")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
The role of erythropoietin stimulating agents in anemic patients with heart failure: solved and unresolved questions
Authors Palazzuoli A, Ruocco G, Pellegrini M, De Gori C, Del Castillo G, Giordano N, Nuti R
Received 29 January 2014
Accepted for publication 14 March 2014
Published 13 August 2014 Volume 2014:10 Pages 641—650
DOI https://doi.org/10.2147/TCRM.S61551
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Alberto Palazzuoli, Gaetano Ruocco, Marco Pellegrini, Carmelo De Gori, Gabriele Del Castillo, Nicola Giordano, Ranuccio Nuti
Department of Internal Medicine and Metabolic Diseases, Cardiology Section, Le Scotte Hospital, University of Siena, Siena, Italy
Abstract: Anemia is a common finding in congestive heart failure (CHF) and is associated with an increased mortality and morbidity. Several conditions can cause depression of erythroid progenitor cells: reduction of iron absorption and reuptake, decreased bone marrow activity, reduced endogenous erythropoietin production, and chronic inflammatory state. Anemia’s etiology in CHF is complex and partially understood; it involves several systems including impaired hemodynamic condition, reduced kidney and bone perfusion, increased inflammatory activity, and neurohormonal overdrive. The use of erythropoiesis stimulating agents (ESAs) such as erythropoietin and its derivatives is recently debated; the last interventional trial seems to demonstrate a neutral or negative effect in the active arm with darbepoetin treatment. The current data is opposite to many single blind studies and previous meta-analysis showing an improvement in quality of life, New York Heart Association class, and exercise tolerance using ESA therapy. These contrasting data raise several concerns regarding the target of hemoglobin levels needing intervention, the exact anemia classification and categorization, and the standardization of hematocrit cutoffs. Some cardiac and systemic conditions (ie, hypertension, atrial fibrillation, prothrombotic status) may predispose to adverse events, and ESA administration should be avoided. To prevent the negative effects, high-dosage and chronic administration should be avoided. Clarification of these items could probably identify patients that may benefit from additional iron or ESA treatment. In this review, we discuss the interventional trials made in anemic heart failure patients, the underlying mechanism of anemia in CHF, and the potential role of ESA in this setting.
Keywords: anemia, iron metabolism, heart failure
Introduction
In a recent US report, the first year mortality after a diagnosis of congestive heart failure (CHF) is about 20%, and only 25% survive 5 years after the first diagnosis.1 The estimated direct and indirect cost of CHF in the US is consistently growing. Anemia is a common condition associated with CHF, as with other unfavorable clinical conditions such as diabetes, malnutrition, chronic kidney disease (CKD), and advanced age. Anemia is related to a poor outcome, and it is an independent risk factor for more severe CHF, hospitalization, and death.2–10 Therefore, it is correlated with reduced quality of life and exercise tolerance, increased New York Heart Association (NYHA) class, and use of high diuretic dosage.
Anemia, regardless of its definition, is common in CHF, being found in 4% to 50% of cases, and its prevalence in CHF may be rising.9,10 The wide range of prevalence depends on several factors: the definition of anemia and its cutoff varies across global societies (World Health Organization versus National Kidney Foundation); different inclusion criteria in multicenter studies; and poor information about etiology and pathogenesis.10,11 Few studies detailed the specific causes of anemia in the heart failure (HF) setting; most gave different results regarding its etiology and described anemia as a chronic inflammatory condition without explaining the precise determinant. Because of the current confusion in the classification and definition of anemia, interventional trials reported different results, thus demonstrating a need to understand whether ESA treatment is dangerous, in which patients should it be avoided, and which treatment is the best therapeutic option for anemic CHF patients.
Underlying causes of anemia in CHF
The mechanisms of anemia development in patients with CHF have not been entirely elucidated, and several factors with different impact and timing may be simultaneously involved. Although anemia is common in patients with CHF, only few studies have investigated the topic of the causes of anemia in CHF. The principal determinants of anemia are to due to hemodynamic impairment with consequent water and salt retention, kidney dysfunction due to reduced renal flow, tubuloglomerular damage. The resultant reduction in erythropoietin (EPO) production, deranged bone metabolism, reduced nutritional absorption due to gastrointestinal edema, and systemic proinflammatory status. It is possible to distinguish two main pathophysiological mechanisms in acute and chronic HF. In acute status the prevalent mechanism could be recognized in the increase of intra- and extracellular plasma content due to hydrosaline retention and fluid overload (false anemia); whereas, in the chronic state, CKD and renal blood flow redistribution are the most prevalent mechanisms that leads to relative reduction in erythropoietin production.3
Iron deficiency
The prevalence of iron deficiency in patients with HF is variable, from 20% to 50%.12 In a recent study,13 43% of patients had either low serum iron (<8 μmol/L) or ferritin (<30 μg/L), but microcytic anemia was observed in only 6% of patients. In contrast, Nanas et al14 found depleted iron stores in the bone marrow of 73% of patients despite normal serum iron, ferritin, and EPO. In normal conditions, iron is absorbed by the enterocytes of the gastrointestinal mucosa from diet intake, and it is also recycled from red blood cells and moved from storage sites in the liver. Nearly all of the iron available for erythropoiesis originates from the recycling of senescent red blood cells carried out by macrophages and reticuloendothelial cells. Thereafter, iron is transported from the inside of the cells to the outside by the carrier protein ferroportin-1 and carried on by the iron-binding blood plasma glycoprotein transferrin.15–17 During HF stages, inflammatory cytokines, particularly hepcidin, cause a reduction in iron absorption by blocking ferroportin-1 in the basolateral membrane of enterocytes, with consequent inhibition of iron absorption in the bowel. In addition to this effect, hepcidin blocks ferroportin-1 in the macrophages and reticuloendothelial cells, preventing iron from being exported from the inside of the cells and consequently leads to a reduction in transferrin uptake.17,18 Intravenous (IV) iron therapy has been shown to increase serum hemoglobin (Hb) levels (from 11.2±0.7 to 12.6±1.2 g/dL, P=0.0007) as well as improve exercise tolerance, quality of life, and functional class.19 Vitamin B12 and folate deficiency do not have a significant role in the pathogenesis of anemia in patients with HF.
