catalytic SET domain, to allow transcriptional activation. Collectively, NSD3 can become a vital oncogene related to leukemogenesis when it is fused to NUP98 or bound to BRD4 and CHD8.
NSD3 in breast cancer
Angrand et al5 showed that NSD3 is amplified in primary tumors and cell lines from breast carcinoma. Zhou et al17 indicated that NSD3-long isoform may obviously suppress cell proliferation and invasion capacity through affecting the expression of cell cycle regulator E2F2. However, Yang et al18 found that knockdown of NSD3 via shRNA in breast cancer cells amplified 8p11-12 can dramatically inhibit cell growth and survival. The regulatory factors of WNT signaling, iroquois homeobox 3 (IRX3), and TBL1X are markedly promoted due to an enhanced expression of NSD3, accompanied by the decreased expression of a negative factor SFRP1. Irish et al19 confirmed that the overexpression of NSD3 in SUM-44 breast cancer cells is highly correlated with the overexpression of ESR1 mRNA and ERα protein, which are transcriptionally active in luminal breast cancer. In summary, these studies illustrate that NSD3 may be a candidate tumor suppressor or an activator in different genetic backgrounds. After analyzing the relationship between the NSD3 expression level and overall patient survival, Liu et al20 reported that higher expression patients have an HR of 1.659 compared with lower expression ones in breast cancer. Turner-Ivey et al21 observed that targeted expression of NSD3 in FVB mice develops mammary gland hyperplasias, dysplasias, carcinoma in situ, and mammary carcinomas by the age of 40 weeks. Moreover, functional differentiation in the mammary gland and developmental growth of the offspring are distinctly limited because of overexpressed NSD3. Aside from those experimental researches, Chen et al identified NSD3 as a bona fide transforming breast cancer oncogene through analyzing the gene amplifications in TCGA data sets derived from human specimens.22 Taken together, NSD3 is considered as a novel putative driving oncogene and a promising therapeutic approach in breast cancer patients.
NSD3 in nuclear protein in testis (NUT) midline carcinoma (NMC)
NMC is an exceedingly rare and poorly differentiated squamous cell carcinoma with a median survival of 6.7 months for the lack of standard treatment algorithms so far.23 It was reported that the NUT gene may fuse to NSD3 in NMC, which is known to typically harbor BRD4/3-NUT fusion oncoprotein. French et al24 showed that the protein encoded by NSD3–NUT fusion oncogene is necessary and sufficient for blocking differentiation and maintaining proliferation in NMC cells. Nevertheless, BET inhibitors, JQ1, are able to induce differentiation and arrest proliferation in 1,221 cells expressing NSD3–NUT fusion, which implies that NSD3–NUT complexes may need to utilize the chromatin-reading function of BRD4. Then, Kuroda et al first described the cytological features of lung NMC with the NSD3–NUT rearrangements and suggested that overt pearl formation including a dyskeratocyte, stratification, and cytoplasmic fine vacuoles is helpful to make a diagnosis of NMC.25,26 In addition, high levels of NUT are only unveiled in the undifferentiated NMC cells, but NUT expression is faint or absent in the keratinizing cells.27 Taking those into consideration, NSD3 is certainly a prominent part of the NSD3–NUT complexes triggering the oncogenesis of NMC.
NSD3 in lung, pancreatic, and bladder cancers
Coupling bioinformatics analysis with knockdown studies, Tonon et al28 identified NSD3 as a prime target of the 8p11-12 amplification event in both non-small cell lung cancer and pancreatic ductal adenocarcinoma. Mahmood et al29 further confirmed that NSD3 can promote the cell viability of small-cell lung cancer and pancreatic ductal adenocarcinoma both in anchorage-dependent and anchorage-independent conditions. Of note, the potential downstream genes targeted by NSD3 in these two cancer types are extraordinarily different from those in breast cancer. Kang et al30 indicated that the expression of NSD3 is significantly elevated in the tissues of lung adenocarcinoma and bladder cancer. After the depletion of NSD3, cell proliferation is effectively reduced and the expressions of cell cycle enhancers, cyclin G1 (CCNG1) and NEK7, are decreased via expression profile analysis, which imply that NSD3 appears to be a critical player in the G2/M transition. Besides, Li et al31 uncovered another probable role of NSD3 in activating the oncogenic function of MYC gene by bridging MYC with BRD4 in lung cancer. The BRD4–NSD3–MYC pathway may be a newly therapeutic strategy, which successfully associates tumor activators with druggable targets. And in pancreatic adenocarcinoma, Mann et al32 conducted a mutagenic screen using Sleeping Beauty (SB) in mice and showed that NSD3 can cooperate with oncogenic Kras to result in tumorigenesis, whereas the exact epigenetic mechanism of how dysregulation of NSD3 may lead to lung, pancreatic, and bladder cancers remains to be elucidated.
