")
Back to Journals » Drug Design, Development and Therapy » Volume 8
The role of health technology assessment bodies in shaping drug development
Received 25 July 2014
Accepted for publication 9 September 2014
Published 10 November 2014 Volume 2014:8 Pages 2273—2281
DOI https://doi.org/10.2147/DDDT.S49935
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Oriana Ciani,1,3 Claudio Jommi1,2
1Centre for Research on Health and Social Care Management (CERGAS), Bocconi University, Milan, Italy; 2Department of Pharmaceutical Sciences, Università del Piemonte Orientale, Novara, Italy; 3University of Exeter Medical School, Exeter, UK
Abstract: The use of health technology assessment (HTA) to inform policy-making is established in most developed countries. Compared to licensing agencies, HTA agencies have different interests and, therefore, different evidence requirements. Criteria for coverage or reimbursement decisions on pharmaceutical compounds vary; however, it is common to include, as part of the HTA, a comparative effectiveness evaluation. This type of clinical data might go beyond that required for market authorization, thus creating an additional evidence gap between the regulatory and the reimbursement submission. The relevance of submissions to HTA agencies is consistently increasing in a pharmaceutical company’s perspective, as market prospects are strongly influenced by third-party payers’ coverage. In this study, we aim to describe current HTA activities with a potential impact throughout the drug development process of pharmaceuticals, with a comparative emphasis on the systems in place in Italy and in the UK. Based on an extensive literature and website review, we identified three major classes of HTA activities, beyond mainstream HTA, with the potential to influence the drug development program: 1) horizon scanning and early HTA; 2) bipartite and tripartite early dialogue between manufacturers, regulators, and HTA assessors; and 3) managed market entry agreements. From early stages of clinical research up to postauthorization studies, there is a trend toward increased collaboration between parties, anticipation of market access evidence collection, and postmarketing risk-sharing. Heterogeneity of HTA practices increases the complexity of the market access environment. Overall, there are signals that market access departments are gaining importance in the pharmaceutical companies, but there is still a lack of evidence and reporting on how the increasing relevance of HTA has reshaped the way clinical development is designed and managed.
Keywords: health technology assessment, horizon scanning, scientific advice, risk sharing, drug development
Introduction
During the last decades, governments across many jurisdictions have been faced by the challenge of controlling increasing health care expenditure, partly due to new medical technologies, within a context of dwindling budgets, exacerbated by the recent economic and financial crisis. As a result, demand for evidence and justification of value are increasingly a requirement to support funding, coverage, and reimbursement decisions or price negotiation about health technologies.1 The use of health technology assessment (HTA) to inform policy-making is established in North America, Australia, in many European countries, and is rapidly growing in Latin America and Asia.2–6 HTA has been described as the multidisciplinary process that summarizes information about several dimensions (eg, clinical, ethical, economic) related to the use of a health technology with the aim to achieve the best value in the allocation of public resources.7 In countries where HTA is in place, third-party payers, pricing and reimbursement agencies, or HTA bodies rely upon HTA to determine the reimbursement status of a drug (eg, listing in the local formulary), to issue guidance about the appropriate use of the technology within the health care system, and to support the price negotiation process.8 According to the remit of the organization and the structure of the health care system, this role is played at the central/national level or regional/provincial level.9
Compared to licensing agencies that are focused on the risk-benefit profile of a product, evaluated in a rigorously controlled setting, typically randomized controlled trials, with high attention on internal validity, safety, efficacy, and manufacturing, HTA agencies have different interests and, therefore, different evidence requirements.10 Criteria for coverage or reimbursement decisions on pharmaceutical compounds vary across countries; however, it is common to include, as part of the HTA, a comparative effectiveness evaluation.11 Demonstrating relative effectiveness in addition to efficacy means providing evidence that the intervention has beneficial health outcomes under “real-world” conditions and in comparison with the local standard of care. The most stringent criteria of effectiveness versus efficacy are often met in a subgroup of the licensed-approved population; therefore, the HTA report has to focus on the appropriate target population identified from the reimbursement agencies’ perspective.12 In terms of health outcomes, final patient-relevant outcomes, which are outcomes that reflect how patients feel, function, or survive,13 are preferred over surrogate or intermediate outcomes (eg, bone mineral density, ejection fraction) by pricing and reimbursement agencies.14 This type of clinical data might go beyond that required for market authorization, thus creating an additional evidence gap between the regulatory and the reimbursement submission. In many cases, the information required at the market access stage also includes economic data, presented as cost-effectiveness analyses,12 budget impact analyses, or other types of economic evaluation.
Although submissions to HTA agencies do not have the same legal status as submissions to regulatory bodies, their relevance is consistently increasing in a pharmaceutical company’s perspective, as market prospects are strongly influenced by third-party payers’ coverage. This is because market access, together with market authorization, is often regarded as an important goal of the drug development program,15 and technology appraisal is playing a significant role in opening or hindering market access for many innovative products.
A standard representation of the drug development process is given in Figure 1, where a simplified sequence of stages from basic research, discovery, and preclinical research, through clinical and postmarketing development is illustrated.