Chronic kidney disease
CKD is common in CHF, and it is associated with reduced production of EPO in the kidney. The renal damage is due mainly to hemodynamic and parenchymal derangements. Hemodynamic alteration consists of reduced renal blood flow and intrarenal blood flow redistribution that causes hypoxic renal damage; parenchymal injury is due to tubulointerstitial fibrosis, tubular loss, and glomerulosclerosis.
The adverse liaison linking heart failure with anemic status is the impaired endogenous EPO synthesis and tissue resistance.20–22 EPO is a glycoprotein hormone produced primarily in the kidney by specialized peritubular fibroblasts. It regulates erythroid cell proliferation in the bone marrow in response to tissue hypoxia. The primary stimulus for EPO production is reduced oxygen tension that induces the transcription of the EPO gene. This in turn stimulates erythroid cell proliferation and differentiation. Oxygen delivery into the kidney is determined by renal blood flow, hematocrit, and the PO2 of the Hb oxygen-dissociation curve. Conversely, oxygen consumption is determined by proximal tubular sodium reabsorption and the glomerular filtration rate. Both hemodynamic and parenchymal distortion may contribute to a reduction in EPO production although recruited endogenous levels do not explain the degree of anemia in HF patients.23
Inflammatory status
Inflammatory cytokines elaborated in CHF, such as tumor necrosis factor alpha and interleukin-6, can cause anemia by reducing EPO production in the kidney and its activity in the bone marrow and by eliciting EPO resistance at the peripheral tissue level.24 All these occurrences are mediated by a specific peptide produced by the liver acting as in the erythroid metabolism as in the iron absorption and reuptake: hepcidin. It proceeds by blunting EPO and erythroid progenitor cell production at bone level; therefore, it raises tissue EPO resistance by reducing EPO gene expression, perturbing EPO signal transduction, and downregulation of EPO receptors.25 As previously reported, hepcidine inhibits the protein ferroportin-1, leading to a reduction in gastrointestinal iron absorption as in iron release from its storage (macrophages and reticuloendothelial cells).17 This results in low serum iron and decreased delivery of iron to the bone marrow, and thus iron deficiency anemia (the so called functional iron deficiency anemia). This inflammatory-induced anemia is probably the main cause of anemia in CHF and the anemia of chronic disease.17,24–26
Use of angiotensin converting enzyme inhibitors and beta blockers
Since angiotensin is a stimulator of EPO production and erythropoiesis, angiotensin converting enzyme inhibitors and beta blockers can cause reduced production of EPO and reduced activity of EPO in the bone marrow. Angiotensin converting enzyme inhibitors also increase the levels of erythropoietic inhibitors in the blood, further inhibiting erythropoiesis. Peritubular cells producing endogenous EPO receive sympathetic innervations; therefore, a specific subunit beta 2 receptor shares similar chemical heterodimeric characteristics with the EPO receptors. These two conditions together could explain the reduced EPO production and resistance in patients undergoing beta blocker therapy.27,28
Hemodilution
It has been suggested that the anemia in CHF is secondary, in many cases, to hemodilution.12,29,30 This has recently been further investigated.31 It was found that a red cell deficit was present in 88% of CHF patients with anemia and diastolic CHF and in 59% of those with systolic CHF. All systolic CHF patients and 71% of the diastolic CHF patients also had expanded plasma volume. This study suggests that there is a true red cell deficit in the majority of anemic CHF patients.