NSD3 in squamous cell carcinoma of the head and neck (SCCHN), high-grade serous carcinoma (HGSC), and osteosarcoma
Saloura et al33 found that high NSD3 expression is implicated in poor grade and heavy smoking history in SCCHN. Furthermore, the transcription of cell cycle genes CDC6 and CDK2 contributing to the transition from G1 to S phase is directly modulated by the enhancement of H3K36 di-methylation. Later, they showed that NSD3 can mono-methylate lysine 721 of EGFR to promote activation of the extracellular regulated protein kinases cascade, which interacts with proliferating cell nuclear antigen to accelerate DNA replication and enhances cell cycle progression in SCCHN cells.34 In pelvic HGSC of gynecological (tubo-ovarian or endometrial) origin, Jones and Lin35 insisted that the amplification of NSD3–BRD4–CHD8 axis is closely correlated with worse prognosis and survival compared with nonamplified cases, depending on the retrospective analysis of TCGA cancer cohorts. More recently, Liu et al36 reported that silencing of NSD3 distinctly reduces cell viability and survival, along with increasing the percentage of cells in the G2/M phase and inducing cell apoptosis in osteosarcoma. In addition, 549 NSD3-regulated genes (244 upregulated and 305 downregulated) are identified via RNA-seq analysis. Hence, these data indicate that NSD3 possesses crucial effects in cell cycle progression.
Conclusion and future perspective
This review highlights the biological functions of NSD3 in a subset of cancers. Based on these findings, NSD3 is perceived mostly as an important adaptor protein to underline its indispensable part of oncogenic complexes. For instance, the NUP98–NSD3 fusion and the NSD3–NUT fusion are involved in chromatin remodeling and transcriptional regulation leading to oncogenesis. Also, the NSD3–BRD4–CHD8 axis and the BRD4–NSD3–MYC axis may sustain oncogenic transcriptional programs because their amplification is highly associated with worse overall survival and progression-free survival in patients. Besides, NSD3 can directly target different downstream genes that play central roles in various cellular processes (Figure 2).
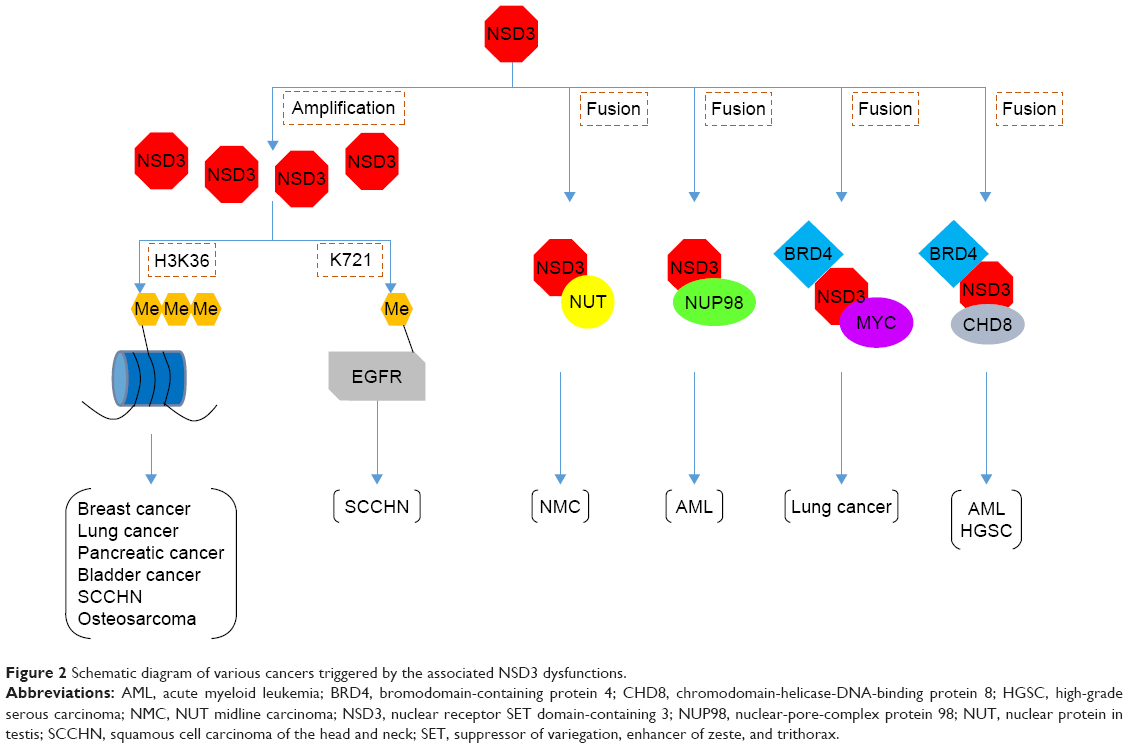 | Figure 2 Schematic diagram of various cancers triggered by the associated NSD3 dysfunctions. |
As NSD3 may represent an effective and valuable target in the epigenetic therapy for patients with aberrant expression of HMTase, further investigation is urgently required to clarify the precise signaling pathways of NSD3 in cancer development and progression. Unfortunately, no inhibitors specifically targeting NSD3 have been found till date. However, in consideration of the BET inhibitors, JQ1, which have been applied in the clinical treatment of certain cancer, we believe that the inhibitors of NSD3 can offer newly promising therapeutic opportunities to cure and prevent human cancers in the near future.
Acknowledgments
This study was supported by the National Science Foundation of Jiangsu Province (grant no BK20150251) and Youth Medical Talent Project of Jiangsu Province (QNRC2016292).
Disclosure
