")
Back to Journals » Drug Design, Development and Therapy » Volume 16
The Role of Glucagon-Like Peptide-1 Receptor Agonists (GLP-1 RA) in Diabetes-Related Neurodegenerative Diseases
Authors Cheng D, Yang S, Zhao X, Wang G
Received 5 November 2021
Accepted for publication 18 February 2022
Published 14 March 2022 Volume 2022:16 Pages 665—684
DOI https://doi.org/10.2147/DDDT.S348055
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Dihe Cheng, Shuo Yang, Xue Zhao, Guixia Wang
Department of Endocrinology and Metabolism, The First Hospital of Jilin University, Changchun, 130021, People’s Republic of China
Correspondence: Guixia Wang; Xue Zhao, Department of Endocrinology and Metabolism, The First Hospital of Jilin University, Changchun, 130021, People’s Republic of China, Tel +86 15843081103 ; +86 18744014213, Email [email protected]; [email protected]
Abstract: Recent clinical guidelines have emphasized the importance of screening for cognitive impairment in older adults with diabetes, however, there is still a lack of understanding about the drug therapy. Glucagon-like peptide 1 receptor agonists (GLP-1 RAs) are widely used in the treatment of type 2 diabetes and potential applications may include the treatment of obesity as well as the adjunctive treatment of type 1 diabetes mellitus in combination with insulin. Growing evidence suggests that GLP-1 RA has the potential to treat neurodegenerative diseases, particularly in diabetes-related Alzheimer’s disease (AD) and Parkinson’s disease (PD). Here, we review the molecular mechanisms of the neuroprotective effects of GLP-1 RA in diabetes-related degenerative diseases, including AD and PD, and their potential effects.
Keywords: glucagon-like peptide-1, diabetes mellitus, Alzheimer’s disease, Parkinson’s disease, cognition
Introduction
Type 2 diabetes mellitus is a group of metabolic diseases characterized by hyperglycemia caused by relatively insufficient insulin secretion. It is estimated that about 415 million people had diabetes in 2015, and this number may continue to rise to 642 million by 2040.1 Diabetes-related neurodegenerative disease (ND) is of particular importance due to the cognitive impairment it causes in older patients with type 2 diabetes. The risk of incident mild cognitive impairment (up to 60%) and dementia (50–100%) is higher in patients with type 2 diabetes than in those without.2 Recent clinical guidelines have emphasized the importance of screening for cognitive impairment in older adults with diabetes;3 however, there is still a lack of understanding about drug therapy. Therefore, an urgent goal is to develop effective neuroprotective drugs that act on the common mechanisms of diabetes-related NDs, thereby slowing the disease progression.
Glucagon-like peptide 1 (GLP-1) is a 30-amino-acid peptide hormone produced in intestinal epithelial endocrine L-cells by the processing of proglucagon.4 GLP-1 is widely used in the treatment of type 2 diabetes because it not only controls blood glucose but may also reduce body weight. Future uses of GLP-1 may also include the treatment of obesity, as well as the adjunctive treatment of type 1 diabetes mellitus in combination with insulin.5 Natural GLP-1 degrades within 2–3 min in circulation, thus greatly limiting its effects. Various GLP-1 receptor agonists (GLP-1 RAs) have been developed to provide long-term effects. GLP-1 RA functions by activating the GLP-1 receptor (GLP-1R), and GLP-1R is widely located throughout the brain.6,7 The ability of GLP-1 and its agonists to cross the blood–brain barrier8–10 suggests its therapeutic potential for NDs. A large number of studies have demonstrated the neuroprotective ability of GLP-1 RA, resulting in the improvement of cognitive and non-cognitive dysfunction of the central nervous system (CNS).
The proposed mechanisms of diabetes-related NDs include cerebral insulin resistance (IR), vascular endothelial dysfunction, inflammation, blood–brain barrier injury, white matter disease of vascular origin, demyelination and axonal loss, and peroxidative membrane injury.11 Among these mechanisms, brain IR may play a primary role, and it is worth noting that neurologic complications may already occur with prediabetes IR.12 Oxidative stress,13 mitochondrial dysfunction,14 and endoplasmic reticulum (ER) stress15 are all involved in NDs induced by brain IR. In this review, we discuss the accumulating evidence concerning the effects of GLP-1 RA in diabetes-related NDs.
The GLP-1 RA and Its Relationship with Brain Insulin Resistance
Brain IR can be defined as the failure of brain cells to respond to insulin, and the lack of response may be due to the downregulation of insulin receptors, an inability of insulin receptors to bind insulin, or faulty activation of the insulin signaling cascade.16 Insulin receptors are distributed throughout the brain, but have the highest concentration in the olfactory bulb, cerebral cortex, hypothalamus, hippocampus, and cerebellum.17 Insulin binds to the insulin receptor, phosphorylates the insulin receptor substrate (IRS), activates the phosphoinositide-3 kinase (PI3K) and mitogen-activated kinase (MAPK) pathways, and modifies the activity of several downstream effectors. PI3K activates protein kinase B (Akt), which inactivates several important substrate proteins, such as glycogen synthase kinase 3β (GSK-3β)18 and forkhead box O,19 and activates mammalian target of rapamycin (mTOR).20 As a result, it modulates some cellular processes, such as cell survival, proliferation, apoptosis, protein synthesis, inflammation, ER stress, mitochondrial function, and autophagy in neurodegenerative disorders.18–20 Akt also promotes B-cell lymphoma 2 and B-cell lymphoma extra-large transcription by activating cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB).21 Thereafter, it regulates learning, memory,22 and neuron survival.23 MAPK regulates various cellular activities including proliferation, differentiation, apoptosis or survival, inflammation, and innate immunity.24 Impairment of insulin signaling is common in diabetes-related NDs.
GLP1-R is a class B G protein-coupled receptor,25 and its expression has been reported in the cerebral cortex, especially the occipital and frontal lobes, hypothalamus, and thalamus, whereas lower levels are found in the caudate putamen, globus pallidus, and hippocampus.21 GLP-1 and its RA cross the blood–brain barrier, with exendin-4 considered as one of the best based on the rate of brain influx, percentage of reaching the brain that accumulates in the brain parenchyma, and percentage of the systemic dose taken per gram of brain tissue.8 Small amounts of GLP-1 may also be produced by preproglucagon neurons, located in the nucleus tractus solitarii of the brainstem26,27 and projected to other brain regions, such as the nuclei of the hypothalamus, including the arcuate and paraventricular nuclei.28 In the case of diabetes or obesity-related IR, GLP-1 secretion in the brain29 and peripheries30 may be impaired, which may contribute to the pathogenic change in neurodegeneration and cognitive decline; however, exogenous GLP-1 may help treat these diseases. When GLP-1 binds to the receptor, adenosine cyclase is activated and intracellular cAMP increases, thereby activating protein kinase A (PKA) and PI3K. The downstream pathways are mainly the PI3K and MAPK pathways; hence, the GLP-1 signaling and insulin signaling pathways are similar and partially overlapping (Figure 1).
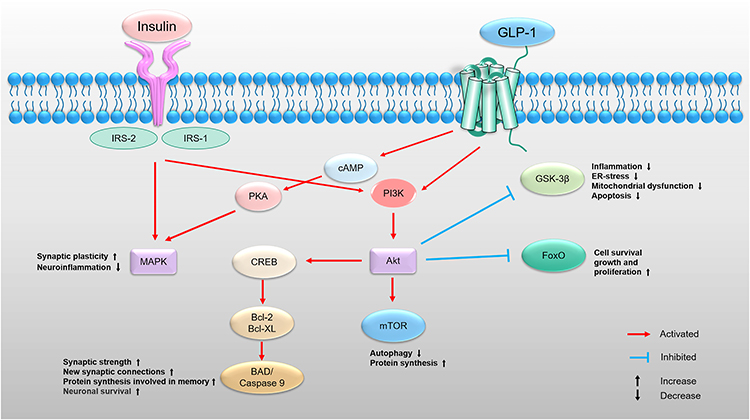 |
Figure 1 Insulin and GLP-1-dependent intracellular signal transduction pathways are similar. Insulin binds to the insulin receptor and further activates the PI3K/Akt and MAPK pathways signaling. PI3K/Akt pathway modulates some cellular processes, such as cell survival, proliferation, apoptosis, protein synthesis, inflammation, ER stress, mitochondrial function, autophagy, synaptic strength in neurodegenerative disorders. MAPK pathway regulates various cellular activities including synaptic plasticity and neuroinflammation. When GLP-1 binds to the GLP-1 receptor, adenosine cyclase is activated and intracellular cAMP increases, thereby activating PKA and PI3K. The downstream pathways are mainly the PI3K and MAPK pathways; hence, the GLP-1 signaling and insulin signaling pathways are similar and partially overlapping. Abbreviations: IRS, insulin receptor substrate; PI3K, phosphoinositide-3 kinase; Akt, protein kinase B; ER, endoplasmic reticulum; GSK-3β, glycogen synthase kinase 3β; FoxO, forkhead box O; mTOR, mammalian target of rapamycin; CREB, cAMP-response element binding protein; Bcl-2, B-cell lymphoma 2; Bcl-XL, B-cell lymphoma extra-large; BAD, (Bcl-2) antagonist of death; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; MAPK, mitogen activated kinase. |
Consequently, exogenous drugs that act on GLP-1Rs increase insulin sensitivity, possibly because GLP-1R stimulation compensates for some of the impaired insulin signaling. Among these drugs, liraglutide has been reported to have neuroprotective effects by ameliorating damage to the insulin pathway. In vitro experiments proved that it reversed the phosphorylation status of IRS1, Akt, and GSK-3β and reduced beta-amyloid formation and tau hyperphosphorylation in the human neuroblastoma cell line, SH-SY5Y.31 In vivo experiments proved that liraglutide prevented the dysregulation of Akt and GSK-3β and Alzheimer-associated tau phosphorylation in the brains of diabetic mice.32,33 Besides, it prevents the loss of brain insulin receptors in an Alzheimer’s disease (AD) model.34 Exenatide also has a similar effect on impaired insulin signaling pathways.35,36
The GLP-1 RA and Mitochondrial Dysfunction and/or Oxidative Stress
Mitochondria are the main energy production systems of most eukaryotic cells and are responsible for energy conversion, tricarboxylic acid cycle, oxidative phosphorylation, calcium storage, etc. Mitochondrial dysfunction has negative effects on the body and is believed to be an important factor in aging and disease. It has been found that insulin receptor knockout mice show reduced mitochondrial oxidative phosphorylation activity.37 Abnormal mitochondrial calcium transport was observed in the myocardium and visceral adipose tissue of obese mice.38,39 In the hippocampal tissue of type 2 diabetic mice, the expression of mitochondrial dynamin-related protein 1 (Drp1) increased, whereas inhibition of Drp1 restored neuronal function.40 In diabetic models, peroxisome proliferator-activated receptor c coactivator 1a (PGC-1a), an important factor in diabetic mitochondrial biosynthesis, is often found to be abnormally expressed, whereas PGC-1a is critical for synaptic growth and CNS function. Reduced levels of the mitochondrial autophagy-associated protein Parkin in the substantia nigra may contribute to the development of Parkinson’s disease (PD) in db/db mice and high-fat diet-induced diabetic mice.41 As discussed above, mitochondrial dysfunction (mitochondrial bioenergetics, calcium buffering) and mitochondrial quality control systems (mitochondrial dynamics, mitophagy, mitochondrial biogenesis) may be involved in the pathological mechanisms of diabetes-related NDs.
Oxidative stress refers to a state of imbalance between oxidation and antioxidant effects in the body, favoring oxidation, leading to inflammatory infiltration of neutrophils, increased protease secretion, and production of a large number of oxidative intermediates. Mitochondria are key sites for aerobic metabolism and reactive oxygen species (ROS) production in cells and are also one of the most important organelles related to oxidative stress. Some experts believe that cerebral IR is the result of ceramide accumulation in brain tissue, and ROS overproduction occurs due to metabolic abnormalities accompanying peripheral IR and impaired mitochondrial activity in the IR brain.13 Studies have shown that ROS can cause age-related synaptic loss and ultimately cognitive impairment,42 where ROS interactions with inflammation may play a role. Oxidative products, including lipid and protein oxidation, are promoters of brain inflammation.43 Nuclear transcription factor-κB (NF-κB) Inflammatory pathway signaling plays a key role in regulating the amount of ROS in the cell.44 Excessive ROS can inhibit IRS1 activation by activating inflammation-related protein kinase C, inhibitor kappa B kinase β (IKKβ), c-Jun N-terminal kinase (JNK), and p38 MAPK, thereby aggravating IR,45 creating a vicious cycle.