Gastrointestinal absorption and bleeding
Gastrointestinal absorption and bleeding due to aspirin, warfarin-like agents, malignant tumors, polyps, esophagitis, or reduced iron absorption resulting from atrophic gastritis are common causes of anemia in HF. Therefore, during refractory HF stages characterized by fluid overload and redistribution, with an increase in abdominal fluid component and pressure, there is abnormal bowel absorption due to mucosae edema, venous congestion, and reduced lymphatic drainage (Figure 1).32
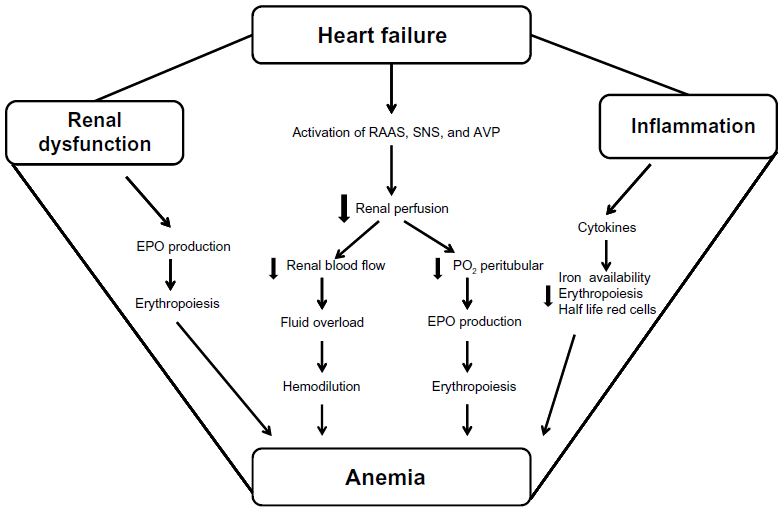 | Figure 1 Relationship among anemia renal dysfunction and heart failure: possible mechanisms of reduced and blunted erythropoietin production. |
Correction of anemia in CHF
The strong association of anemia with poor clinical status and high mortality rates led to increasing exploration of the treatment of anemia as a therapeutic target in HF patients. Only three options are currently available to increase Hb: blood transfusion, iron infusion, and erythropoiesis stimulating agent (ESA) administration. Although blood transfusion can be used as an acute therapy, it is associated with many risks and provides only temporary benefits.33 According to the American guidelines,33 blood transfusion is recommended for Hb levels in the range from 6–8 g/dL, thus the clinical utility of hemotransfusion for higher Hb levels is not well defined. However, blood transfusion is associated with several adverse effects such as suppression of the immune system, increased infection risk, iron overload, and sensitization to human leukocyte antigens.34 For these reasons, transfusion should be avoided for long-term treatment and may be considered only in the acute phase in severe anemia status.
Iron supplementation therapy
Given the high prevalence of iron deficiency in patients with HF and the close association between iron deficiency and HF prognosis, the treatment of iron deficiency has gained increasing attention. Iron is essential not only for erythropoiesis but also for several enzyme-linked bioenergetic processes in skeletal muscles and in the Krebs cycle. However, iron therapy at high doses has severe side effects, such as a greater oxidative stress on the endothelial tissue and atherosclerosis progression. Moreover, iron supplementation may have potential side effects such as anaphylaxis, increased risk of developing bacteremia, and shock. Several observational and uncontrolled trials35–37 have shown a promising association between IV iron replacement and improved symptoms in HF patients. To date, there are only a few randomized, controlled trials evaluating the efficacy of iron supplementation alone, without ESA, on HF outcomes. The number of randomized, controlled trials evaluating the role of iron supplementation is exiguous, and none of them assessed hard clinical end points of HF hospitalization or death as primary end points. Three recent studies of IV iron administration in anemic CHF patients, two uncontrolled studies and a double-blind placebo-controlled study, have shown improved Hb, left ventricular ejection fraction (LVEF), NYHA class, quality of life, left ventricular ejection fraction (LVEF), NYHA class, quality of life, and exercise capacity reducing BNP, C-reactive protein (CRP) and hospitalization. Another small, randomized controlled trial, the Ferric Iron Sucrose in Heart Failure Trial,13 evaluated the effect of 16 weeks of IV iron sucrose in patients with iron deficiency (ferritin G 100 ng/mL or ferritin 100–300 ng/mL with transferrin saturation 20%) and found improvement in functional capacity and symptoms. In the largest controlled study, the Ferric Carboxymaltose Assessment In Patients With Iron Deficiency And Chronic Heart Failure With And Without Anemia Trial,38 which recruited 459 patients with HF and iron deficiency (defined as ferritin G 100 ng/mL, or between 100–299 ng/mL and transferrin saturation <20%), the effect of IV ferric carboxymaltose or saline was evaluated. The active arm demonstrated a significant improvement in symptoms and NYHA class, although mortality rate did not differ between treatment and placebo groups. Oral iron is inexpensive and effective, but compliance is often limited by significant gastrointestinal side effects; up to 70% of patients were not taking oral iron as prescribed in clinical practice. In addition, various malabsorptive states such as decreased bowel drainage, CKD, and fluid redistribution may decrease a patient’s ability to absorb oral iron. One such trial, Iron Supplementation in Heart Failure Patients With Anemia, is now in progress, and it compares IV iron, oral iron supplementation, and placebo.39
ESAs in heart failure
Several small studies in HF have tested the effects of increasing Hb with ESAs. Such treatments have been reported in patients with CHF in whom the anemia has been treated with ESAs such as EPO or its derivative, the longer acting darbepoetin, alone or with the addition of either oral or IV iron. In the first study, Silverberg et al40 described the effect of epoetin alfa and IV iron in 26 patients with anemia and HF. An increase in Hb was associated with an improvement in NYHA functional class and LVEF and a decrease in diuretic dose and hospitalization. Similar results were reported 1 year later by the same authors in a randomized, open label trial in which EPO significantly decreased hospitalization and worsened NYHA class. Mancini et al41 conducted a patient-blind, randomized study in 23 HF patients with anemia. Patients were randomized to epoetin alfa plus oral iron and folate or placebo. After 3 months, EPO therapy was associated with significant increase in Hb values from 11.0±0.5 to 14.3±1.0 g/dL, with a concomitant increase in 6-minute walk distance and peak oxygen uptake (11.0±1.8 to 12.7±2.8 mL/minute per kilogram; P<0.05). The increases in Hb levels were linearly associated with the increase in peak oxygen uptake.
More recently, our group reported the effects of epoetin beta and oral iron on LV structure and function, pulmonary artery pressure, and BNP in 51 anemic HF patients with CKD.42 After 1 year, Hb increased, LV dimensions, volume, and mass decreased, and LVEF increased in the epoetin group, with no change in the control group. The pulmonary artery pressure and BNP blood levels also decreased as well as the need for hospitalization in patients treated with EPO.