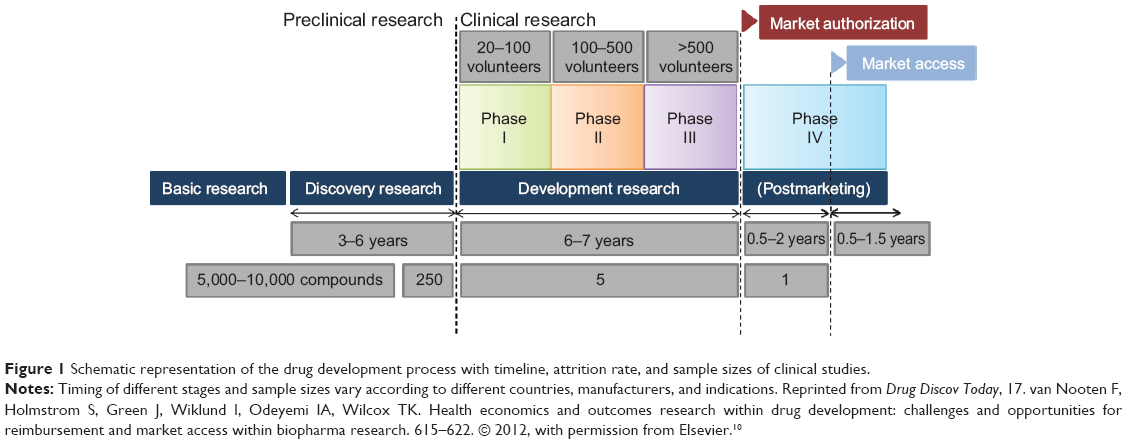 | Figure 1 Schematic representation of the drug development process with timeline, attrition rate, and sample sizes of clinical studies. |
In order to overcome the challenges posed by the “fourth hurdle,”16,17 the manufacturers need to plan in advance for the evidence requirement and assessment for coverage and reimbursement. Moreover, while in the past postmarketing risk of failure of pharmaceuticals was sustained by payers, in recent years, managed market entry agreements (MMEAs) have been introduced. Under such agreements, the risk of a negative risk-benefit profile or excessive burden is, at least partially, covered by the industry; therefore, the drug development cycle has to evolve to provide data for this purpose.
In this study, we aim to describe current HTA activities, with comparative emphasis on the systems in place in Italy and in the UK, with the potential to impact on the drug development path of pharmaceuticals.
Methods
We accessed the scientific literature as of June 2014 via PubMed, using two relevant keywords (ie, “HTA” and “drug development”), to build the background for this study. As we referred to institutionalized HTA activities,18 we identified two HTA bodies, the National Institute of Health and Care Excellence (NICE) in the UK19 and the Italian Drug Agency (AIFA [Agenzia Italiana del Farmaco]) in Italy20 and accessed their institutional websites to collect information according to a predefined template. The two agencies were selected because, 1) they are operating in large and dynamic pharmaceutical markets, 2) they have both introduced an early advice program on HTA with relevant guidance published on institutional websites, and 3) they present different models of price and reimbursement. In the UK, there is no formal negotiation of price and reimbursement status between the agency and the manufacturer, which is instead indirectly regulated through the Pharmaceutical Price Regulation Scheme. Prioritized drugs are subject to an appraisal process based on clinical and cost-effectiveness data, managed by NICE and the Scottish Medicine Consortium, that produces a positive (fully or restricted) or negative recommendation. In Italy, price and reimbursement are simultaneously negotiated by AIFA and the pharmaceutical company: disease relevance (ie, target population), place in therapy, added therapeutic value, and drug budget impact, together with prices in other countries, are the most important parameters considered, whereas cost-effectiveness does not play an important role.21
Results
The development of HTA to inform the coverage and reimbursement decisions about new drug therapies has necessarily contributed to the streamlining of the drug development program. In particular, the emphasis on the incremental socioeconomic value to be established on new medical technologies has fostered the adoption and relevance of health economics and outcomes research through the drug development process.10 As outlined earlier, mainstream HTA activities and reports demand more additional evidence than do regulatory dossiers. Effectiveness under “real-world” conditions and in comparison with the current standard of care, patient-relevant outcomes, patient-reported outcomes, and health-related quality of life, economic data, and, albeit with various definitions, innovation, are usually required in a value dossier for coverage and reimbursement agencies. In order to address these needs, manufacturers have strategically started to secure time and funding during the clinical development program to integrate health economics and outcomes research endpoints in clinical studies, to develop, validate, and use patient-reported outcomes tools (eg, dermatology life quality index, psoriasis area, and severity index), to run burden of illness studies and build cost-effectiveness models or budget impact analyses.
Most importantly, on top of mainstream HTA, our review identified other activities, with lower levels of diffusion to date, that have the potential to influence the drug development process of a new medical technology. These are described in the following sections.
Horizon scanning and early HTA
Horizon scanning systems (HSSs) were introduced as part of the broad cycle of HTA in order to systematically assess the potential impact of new and emerging technologies to anticipate policy development, access, and provision of health services.22 For this reason, they are also called “early-warning” or “early-awareness” systems.
Since 2006, the vast majority of topics for technology appraisals at NICE are identified by the National Institute for Health Research Horizon Scanning Centre, based at the University of Birmingham.23 Technologies are prioritized based on the expected health benefit or financial impact, inappropriate use, and variation of use across the country and effect on other health-related policies, such as reduction in health inequalities. This process allows notification of relevant new drugs at NICE, 20 months before the market authorization.