The authors report no conflicts of interest in this work.
References
Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13(2):115–126. | ||
Vougiouklakis T, Hamamoto R, Nakamura Y, Saloura V. The NSD family of protein methyltransferases in human cancer. Epigenomics. 2015;7(5):863–874. | ||
Lucio-Eterovic AK, Singh MM, Gardner JE, Veerappan CS, Rice JC, Carpenter PB. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc Natl Acad Sci U S A. 2010;107(39):16952–16957. | ||
Morishita M, di Luccio E. Cancers and the NSD family of histone lysine methyltransferases. Biochim Biophys Acta. 2011;1816(2):158–163. | ||
Angrand PO, Apiou F, Stewart AF, Dutrillaux B, Losson R, Chambon P. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics. 2001;74(1):79–88. | ||
Stec I, van Ommen GJ, den Dunnen JT. WHSC1L1, on human chromosome 8p11.2, closely resembles WHSC1 and maps to a duplicated region shared with 4p16.3. Genomics. 2001;76(1–3):5–8. | ||
He C, Li FD, Zhang JH, Wu JH, Shi YY. The methyltransferase NSD3 has chromatin-binding motifs, PHD5–C5HCH, that are distinct from other NSD (nuclear receptor SET domain) family members in their histone H3 recognition. J Biol Chem. 2013;288(7):4692–4703. | ||
Bennett RL, Swaroop A, Troche C, Licht JD. The role of nuclear receptor-binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harb Perspect Med. 2017;7(6):a026708. | ||
Allali-Hassani A, Kuznetsova E, Hajian T, et al. A basic post-SET extension of NSDs is essential for nucleosome binding in vitro. J Biomol Screen. 2014;19(6):928–935. | ||
Morishita M, Mevius D, di Luccio E. In vitro histone lysine methylation by NSD1, NSD2/MMSET/WHSC1 and NSD3/WHSC1L. BMC Struct Biol. 2014;14:25. | ||
Wu H, Zeng H, Lam R, et al. Structural and histone binding ability characterizations of human PWWP domains. PLoS One. 2011;6(6):e18919. | ||
Vermeulen M, Eberl HC, Matarese F, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142(6):967–980. | ||
Kim SM, Kee HJ, Eom GH, et al. Characterization of a novel WHSC1-associated SET domain protein with H3K4 and H3K27 methyltransferase activity. Biochem Biophys Res Commun. 2006;345(1):318–323. | ||
Rosati R, La Starza R, Veronese A, et al. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood. 2002;99(10):3857–3860. | ||
Taketani T, Taki T, Nakamura H, Taniwaki M, Masuda J, Hayashi Y. NUP98-NSD3 fusion gene in radiation-associated myelodysplastic syndrome with t(8;11)(p11;p15) and expression pattern of NSD family genes. Cancer Genet Cytogenet. 2009;190(2):108–112. | ||
Shen C, Ipsaro JJ, Shi JW, et al. NSD3-short is an adaptor protein that couples BRD4 to the CHD8 chromatin remodeler. Mol Cell. 2015;60(6):847–859. | ||
Zhou ZL, Thomsen R, Kahns S, Nielsen AL. The NSD3L histone methyltransferase regulates cell cycle and cell invasion in breast cancer cells. Biochem Biophys Res Commun. 2010;398(3):565–570. | ||
Yang ZQ, Liu G, Bollig-Fischer A, Giroux CN, Ethier SP. Transforming properties of 8p11-12 amplified genes in human breast cancer. Cancer Res. 2010;70(21):8487–8497. | ||