The randomized, placebo-controlled trial carried out by Ponikowski et al43 in which 41 patients with HF anemia were treated with darbepoetin-alpha once every 2 weeks for 26 weeks showed an increase in Hb values with concomitant improvement in quality of life as evaluated by Patient’s Global Assessment; however, no differences were revealed regarding peak VO2, exercise duration, BNP, and renal function between treatment groups. Despite the beneficial effects shown in these trials, the sample sizes of the studies were also relatively small, preventing far-reaching applications. Moreover, none of the smaller randomized, controlled trials evaluated hard clinical outcomes such as all-cause mortality or HF hospitalization as primary end points, and they were limited by small sample size, limited duration, and lacked double-blind study design. On the other hand, in another single randomized, double-blind, placebo-controlled trial44 involving 165 HF patients with anemia, LVEF <40%, and Hb 9.0–12.5 g/dL, subcutaneous treatment with darbepoetin alpha every 2 weeks for 26 weeks at a starting weight-adjusted dose of 0.75 μg/kg (n=56) or a fixed dose of 50 μg (n=54) (placebo group n=55) gradually achieved and maintained a target Hb of 14.0±1.0 g/dL. The rate of Hb rise was equivalent between darbepoetin alpha weight-based and fixed dose groups. There were nonsignificant improvements in the combined darbepoetin alfa group versus placebo for 6-minute walk distance (P=0.074) and Patient’s Global Assessment score (P=0.057). There was a significant improvement in the Kansas City Cardiomyopathy Questionnaire total symptom score without change in NYHA class, LVEF, and the Minnesota Living With Heart Failure Questionnaire score. However, in the two larger multicenter double-blind, placebo-controlled studies using darbepoetin (Study of Anemia in Heart Failure Trial [STAMINA-HF] and Reduction of Events With Darbepoetin Alfa in Heart Failure [RED-HF] Trial),45,46 no significant improvement was found in either NYHA class, hospitalization, or death. In the STAMINA-HF Trial, 319 HF patients with LV systolic dysfunction (LVEF <40%) and Hb >9–12.5 g/dL were randomized to placebo or darbepoetin alpha every 2 weeks for 1 year (target Hb, 14.0±1.0 g/dL). The primary end point was a change from baseline to week 27 in treadmill exercise time. Secondary end points were a change from baseline to week 27 in NYHA class and quality of life. An additional, prespecified efficacy analysis included the time of death by any cause or first HF-related hospitalization by 1 year. At baseline, the median (interquartile range) Hb was 11.4 (10.9–12.0) g/dL. At week 27, darbepoetin alpha treatment increased median (interquartile range) Hb by 1.8 (1.1–2.5) g/dL. Despite this trend, darbepoetin treatment did not significantly improve exercise duration, NYHA class, or quality of life score compared with placebo. A difference, although nonsignificant, toward a lower risk of all-cause mortality and first HF hospitalization was observed in the darbepoetin arm versus the placebo (hazard ratio 0.68; 95% confidence interval 0.43–1.08; P=0.10). The incidence of other adverse events including development of hypertension and myocardial infarction was similar in the two groups.45
In the largest multicenter randomized, controlled trial (RED-HF) enrolling 2,278 patients with systolic HF (LVEF ≤40%, NYHA class ≥2) and anemia (Hb 9–12 g/dL), the darbepoetin alfa dose was adjusted to achieve a goal Hb of ≥13 g/dL. After a median follow up of 28 months, Hb increased from 11.2–13.0 g/dL in the active arm, and it remained unchanged in the placebo group. There was no significant difference between the darbepoetin alfa and placebo groups for the primary composite outcome of death from any cause or first HF hospitalization (50.7% versus 49.5%, respectively, with a hazard ratio of 1.01; 95% confidence interval 0.90–1.13; P=0.87). Therefore, no benefit was found regarding all other secondary outcomes including all-cause mortality, cardiovascular death, HF-related hospitalization, and coronary events. Despite these neutral effects of darbepoietin, an increased rate of thromboembolic events was seen in the treated patients compared with placebo (13.5% versus 10.0%; P=0.01). In line with the above data, an increased risk of stroke in the darbepoetin alfa group has been revealed (5.4% versus 3.9%) (Table 1).46