In Italy, AIFA does not have an HSS in place. In 2008, this institution co-founded a project, named Italian Horizon Scanning Project (IHSP), which is still part of the European Network of Horizon Scanning (HS) Centres.24 The IHSP produces an annual list of drugs that are expected to be launched within 3 years; every 6 months, a report on the epidemiology of the target population, the clinical evidence, and the comparators of listed drugs is released and a more detailed report on prioritized drugs is released 12 months before their expected market launch. Prioritization depends on the burden of disease, the therapeutic value, and the expected impact on drugs expenditure. Despite the fact that AIFA has co-founded IHSP, there is no evidence on whether and how it has relied on the results of this project.
Murphy et al25 described an effective HSS as one that identifies innovations likely to have a significant impact and disseminates in a timely manner the information relevant to the needs of the customers, so as to enable appropriate decision making, facilitate appropriate adoption, and identify further research requirements. These characteristics are summarized in a set of five primary (ie, relevance of information, independence, sufficient staff and funds, clearly defined pathway for the outputs to reach decision-makers, and defined customers) and eleven secondary indicators (eg, international collaboration, centrally coordinated, where tasks can be distributed among the participants, collaboration with industry to obtain information). They were used to assess the 12 European Network of HS Centres member agencies,26 with some degree of variability in the presence of these desirable features. According to the definition, HSSs are primarily intended to support governments and health authorities’ decision-making and planning to anticipate the impact of emerging technologies within the health care system. However, HSSs may also contribute to support key decisions made by the manufacturers during the product development process. Although experiences have been reported mainly on diagnostics and devices,27–29 HSSs may help the drug developers to anticipate possible barriers to the downstream market access decision and to proactively work to overcome those by shaping the market and engaging stakeholders and key opinion leaders.
Ijzerman and Steuten30 proposed a conceptual framework for “early HTA” as a tool to inform mainly the industry. Early HTA is identified as the early assessment of safety, effectiveness, and cost-effectiveness profiles of new medical technologies based on evidence mainly derived from bench and animal tests, early clinical experience, and previous generations of the technology.31 This tool is certainly more suitable for the evaluation of medical devices than drugs32 and the extent to which it influences product development decisions is to date not known. Moreover, although its denomination seems to fall under the broad HTA umbrella definition, it is rarely used by HTA bodies and it is being developed by consultants or manufacturers to support industry and investors’ decisions on design and management of the technology, regulatory, and reimbursement strategies.30
Bipartite and tripartite scientific advice
Some reimbursement agencies have set up in recent years early scientific advice (SA) services to pharmaceutical companies in order to outline, for specific submissions, the expected evidence base that could lead to optimal market access outcomes.
NICE was the first agency to set up HTA product-specific SA in 2009 (Table 1).33 The objective was to help product developers generate the evidence that is relevant for technology appraisal, with the aim of gaining more robust data for the reimbursement perspective, reducing the uncertainty at the time of the appraisal, and therefore, the length of decision-making and patients’ time to access the therapies. The SA services given relates with a broad range of issues concerning the strategic development plans for postregulatory evaluation rather than advice on regulatory requirements or a preevaluation.34 The content of the SA pertains to research design considerations (eg, study population, follow-up duration, appropriate comparators, endpoints, and type of study), approaches to economic analysis and generation of quality-adjusted life years, interpretation of guidelines, and other methodological issues. As the aim is that of making an impact on the strategic development and evidence generation plans, it is recommended to seek the advice when there is still time for the companies to amend the design of the pivotal trials, ie, when Phase III study design is ongoing. At NICE, the SA is available as a fee-for-service consultation to manufacturers, and it is provided through a structured process. The company seeking advice has to contact the unit up to 9 months before the actual meeting and prepare a predefined briefing book with all relevant information for the advisors and a set of clearly stated questions accompanied by the respective manufacturer’s position on them.35 After about 12 weeks of indirect exchange, a 4-hour face-to-face meeting is held where NICE staff and companies’ representatives engage in a discussion around the questions on the agenda. After this meeting, a report is drafted and sent to the company for follow-up. As of October 2013, 77 SA requests from 26 companies were recorded at NICE; of these 17 (22%) were withdrawn, possibly because final agreement on the contract between client and agency was not reached.36 Of the remaining technologies, the majority (26 out of 60, 43%) were in the trial progression state, 17 (28%) had completed the trials, and only one had completed the technology appraisal process, gaining a positive recommendation. However, as Osipenko noted, HTA advice was sought for that particular drug when the Phase III trial design was locked, thus limiting the likelihood of the favorable reimbursement decision being due to the HTA advice provided.36 In addition to the standard bipartite (ie, between HTA body and manufacturer) SA process, this agency also provides feedback on the drug development plan from the payers’ perspective alongside the Medicines and Healthcare Products Regulatory Agency and the European Medicines Agency (EMA).