In the nervous system, the regulatory effect of GLP-1 RA on mitochondrial function and oxidative stress is involved in the remission of diabetes-related NDs. In diabetes-related AD, GLP-1 promotes mitochondrial biogenesis and the antioxidant system by regulating the PGC-1a signaling pathway in vivo to directly reverse tau hyperphosphorylation.46 GLP-1(9-36) (amide) reduced elevated levels of mitochondrial-derived ROS in the hippocampus of AD model (APP/PS1) mice.47 Exendin-4 significantly increased amyloid β protein (Aβ)-induced reduction in mitochondrial function, integrity, respiratory control rate, and mitochondrial P/O ratio in all brain regions and decreased Aβ-induced increase in the mitochondrial complex enzyme-I, IV, and V activities in all brain regions.36 Exenatide also improved hippocampal mitochondrial morphology and dynamics and reduced oxidative stress in the hippocampus of AD model (5xFAD) mice.48
The mechanism by which GLP-1 RA regulates mitochondrial function and oxidative stress has not been well elucidated. GLP-1 signaling may improve mitochondrial biogenesis via PGC-1a/nuclear respiratory factor-1/mitochondrial transcription factor A signaling regulated by adiponectin/ adenosine 5‘-monophosphate (AMP)-activated protein kinase (AMPK)49 and elevates the expression of NAD-dependent protein deacetylase sirtuin 1 (SIRT1), which increases the expression of Parkin, leading to mitophagy activation.50 Evidence strongly suggests that GLP-1 increases ER-mitochondria communication, resulting in higher mitochondrial activity.51 Upregulating SIRT3 expression and activation of the extracellular signal-regulated kinase-Yes-associated protein (ERK-Yap) signaling pathway, as well as the CREB/adiponectin axis may also be involved in the protection of mitochondria by GLP-1.52,53 The improvement of antioxidant stress through GLP-1 signaling seems to be related to the activation of the GLP-1 R/cAMP/PKA signaling pathway and nuclear factor erythroid 2-related factor 2/heme oxygenase 1 signaling pathway.54,55
The GLP-1 RA and Endoplasmic Reticulum (ER) Stress
The ER is the basic organelle for the synthesis of a series of important biological molecules, such as proteins, lipids (such as triglycerides), and carbohydrates. ER stress refers to the activation of ER responses, such as the unfolded protein response (UPR) and apoptosis signaling pathway, through the accumulation of misfolded and unfolded proteins and the disorder of calcium balance after various stress agents are applied to cells. The early role of UPR is to reduce translation to lessen the need for new protein folding, degrade unfolded proteins to minimize damage, and increase the expression of chaperone proteins to assist protein folding. The UPR is thought to promote cell homeostasis. However, if this mechanism persists, it may lead to different metabolic diseases56 and NDs.57 UPR is mainly involved in the activation of three transmembrane proteins, inositol-requiring enzyme 1 (IRE-1), activating transcription factor 6 (ATF6), and protein kinase R (PKR)-like ER kinase (PERK). Normally, these three proteins are associated with luminal binding immunoglobulin protein (BiP), also known as 78-kDa glucose-regulating protein (GRP78), and are inactive. Under stress conditions, BiP is released and thus activates the IRE-1, ATF6, and PERK signaling cascades.58
ER stress plays a role in the occurrence and development of diabetes and IR in peripheral tissues such as the pancreas, liver, adipose tissues, and skeletal muscle.58–63 Although some of the effects of ER stress are tissue-specific, there are some commonalities in the damage to insulin signaling. Under the action of unhealthy metabolic factors (obesity, diabetes), ER stress is initiated, and IRE-1 is activated, which in turn leads to the phosphorylation of IRS1 at the serine 307 residue by activating JNK, thereby impairing insulin signaling.64–66 A similar pattern was observed in the brain. Evidence indicates that ER stress was increased, thereby resulting in impaired insulin receptor signaling in the hippocampus and frontal cortex of obese rats, which is also caused by the activation of JNK.67 Therefore, diabetes and ER stress are vicious cycles in the brain, and IR is the key link. ER stress is also involved in the degenerative brain changes caused by diabetes. Elevated expression of ER stress markers, including GRP78, ATF-6, X-box binding protein-1, C/EBP homologous protein (CHOP), and phospho-Jun N-terminal kinase (p-JNK), was evident in the hippocampal CA1 of diabetic rats,68 which may ultimately affect synaptic plasticity.
ER stress has always been considered a result of NDs, but previous studies have shown that it is a more complex process by interfering with UPR to affect disease progression.69 GLP-1 RA has been shown to interfere with UPR to protect against NDs. Liraglutide treatment reduced neuroinflammation and ameliorated ER stress in the inferior olive of the aged Wolfram syndrome rat model.70 Moreover, it can prevent the disease before the appearance of metabolic symptoms.71 Liraglutide may engage Akt and signal transducer and activator of transcription 3 signaling to favor adaptive responses and shift cell fate from apoptosis to survival under chronic ER stress conditions in nerve cells.72 Our team used palmitic acid stimulation to induce neuronal IR, confirming that ER stress is involved in the functional damage of neurons induced by IR, and exendin significantly alleviates both ER stress and neuronal damage (data not shown).
However, it is not clear how GLP-1 RA regulates ER stress. Inhibiting the PI3K/Akt signaling pathway may eliminate the protective effect of GLP-1R by increasing ER stress, suggesting that this pathway may be involved in the effect of GLP-1 on ER stress.73 Besides, the PKA pathway may also be involved in GLP-1, attenuating the ER stress signaling pathway and protecting cells from apoptosis.74–76 Evidence suggests that PKA-dependent protection of GLP-1 is mediated through enhanced ATF4-CHOP-growth arrest and DNA damage inducible gene 34 (GADD34) signaling, resulting in eukaryotic initiation factor 2 alpha dephosphorylation and translational recovery.76 However, some researchers believe that exendin-4 protects β-cells against free fatty acids and salubrinal-induced ER stress and apoptosis, not through ATF4-CHOP- GADD34 feedback signaling but through enhancing cellular defense mechanisms (eg, BiP, Bcl-2, and JunB).77 In addition, other studies have investigated the mechanism by which GLP-1 regulates ER stress. Exendin-4 enhances the binding of heat shock factor 1 to the promoter of heat shock protein (HSP) genes through SIRT1-mediated deacetylation, which then increased the expression of molecular chaperones HSP70 and HSP40 to alleviate lipid-induced hepatic ER stress.78 ER oxidoreductase mediates the inhibitory effects of exendin-4 on ER stress, ameliorating hyperhomocysteinemia-induced endothelial dysfunction.79 ER protein 46, a new member of the thioredoxin family, highly expressed in pancreatic β-cells, may mediate GLP-1 regulation of ER stress and thus increase the protection of pancreatic β cells.80,81 These studies suggest the complexity of GLP-1 RA in regulating ER stress.
The GLP-1 RA and Central Nervous System (CNS) Inflammation
It is well known that CNS inflammation plays a major role in the pathophysiology of NDs.82 In type 2 diabetes-associated cognitive impairment animal models and high-glucose in vitro studies, neuroinflammatory markers, such as IL-1 β, TNF-α, IL-6, and MCP-1, and inflammatory responses, such as toll-like receptor 4, cyclooxygenase 1 (COX1), COX2, NF-κB, leukocyte common antigen, and inducible nitric oxide synthase were increased in the brain.83–88 Among participants with dementia and AD pathology, type 2 diabetes had a significantly positive relationship with JNK.89 In vitro, high glucose increased the expression of inflammasome recombinant NLR Family, pyrin domain containing protein 3 (NLRP3) markers in hippocampal cells.90 Besides, CNS inflammation is an immune response mediated by microglia and astrocytes. Evidence demonstrates that acute glucose fluctuation forms the stress that alters microglial activity (eg, inflammatory activation or self-degradation), which may be one of the mechanisms of cognitive deterioration in diabetic patients.91 Diabetic mice also show astrocyte changes in the hippocampus.92,93 Since astrocytes are important neuronal support cells, astrocyte changes may aggravate the dysfunction of neuronal function. Our team previously designed an IR model induced by palmitic acid in vitro and established a neuron-microglia-astrocyte co-culture system, confirming that IR induced microglial activation, and the secretion of cytokines were significantly increased. This study also confirmed that activated microglia can activate the NF-κB pathway in astrocytes, activate astrocytes, and reduce support for neurons (data not shown). However, the association between hyperglycemia and neuroinflammation is not clearly understood. Some researchers have suggested that oxidative stress-mediated mitochondrial dysfunction stimulates the upregulation of mitochondrial HSP60 and ultimately initiates diabetes-induced inflammatory pathways by activating pattern recognition receptors.94
GLP-1 RA has been shown to exert anti-inflammatory effects in the CNS. Under inflammatory conditions in vitro, GLP-1 suppressed the secretion of TNF-α-associated cytokines and chemokines in BV-2 microglia.95 Liraglutide also decreased activated microglia and astrocyte load in the brain induced by chronic inflammation in mice.96 Besides, liraglutide treatment prevented the neuroinflammatory process, promoting the production of anti-inflammatory molecules such as IL10, TGFβ, and arginase 1.97 In a model of lipopolysaccharide (LPS)-induced inflammation, liraglutide inhibited the polarization of pro-inflammatory microglia and promoted the polarization of anti-inflammatory microglia, diminished inflammatory cytokine expression, and decreased NF-κB pathway activation.98 Similarly, exendin-4 also decreased the mRNA levels of IL-1β and TNF-α in LPS-stimulated microglia, and significantly attenuated the activation of the NF-κB signaling pathway.99 The anti-inflammatory effect of GLP-1 also occurs in diabetes-induced neuroinflammation. During exendin-4 treatment, IL- 1b was transiently increased in normoglycemic mice and decreased in hyperglycemic mice.88 Liraglutide also protects astrocytes against advanced glycation end product (AGE)-induced TNF-α and IL-1β secretion.100
There are several possible mechanisms by which GLP-1 RA regulates neuroinflammation. First, GLP-1 RA inhibited LPS-induced IL-1β mRNA expression, whereas adenylate cyclase inhibitor preconditioning inhibited this effect, suggesting that cAMP mediated its anti-inflammatory effect.101 The cAMP/ PKA pathway is also involved in the protection of astrocytes from AGE-induced inflammatory response.100 Second, the anti-inflammatory effects of GLP-1 RA are partially mediated by its metabolite in a phosphorylated AMPK-dependent manner. Therapies that inhibit GLP-1 degradation may weaken the metabolite-mediated effects.102
The GLP-1 RA and Neurogenesis
Neurogenesis is a complete process in which neural stem cells (NSCs) proliferate, undergo balanced and imbalanced division to become directed progenitor cells and gradually migrate to functional areas, undergo plasticity changes, and establish synaptic connections with other neurons to generate neural function. Adult neurogenesis is generated mainly in two parts of the brain: the subventricular zone of the lateral ventricle and the subgranular zone of the dentate gyrus in the hippocampus. The integration of adult-born neurons into the circuitry of the adult hippocampus suggests an important role for adult hippocampal neurogenesis in learning and memory.103 The insulin/insulin-like growth factor (IGF) pathway promotes NSCs proliferation, differentiation, and survival;104 therefore, impaired insulin signaling may affect neurogenesis. The diabetes-induced reduction of neurogenesis in the hippocampal dentate implies a potential mechanism for diabetes-related cognitive decline.105–110 A possible explanation is that pro-inflammatory factors in type 2 diabetes compromise endothelial caveolin-1, a major membrane intrinsic protein in the caveolae on the cell surface, leading to vascular dysfunction, affecting neurogenesis, and subsequently leading to AD.111 IKKβ/NF-κB-mediated impairment112 and γ-aminobutyric acid and glutamate transporter systems113 may also be involved in diabetes-induced damage of neurogenesis.
Enhancing the GLP-1R signaling pathway leads to the proliferation of neuronal cells114 and neuronal differentiation.115 In severely obese and insulin-resistant mice, liraglutide elicits beneficial effects on metabolic control and synaptic plasticity and improves hippocampal neurogenesis.116
The mechanism by which GLP-1 RA influences neurogenesis remains unclear. One possible mechanism is, in part, through increased expression of mammalian achaete–scute homologue 1 (Mash1), which plays an important role in neuronal differentiation and is believed to improve hippocampal neurogenesis. Active Akt increased the protein levels and transactivation activity of Mash1,117 suggesting the potential function of the GLP-1-activated PI3K-Akt pathway.