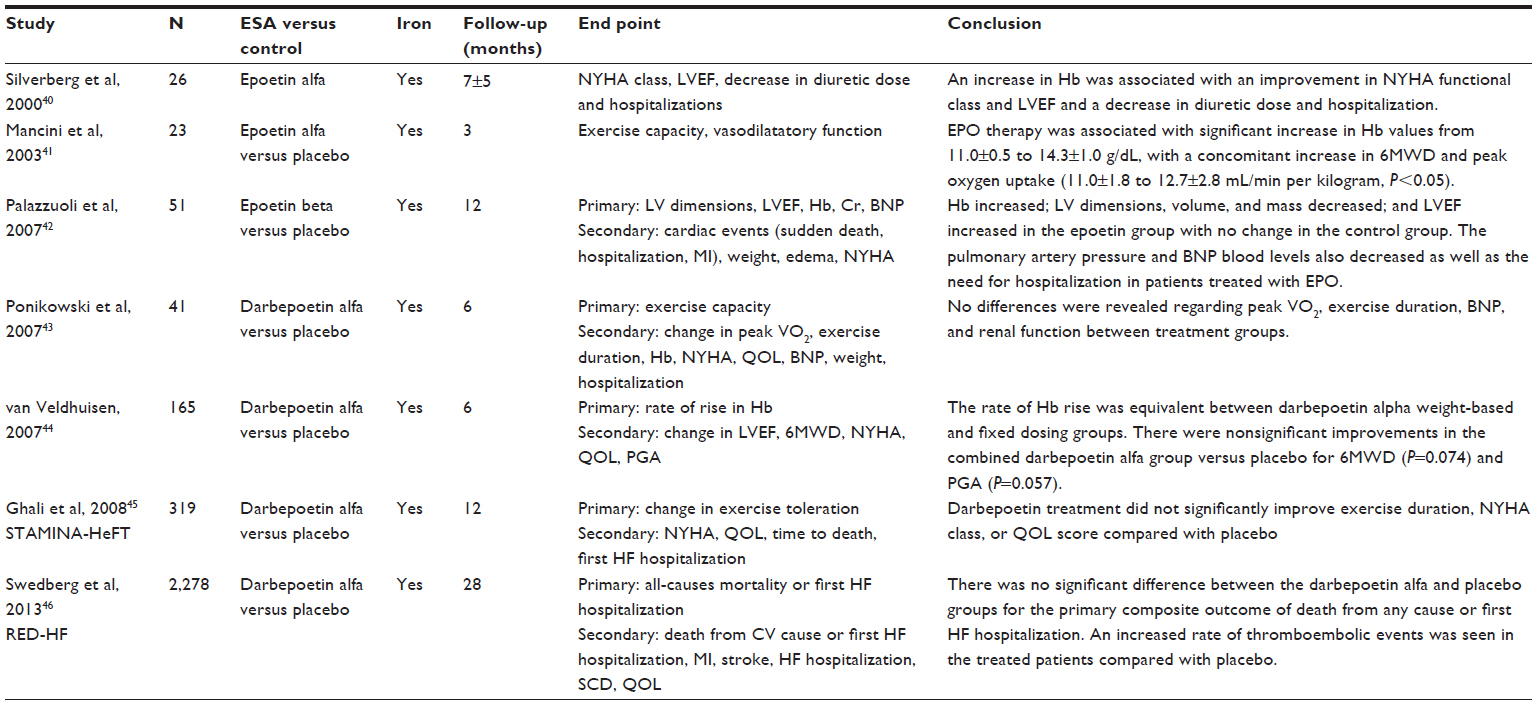 | Table 1 Randomized, controlled trials on ESA administration in patients with anemia and heart failure |
Two previous meta-analysis, not including RED-HF data, demonstrated that patients treated with ESAs had a significant increase in quality of life, exercise tolerance, and NYHA class associated with lower risk of CHF hospitalization. Significant differences in mortality and risk of adverse events were found between the ESA and placebo groups, with a reduction of mortality risk and adverse events in the ESA-treated patients.47,48 A further meta-analysis including RED-HF data is mandatory to better define beneficial/adverse effects ratio by ESA administration. The differences among all of these reports may be partially explained by use of different protocols, various ESA administration and dosage, and different degrees of anemia.
Potential risks of ESA therapy
Correcting anemia in HF is a tradeoff between the favorable effects of improving oxygen delivery and the unfavorable effects of increasing viscosity, blood pressure, and thrombocytosis. In this sense, many studies have shown that ESA therapy is associated with an increased prevalence and severity of cardiovascular events, including myocardial infarction, stroke, and venous thromboembolism.49–53
EPO can potentially increase the platelet count.51,54–56 This action can be related both to an indirect effect of EPO leading to iron deficiency and a direct effect of the hormone49,51 that enhances the action of thrombopoietin, and thus increases platelet activation. Correction of iron deficiency with IV iron in EPO-treated dialysis patients reduces the platelet count significantly,57 thus thrombocytosis may be one of the missing links in causing the increased incidence of cardiovascular events associated with EPO in CKD and CHF. Iron deficiency also increases oxidative stress.58 These effects may be due to an increase in platelet aggregation by effects on proteins C and S as well as endothelial activation.
A recent meta-analysis has shown that EPO therapy is associated with a trend of increased risk of venous thrombosis and cerebrovascular events in four studies in patients with Hb target levels of 12.5–15.0 g/dL.59 However, no difference between ESA and control groups was shown for patients with lower Hb levels.59
Biological effects of ESA beyond erythropoiesis
Several studies have confirmed that ESAs decrease experimental infarct size, reduce hypoxic injury, prevent myocyte apoptosis, and mobilize endothelial progenitor cells (EPCs) independently from Hb levels. These beneficial effects of ESAs appear unrelated to their effect on Hb increase, and they occur even when administered at low dosage. The usefulness of ESAs in CHF has been shown in animal studies, where their use in CHF after a myocardial infarction or after cardiac damage from other causes, with or without improving the Hb, improved endothelial dysfunction, increased neovascularization of heart muscle, reduced apoptosis of the cardiomyocytes, oxidative stress, inflammation, fibrosis, hypoxic damage, and prevented functional impairment of the heart.60–62 At least part of these effects is related to the increase in the number and activity of endothelial and vascular growth factors due to raised activity of EPCs from the bone marrow.60–62 In CHF,63 as in CKD,64 there is a relative reduction in the number of capillaries to the number of myocytes; therefore, neovascularization and other processes activated by ESA may improve the microvessel number into myocardial tissue. In an animal study, Westenbrink et al12 observed that neovascularization induced by EPO in HF following myocardial infarction was induced by several mechanisms including EPC recruitment from the bone marrow and increased myocardial expression of vascular endothelial growth factor (Figure 2). That study showed, in particular, that EPO’s effects appear 3 weeks after myocardial infarction and increased the mobilization, myocardial homing, and vascular incorporation of EPCs.