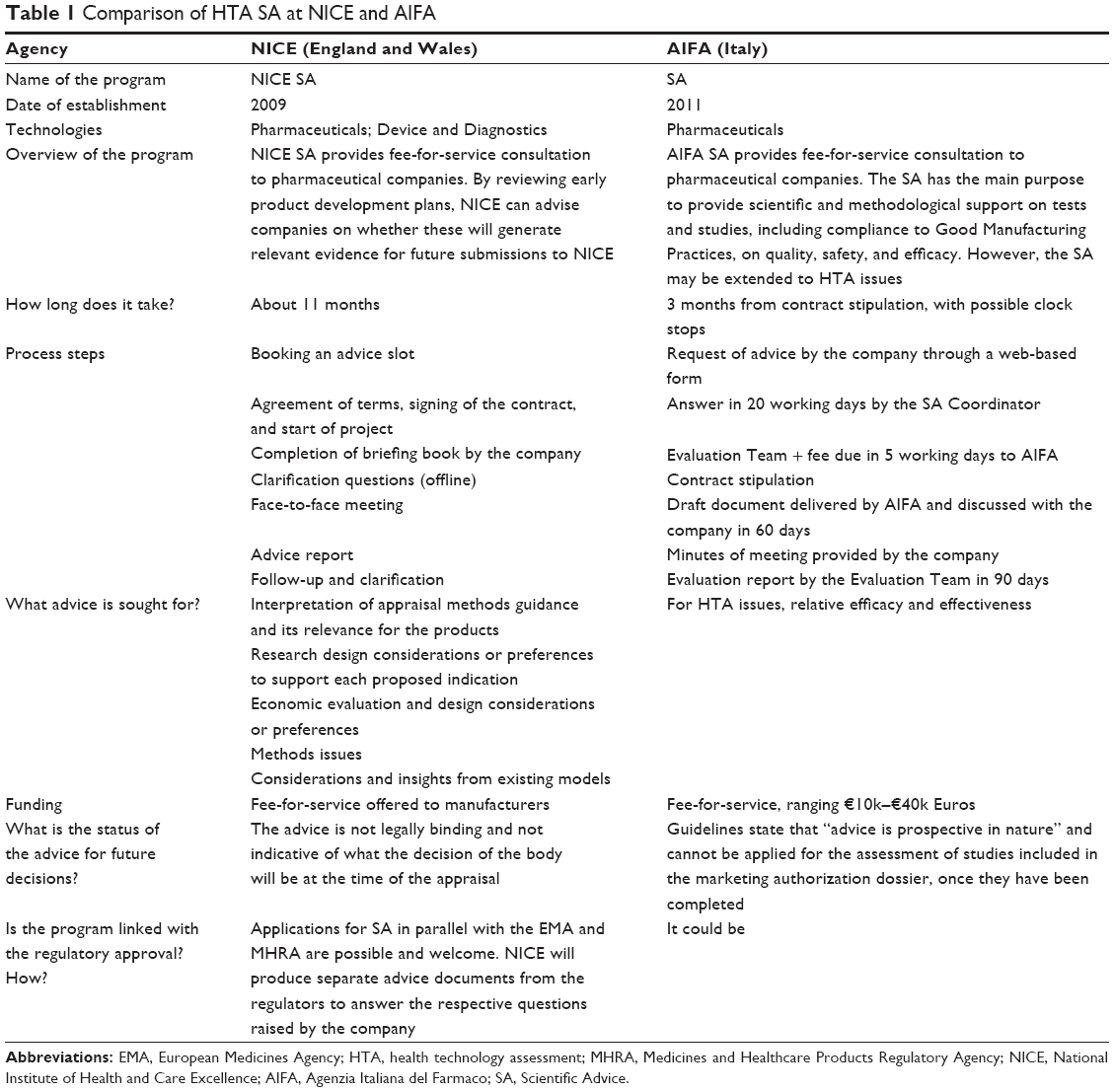 | Table 1 Comparison of HTA SA at NICE and AIFA |
In 2011, AIFA introduced both an SA and an HTA Advice (SA-HTA) for regulatory and HTA purposes in Italy (Table 1).37 The guidelines for applicants to both processes were published in November 2012 and are available on the institutional website. As it is possible to ask for both advices jointly, this document refers to preclinical and clinical development for marketing authorization and production issues. The HTA aspects are generally related to relative efficacy and relative effectiveness evaluation. The SA-HTA advice is generally asked for a single product in early stages of the product development, but it may be requested also for broader therapeutic classes. Issues subject to advice include the most appropriate comparator(s), the endpoints used, and the acceptability of indirect comparison. In addition, due to the relevance of disease severity and drug budget impact in price/reimbursement negotiation, the advice sought might often relate to the target population. Applicants wishing to apply should fill in a predefined form, the rationale for the request, whether other advices have been received by other European Agencies, and the specific questions asked. Within 20 working days, the SA Coordinator notifies the applicant by email regarding whether the request for SA is acceptable or not. After 5 working days, the AIFA Evaluation Team is defined and the contract is drafted including the fee due to AIFA, which ranges from €10k to €40k, according to the questions asked. The answers by the Evaluation Team are collected in a report due in 90 days from the signing of the contract between the agency and the company. A preliminary version of the report is discussed after 60 days with the company, within a meeting or a teleconference. The minutes of the meeting are expected by the company and should be validated by the Evaluation Team. The current uptake of this service by manufacturers in Italy is currently not publicly available. A representative of the agency reports that 21 advices have been processed in 2011–2013: eleven (52%) for cancer and autoimmune diseases, five (24%) for neurologic diseases, four (19%) for infectious diseases, and one (5%) for hormone-related therapies.38 They generally refer to products in Phase II (likely to be authorized after Phase II) and Phase III.