GLP-1 RA and Synaptic Plasticity
Synaptic plasticity refers to the adjustable strength of connections between nerve cells, known as synapses. It is widely recognized that diabetes affects hippocampal synaptic plasticity, and this disruption in synaptic plasticity is related to cognition.118 Reisi et al reported that both presynaptic and postsynaptic components are involved in diabetes-induced damage to synaptic plasticity.119 In animal models of diabetes with cognitive impairment, synaptic plasticity was impaired in the two experimental forms of long-term enhancement (LTP)120–124 and long-term depression (LTD).125,126 Moreover, the ultrastructure of hippocampal synapses is destroyed,127 thereby reducing the hippocampal dendritic spine density.123 Besides, synaptic plasticity-related proteins, including CREB, pCREB, brain-derived neurotrophic factor (BDNF), and activity-regulated cytoskeleton (Arc) proteins, are significantly reduced.128 Our previous data showed that central IR may significantly affect the expression of synaptic plasticity proteins, such as postsynaptic density protein-95 (PSD95), Arc, synapsin1, BDNF, resulting in the impaired synaptic plasticity of neurons and decreased learning and memory ability (data not shown). Sasaki-Hamada showed that disruption of synaptic plasticity occurs in the prediabetes stage, when glucose tolerance is impaired.129 Further, the onset age130 and duration of diabetes mellitus131 may have some influence on synaptic plasticity. However, short-term acute changes in glucose concentrations may not directly contribute to the synaptic plasticity associated with diabetes, unless extremely severe.132
Glutamate receptors, including the amino-3-hydroxy-5-methyl-4-isoxazolepro-pionicacid (AMPA) and N-methyl-D-aspartate (NMDA) receptors, mediate excitatory synaptic transmission in the CNS, and its expression in the postsynaptic membrane is associated with LTP and LTD and is involved in the regulation of learning and memory activities. Abnormal regulation of glutamatergic receptors appears to participate in diabetes-induced impairment of synaptic plasticity.120,122,125,133,134 In addition, insulin signaling is important for synaptic plasticity. IR β-subunit heterozygous mice135 and complete disruption of IRS2 in mice136 impaired the LTP of synaptic transmission in the hippocampus. Histone deacetylases (HDAC2), a member of the HDAC family, is correlated with insulin signaling components in postsynaptic glutamatergic neurons of the adult mouse hippocampus, and hyperactivity of the HDAC system (including HDAC2) may result in the suppression of the insulin signaling system and consequent disruption of synaptic plasticity in type 2 diabetes.137
GLP-1 RA has a definite effect on improving synaptic plasticity. Exendin-4 inhibits the reduction of LTP in the brain of a mouse fed a high-fat diet138 and significantly increased the phosphorylation level of CREB and the expression level of BDNF. Furthermore, exendin-4 increased the membrane protein levels of the AMPA receptor GluR1 subunit and PSD-95.139 Liraglutide also rescued the deleterious effects of a high-fat diet on hippocampal LTP of neurotransmission140 and enhanced the number of hippocampal and cortical synapses in AD model mice.141
The effect of GLP-1 RA on synaptic plasticity is partly due to the GLP-1R. In a GLP-1R knockout mouse model, LTP in the CA1 area of the hippocampus was severely impaired.142 In addition, GLP-1 RA upregulated neurotrophic tyrosine kinase receptor type 2 and mTOR genes in the hippocampus of high-fat-fed mice, which are involved in regulating synaptic plasticity and LTP.143 By modulating calcium responses to glutamate and membrane depolarization,144 and AMPA receptors,139,145 GLP-1 RA may play important roles in regulating neuronal plasticity.
GLP-1 RA in Diabetes-Related AD and PD
AD is an insidiously progressive ND clinically characterized by memory impairment, aphasia, apraxia, agnosia, impaired executive function, personality and behavior changes, and other comprehensive dementia manifestations. PD is another common degenerative disease, and the main pathology is the degeneration and death of dopaminergic (DA) neurons in the substantia nigra of the midbrain, which leads to a significant decrease in DA content in the striatum. Both AD and PD are NDs associated with diabetes; however, the current treatment of these two diseases still focuses on the improvement of symptoms. It is necessary to better understand their mechanisms to obtain better medications on their pathogenesis.
Diabetes and Alzheimer’s Disease
Diabetes is closely linked to AD, and a meta-analysis showed that patients with diabetes had a significantly higher incidence of AD than in those without diabetes (relative risk [RR], 1.53; 95% CI, 1.42–1.63),146 suggesting that diabetes may promote the development of AD. Insulin acts on β-site amyloid precursor protein cleaving enzyme 1 and γ-secretase to regulate Aβ levels and degrades excess Aβ by modulating insulin-degrading enzyme.147 Activation of insulin signaling pathway PI3k/Akt leads to Ser9 phosphorylation of GSK3β and its impaired kinase activity leads to phosphorylation of tau.148 Hence, IR promotes the pathology of AD by reducing amyloid clearance and increasing tau hyperphosphorylation neurofibrillary tangles,149 both of which are associated with cognitive impairment.150,151
It is widely accepted that changes in the mitochondria are involved in the development of AD. The manifestations of mitochondrial dysfunction in AD mainly include increased oxidative stress and ROS production,152,153 mitochondrial DNA damage,154 mitochondrial respiratory injury,155 and calcium abnormalities.156 Mitophagy,157 mitochondrial dynamics,158 and mitochondrial biogenesis158 are also affected in patients with AD, ultimately resulting in the accumulation of dysfunctional mitochondria and mitochondrial fragmentation. Mitochondrial damage may not only be the common pathological mechanism of diabetes and AD, but also the key point of crosstalk between them. Two AD-related markers, Aβ-production and tau phosphorylation induced by IR, may be the upstream mechanisms of AD-related mitochondrial damage.159–161 In addition, mitochondrial damage may be a contributing factor to the progression of diabetes to AD, as exposure to Aβ Increases the vulnerability of brain mitochondria in diabetic rats.162
ER stress plays a complex role in the control of neuronal survival, amyloid cascade, neurodegeneration, and synaptic function in AD.163 In vitro, the abovementioned ER stress/JNK/IRS1 pathway was involved in Aβ1-42 oligomer-induced tau hyperphosphorylation, which may indicate that IR promotes the role of ER stress in AD.164 de la Monte et al suggested that in AD, a triangulated Mal-signaling network initiated by the brain’s insulin/IGF resistance is transmitted through the ceramides and ER stress homeostasis disorder, which in turn promotes IR.165
Studies have established that inflammation contributes to the pathogenesis of AD. In AD, Aβ damages microglia, produces inflammatory cytokines and chemokines, and affects surrounding CNS resident cells (astrocytes, oligodendrocytes, and neurons), which may aggravate tau pathology and ultimately lead to neurodegeneration and neuron loss.166 Inflammation may also be a potential link between diabetes and AD. Takeda et al crossed Alzheimer transgenic mice (APP23) with two types of diabetic mice (ob/ob and NSY mice) and found a significant increase in IL-6 in the brains of hybrid mice fed with a high-fat diet.167 In addition, it has been shown that feeding AD model mice (triple transgenic AD [3xTgAD]) a high-fat diet may increase the activation of microglia.168 These studies suggest that diabetes mellitus and a high-fat diet may exacerbate AD inflammation.
Neurogenesis is defective in the AD model, which is characterized by decreased proliferation and differentiation, diminished neuronal maturity, and reduced survival, before processes that may secondarily affect neurogenesis, such as neuronal loss, amyloid deposition, and inflammation.169 Chronic hyperglycemia decreases the complexity and differentiation of 3xTg-AD newborn neurons and depressed synaptic facilitation, accompanied by defective hippocampal-dependent memory,109 suggesting that diabetes promotes changes in AD neurogenesis that ultimately exacerbates cognitive impairment.
Diabetes and Parkinson’s Disease
A systematic review and meta-analysis suggested that diabetes was a risk factor for PD (RR = 1.37, 95% CI, 1.21–1.55; P < 0.0001).170 Diabetes may exacerbate the progression of PD, including cognitive impairment and axial motor symptoms.171 IR is still the key link between diabetes and PD and may impair nigrostriatal dopamine function,172 exacerbate nigrostriatal DA depletion,173 and enhance cognitive impairment174 and behavioral abnormalities.175
Mitochondrial dysfunction is a defect in the early stage of PD and mainly includes impairment of the mitochondrial electron transport chain, alterations in mitochondrial morphology and dynamics, mutations in mitochondrial DNA, and anomalies in calcium homeostasis,176 which are closely related to the PD phenotype. Mitochondrial damage may also be the reason why diabetes-related IR promotes the development of PD. In vitro, in differentiated human DA neurons, IR was associated with increased α-synuclein and ROS levels, as well as mitochondrial depolarization, which may be mediated by polo-like kinase-2.177 In vivo, mitochondrial dynamics-related factor Parkin was significantly reduced in the substantia nigra of a mice fed a high-fat diet and a diabetic mice, leading to the accumulation of Parkin-interacting substrate and the reduction of PGC-1α.41 Also, high glucose levels may modulate Parkin/PINK1-mediated mitochondrial autophagy in DA cells through the thioredoxin-interacting protein.178 All the above mechanisms suggest that diabetes-related metabolic factors may promote PD by regulating the mitochondria.
All branches of the UPR in ER stress are likely implicated in PD etiology.179 At present, studies on whether diabetes and IR aggravate ER stress in PD are few. However, given the ubiquity of ER stress-related pathways and IR crosstalk mentioned above, diabetes-related IR is likely to be involved in the generation of ER stress in PD.
Similar to AD, neuroinflammation is involved in the degeneration of DA neurons, which is mainly mediated by activated glial cells and surrounding immune cells. This cellular response may eventually lead to the death of DA cells, leading to disease progression.180 A study that used 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) to mimic PD-like neural injury found that neuroinflammation is aggravated in the midbrain of type 2 diabetes mice, who are more susceptible to the neurotoxicity induced by MPTP.181 This may indicate that diabetes exacerbates DA neuronal degeneration during the progression of PD, which may be mediated by neuroinflammation.
Adult neurogenesis is severely affected in PD, although the exact mechanisms and effects of these changes are not fully understood, there may be a dynamic interaction between them and PD-related pathology.182 Although it is not clear whether there is crosstalk between diabetes and PD in neurogenesis, similar pathophysiological changes may indicate a close association between the two diseases.
GLP-1 RA Show Effects
Some similar or overlapping mechanisms certainly exist between diabetes and AD or PD, such as mitochondrial dysfunction, oxidative stress, and inflammation, that may strengthen their correlation. Furthermore, these mechanisms may underlie the use of the diabetes drug GLP-1 RA to treat AD and PD. In some AD and PD models, a considerable number of studies have clarified the role of GLP-1 RA in these cellular processes. However, GLP-1 RA also has some disease-specific effects in AD and PD, such as reduced Aβ levels, tau hyperphosphorylation in AD, and reduced α-synuclein pathology and DA neuronal loss in PD, suggesting that GLP-1 RA has a strong neuroprotective function (Tables 1 and 2).