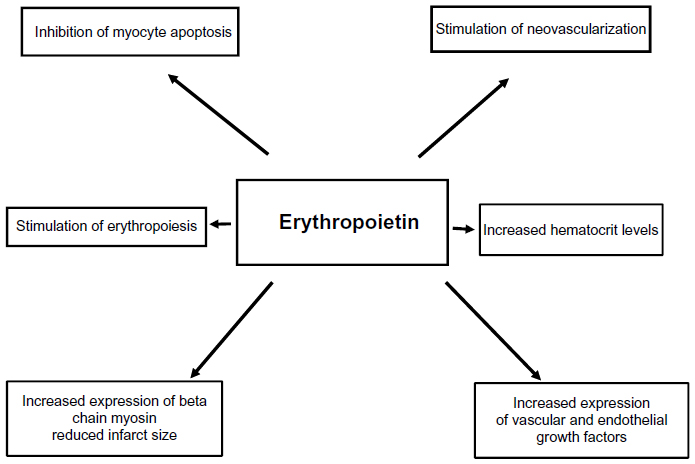 | Figure 2 ESA effects beyond erythropietic capacity: potential endothelial and intracellular signaling effects. |
Open questions and future directions
In HF patients with moderate-to-severe anemia (Hb <11 g/dL), current guidelines of the US Food and Drug Administration and of the National Kidney Foundation65,66 state that dosing should be individualized to achieve and maintain Hb levels within the range of 10–12 g/dL. Currently there are no clinical data supporting the use of ESAs to improve the outcome in HF patients. The 2012 guidelines of the European Society of Cardiology67 underlined that anemia is a frequently associated condition in HF patients; however, they do not indicate the best modality correction. Due to the absence of definitive evidence, ESAs administration effects are unknown. The same guidelines focused attention on the importance of iron deficiency and its correction for quality of life and NYHA class improvement.67 However, among traditional therapies, the use of ESAs alone or together with iron represents the only potential option. Given the possible risks of worsening hypertension and thrombosis observed in the last study, the recent guidelines of American College of Physicians68 conclude against the use of ESAs in patients with mild-to-moderate anemia. On the other hand, IV iron administration appears to improve quality of life and exercise tolerance; for these reasons, it is accepted for reducing anemic status. Nevertheless, we know from epidemiological data that iron deficiency in HF, when defined as a serum ferritin below 100 g/L or a serum ferritin 100–299 g/L and transferrin <20%, is found in about half of the CHF patients, so less than 50% of patients could potentially benefit from this treatment. Thus, an enormous question emerges from the epidemiological data: how can we treat the other residual patients? In our opinion, the “end” of ESA administration in anemic HF patients is long to be realized for several reasons: 1) we do not have other treatment options, particularly in patients without iron metabolism disorder; 2) most patients with anemic HF have a concomitant renal dysfunction, and the use of ESAs is commonly accepted in the nephrological setting, even if a recent trial showed negative results (Trial to Reduce cardiovascular Events with Aranesp Therapy [TREAT]);69 3) the starting Hb level in larger trials was 11.5 g/dL and is not a correct target to begin ESA treatment; in the same studies 30% of the population had atrial fibrillation and 70% had hypertension, both conditions that predispose to embolic events; 4) the mean dosage of darbepoetin used was high, particularly considering the baseline hematocrit values; 5) in different studies, ESAs were sometimes administered together with iron in different dosages and different modalities – some oral and some IV – and this represent a great bias; 6) reports using low ESA dosage confirmed the more positive results. The latter point is in line with experimental studies in which low ESA administration was related to increased vascular and endothelial grow factors, increased expression of heavy chain of myoglobin as well as nitric oxide liberation. All these doubts and concerns deserve further investigations to address the optimal Hb intervention target, the clinical characteristic of patients, the comorbidities in which ESAs should be avoided, the etiology of anemia, and better classification of iron dysfunction. When the literature responds to all these points, we should obtain a definitive answer about the role of ESAs and their tailored dosage and rate of administration in HF with anemia. At present, we can assert that the use of high ESA dosage is potentially dangerous in subjects with a definite embolic risk. The use of the same agents at low dosage in patients with low embolic profile could still have a rationale.
Conclusion
The use of ESAs has recently demonstrated negative effects in anemic patients with HF. To prevent the negative effects, high dosage and chronic administration should be avoided. Nevertheless, few studies investigated the specific causes of anemia, providing different results regarding its etiology. Understanding the precise causes of anemia, acquiring greater knowledge of erythroid metabolism dysfunction, and elucidating the optimal Hb intervention target would facilitate more precise answers regarding the exact role of ESAs in this setting.
Disclosure
The authors report no conflicts of interest in this work.