The so-called tripartite “early dialogue” between regulators, HTA organizations, and industry is gaining attention due to the potential advantages attached to it. First and foremost, it may reduce delays to patient access as the advice for different data needs for regulators and HTA bodies is sought in parallel rather than in sequence. Second, it provides an opportunity to learn about HTA requirements at an early stage and to discuss divergent data needs with all parties around the same table, with the aim of minimizing discrepancies and identifying trade-offs.39 For instance, when discussing the comparator for a novel therapy for chronic obstructive pulmonary disease during a parallel HTA-EMA SA session, the regulator agreed with the proposed licensed comparator while the HTA agency wanted to compare the value of the new therapy to what it will replace in practice, even if the alternative was not licensed for that use. The solution agreed was to introduce a new arm in the pivotal study to include both options.40 Thirty procedures have undergone the parallel HTA-EMA SA process so far; of those, 23 are now finalized. They include drugs for several conditions: diabetes, Alzheimer’s disease, oncology, asthma, rheumatoid arthritis, osteoporosis, food allergies, and orphan conditions.40 The HTA advice in the tripartite early dialogue may come from one or more reimbursement agencies, often represented through the European Network for HTA (EUnetHTA).41 As different HTA bodies may focus on different dimensions (eg, relative clinical effectiveness versus cost-effectiveness), this multi-HTA dialogue aims at supporting the company in finding a balance between perspectives from licensing body and HTA agencies in different countries as well as its own international product development program.42 Most of the time, the HTA body involved in the EMA-HTA parallel SA has been NICE, followed by the German Federal Joint Committee (G-BA) and AIFA.43
HTA role on postmarketing development
Linking between licensing and market access evidence requirements is also being extended to the postmarketing phase, with the aim of creating continuum evidence generation requirements relevant for registration, reimbursement, and postauthorization research.44 Insufficient evidence and uncertainty at market launch and the need of limiting the budget impact of new drugs have increased the number of MMEAs. The taxonomy of these agreements has been illustrated in a recent review.45,46 MMEAs may be 1) financial based or outcome based, when the price, the reimbursement status, or both depend on the financial and clinical impact, respectively, and 2) population based or patient based, when the performance is measured on the population as an aggregate or on each individual patient (Table 2). Population- and outcome-based agreements, sometimes called coverage with evidence development, generally rely on new observational studies or randomized controlled trials, whereas individual- and outcome-based agreements are usually based on registries.47
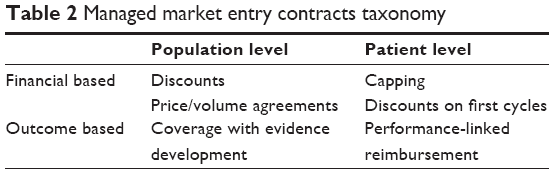 | Table 2 Managed market entry contracts taxonomy |
Both payers and the industry may take advantage of MMEAs.48 In a situation where the alternative would be not reaching drugs reimbursement at the price requested by the industry, MMEAs allow the product to be accessible to the patients, the companies to leave the list price in the price-corridor defined by the headquarters, and at the same time, to improve their reputation by making drugs accessible and sharing the risk of clinical failure and financial impact with payers. On the other hand, payers have the advantage of prioritizing drugs, collecting real-world evidence, and applying, in the case of outcome-based agreements, value-based price and reimbursement. The system as a whole takes the advantage of avoiding the risk that the drug is not available.
Outcome-based agreements are more consistent with value-based pricing and reimbursement, and have the potential to influence the drugs development program differently from financial-based agreements. However, they are more complex to manage. The work of a dedicated task force at the International Society of Pharmacoeconomics and Outcome Research has recently disentangled important issues deriving from these agreements, including their desirability, design, governance, implementation, and impact evaluation,47 all essential factors for a successful contract. For example, the coverage with evidence development contract on drugs for multiple sclerosis in the UK has partially failed49 due to important delays in patients’ recruitment and data analysis, and, on top of them, incompleteness of the contract.47 These agreements are highly desirable if the drug has an important budget impact (high unit price and/or volume); the level of uncertainty on benefits is significant, but the expected benefit is so high that the opportunity cost of delaying access to get more evidence would not be acceptable.