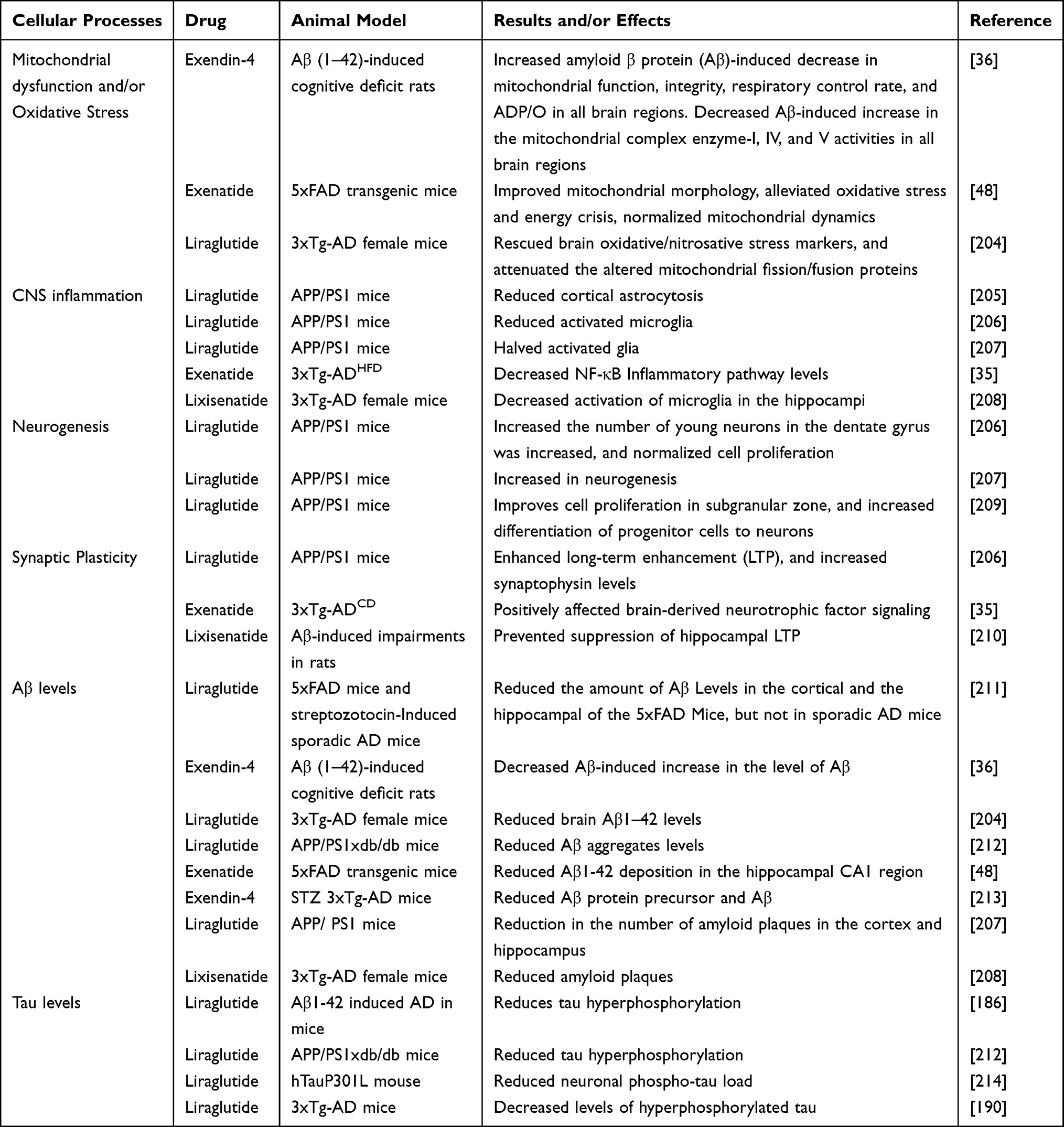 |
Table 1 Effects of GLP-1 Receptor Agonists in Models of AD: Data from Animal Experimental Models |
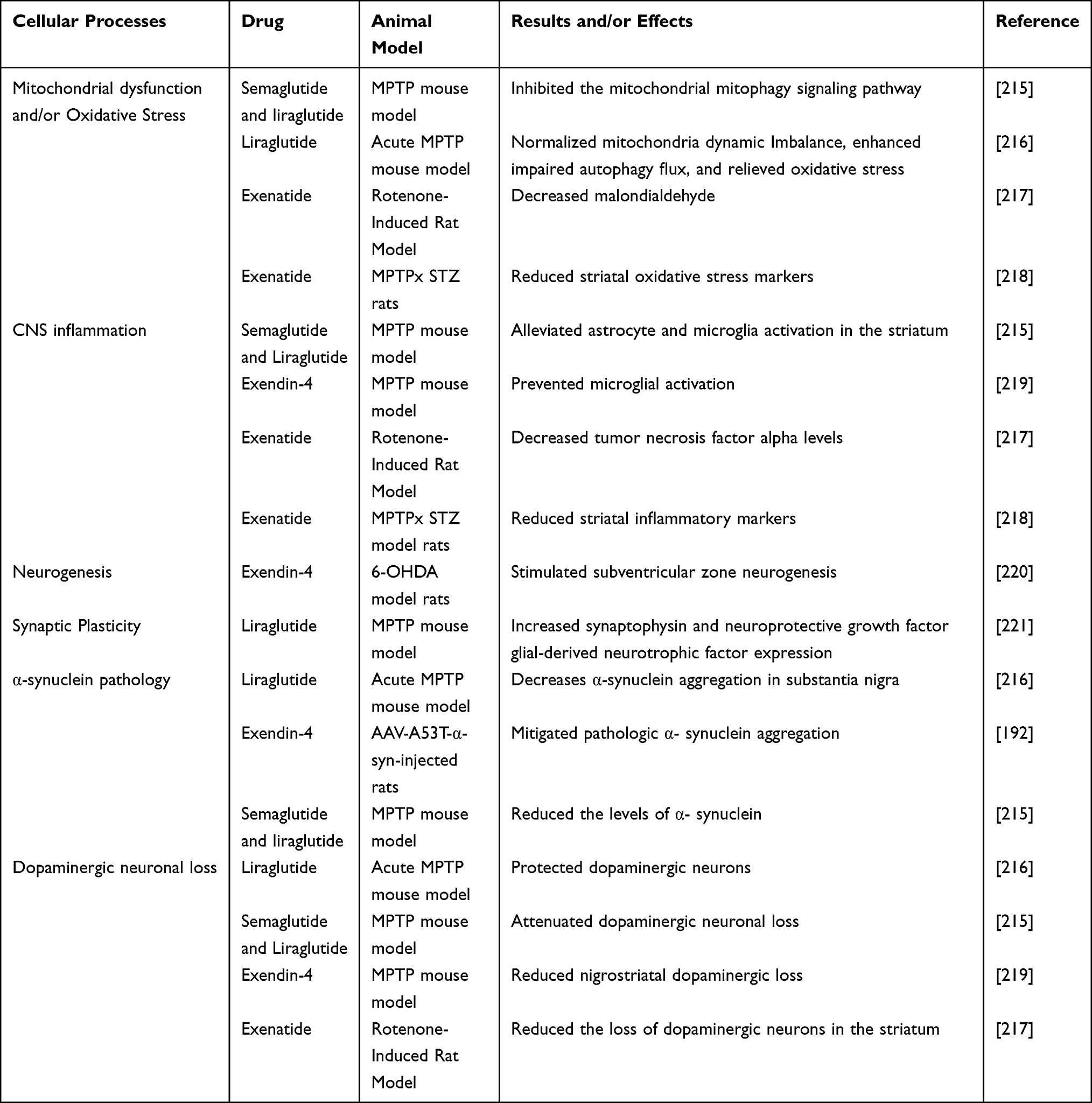 |
Table 2 Effects of GLP-1 Receptor Agonists in Models of PD: Data from Animal Experimental Models |
The mechanism of GLP-1 RA on Aβ is not clear. One possibility is that amyloid precursor protein (βAPP) binds to GLP-1 as a G-protein-coupled receptor, resulting in reduced βAPP synthesis.183 GLP-1 RA reduces tau phosphorylation not only in the AD model, but also in diabetes.32,33,184,185 However, the mechanism by which GLP-1 RA reduces tau hyperphosphorylation may be complex. It has been reported that GLP-1 RA reduces tau phosphorylation through Akt and GSK-3β, a pathway related to insulin signaling, which also confirmed that insulin resistance is the key to tau phosphorylation.186–188 It has also been reported that the effects of liraglutide on decreasing the hyperphosphorylation of tau by enhancing O-glycosylation of neuronal cytoskeleton protein, improving the JNK and ERK signaling pathway.189,190 In addition, the mitochondrial PGC-1α signaling pathway is also the mechanism of GLP-1 RA to protect neurons from tau hyperphosphorylation, indicating that mitochondrial dysregulation has crosstalk with tau pathology.46 The common mechanism of GLP-1 RA reduction in Aβ overproduction and tau hyperphosphorylation may be the restoration of protein phosphatase 2A activity and inhibition of β- and γ- secretase.191 The mechanism of GLP-1 RA to improve dopaminergic degeneration and pathological α -synuclein aggregation in the PD model may involve inhibiting the PI3K/Akt/mTOR pathway192 and enhancing AMPK/PGC-1a signaling pathway.193
Although some results have been achieved in animal models, clinical studies on GLP-1 RA remain limited. This review summarizes some of the clinical data on GLP-1 RA (Table 3), with some exciting results. But some studies failed to find efficacy due to early termination194 or some studies may fail to find drug effects due to short follow-up time or low statistical thresholds.195 Several systematic reviews or meta-analyses have attempted to comprehensively summarize the clinical data of GLP-1 RA in the treatment of AD or PD, but the results are still inconclusive,196–200 possibly because some clinical studies are still ongoing and require ongoing attention.201,202 Due to the limitations of clinical studies, the evaluation of drugs needs to rely on some new examination methods, such as imaging.203 There is reason to believe that with the continuous improvement of technology, the judgment of drug efficacy will be easier and more diversified.
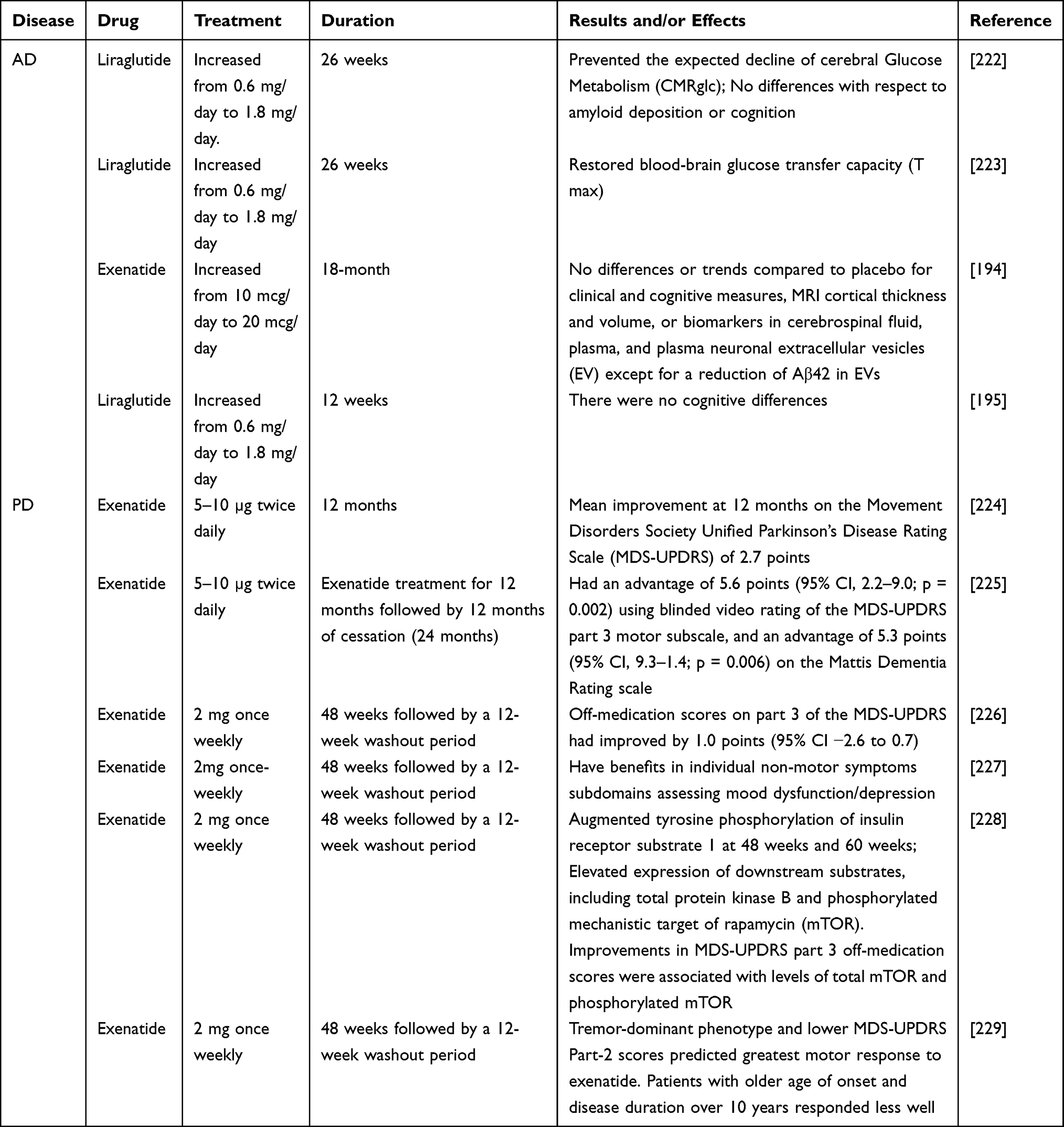 |
Table 3 Clinical Trials of GLP-1 Receptor Agonists in AD and PD |
Conclusion
In recent years, clinical guidelines have begun to emphasize the importance of diabetes-related NDs and their risk of cognitive impairment, despite widespread concerns. Diabetes and related NDs share common mechanisms, such as central IR, oxidative stress, and inflammation, which underlie their crosstalk, which has also inspired the investigation of hypoglycemic agents, particularly GLP-1 RA, as potential treatments for diabetes and related NDs.
This review describes in detail the beneficial effects of GLP-1 RA on the central pathological mechanisms of diabetes and related degenerative diseases. However, the role of GLP-1 RA in the body is complex. First, GLP-1 RA has been shown to have powerful hypoglycemic effects, and the influence of blood glucose on these mechanisms cannot be ruled out. Second, IR exists in the brains of patients with diabetes mellitus, AD, and PD and is also a factor affecting these mechanisms. Therefore, it is not clear whether GLP-1 RA directly improves mitochondrial function, reduces ER stress, and reduces neuroinflammation, or indirectly improves these mechanisms by lowering blood glucose and improving IR. Further studies are needed to confirm the central protective effect of GLP-1 RA, and further clinical trials should be actively conducted.
Abbreviations
Aβ, amyloid β protein; AD, Alzheimer’s disease; AGE, advanced glycation end product; Akt, protein kinase B; AMPA, amino-3-hydroxy-5-methyl-4-isoxazolepro-pionicacid; AMP, adenosine 5‘-monophosphate; AMPK, AMP-activated protein kinase; Arc, activity-regulated cytoskeleton; ATF6, activating transcription factor 6; βAPP, amyloid precursor protein; BDNF, brain-derived neurotrophic factor; BiP, binding immunoglobulin protein; CHOP, C/EBP homologous protein; CNS, central nervous system; cAMP, cyclic adenosine monophosphate; COX, cyclooxygenase; CREB, cAMP response element-binding protein; DA, dopaminergic; Drp1, dynamin-related protein 1; ER, endoplasmic reticulum; ERK-Yap, extracellular signal-regulated kinase-Yes-associated protein; GADD34, growth arrest and DNA damage inducible gene 34; GLP-1, glucagon-like peptide 1; GLP-1 RA, GLP-1 receptor agonists; GLP-1R, GLP-1 receptor; GRP78, 78-kDa glucose-regulating protein; GSK-3β, glycogen synthase kinase 3β; HDAC, histone deacetylase; HSP, heat shock protein; IGF, insulin-like growth factor; IKKβ, inhibitor kappa B kinase β; IR, insulin resistance; IRE-1, inositol-requiring enzyme 1; IRS, insulin receptor substrate; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; LTD, long-term depression; LTP, long-term enhancement; MAPK, mitogen-activated kinase; Mash1, mammalian achaete–scute homologue 1; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; mTOR, mammalian target of rapamycin; ND, neurodegenerative disease; NLRP3, recombinant NLR Family, pyrin domain containing protein 3; NMDA, N-methyl-D-aspartate; NSCs, neural stem cells; NF-κB, nuclear transcription factor-κB; PD, Parkinson’s disease; PERK, PKR-like ER kinase; PGC1a, peroxisome proliferator-activated receptor c coactivator 1a; PI3K, phosphoinositide-3 kinase; p-JNK, phospho-Jun N-terminal kinase; PKA, protein kinase A; PSD95, postsynaptic density protein-95; PKR, protein kinase R; ROS, reactive oxygen species; RR, relative risk; SIRT1, NAD-dependent protein deacetylase sirtuin 1; UPR, unfolded protein response; 3xTgAD, triple transgenic AD.