References
Lloyd-Jones D, Adams R, Carnethon M, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119(3):480–486. | |
Silverberg DS, Wexler D, Iaina A, Schwartz D. The role of correction of anaemia in patients with congestive heart failure: a short review. Eur J Heart Fail. 2008;10(9):819–823. | |
Anand IS. Anemia and chronic heart failure. J Am Coll Cardiol. 2008; 52(7):501–511. | |
Mitchell JE. Emerging role of anemia in heart failure. Am J Cardiol. 2007;99(6B):15D–20D. | |
Silverberg DS, Wexler D, Iaina A, Schwartz D. The interaction between heart failure and other heart diseases, renal failure, and anemia. Semin Nephrol. 2006;26(4):296–306. | |
Tang YD, Katz SD. Anemia in chronic heart failure: prevalence, etiology, clinical correlates, and treatment options. Circulation. 2006;113(20):2454–2461. | |
Lindenfeld JA. Prevalence of anemia and the effects on mortality in patients with heart failure. Am Heart J. 2005;149(3):391–401. | |
Groenveld HF, Januzzi JL, Damman K, et al. Anemia and mortality in heart failure patients: a systematic review and meta-analysis. J Am Coll Cardiol. 2008;52(10):818–827. | |
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Secular trends in renal dysfunction and outcomes in hospitalized heart failure patients. J Card Fail. 2006;12(4):257–262. | |
Dunlay SM, Weston SA, Redfield MM, Killian JM, Roger VL. Anemia and heart failure: a community study. Am J Med. 2008;121(8):726–732. | |
Nemeth E. Iron regulation and erythropoiesis. Curr Opin Hematol. 2008;15(3):169–175. | |
Westenbrink BD, Visser FW, Voors AA, et al. Anaemia in chronic heart failure is not only related to impaired renal perfusion and blunted erythropoietin production, but to fluid retention as well. Eur Heart J. 2007;28(2):166–171. | |
Okonko DO, Grzeslo A, Witkowski T, et al. Effect of intravenous iron sucrose on exercise tolerance in anemic and nonanemic patients with symptomatic chronic heart failure and iron deficiency FERRIC-HF: a randomized, controlled, observer-blinded trial. J Am Coll Cardiol. 2008;51(2):103–112. | |
Nanas JN, Matsouka C, Karageorgopoulos D, et al. Etiology of anemia in patients with advanced heart failure. J Am Coll Cardiol. 2006;48(12):2485–2489. | |
Westenbrink BD, Voors AA, van Veldhuisen DJ. Is anemia in chronic heart failure caused by iron deficiency? J Am Coll Cardiol. 2007;49(23):2301–2302; author reply 2302. | |
Weiss G. Iron metabolism in the anemia of chronic disease. Biochim Biophys Acta 2009;1790(7):682–693. | |
Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005;352(10):1011–1023. | |
Jankowska EA, von Haehling S, Anker SD, Macdougall IC, Ponikowski P. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. Eur Heart J. 2013;34:816–829. | |
Bolger AP, Bartlett FR, Penston HS, et al. Intravenous iron alone for the treatment of anemia in patients with chronic heart failure. J Am Coll Cardiol. 2006;48(6):1225–1227. | |
Volpe M, Tritto C, Testa U, et al. Blood levels of erythropoietin in congestive heart failure and correlation with clinical, hemodynamic, and hormonal profiles. Am J Cardiol. 1994;74(5):468–473. | |
van der Meer P, Voors AA, Lipsic E, Smilde TD, van Gilst WH, van Veldhuisen DJ. Prognostic value of plasma erythropoietin on mortality in patients with chronic heart failure. J Am Coll Cardiol. 2004;44(1):63–67. | |
George J, Patal S, Wexler D, et al. Circulating erythropoietin levels and prognosis in patients with congestive heart failure: comparison with neurohormonal and inflammatory markers. Arch Intern Med. 2005;165(11):1304–1309. | |
Palazzuoli A, Antonelli G, Nuti R. Anemia in cardio-renal syndrome: clinical impact and pathophysiologic mechanisms. Heart Fail Rev. 2011;16(6):603–607. | |
Weiss G, Meusburger E, Radacher G, Garimorth K, Neyer U, Mayer G. Effect of iron treatment on circulating cytokine levels in ESRD patients receiving recombinant human erythropoietin. Kidney Int. 2003;64(2):572–578. | |
Macdougall IC, Cooper AC. Erythropoietin resistance: the role of inflammation and pro-inflammatory cytokines. Nephrol Dial Transplant. 2002;17(Suppl 11):39–43. | |
Rauchhaus M, Doehner W, Francis DP, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102(25):3060–3067. | |
Abdulla J, Abildstrom SZ, Christensen E, Kober L, Torp-Pedersen C. A meta-analysis of the effect of angiotensin-converting enzyme inhibitors on functional capacity in patients with symptomatic left ventricular systolic dysfunction. Eur J Heart Fail. 2004;6(7):927–935. | |
Australia/New Zealand Heart Failure Research Collaborative Groupa (no authors listed). Randomized placebo-controlled trial of carvedilol in patients with congestive heart failure due to ischemic heart disease. Australia/New Zealand Heart Failure Research Collaborative Group. Lancet. 1997;349(9049):374–380. | |
Androne AS, Katz SD, Lund L, et al. Hemodilution is common in patients with advanced heart failure. Circulation. 2003;107(2):226–229. | |
Adlbrecht C, Kommata S, Hülsmann M, et al. Chronic heart failure leads to an expanded plasma volume and pseudoanaemia, but does not lead to a reduction in the body’s red cell volume. Eur Heart J. 2008;29(19):2343–2350. | |