The English and Italian approach to MMEAs is different. The UK has mainly adopted a dichotomous approach at market launch: the drug is recommended (or recommended for a restricted target) or not recommended. In order to make accessible drugs that are not cost-effective at list price, several strategies are in place: 1) companies have been allowed to require a patient access scheme: 31 of the 44 patient access schemes are discounts on list prices,50 2) thresholds on incremental cost-effectiveness ratio have been increased for some drugs categories (eg, for end-of-life treatments, indicated for patients with a short life expectancy – normally <24 months – small patient population and with sufficient evidence that the treatment offers an extension to life of at least 3 months, compared to current National Health Service treatment),51 3) a cancer drugs fund has been created to partially cover cancer drugs that have not been recommended (eg, aflibercept as a second-line treatment of metastatic colorectal cancer) – or are waiting to be appraised (eg, pertuzumab as first-line treatment of locally advanced or metastatic breast cancer).52
While in the UK, apart from patient access schemes, access is mostly driven by cost-effectiveness at market launch and many drugs have not been recommended, in Italy most of the new drugs are approved with an MMEA (sometimes with multiple MMEAs), and outcome-based contracts have been implemented, particularly on an individual basis, to oncological drugs: 25 drugs have been subject to such contracts to date. For example, everolimus has been approved with a performance-linked reimbursement contract that requires a payback to hospitals from Novartis should patients not respond after 3/6 months of treatment for advanced renal carcinoma/human epidermal growth factor receptor-2–negative advanced breast cancer and primitive neuroectodermal tumor, respectively. Finally, registries on 46 drugs have been created to carry out the 25 outcome-based contracts and, under a broader perspective, to review the prescribing behavior for expensive drugs.53
Discussion
This study highlights particular HTA activities that are spreading in most developed countries with the potential of significantly influencing the drug development paths of new pharmaceuticals. From early stages of clinical research up to postauthorization studies, there is a trend toward increased collaboration between parties,54 like regulators, developers, and HTA assessors, to improve the evidence profile of new drugs, to anticipate patients’ access to innovative therapies, and to reduce clinical and economic risks deriving from an incomplete knowledge profile at the time of the launch.44 Comprehensiveness and inclusiveness are acknowledged in a new paradigm, known as constructive HTA, which, in addition to traditional HTA, emphasizes the technology dynamics elements and the thorough assessment and involvement of multiple stakeholders in the production and evaluation of different types of new evidence.55,56 Another emerging feature is the anticipation of market access evidence collection to early stages of development and the increasing importance of postmarketing real-world evidence.
It is accepted that the evidence required for marketing authorization and market access is different: comparative evidence and cost consequences are gaining importance in the eyes of payers, and payers are more and more important for market access. While licensing and regulatory postmarketing risk plans are quite homogeneous across regulators in different jurisdictions (eg, EMA, Food and Drug Administration in the US), payers may have different preferences on comparative clinical and economic evidence, despite the effort of the European Network for HTA collaboration to develop common methodologies and practices for HTA across European member states. Hence, the companies are offered the currently available opportunities for bipartite and tripartite early dialogue between regulators and HTA assessors to identify product development plans that satisfy both interlocutors. Companies’ representatives who took part in the SA process generally agree that the advice given is reasonable and constructive; some declare to have modified the development program as a result of the consultation.15,36,57 However, based on the current available experience and poor reporting of it, it is generally difficult to establish whether the modification and which modification on the drug development plan occurs as a direct consequence of the HTA advice.57 Beyond gaining a reimbursement agency’s insight on the strategic development plan and optimizing the evidence generation program accordingly, other more subtle factors for the manufacturers to apply could be an expectation of a later-facilitated technology appraisal, the collection of an informal endorsement on the evidence program, or of the agency perspective on the development program, regardless of the intention to access that specific market.36,57
In terms of interactions between different players, models are still different in terms of the importance played by early interaction and postlaunch risk-sharing agreements, the level of transparency of this interaction, and the object of this interaction. Our research focused on two case studies that share a rather good level of transparency in the way premarketing advice is managed, but follow different appraisal and recommendation rules. As already mentioned, heterogeneity of HTA practices around Europe increases the complexity of the market access environment and future European projects’ collaboration on early dialogue, such as the Shaping European Early Dialogue program, are needed to address this issue.41
Overall, there are concrete signals that Market Access departments and activities are gaining importance in the pharmaceutical companies, but there is still a lack of evidence and reporting on how the increasing relevance of HTA has reshaped the way clinical development is designed and managed. This may strongly depend on the importance played by market access issues compared with marketing approval and market access barriers: the more these barriers are diffused in countries where market potentials are high and growing, the more companies are expected to design clinical development considering market issues also.58
As our objective was to illustrate HTA activities potentially influencing the drug development path of pharmaceuticals, we performed a review of information available on institutional websites or published documents. An interesting extension of the present work would be the collection of primary data and insights from manufacturers to measure the actual extent of the impact such HTA activities are having on their business. In conclusion, further research on how HTA and regulatory authorities’ practices develop to manage evolving knowledge and on how manufacturers respond with an efficient and adequate drug development program, both with the ultimate interest of patients’ benefit, is welcome.
Disclosure
The authors declare no conflicts of interest.