Acknowledgments
This work was supported by the grant from the National Natural Science Fund of China (81970687, 81670732 belonging to Guixia Wang; 81900726 belonging to Xue Zhao); the grant from the Engineering Research Center of Individualized Accurate Diagnosis and Treatment of Jilin Province Science and Technology Department Project (20170623005TC); the grant from Development and Reform Commission Innovation Project (2017C019); the grant from Department Science and Technology Department of Jilin Province (20190901006JC belonging to Guixia Wang; 20210101439JC belonging to Xue Zhao); the grant from Research Fund of the First Hospital of Jilin University (2021-zl-01 belonging to Xue Zhao) and the grant from Jilin Medical and Health Talent Project (JLSWSRCZX2021-081 belonging to Xue Zhao).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chatterjee S, Khunti K, Davies MJ. Type 2 diabetes. Lancet (London, England). 2017;389(10085):2239–2251. doi:10.1016/S0140-6736(17)30058-2
2. Srikanth V, Sinclair AJ, Hill-Briggs F, Moran C, Biessels GJ. Type 2 diabetes and cognitive dysfunction-towards effective management of both comorbidities. Lancet Diabetes Endocrinol. 2020;8(6):535–545. doi:10.1016/S2213-8587(20)30118-2
3. Biessels GJ, Whitmer RA. Cognitive dysfunction in diabetes: how to implement emerging guidelines. Diabetologia. 2020;63(1):3–9. doi:10.1007/s00125-019-04977-9
4. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–1439. doi:10.1152/physrev.00034.2006
5. Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(12):728–742. doi:10.1038/nrendo.2012.140
6. Beiroa D, Imbernon M, Gallego R, et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes. 2014;63(10):3346–3358. doi:10.2337/db14-0302
7. Cork SC, Richards JE, Holt MK, Gribble FM, Reimann F, Trapp S. Distribution and characterisation of Glucagon-like peptide-1 receptor expressing cells in the mouse brain. Mol Metab. 2015;4(10):718–731. doi:10.1016/j.molmet.2015.07.008
8. Salameh TS, Rhea EM, Talbot K, Banks WA. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol. 2020;180:114187. doi:10.1016/j.bcp.2020.114187
9. Kastin AJ, Akerstrom V. Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obes Relat Metab Disord. 2003;27(3):313–318. doi:10.1038/sj.ijo.0802206
10. Hunter K, Hölscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012;13:33. doi:10.1186/1471-2202-13-33
11. Biessels GJ, Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. 2018;14(10):591–604. doi:10.1038/s41574-018-0048-7
12. Luchsinger JA. Adiposity, hyperinsulinemia, diabetes and Alzheimer’s disease: an epidemiological perspective. Eur J Pharmacol. 2008;585(1):119–129. doi:10.1016/j.ejphar.2008.02.048
13. Maciejczyk M, Żebrowska E, Chabowski A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int J Mol Sci. 2019;20(4):874. doi:10.3390/ijms20040874
14. Cheng H, Gang X, Liu Y, Wang G, Zhao X, Wang G. Mitochondrial dysfunction plays a key role in the development of neurodegenerative diseases in diabetes. Am J Physiol Endocrinol Metab. 2020;318(5):E750–e764. doi:10.1152/ajpendo.00179.2019
15. Sims-Robinson C, Bakeman A, Glasser R, Boggs J, Pacut C, Feldman EL. The role of endoplasmic reticulum stress in hippocampal insulin resistance. Exp Neurol. 2016;277:261–267. doi:10.1016/j.expneurol.2016.01.007
16. Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168–181. doi:10.1038/nrneurol.2017.185
17. Banks WA, Owen JB, Erickson MA. Insulin in the brain: there and back again. Pharmacol Ther. 2012;136(1):82–93. doi:10.1016/j.pharmthera.2012.07.006
18. Golpich M, Amini E, Hemmati F, et al. Glycogen synthase kinase-3 beta (GSK-3β) signaling: Implications for Parkinson’s disease. Pharmacol Res. 2015;97:16–26. doi:10.1016/j.phrs.2015.03.010
19. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813(11):1978–1986. doi:10.1016/j.bbamcr.2011.03.010
20. Perluigi M, Di Domenico F, Butterfield DA. mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis. 2015;84:39–49. doi:10.1016/j.nbd.2015.03.014
21. Bassil F, Fernagut PO, Bezard E, Meissner WG. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: targets for disease modification? Prog Neurobiol. 2014;118:1–18. doi:10.1016/j.pneurobio.2014.02.005
22. Carlezon WA
23. Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. doi:10.1146/annurev.neuro.20.1.245
24. Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: an update. Arch Toxicol. 2015;89(6):867–882. doi:10.1007/s00204-015-1472-2
25. Wu F, Yang L, Hang K, et al. Full-length human GLP-1 receptor structure without orthosteric ligands. Nat Commun. 2020;11(1):1272. doi:10.1038/s41467-020-14934-5
26. Llewellyn-Smith IJ, Reimann F, Gribble FM, Trapp S. Preproglucagon neurons project widely to autonomic control areas in the mouse brain. Neuroscience. 2011;180:111–121. doi:10.1016/j.neuroscience.2011.02.023
27. Hisadome K, Reimann F, Gribble FM, Trapp S. Leptin directly depolarizes preproglucagon neurons in the nucleus tractus solitarius: electrical properties of glucagon-like Peptide 1 neurons. Diabetes. 2010;59(8):1890–1898. doi:10.2337/db10-0128
28. Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience. 1997;77(1):257–270. doi:10.1016/S0306-4522(96)00434-4
29. Kappe C, Tracy LM, Patrone C, Iverfeldt K, Sjöholm Å. GLP-1 secretion by microglial cells and decreased CNS expression in obesity. J Neuroinflammation. 2012;9:276. doi:10.1186/1742-2094-9-276
30. Laakso M, Zilinskaite J, Hansen T, et al. Insulin sensitivity, insulin release and glucagon-like peptide-1 levels in persons with impaired fasting glucose and/or impaired glucose tolerance in the EUGENE2 study. Diabetologia. 2008;51(3):502–511. doi:10.1007/s00125-007-0899-2
31. Jantrapirom S, Nimlamool W, Chattipakorn N, et al. Liraglutide Suppresses Tau Hyperphosphorylation, Amyloid Beta Accumulation through Regulating Neuronal Insulin Signaling and BACE-1 Activity. Int J Mol Sci. 2020;21(5):1725. doi:10.3390/ijms21051725
32. Ma DL, Chen FQ, Xu WJ, Yue WZ, Yuan G, Yang Y. Early intervention with glucagon-like peptide 1 analog liraglutide prevents tau hyperphosphorylation in diabetic db/db mice. J Neurochem. 2015;135(2):301–308. doi:10.1111/jnc.13248
33. Yang Y, Zhang J, Ma D, et al. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J Alzheimers Dis. 2013;37(3):637–648. doi:10.3233/JAD-130491
34. Batista AF, Forny-Germano L, Clarke JR, et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J Pathol. 2018;245(1):85–100. doi:10.1002/path.5056
35. Bomba M, Granzotto A, Castelli V, et al. Exenatide reverts the high-fat-diet-induced impairment of BDNF signaling and inflammatory response in an animal model of Alzheimer’s disease. J Alzheimers Dis. 2019;70(3):793–810. doi:10.3233/JAD-190237
36. Garabadu D, Verma J. Exendin-4 attenuates brain mitochondrial toxicity through PI3K/Akt-dependent pathway in amyloid beta (1-42)-induced cognitive deficit rats. Neurochem Int. 2019;128:39–49. doi:10.1016/j.neuint.2019.04.006
37. Kleinridders A, Cai W, Cappellucci L, et al. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc Natl Acad Sci U S A. 2015;112(11):3463–3468. doi:10.1073/pnas.1500877112
38. Fauconnier J, Lanner JT, Zhang SJ, et al. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of ob/ob mice. Diabetes. 2005;54(8):2375–2381. doi:10.2337/diabetes.54.8.2375
39. Wright LE, Vecellio Reane D, Milan G, et al. Increased mitochondrial calcium uniporter in adipocytes underlies mitochondrial alterations associated with insulin resistance. Am J Physiol Endocrinol Metab. 2017;313(6):E641–e650. doi:10.1152/ajpendo.00143.2016
40. Huang S, Wang Y, Gan X, et al. Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes. 2015;64(5):1728–1742. doi:10.2337/db14-0758
41. Khang R, Park C, Shin JH. Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience. 2015;294:182–192. doi:10.1016/j.neuroscience.2015.03.017
42. Neniskyte U, Gross CT. Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat Rev Neurosci. 2017;18(11):658–670. doi:10.1038/nrn.2017.110
43. Verdile G, Keane KN, Cruzat VF, et al. Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm. 2015;2015:105828. doi:10.1155/2015/105828
44. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103–115. doi:10.1038/cr.2010.178
45. Di Meo S, Iossa S, Venditti P. Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol. 2017;233(1):R15–r42. doi:10.1530/JOE-16-0598
46. An FM, Chen S, Xu Z, et al. Glucagon-like peptide-1 regulates mitochondrial biogenesis and tau phosphorylation against advanced glycation end product-induced neuronal insult: studies in vivo and in vitro. Neuroscience. 2015;300:75–84. doi:10.1016/j.neuroscience.2015.05.023
47. Ma T, Du X, Pick JE, Sui G, Brownlee M, Klann E. Glucagon-like peptide-1 cleavage product GLP-1 (9-36)amide rescues synaptic plasticity and memory deficits in Alzheimer’s disease model mice. J Neurosci. 2012;32(40):13701–13708. doi:10.1523/JNEUROSCI.2107-12.2012
48. An J, Zhou Y, Zhang M, et al. Exenatide alleviates mitochondrial dysfunction and cognitive impairment in the 5×FAD mouse model of Alzheimer’s disease. Behav Brain Res. 2019;370:111932. doi:10.1016/j.bbr.2019.111932
49. Zhang X, Zhang Z, Zhao Y, et al. Alogliptin, a dipeptidyl peptidase-4 inhibitor, alleviates atrial remodeling and improves mitochondrial function and biogenesis in diabetic rabbits. J Am Heart Assoc. 2017;6(5). doi:10.1161/JAHA.117.005945
50. Qiao H, Ren H, Du H, Zhang M, Xiong X, Lv R. Liraglutide repairs the infarcted heart: the role of the SIRT1/Parkin/mitophagy pathway. Mol Med Rep. 2018;17(3):3722–3734. doi:10.3892/mmr.2018.8371
51. Morales PE, Torres G, Sotomayor-Flores C, et al. GLP-1 promotes mitochondrial metabolism in vascular smooth muscle cells by enhancing endoplasmic reticulum-mitochondria coupling. Biochem Biophys Res Commun. 2014;446(1):410–416. doi:10.1016/j.bbrc.2014.03.004
52. Li J, Li N, Yan S, et al. Liraglutide protects renal mesangial cells against hyperglycemia‑mediated mitochondrial apoptosis by activating the ERK‑Yap signaling pathway and upregulating Sirt3 expression. Mol Med Rep. 2019;19(4):2849–2860. doi:10.3892/mmr.2019.9946
53. Xiong X, Lu W, Qin X, Luo Q, Zhou W. Downregulation of the GLP-1/CREB/adiponectin pathway is partially responsible for diabetes-induced dysregulated vascular tone and VSMC dysfunction. Biomed Pharmacother. 2020;127:110218. doi:10.1016/j.biopha.2020.110218