Irish JC, Mills JN, Turner-Ivey B, et al. Amplification of WHSC1L1 regulates expression and estrogen-independent activation of ERα in SUM-44 breast cancer cells and is associated with ERα over-expression in breast cancer. Mol Oncol. 2016;10(6):850–865. | ||
Liu LX, Kimball S, Liu H, Holowatyj A, Yang ZQ. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget. 2015;6(4):2466–2482. | ||
Turner-Ivey B, Smith EL, Rutkovsky AC, Spruill LS, Mills JN, Ethier SP. Development of mammary hyperplasia, dysplasia, and invasive ductal carcinoma in transgenic mice expressing the 8p11 amplicon oncogene NSD3. Breast Cancer Res Treat. 2017;164(2):349–358. | ||
Chen Y, McGee J, Chen XM, et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One. 2014;9(5):e98293. | ||
Bauer DE, Mitchell CM, Strait KM, et al. Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin Cancer Res. 2012;18(20):5773–5779. | ||
French CA, Rahman S, Walsh EM, et al. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discov. 2014;4(8):928–941. | ||
Kuroda S, Suzuki S, Kurita A, et al. Cytological features of a variant NUT midline carcinoma of the lung harboring the NSD3-NUT fusion gene: a case report and literature review. Case Rep Pathol. 2015;2015:572951. | ||
Suzuki S, Kurabe N, Ohnishi I, et al. NSD3-NUT-expressing midline carcinoma of the lung: first characterization of primary cancer tissue. Pathol Res Pract. 2015;211(5):404–408. | ||
Harms A, Herpel E, Pfarr N, et al. NUT carcinoma of the thorax: case report and review of the literature. Lung Cancer. 2015;90(3):484–491. | ||
Tonon G, Wong KK, Maulik G, et al. High-resolution genomic profiles of human lung cancer. Proc Natl Acad Sci U S A. 2005;102(27):9625–9630. | ||
Mahmood SF, Gruel N, Nicolle R, et al. PPAPDC1B and WHSC1L1 are common drivers of the 8p11-12 amplicon, not only in breast tumors but also in pancreatic adenocarcinomas and lung tumors. Am J Pathol. 2013;183(5):1634–1644. | ||
Kang D, Cho HS, Toyokawa G, et al. The histone methyltransferase Wolf-Hirschhorn syndrome candidate 1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes Chromosomes Cancer. 2013;52(2):126–139. | ||
Li ZG, Ivanov AA, Su RN, et al. The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat Commun. 2017;8:14356. | ||
Mann KM, Ward JM, Yew CC, et al; Australian Pancreatic Cancer Genome Initiative. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2012;109(16):5934–5941. | ||
Saloura V, Vougiouklakis T, Zewde M, et al. WHSC1L1 drives cell cycle progression through transcriptional regulation of CDC6 and CDK2 in squamous cell carcinoma of the head and neck. Oncotarget. 2016;7(27):42527–42538. | ||
Saloura V, Vougiouklakis T, Zewde M, et al. WHSC1L1-mediated EGFR mono-methylation enhances the cytoplasmic and nuclear oncogenic activity of EGFR in head and neck cancer. Sci Rep. 2017;7:40664. | ||
Jones DH, Lin DI. Amplification of the NSD3–BRD4–CHD8 pathway in pelvic high-grade serous carcinomas of tubo-ovarian and endometrial origin. Mol Clin Oncol. 2017;7(2):301–307. | ||
Liu ZW, Piao LH, Zhuang M, et al. Silencing of histone methyltransferase NSD3 reduces cell viability in osteosarcoma with induction of apoptosis. Oncol Rep. 2017;38(5):2796–2802. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.