References
Sorenson C, Drummond M, Kanavos P. Ensuring Value for Money in Health Care. Bodmin: European Observatory on Health Systems and Policies; 2008. | ||
Oortwijn W, Mathijssen J, Banta D. The role of health technology assessment on pharmaceutical reimbursement in selected middle-income countries. Health Policy. 2010;95:174–184. | ||
Banta D. Health technology assessment in Latin America and the Caribbean. Int J Technol Assess Health Care. 2009;25(suppl 1):253–254. | ||
Franken M, Le Polain M, Cleemput I, Koopmanschap M. Similarities and differences between five European drug reimbursement systems. Int J Technol Assess Health Care. 2012;28:349–357. | ||
Banta D, Oortwijn W. Health technology assessment and health care in the European Union. Int J Technol Assess Health Care. 2000;16:626–635. | ||
Oortwijn W, Broos P, Vondeling H, Banta D, Todorova L. Mapping of health technology assessment in selected countries. Int J Technol Assess Health Care. 2013;29:424–434. | ||
Kristensen F. EUnetHTA and health policy-making in Europe. Eurohealth (Lond). 2006;12:36–38. | ||
Drummond MF, Schwartz JS, Jönsson B, et al. Key principles for the improved conduct of health technology assessments for resource allocation decisions. Int J Technol Assess Health Care. 2008;24:244–258. | ||
Ciani O, Tarricone R, Torbica A. Diffusion and use of health technology assessment in policy making: what lessons for decentralised healthcare systems? Health Policy. 2012;108:194–202. | ||
van Nooten F, Holmstrom S, Green J, Wiklund I, Odeyemi IA, Wilcox TK. Health economics and outcomes research within drug development: challenges and opportunities for reimbursement and market access within biopharma research. Drug Discov Today. 2012;17:615–622. | ||
Luce BR, Drummond M, Jönsson B, et al. EBM, HTA, and CER: clearing the confusion. Milbank Q. 2010;88:256–276. | ||
National Institute for Health and Care Excellence. Guide to the Methods of Technology Appraisal; 2013. National Institute for Health and Care Excellence; [updated 2014]. Available from: http://www.nice.org.uk/About/What-we-do/Our-Programmes/NICE-guidance/NICE-technology-appraisal-guidance. Accessed June 30, 2014. | ||
Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. | ||
Velasco Garrido M, Mangiapane S. Surrogate outcomes in health technology assessment: an international comparison. Int J Technol Assess Health Care. 2009;25:315–322. | ||
Backhouse ME, Wonder M, Hornby E, Kilburg A, Drummond M, Mayer FK. Early dialogue between the developers of new technologies and pricing and reimbursement agencies: a pilot study. Value Health. 2011;14:608–615. | ||
Taylor RS, Drummond MF, Salkeld G, Sullivan SD. Inclusion of cost effectiveness in licensing requirements of new drugs: the fourth hurdle. BMJ. 2004;329:972–975. | ||
Rogowski WH, Hartz SC, John JH. Clearing up the hazy road from bench to bedside: a framework for integrating the fourth hurdle into translational medicine. BMC Health Serv Res. 2008;8:194. | ||
WHO. Institutionalization of Health Technology Assessment: Report on a WHO Meeting, Bonn; 2001. June 30–July 1, 2000; Copenhagen: World Health Organization Regional Office for Europe. | ||
About NICE [homepage on the Internet]. National Institute for Health and Care Excellence; [updated 2014]. Available from: http://www.nice.org.uk/aboutnice/about_nice.jsp. Accessed June 30, 2014. | ||
Agenzia Italiana del Farmaco. [homepage on the Internet]. Agenzia Italiana del Farmaco; [updated 2014]. Available from: http://www.agenziafarmaco.com/en. Accessed June 30, 2014. | ||
Fattore G, Jommi C. The last decade of Italian pharmaceutical policy: instability or consolidation? Pharmacoeconomics. 2008;26:5–15. | ||
Velasco-Garrido M, Busse R. Health Technology Assessment. An Introduction to Objectives, Role of Evidence, and Structure in Europe. Brussels: European Observatory on Health Systems and Policies; 2005. | ||
Horizon Scanning Centre [homepage on the Internet]. Birmingham: NIHR Horizon Scanning Centre; [updated 2014]. Available from: http://www.hsc.nihr.ac.uk/about-us/. Accessed June 30, 2014. | ||
Joppi R, Dematte L, Menti AM, Pase D, Poggiani C, Mezzalira L. The Italian Horizon Scanning Project. Eur J Clin Pharmacol. 2009;65:775–781. | ||
Murphy K, Packer C, Stevens A, Simpson S. Effective early warning systems for new and emerging health technologies: developing an evaluation framework and an assessment of current systems. Int J Technol Assess Health Care. 2007;23:324–330. | ||
EuroScan. International Network [homepage on the Internet]. NIHR Horizon Scanning Centre; [updated 2014]. Available from: http://euroscan.org.uk/. Accessed June 30, 2014. | ||
Storz P, Kolpatzik K, Perleth M, Klein S, Haussler B. Future relevance of genetic testing: a systematic horizon scanning analysis. Int J Technol Assess Health Care. 2007;23:495–504. | ||
Retel VP, Hummel MJ, van Harten WH. Review on early technology assessments of nanotechnologies in oncology. Mol Oncol. 2009;3:394–401. | ||
O’Malley SP, Jordan E. Horizon scanning of new and emerging medical technology in Australia: its relevance to Medical Services Advisory Committee health technology assessments and public funding. Int J Technol Assess Health Care. 2009;25:374–382. | ||
Ijzerman MJ, Steuten LM. Early assessment of medical technologies to inform product development and market access: a review of methods and applications. Appl Health Econ Health Policy. 2011;9:331–347. | ||
Pietzsch JB, Pate-Cornell ME. Early technology assessment of new medical devices. Int J Technol Assess Health Care. 2008;24:36–44. | ||
Markiewicz K, van Til JA, MJ IJ. Medical devices early assessment methods: systematic literature review. Int J Technol Assess Health Care. 2014;30:137–146. | ||