54. Zhang WY, Hu XF, Wan N, et al. Protective effect of the glucagon-like peptide-1 analogue liraglutide on carbon tetrachloride-induced acute liver injury in mice. Biochem Biophys Res Commun. 2019;514(2):386–392. doi:10.1016/j.bbrc.2019.04.160
55. Chang G, Liu J, Qin S, et al. Cardioprotection by exenatide: a novel mechanism via improving mitochondrial function involving the GLP-1 receptor/cAMP/PKA pathway. Int J Mol Med. 2018;41(3):1693–1703. doi:10.3892/ijmm.2017.3318
56. Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. J Biol Chem. 2014;289(3):1203–1211. doi:10.1074/jbc.R113.534743
57. Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13(8):477–491. doi:10.1038/nrneurol.2017.99
58. Salvadó L, Palomer X, Barroso E, Vázquez-Carrera M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol Metab. 2015;26(8):438–448. doi:10.1016/j.tem.2015.05.007
59. Tsiotra PC, Tsigos C. Stress, the endoplasmic reticulum, and insulin resistance. Ann N Y Acad Sci. 2006;1083:63–76. doi:10.1196/annals.1367.007
60. Rieusset J. Contribution of mitochondria and endoplasmic reticulum dysfunction in insulin resistance: distinct or interrelated roles? Diabetes Metab. 2015;41(5):358–368. doi:10.1016/j.diabet.2015.02.006
61. Villalobos-Labra R, Subiabre M, Toledo F, Pardo F, Sobrevia L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol Aspects Med. 2019;66:49–61. doi:10.1016/j.mam.2018.11.001
62. Rocha M, Diaz-Morales N, Rovira-Llopis S, et al. Mitochondrial dysfunction and endoplasmic reticulum stress in diabetes. Curr Pharm Des. 2016;22(18):2640–2649. doi:10.2174/1381612822666160209152033
63. Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science (New York, NY). 2004;306(5695):457–461. doi:10.1126/science.1103160
64. Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799. doi:10.1038/srep00799
65. Nakatani Y, Kaneto H, Kawamori D, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280(1):847–851. doi:10.1074/jbc.M411860200
66. Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science (New York, NY). 2000;287(5453):664–666. doi:10.1126/science.287.5453.664
67. Liang L, Chen J, Zhan L, et al. Endoplasmic reticulum stress impairs insulin receptor signaling in the brains of obese rats. PLoS One. 2015;10(5):e0126384. doi:10.1371/journal.pone.0126384
68. Wang Z, Huang Y, Cheng Y, et al. Endoplasmic reticulum stress-induced neuronal inflammatory response and apoptosis likely plays a key role in the development of diabetic encephalopathy. Oncotarget. 2016;7(48):78455–78472. doi:10.18632/oncotarget.12925
69. Duran-Aniotz C, Cornejo VH, Espinoza S, et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017;134(3):489–506. doi:10.1007/s00401-017-1694-x
70. Seppa K, Toots M, Reimets R, et al. GLP-1 receptor agonist liraglutide has a neuroprotective effect on an aged rat model of Wolfram syndrome. Sci Rep. 2019;9(1):15742. doi:10.1038/s41598-019-52295-2
71. Toots M, Seppa K, Jagomäe T, et al. Preventive treatment with liraglutide protects against development of glucose intolerance in a rat model of Wolfram syndrome. Sci Rep. 2018;8(1):10183. doi:10.1038/s41598-018-28314-z
72. Panagaki T, Michael M, Hölscher C. Liraglutide restores chronic ER stress, autophagy impairments and apoptotic signalling in SH-SY5Y cells. Sci Rep. 2017;7(1):16158. doi:10.1038/s41598-017-16488-x
73. Chen J, Xie JJ, Shi KS, et al. Glucagon-like peptide-1 receptor regulates endoplasmic reticulum stress-induced apoptosis and the associated inflammatory response in chondrocytes and the progression of osteoarthritis in rat. Cell Death Dis. 2018;9(2):212. doi:10.1038/s41419-017-0217-y
74. He J, Wang C, Sun Y, et al. Exendin-4 protects bone marrow-derived mesenchymal stem cells against oxygen/glucose and serum deprivation-induced apoptosis through the activation of the cAMP/PKA signaling pathway and the attenuation of ER stress. Int J Mol Med. 2016;37(4):889–900. doi:10.3892/ijmm.2016.2509
75. Wang MD, Huang Y, Zhang GP, et al. Exendin-4 improved rat cortical neuron survival under oxygen/glucose deprivation through PKA pathway. Neuroscience. 2012;226:388–396. doi:10.1016/j.neuroscience.2012.09.025
76. Yusta B, Baggio LL, Estall JL, et al. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4(5):391–406. doi:10.1016/j.cmet.2006.10.001
77. Cunha DA, Ladrière L, Ortis F, et al. Glucagon-like peptide-1 agonists protect pancreatic beta-cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes. 2009;58(12):2851–2862. doi:10.2337/db09-0685
78. Zheng X, Xu F, Liang H, et al. SIRT1/HSF1/HSP pathway is essential for exenatide-alleviated, lipid-induced hepatic endoplasmic reticulum stress. Hepatology (Baltimore, Md). 2017;66(3):809–824. doi:10.1002/hep.29238
79. Cheng CK, Luo JY, Lau CW, et al. A GLP-1 analog lowers ER stress and enhances protein folding to ameliorate homocysteine-induced endothelial dysfunction. Acta Pharmacol Sin. 2021;42(10):1598–1609. doi:10.1038/s41401-020-00589-x
80. Chen DL, Xiang JN, Yang LY. Role of ERp46 in β-cell lipoapoptosis through endoplasmic reticulum stress pathway as well as the protective effect of exendin-4. Biochem Biophys Res Commun. 2012;426(3):324–329. doi:10.1016/j.bbrc.2012.08.072
81. Lampropoulou E, Lymperopoulou A, Charonis A. Reduced expression of ERp46 under diabetic conditions in β-cells and the effect of liraglutide. Metabolism. 2016;65(1):7–15. doi:10.1016/j.metabol.2015.09.011
82. Fakhoury M. Role of Immunity and Inflammation in the Pathophysiology of Neurodegenerative Diseases. Neurodegener Dis. 2015;15(2):63–69. doi:10.1159/000369933
83. Esmaeili MH, Enayati M, Khabbaz Abkenar F, Ebrahimian F, Salari AA. Glibenclamide mitigates cognitive impairment and hippocampal neuroinflammation in rats with type 2 diabetes and sporadic Alzheimer-like disease. Behav Brain Res. 2020;379:112359. doi:10.1016/j.bbr.2019.112359
84. Mehta BK, Singh KK, Banerjee S. Effect of exercise on type 2 diabetes-associated cognitive impairment in rats. Int J Neurosci. 2019;129(3):252–263. doi:10.1080/00207454.2018.1526795
85. Jing GC, Zhang MR, Ji C, Zuo PP, Liu YQ, Gu B. Effect of Chinese herbal compound Naofucong () on the inflammatory process induced by high glucose in BV-2 cells. Chin J Integr Med. 2016;22(11):832–839.
86. Solmaz V, Çınar BP, Yiğittürk G, Çavuşoğlu T, Taşkıran D, Erbaş O. Exenatide reduces TNF-α expression and improves hippocampal neuron numbers and memory in streptozotocin treated rats. Eur J Pharmacol. 2015;765:482–487. doi:10.1016/j.ejphar.2015.09.024
87. Sedky AA, Magdy Y. Reduction in TNF alpha and oxidative stress by liraglutide: Impact on ketamine-induced cognitive dysfunction and hyperlocomotion in rats. Life Sci. 2021;278:119523. doi:10.1016/j.lfs.2021.119523
88. Huang HJ, Chen YH, Liang KC, et al. Exendin-4 protected against cognitive dysfunction in hyperglycemic mice receiving an intrahippocampal lipopolysaccharide injection. PLoS One. 2012;7(7):e39656. doi:10.1371/journal.pone.0039656
89. Taga M, Minett T, Classey J, et al. Metaflammasome components in the human brain: a role in dementia with Alzheimer’s pathology? Brain Pathol. 2017;27(3):266–275. doi:10.1111/bpa.12388
90. Yang X, Chen Y, Zhang W, et al. Association between inflammatory biomarkers and cognitive dysfunction analyzed by MRI in diabetes patients. Diabetes Metab Syndr Obes Targets Ther. 2020;13:4059–4065. doi:10.2147/DMSO.S271160
91. Hsieh CF, Liu CK, Lee CT, Yu LE, Wang JY. Acute glucose fluctuation impacts microglial activity, leading to inflammatory activation or self-degradation. Sci Rep. 2019;9(1):840. doi:10.1038/s41598-018-37215-0
92. Saravia FE, Beauquis J, Revsin Y, Homo-Delarche F, de Kloet ER, De Nicola AF. Hippocampal neuropathology of diabetes mellitus is relieved by estrogen treatment. Cell Mol Neurobiol. 2006;26(4–6):943–957. doi:10.1007/s10571-006-9096-y
93. Nardin P, Zanotto C, Hansen F, et al. Peripheral levels of AGEs and astrocyte alterations in the hippocampus of STZ-diabetic rats. Neurochem Res. 2016;41(8):2006–2016. doi:10.1007/s11064-016-1912-2
94. Liyanagamage D, Martinus RD. Role of Mitochondrial Stress Protein HSP60 in Diabetes-Induced Neuroinflammation. Mediators Inflamm. 2020;2020:8073516. doi:10.1155/2020/8073516
95. Yoon G, Kim YK, Song J. Glucagon-like peptide-1 suppresses neuroinflammation and improves neural structure. Pharmacol Res. 2020;152:104615. doi:10.1016/j.phrs.2019.104615
96. Parthsarathy V, Hölscher C. The type 2 diabetes drug liraglutide reduces chronic inflammation induced by irradiation in the mouse brain. Eur J Pharmacol. 2013;700(1–3):42–50. doi:10.1016/j.ejphar.2012.12.012
97. Diz-Chaves Y, Toba L, Fandiño J, González-Matías LC, Garcia-Segura LM, Mallo F. The GLP-1 analog, liraglutide prevents the increase of proinflammatory mediators in the hippocampus of male rat pups submitted to maternal perinatal food restriction. J Neuroinflammation. 2018;15(1):337. doi:10.1186/s12974-018-1370-7
98. Liao T, Zhang SL, Yuan X, et al. Liraglutide lowers body weight set point in DIO rats and its relationship with hypothalamic microglia activation. Obesity (Silver Spring, Md). 2020;28(1):122–131. doi:10.1002/oby.22666
99. Lee CH, Jeon SJ, Cho KS, et al. Activation of glucagon-like peptide-1 receptor promotes neuroprotection in experimental autoimmune encephalomyelitis by reducing neuroinflammatory responses. Mol Neurobiol. 2018;55(4):3007–3020. doi:10.1007/s12035-017-0550-2
100. Bao Y, Jiang L, Chen H, Zou J, Liu Z, Shi Y. The neuroprotective effect of liraglutide is mediated by glucagon-like peptide 1 receptor-mediated activation of cAMP/PKA/CREB pathway. Cell Physiol Biochem. 2015;36(6):2366–2378. doi:10.1159/000430199
101. Iwai T, Ito S, Tanimitsu K, Udagawa S, Oka J. Glucagon-like peptide-1 inhibits LPS-induced IL-1beta production in cultured rat astrocytes. Neurosci Res. 2006;55(4):352–360. doi:10.1016/j.neures.2006.04.008
102. Hou J, Manaenko A, Hakon J, Hansen-Schwartz J, Tang J, Zhang JH. Liraglutide, a long-acting GLP-1 mimetic, and its metabolite attenuate inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2012;32(12):2201–2210. doi:10.1038/jcbfm.2012.133
103. Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11(5):339–350. doi:10.1038/nrn2822
104. Spéder P, Liu J, Brand AH. Nutrient control of neural stem cells. Curr Opin Cell Biol. 2011;23(6):724–729. doi:10.1016/j.ceb.2011.08.004
105. Zhang WJ, Tan YF, Yue JT, Vranic M, Wojtowicz JM. Impairment of hippocampal neurogenesis in streptozotocin-treated diabetic rats. Acta Neurol Scand. 2008;117(3):205–210. doi:10.1111/j.1600-0404.2007.00928.x