About NICE Scientific Advice. National Institute for Health and Care Excellence; [updated 2014]. Available from: http://www.nice.org.uk/about/What-we-do/Scientific-advice. Accessed June 30, 2014. | ||
What advice can be requested? National Institute for Health and Care Excellence; [updated 2014]. Available from: http://www.nice.org.uk/About/What-we-do/Scientific-advice/Find-out-what-advice-can-be-requested. Accessed June 30, 2014. | ||
Briefing Book Guidance for Company; 2014. National Institute for Health and Care Excellence; [updated 2014]. Available from: http://www.nice.org.uk/Media/Default/About/what-we-do/Scientific-advice/Scientific-Advice-BB-Guidance.pdf. Accessed June 30, 2014. | ||
Osipenko L. NICE scientific advice: an HTA agency perspective. In: ISPOR 16th Annual European Congress. Dublin; 2013. | ||
Scientific Advice. Agenzia Italiana del Farmaco; [updated 2014]. Available from: http://www.agenziafarmaco.gov.it/it/content/scientific-advice. Accessed June 30, 2014. | ||
Siviero P. Le applicazioni in Italia ai farmaci oncologici: quali prospettive in futuro. Economia del Farmaco e delle Tecnologie Sanitarie; June 23, 2014; Novara, Italy. Italian. | ||
EMA. EMA-HTA workshop: bringing together stakeholders for early dialogue in medicines development. November 26, 2013; London, UK. | ||
Regnstrom J. HTAs and EMA working together: 23 parallel scientific advice procedures later – what have we learned? In: DIA 26th EuroMeeting; 2014; Vienna, Austria. | ||
Pavlovic M. Early engagement between manufacturers, HTA assessors and regulators. Learning from the past to guide the future. A EUnetHTA perspective. In: ISPOR 16th Annual European Congress; November 3–6, 2013; Dublin, Ireland. | ||
Wonder M, Backhouse ME, Hornby E. Early scientific advice obtained simultaneously from regulators and payers: findings from a pilot study in Australia. Value Health. 2013;16:1067–1073. | ||
Industry Perspective on Advice Models. Where are we now, and where should we go? Results of the EFPIA Industry Survey; [updated 2013]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2013/11/WC500155674.pdf. Accessed June 30, 2014. | ||
Patarnello F, Recchia G. Ricerca post-autorizzativa, registri e sviluppo del farmaco. [Post-authorization research, registries, and drug development]. Recenti Prog Med. 2013;104:275–279. Italian. | ||
Morel T, Arickx F, Befrits G, et al. Reconciling uncertainty of costs and outcomes with the need for access to orphan medicinal products: a comparative study of managed entry agreements across seven European countries. Orphanet J Rare Dis. 2013;8:198. | ||
Carlson JJ, Sullivan SD, Garrison LP, Neumann PJ, Veenstra DL. Linking payment to health outcomes: a taxonomy and examination of performance-based reimbursement schemes between healthcare payers and manufacturers. Health Policy. 2010;96:179–190. | ||
Garrison LP Jr, Towse A, Briggs A, et al. Performance-based risk-sharing arrangements-good practices for design, implementation, and evaluation: report of the ISPOR good practices for performance-based risk-sharing arrangements task force. Value Health. 2013;16:703–719. | ||
Adamski J, Godman B, Ofierska-Sujkowska G, et al. Risk sharing arrangements for pharmaceuticals: potential considerations and recommendations for European payers. BMC Health Serv Res. 2010;10:153. | ||
Raftery J. Multiple sclerosis risk sharing scheme: a costly failure. BMJ. 2010;340:c1672. | ||
National Institute for Health and Care Excellence. List of Technologies with Approved Patient Access Schemes. National Institute for Health and Care Excellence; [updated 2014]. Available from: https://www.nice.org.uk/About/What-we-do/Patient-access-schemes-liaison-unit/List-of-technologies-with-approved-Patient-Access-Schemes. Accessed June 30, 2014. | ||
National Institute for Health and Care Excellence. Appraising Life-Extending, End of Life Treatments. National Institute for Health and Care Excellence; [updated 2009]. Available from: http://www.nice.org.uk/guidance/gid-tag387/resources/appraising-life-extending-end-of-life-treatments-paper2. Accessed June 30, 2014. | ||
National Cancer Drugs Fund List. National Institute for Health and Care Excellence; [updated 2014]. Available from: http://www.england.nhs.uk/wp-content/uploads/2014/02/ncdf-list-feb-14-fin.pdf. Accessed June 30, 2014. | ||
Agenzia Italiana del Farmaco. Registri Farmaci sottoposti a monitoraggio; 2014. Agenzia Italiana del Farmaco; [updated 2014]. Available from: http://www.agenziafarmaco.gov.it/it/content/registri-farmaci-sottoposti-monitoraggio. Accessed June 30, 2014. Italian. | ||
Henshall C, Mardhani-Bayne L, Fronsdal KB, Klemp M. Interactions between health technology assessment, coverage, and regulatory processes: emerging issues, goals, and opportunities. Int J Technol Assess Health Care. 2011;27:253–260. | ||
Douma KF, Karsenberg K, Hummel MJ, Bueno-de-Mesquita JM, van Harten WH. Methodology of constructive technology assessment in health care. Int J Technol Assess Health Care. 2007;23:162–168. | ||
Poulin P, Austen L, Scott CM, et al. Introduction of new technologies and decision making processes: a framework to adapt a Local Health Technology Decision Support Program for other local settings. Med Devices (Auckl). 2013;6:185–193. | ||
Grueger J. Scientific advice from regulators and HTA: industry perspective. In: ISPOR 16th Annual European Congress; November 3–6, 2013; Dublin, Ireland. | ||
Jommi C, Otto M, Armeni P, De Luca C. Market access management by pharmaceutical companies in a complex environment: the Italian case study. J Med Markting. 2012;12:10. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.