106. Wang SH, Sun ZL, Guo YJ, Yuan Y, Yang BQ. Diabetes impairs hippocampal function via advanced glycation end product mediated new neuron generation in animals with diabetes-related depression. Toxicol Sci. 2009;111(1):72–79. doi:10.1093/toxsci/kfp126
107. Lang BT, Yan Y, Dempsey RJ, Vemuganti R. Impaired neurogenesis in adult type-2 diabetic rats. Brain Res. 2009;1258:25–33. doi:10.1016/j.brainres.2008.12.026
108. Han H, Wu LM, Han MX, Yang WM, Wang YX, Fang ZH. Diabetes impairs spatial learning and memory and hippocampal neurogenesis via BDNF in rats with transient global ischemia. Brain Res Bull. 2016;124:269–277. doi:10.1016/j.brainresbull.2016.05.011
109. Ferreiro E, Lanzillo M, Canhoto D, et al. Chronic hyperglycemia impairs hippocampal neurogenesis and memory in an Alzheimer’s disease mouse model. Neurobiol Aging. 2020;92:98–113. doi:10.1016/j.neurobiolaging.2020.04.003
110. Bonds JA, Shetti A, Stephen TKL, Bonini MG, Minshall RD, Lazarov O. Deficits in hippocampal neurogenesis in obesity-dependent and -independent type-2 diabetes mellitus mouse models. Sci Rep. 2020;10(1):16368. doi:10.1038/s41598-020-73401-9
111. Bonds JA, Shetti A, Bheri A, et al. Depletion of caveolin-1 in type 2 diabetes model induces Alzheimer’s disease pathology precursors. J Neurosci. 2019;39(43):8576–8583. doi:10.1523/JNEUROSCI.0730-19.2019
112. Li J, Tang Y, Cai D. IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol. 2012;14(10):999–1012. doi:10.1038/ncb2562
113. Wakabayashi T, Hidaka R, Fujimaki S, Asashima M, Kuwabara T. Diabetes impairs Wnt3 protein-induced neurogenesis in olfactory bulbs via glutamate transporter 1 inhibition. J Biol Chem. 2016;291(29):15196–15211. doi:10.1074/jbc.M115.672857
114. Li Y, Tweedie D, Mattson MP, Holloway HW, Greig NH. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J Neurochem. 2010;113(6):1621–1631. doi:10.1111/j.1471-4159.2010.06731.x
115. Luciani P, Deledda C, Benvenuti S, et al. Differentiating effects of the glucagon-like peptide-1 analogue exendin-4 in a human neuronal cell model. Cell Mol Life Sci. 2010;67(21):3711–3723. doi:10.1007/s00018-010-0398-3
116. Porter WD, Flatt PR, Hölscher C, Gault VA. Liraglutide improves hippocampal synaptic plasticity associated with increased expression of Mash1 in ob/ob mice. Int J Obes. 2013;37(5):678–684. doi:10.1038/ijo.2012.91
117. Oishi K, Watatani K, Itoh Y, et al. Selective induction of neocortical GABAergic neurons by the PDK1-Akt pathway through activation of Mash1. Proc Natl Acad Sci U S A. 2009;106(31):13064–13069. doi:10.1073/pnas.0808400106
118. Kamal A, Biessels GJ, Duis SE, Gispen WH. Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: interaction of diabetes and ageing. Diabetologia. 2000;43(4):500–506. doi:10.1007/s001250051335
119. Reisi P, Alaei H, Babri S, Sharifi MR, Mohaddes G, Soleimannejad E. Determination of the extracellular basal levels of glutamate and GABA at dentate gyrus of streptozotocin-induced diabetic rats. Pathophysiology. 2009;16(1):63–66. doi:10.1016/j.pathophys.2009.03.003
120. Yin H, Wang W, Yu W, et al. Changes in synaptic plasticity and glutamate receptors in type 2 diabetic KK-Ay mice. J Alzheimers Dis. 2017;57(4):1207–1220. doi:10.3233/JAD-160858
121. Yang J, Song Y, Wang H, et al. Insulin treatment prevents the increase in D-serine in hippocampal CA1 area of diabetic rats. Am J Alzheimers Dis Other Demen. 2015;30(2):201–208. doi:10.1177/1533317514545379
122. Valastro B, Cossette J, Lavoie N, Gagnon S, Trudeau F, Massicotte G. Up-regulation of glutamate receptors is associated with LTP defects in the early stages of diabetes mellitus. Diabetologia. 2002;45(5):642–650. doi:10.1007/s00125-002-0818-5
123. Stranahan AM, Norman ED, Lee K, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18(11):1085–1088. doi:10.1002/hipo.20470
124. Yang Y, Gao L. Celecoxib alleviates memory deficits by downregulation of COX-2 expression and upregulation of the BDNF-TrkB signaling pathway in a diabetic rat model. J Mol Neurosci. 2017;62(2):188–198. doi:10.1007/s12031-017-0922-0
125. Sacai H, Sasaki-Hamada S, Sugiyama A, et al. The impairment in spatial learning and hippocampal LTD induced through the PKA pathway in juvenile-onset diabetes rats are rescued by modulating NMDA receptor function. Neurosci Res. 2014;81-82:55–63. doi:10.1016/j.neures.2014.02.002
126. Pipatpiboon N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. PPARγ agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology. 2012;153(1):329–338. doi:10.1210/en.2011-1502
127. Yan W, Pang M, Yu Y, et al. The neuroprotection of liraglutide on diabetic cognitive deficits is associated with improved hippocampal synapses and inhibited neuronal apoptosis. Life Sci. 2019;231:116566. doi:10.1016/j.lfs.2019.116566
128. Zhong Y, Zhu Y, He T, Li W, Yan H, Miao Y. Rolipram-induced improvement of cognitive function correlates with changes in hippocampal CREB phosphorylation, BDNF and Arc protein levels. Neurosci Lett. 2016;610:171–176. doi:10.1016/j.neulet.2015.09.023
129. Sasaki-Hamada S, Hojo Y, Koyama H, Otsuka H, Oka J. Changes in hippocampal synaptic functions and protein expression in monosodium glutamate-treated obese mice during development of glucose intolerance. Eur J Neurosci. 2015;41(11):1393–1401. doi:10.1111/ejn.12891
130. Sasaki-Hamada S, Sacai H, Oka JI. Diabetes onset influences hippocampal synaptic plasticity in streptozotocin-treated rats. Neuroscience. 2012;227:293–304. doi:10.1016/j.neuroscience.2012.09.081
131. Tekkök S, Krnjević K. Diabetes mellitus preserves synaptic plasticity in hippocampal slices from middle-aged rats. Neuroscience. 1999;91(1):185–191. doi:10.1016/S0306-4522(98)00662-9
132. Youssef FF, Manswell S, Homeward L. Effect of acute changes in glucose concentration on neuronal activity and plasticity in the rat hippocampus. West Indian Med J. 2009;58(5):410–416.
133. Trudeau F, Gagnon S, Massicotte G. Hippocampal synaptic plasticity and glutamate receptor regulation: influences of diabetes mellitus. Eur J Pharmacol. 2004;490(1–3):177–186. doi:10.1016/j.ejphar.2004.02.055
134. Chabot C, Massicotte G, Milot M, Trudeau F, Gagné J. Impaired modulation of AMPA receptors by calcium-dependent processes in streptozotocin-induced diabetic rats. Brain Res. 1997;768(1–2):249–256. doi:10.1016/S0006-8993(97)00648-3
135. Nisticò R, Cavallucci V, Piccinin S, et al. Insulin receptor β-subunit haploinsufficiency impairs hippocampal late-phase LTP and recognition memory. Neuromolecular Med. 2012;14(4):262–269. doi:10.1007/s12017-012-8184-z
136. Martín ED, Sánchez-Perez A, Trejo JL, et al. IRS-2 deficiency impairs NMDA receptor-dependent long-term potentiation. Cereb Cortex. 2012;22(8):1717–1727.
137. Yao ZG, Liu Y, Zhang L, et al. Co-location of HDAC2 and insulin signaling components in the adult mouse hippocampus. Cell Mol Neurobiol. 2012;32(8):1337–1342. doi:10.1007/s10571-012-9859-6
138. Wang M, Yoon G, Song J, Jo J. Exendin-4 improves long-term potentiation and neuronal dendritic growth in vivo and in vitro obesity condition. Sci Rep. 2021;11(1):8326. doi:10.1038/s41598-021-87809-4
139. Ohtake N, Saito M, Eto M, Seki K. Exendin-4 promotes the membrane trafficking of the AMPA receptor GluR1 subunit and ADAM10 in the mouse neocortex. Regul Pept. 2014;190–191:1–11. doi:10.1016/j.regpep.2014.04.003
140. Porter DW, Kerr BD, Flatt PR, Holscher C, Gault VA. Four weeks administration of Liraglutide improves memory and learning as well as glycaemic control in mice with high fat dietary-induced obesity and insulin resistance. Diabetes Obes Metab. 2010;12(10):891–899. doi:10.1111/j.1463-1326.2010.01259.x
141. McClean PL, Hölscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014;76(Pt A):57–67. doi:10.1016/j.neuropharm.2013.08.005
142. Abbas T, Faivre E, Hölscher C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: interaction between type 2 diabetes and Alzheimer’s disease. Behav Brain Res. 2009;205(1):265–271. doi:10.1016/j.bbr.2009.06.035
143. Lennox R, Flatt PR, Gault VA. Lixisenatide improves recognition memory and exerts neuroprotective actions in high-fat fed mice. Peptides. 2014;61:38–47. doi:10.1016/j.peptides.2014.08.014
144. Gilman CP, Perry T, Furukawa K, Grieg NH, Egan JM, Mattson MP. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J Neurochem. 2003;87(5):1137–1144. doi:10.1046/j.1471-4159.2003.02073.x
145. Park SW, Mansur RB, Lee Y, et al. Liraglutide activates mTORC1 signaling and AMPA receptors in rat hippocampal neurons under toxic conditions. Front Neurosci. 2018;12:756. doi:10.3389/fnins.2018.00756
146. Zhang J, Chen C, Hua S, et al. An updated meta-analysis of cohort studies: diabetes and risk of Alzheimer’s disease. Diabetes Res Clin Pract. 2017;124:41–47. doi:10.1016/j.diabres.2016.10.024
147. Jayaraj RL, Azimullah S, Beiram R. Diabetes as a risk factor for Alzheimer’s disease in the Middle East and its shared pathological mediators. Saudi J Biol Sci. 2020;27(2):736–750. doi:10.1016/j.sjbs.2019.12.028
148. Ke YD, Delerue F, Gladbach A, Götz J, Ittner LM. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One. 2009;4(11):e7917. doi:10.1371/journal.pone.0007917
149. Kellar D, Craft S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19(9):758–766. doi:10.1016/S1474-4422(20)30231-3
150. Ciarmiello A, Giovannini E, Riondato M, et al. Longitudinal cognitive decline in mild cognitive impairment subjects with early amyloid-β neocortical deposition. Eur J Nucl Med Mol Imaging. 2019;46(10):2090–2098. doi:10.1007/s00259-019-04409-1
151. Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain. 2017;140(12):3286–3300. doi:10.1093/brain/awx243
152. Bonda DJ, Wang X, Lee HG, Smith MA, Perry G, Zhu X. Neuronal failure in Alzheimer’s disease: a view through the oxidative stress looking-glass. Neurosci Bull. 2014;30(2):243–252. doi:10.1007/s12264-013-1424-x
153. Ahmad W, Ijaz B, Shabbiri K, Ahmed F, Rehman S. Oxidative toxicity in diabetes and Alzheimer’s disease: mechanisms behind ROS/ RNS generation. J Biomed Sci. 2017;24(1):76. doi:10.1186/s12929-017-0379-z
154. Wang J, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem. 2006;96(3):825–832. doi:10.1111/j.1471-4159.2005.03615.x
155. Grimm A, Schmitt K, Eckert A. Advanced mitochondrial respiration assay for evaluation of mitochondrial dysfunction in Alzheimer’s disease. Methods Mol Biol. 2016;1303:171–183.
156. Gibson GE, Thakkar A. Interactions of mitochondria/metabolism and calcium regulation in Alzheimer’s disease: a calcinist point of view. Neurochem Res. 2017;42(6):1636–1648. doi:10.1007/s11064-017-2182-3
157. Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017;40(3):151–166. doi:10.1016/j.tins.2017.01.002
158. Flannery PJ, Trushina E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol Cell Neurosci. 2019;98:109–120. doi:10.1016/j.mcn.2019.06.009
159. Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(8):759–767. doi:10.1093/jnen/60.8.759
160. Pastorino L, Sun A, Lu PJ, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440(7083):528–534. doi:10.1038/nature04543
161. Corsetti V, Florenzano F, Atlante A, et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum Mol Genet. 2015;24(11):3058–3081. doi:10.1093/hmg/ddv059
162. Moreira PI, Santos MS, Moreno AM, Seiça R, Oliveira CR. Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes. 2003;52(6):1449–1456. doi:10.2337/diabetes.52.6.1449
163. Gerakis Y, Hetz C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018;285(6):995–1011. doi:10.1111/febs.14332
164. Zhang X, Tang S, Zhang Q, et al. Endoplasmic reticulum stress mediates JNK-dependent IRS-1 serine phosphorylation and results in Tau hyperphosphorylation in amyloid β oligomer-treated PC12 cells and primary neurons. Gene. 2016;587(2):183–193. doi:10.1016/j.gene.2016.05.018
165. de la Monte SM, Re E, Longato L, Tong M. Dysfunctional pro-ceramide, ER stress, and insulin/IGF signaling networks with progression of Alzheimer’s disease. J Alzheimers Dis. 2012;30 Suppl 2(02):S217–229. doi:10.3233/JAD-2012-111728
166. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–372. doi:10.1038/nrn3880
167. Takeda S, Sato N, Uchio-Yamada K, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107(15):7036–7041. doi:10.1073/pnas.1000645107
168. Knight EM, Martins IV, Gümüsgöz S, Allan SM, Lawrence CB. High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging. 2014;35(8):1821–1832. doi:10.1016/j.neurobiolaging.2014.02.010
169. Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener. 2011;6:85. doi:10.1186/1750-1326-6-85
170. Cereda E, Barichella M, Pedrolli C, et al. Diabetes and risk of Parkinson’s disease: a systematic review and meta-analysis. Diabetes Care. 2011;34(12):2614–2623. doi:10.2337/dc11-1584
171. Giuntini M, Baldacci F, Del Prete E, Bonuccelli U, Ceravolo R. Diabetes is associated with postural and cognitive domains in Parkinson’s disease. Results from a single-center study. Parkinsonism Relat Disord. 2014;20(6):671–672. doi:10.1016/j.parkreldis.2014.02.016
172. Morris JK, Bomhoff GL, Gorres BK, et al. Insulin resistance impairs nigrostriatal dopamine function. Exp Neurol. 2011;231(1):171–180. doi:10.1016/j.expneurol.2011.06.005
173. Morris JK, Bomhoff GL, Stanford JA, Geiger PC. Neurodegeneration in an animal model of Parkinson’s disease is exacerbated by a high-fat diet. Am J Physiol Regul Integr Comp Physiol. 2010;299(4):R1082–1090. doi:10.1152/ajpregu.00449.2010
174. Yang L, Wang H, Liu L, Xie A. The role of insulin/IGF-1/PI3K/Akt/GSK3β signaling in Parkinson’s disease dementia. Front Neurosci. 2018;12:73. doi:10.3389/fnins.2018.00073
175. Sharma S, Taliyan R. High fat diet feeding induced insulin resistance exacerbates 6-OHDA mediated neurotoxicity and behavioral abnormalities in rats. Behav Brain Res. 2018;351:17–23. doi:10.1016/j.bbr.2018.05.025
176. Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol. 2013;106–107:17–32. doi:10.1016/j.pneurobio.2013.04.004
177. Hong CT, Chen KY, Wang W, et al. Insulin resistance promotes Parkinson’s disease through aberrant expression of α-synuclein, mitochondrial dysfunction, and deregulation of the polo-like kinase 2 signaling. Cells. 2020;9(3):740. doi:10.3390/cells9030740
178. Su CJ, Shen Z, Cui RX, et al. Thioredoxin-interacting protein (TXNIP) regulates Parkin/PINK1-mediated mitophagy in dopaminergic neurons under high-glucose conditions: implications for molecular links between Parkinson’s disease and diabetes. Neurosci Bull. 2020;36(4):346–358. doi:10.1007/s12264-019-00459-5
179. Costa CAD, Manaa WE, Duplan E, Checler F. The endoplasmic reticulum stress/unfolded protein response and their contributions to Parkinson’s disease physiopathology. Cells. 2020;9(11):2495. doi:10.3390/cells9112495
180. Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–397. doi:10.1016/S1474-4422(09)70062-6
181. Wang L, Zhai YQ, Xu LL, et al. Metabolic inflammation exacerbates dopaminergic neuronal degeneration in response to acute MPTP challenge in type 2 diabetes mice. Exp Neurol. 2014;251:22–29. doi:10.1016/j.expneurol.2013.11.001
182. Marxreiter F, Regensburger M, Winkler J. Adult neurogenesis in Parkinson’s disease. Cell Mol Life Sci. 2013;70(3):459–473. doi:10.1007/s00018-012-1062-x
183. Perry TA, Greig NH. A new Alzheimer’s disease interventive strategy: GLP-1. Curr Drug Targets. 2004;5(6):565–571. doi:10.2174/1389450043345245
184. Yang Y, Ma D, Xu W, et al. Exendin-4 reduces tau hyperphosphorylation in type 2 diabetic rats via increasing brain insulin level. Mol Cell Neurosci. 2016;70:68–75. doi:10.1016/j.mcn.2015.10.005
185. Xu W, Yang Y, Yuan G, Zhu W, Ma D, Hu S. Exendin-4, a glucagon-like peptide-1 receptor agonist, reduces Alzheimer disease-associated tau hyperphosphorylation in the hippocampus of rats with type 2 diabetes. J Investig Med. 2015;63(2):267–272. doi:10.1097/JIM.0000000000000129
186. Qi L, Ke L, Liu X, et al. Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3β pathway in an amyloid β protein induced Alzheimer disease mouse model. Eur J Pharmacol. 2016;783:23–32. doi:10.1016/j.ejphar.2016.04.052
187. Qi L, Chen Z, Wang Y, et al. Subcutaneous liraglutide ameliorates methylglyoxal-induced Alzheimer-like tau pathology and cognitive impairment by modulating tau hyperphosphorylation and glycogen synthase kinase-3β. Am J Transl Res. 2017;9(2):247–260.
188. Chen S, An FM, Yin L, et al. Glucagon-like peptide-1 protects hippocampal neurons against advanced glycation end product-induced tau hyperphosphorylation. Neuroscience. 2014;256:137–146. doi:10.1016/j.neuroscience.2013.10.038
189. Xiong H, Zheng C, Wang J, et al. The neuroprotection of liraglutide on Alzheimer-like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in mice. J Alzheimers Dis. 2013;37(3):623–635. doi:10.3233/JAD-130584
190. Chen S, Sun J, Zhao G, et al. Liraglutide improves water Maze learning and memory performance while reduces hyperphosphorylation of tau and neurofilaments in APP/PS1/Tau triple transgenic mice. Neurochem Res. 2017;42(8):2326–2335. doi:10.1007/s11064-017-2250-8
191. Zhang Y, Xie JZ, Xu XY, et al. Liraglutide ameliorates hyperhomocysteinemia-induced alzheimer-like pathology and memory deficits in rats via multi-molecular targeting. Neurosci Bull. 2019;35(4):724–734. doi:10.1007/s12264-018-00336-7
192. Bu LL, Liu YQ, Shen Y, et al. Neuroprotection of exendin-4 by enhanced autophagy in a Parkinsonian rat model of α-synucleinopathy. Neurotherapeutics. 2021;18(2):962–978. doi:10.1007/s13311-021-01018-5
193. Ma D, Liu X, Liu J, et al. Long-term liraglutide ameliorates nigrostriatal impairment via regulating AMPK/PGC-1a signaling in diabetic mice. Brain Res. 2019;1714:126–132. doi:10.1016/j.brainres.2019.02.030
194. Mullins RJ, Mustapic M, Chia CW, et al. A pilot study of exenatide actions in Alzheimer’s disease. Curr Alzheimer Res. 2019;16(8):741–752. doi:10.2174/1567205016666190913155950
195. Watson KT, Wroolie TE, Tong G, et al. Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav Brain Res. 2019;356:271–278. doi:10.1016/j.bbr.2018.08.006
196. Vaccari C, Grotto D, Pereira TDV, de Camargo JLV, Lopes LC. GLP-1 and GIP receptor agonists in the treatment of Parkinson’s disease: translational systematic review and meta-analysis protocol of clinical and preclinical studies. PLoS One. 2021;16(8):e0255726. doi:10.1371/journal.pone.0255726
197. Erbil D, Eren CY, Demirel C, Küçüker MU, Solaroğlu I, Eser HY. GLP-1ʹs role in neuroprotection: a systematic review. Brain Inj. 2019;33(6):734–819. doi:10.1080/02699052.2019.1587000
198. Laurindo LF, Barbalho SM, Guiguer EL, et al. GLP-1a: going beyond traditional use. Int J Mol Sci. 2022;23(2):739. doi:10.3390/ijms23020739
199. Mulvaney CA, Duarte GS, Handley J, et al. GLP-1 receptor agonists for Parkinson’s disease. Cochrane Database Syst Rev. 2020;7(7):Cd012990. doi:10.1002/14651858.CD012990.pub2
200. Wang SY, Wu SL, Chen TC, Chuang CS. Antidiabetic agents for treatment of Parkinson’s disease: a meta-analysis. Int J Environ Res Public Health. 2020;17(13):4805.
201. Femminella GD, Frangou E, Love SB, et al. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: study protocol for a randomised controlled trial (ELAD study). Trials. 2019;20(1):191. doi:10.1186/s13063-019-3259-x
202. Egefjord L, Gejl M, Møller A, et al. Effects of liraglutide on neurodegeneration, blood flow and cognition in Alzheimer´s disease - protocol for a controlled, randomized double-blinded trial. Dan Med J. 2012;59(10):A4519.
203. Femminella GD, Edison P. Evaluation of neuroprotective effect of glucagon-like peptide 1 analogs using neuroimaging. Alzheimers Dement. 2014;10(1 Suppl):S55–61. doi:10.1016/j.jalz.2013.12.012
204. Duarte AI, Candeias E, Alves IN, et al. Liraglutide protects against brain amyloid-β(1-42) accumulation in female mice with early Alzheimer’s disease-like pathology by partially rescuing oxidative/nitrosative stress and inflammation. Int J Mol Sci. 2020;21(5):1746. doi:10.3390/ijms21051746
205. Holubová M, Hrubá L, Popelová A, et al. Liraglutide and a lipidized analog of prolactin-releasing peptide show neuroprotective effects in a mouse model of β-amyloid pathology. Neuropharmacology. 2019;144:377–387. doi:10.1016/j.neuropharm.2018.11.002
206. McClean PL, Jalewa J, Hölscher C. Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav Brain Res. 2015;293:96–106. doi:10.1016/j.bbr.2015.07.024
207. Hölscher C. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement. 2014;10(1 Suppl):S47–54. doi:10.1016/j.jalz.2013.12.009
208. Cai HY, Yang JT, Wang ZJ, et al. Lixisenatide reduces amyloid plaques, neurofibrillary tangles and neuroinflammation in an APP/PS1/tau mouse model of Alzheimer’s disease. Biochem Biophys Res Commun. 2018;495(1):1034–1040. doi:10.1016/j.bbrc.2017.11.114
209. Parthsarathy V, Hölscher C. Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PLoS One. 2013;8(3):e58784. doi:10.1371/journal.pone.0058784
210. Cai HY, Hölscher C, Yue XH, et al. Lixisenatide rescues spatial memory and synaptic plasticity from amyloid β protein-induced impairments in rats. Neuroscience. 2014;277:6–13. doi:10.1016/j.neuroscience.2014.02.022
211. Paladugu L, Gharaibeh A, Kolli N, et al. Liraglutide has anti-inflammatory and anti-amyloid properties in streptozotocin-induced and 5xFAD mouse models of Alzheimer’s disease. Int J Mol Sci. 2021;22(2):860. doi:10.3390/ijms22020860
212. Carranza-Naval MJ, Del Marco A, Hierro-Bujalance C, et al. Liraglutide reduces vascular damage, neuronal loss, and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Front Aging Neurosci. 2021;13:741923. doi:10.3389/fnagi.2021.741923
213. Li Y, Duffy KB, Ottinger MA, et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis. 2010;19(4):1205–1219. doi:10.3233/JAD-2010-1314
214. Hansen HH, Barkholt P, Fabricius K, et al. The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy. Brain Res. 2016;1634:158–170. doi:10.1016/j.brainres.2015.12.052
215. Zhang L, Zhang L, Li L, Hölscher C. Semaglutide is neuroprotective and reduces α-synuclein levels in the chronic MPTP mouse model of Parkinson’s disease. J Parkinsons Dis. 2019;9(1):157–171. doi:10.3233/JPD-181503
216. Lin TK, Lin KJ, Lin HY, et al. Glucagon-Like Peptide-1 Receptor Agonist Ameliorates 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) Neurotoxicity Through Enhancing Mitophagy Flux and Reducing α-Synuclein and Oxidative Stress. Front Mol Neurosci. 2021;14:697440. doi:10.3389/fnmol.2021.697440
217. Aksoy D, Solmaz V, Çavuşoğlu T, Meral A, Ateş U, Erbaş O. Neuroprotective effects of eexenatide in a rotenone-induced rat model of Parkinson’s disease. Am J Med Sci. 2017;354(3):319–324. doi:10.1016/j.amjms.2017.05.002
218. Elbassuoni EA, Ahmed RF. Mechanism of the neuroprotective effect of GLP-1 in a rat model of Parkinson’s with pre-existing diabetes. Neurochem Int. 2019;131:104583. doi:10.1016/j.neuint.2019.104583
219. Kim S, Moon M, Park S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J Endocrinol. 2009;202(3):431–439. doi:10.1677/JOE-09-0132
220. Bertilsson G, Patrone C, Zachrisson O, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J Neurosci Res. 2008;86(2):326–338. doi:10.1002/jnr.21483
221. Feng P, Zhang X, Li D, et al. Two novel dual GLP-1/GIP receptor agonists are neuroprotective in the MPTP mouse model of Parkinson’s disease. Neuropharmacology. 2018;133:385–394. doi:10.1016/j.neuropharm.2018.02.012
222. Gejl M, Gjedde A, Egefjord L, et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci. 2016;8:108. doi:10.3389/fnagi.2016.00108
223. Gejl M, Brock B, Egefjord L, Vang K, Rungby J, Gjedde A. Blood-brain glucose transfer in Alzheimer’s disease: effect of GLP-1 analog treatment. Sci Rep. 2017;7(1):17490. doi:10.1038/s41598-017-17718-y
224. Aviles-Olmos I, Dickson J, Kefalopoulou Z, et al. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest. 2013;123(6):2730–2736. doi:10.1172/JCI68295
225. Aviles-Olmos I, Dickson J, Kefalopoulou Z, et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J Parkinsons Dis. 2014;4(3):337–344. doi:10.3233/JPD-140364
226. Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet (London, England). 2017;390(10103):1664–1675. doi:10.1016/S0140-6736(17)31585-4
227. Athauda D, Maclagan K, Budnik N, et al. What effects might exenatide have on non-motor symptoms in Parkinson’s disease: a post hoc analysis. J Parkinsons Dis. 2018;8(2):247–258. doi:10.3233/JPD-181329
228. Athauda D, Gulyani S, Karnati HK, et al. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: a secondary analysis of the exenatide-PD trial. JAMA Neurol. 2019;76(4):420–429. doi:10.1001/jamaneurol.2018.4304
229. Athauda D, Maclagan K, Budnik N, et al. Post hoc analysis of the exenatide-PD trial-factors that predict response. Eur J Neurosci. 2019;49(3):410–421. doi:10.1111/ejn.14096
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.