Abramov D, Cohen RS, Katz SD, Mancini D, Maurer MS. Comparison of blood volume characteristics in anemic patients with low versus preserved left ventricular ejection fractions. Am J Cardiol. 2008;102(8):1069–1072. | |
Hutchinson C, Geissler CA, Powell JJ, Bomford A. Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. Gut. 2007;56(9):1291–1295. | |
Practice guidelines for blood component therapy: a report by the American Society of Anesthesiologists Task Force on Blood Component Therapy. Anesthesiology. 1996;84(3):732–747. | |
Goodnough LT, Brecher ME, Kanter MH, AuBuchon JP. Transfusion medicine. First of two parts – blood transfusion. N Engl J Med. 1999; 340(6):438–447. | |
Toblli JE, Lombraña A, Duarte P, Di Gennaro F. Intravenous iron reduces NT-pro-brain natriuretic peptide in anemic patients with chronic heart failure and renal insufficiency. J Am Coll Cardiol. 2007;50:1657–1665. | |
Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Rerritin (DRIVE) Study. J Am Soc Nephrol. 2007;18(3):975–984. | |
Kapoian T, O’Mara NB, Singh AK, et al. Ferric gluconate reduces epoetin requirements in hemodialysis patients with elevated ferritin. J Am Soc Nephrol. 2008;19(2):372–379. | |
Anker SD, Comin Colet J, Filippatos G, et al; FAIR-HF Trial Investigators. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med. 2009;361(25):2436–2448. | |
Beck-da-Silva L, Piardi D, Soder S, et al. IRON-HF study: a randomized trial to assess the effects of iron in heart failure patients with anemia. Int J Cardiol. 2013;168(4):3439–3442. | |
Silverberg DS, Wexler D, Blum M, et al. The use of subcutaneous erythropoietin and intravenous iron for the treatment of the anemia of severe, resistant congestive heart failure improves cardiac and renal function and functional cardiac class, and markedly reduces hospitalizations. J Am Coll Cardiol. 2000;35(7):1737–1744. | |
Mancini DM, Katz SD, Lang CC, LaManca J, Hudaihed A, Androne AS. Effect of erythropoietin on exercise capacity in patients with moderate to severe chronic heart failure. Circulation. 2003;107(2):294–299. | |
Palazzuoli A, Silverberg DS, Iovine F, et al. Effects of beta-erythropoietin treatment on left ventricular remodeling, systolic function, and B-type natriuretic peptide levels in patients with the cardiorenal anemia syndrome. Am Heart J. 2007;154(4):645.e9–645.15. | |
Ponikowski P, Anker SD, Szachniewicz J, et al. Effect of darbepoetin alpha on exercise tolerance in anemic patients with symptomatic chronic heart failure: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2007;49(7):753–762. | |
van Veldhuisen DJ, Dickstein K, Cohen-Solal A, et al. Randomized, double-blind, placebo-controlled study to evaluate the effect of two dosing regimens of darbepoetin alfa in patients with heart failure and anaemia. Eur Heart J. 2007;28(18):2208–2216. | |
Ghali JK, Anand IS, Abraham WT, et al; Study of Anemia in Heart Failure Trial (STAMINA-HeFT) Group. Randomized double-blind trial of darbepoetin alfa in patients with symptomatic heart failure and anemia. Circulation. 2008;117(4):526–535. | |
Swedberg K, Young JB, Anand IS, et al; RED-HF Committees; RED-HF Investigators. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med. 2013;368(13):1210–1219. | |
van der Meer P, Groenveld HF, Januzzi JL Jr, Van Veldhuisen DJ. Erythropoietin treatment in patients with chronic heart failure: a meta-analysis. Heart. 2009;95(16):1309–1314. | |
Ngo K, Kotecha D, Walters JA, et al. Erythropoiesis-stimulating agents for anemia in chronic heart failure patients. Cochrane Database Syst Rev. 2010;(1):CD007613. | |
Turner SJ, Ketch TR, Gandhi SK, Sane DC. Routine hematologic clinical tests as prognostic markers in patients with acute coronary syndromes. Am Heart J. 2008;155(5):806–816. | |
Keung YK, Owen J. Iron deficiency and thrombosis: literature review. Clin Appl Thromb Hemost. 2004;10(4):387–391. | |
Vaziri ND. Anemia and anemia correction: surrogate markers or causes of morbidity in chronic kidney disease? Nat Clin Pract Nephrol. 2008;4(8):436–445. | |
Franchini M, Targher G, Montagnana M, Lippi G. Iron and thrombosis. Ann Hematol. 2008;87(3):167–173. | |
Davì G, Patrono C. Platelet activation and atherosclerosis. N Engl J Med. 2007;357(24):2482–2494. | |
Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Int Med. 1989;111(12):992–1000. | |
Beguin Y, Loo M, R’Zik S, et al. Effect of recombinant human erythropoietin on platelets in patients with anemia of renal failure: correlation of platelet count with erythropoietic activity and iron parameters. Eur J Haematol. 1994;53(5):265–270. | |
Stohlawetz PJ, Dzirlo L, Hergovich N, et al. Effects of erythropoietin on platelet reactivity and thrombopoiesis in humans. Blood. 2000;95(9):2983–2989. | |
Dahl NV, Henry DH, Coyne DW. Thrombosis with erythropoietic stimulating agents-does iron-deficient erythropoiesis play a role? Semin Dial. 2008;21(3):210–211. | |
Nagababu E, Gulyani S, Earley CJ, Cutler RG, Mattson MP, Rifkind JM. Iron-deficiency anaemia enhances red blood cell oxidative stress. Free Radic Res. 2008;42(9):824–829. | |
Rozen-Zvi B, Gafter-Gvili A, Paul M, Leibovici L, Shpilberg O, Gafter U. Intravenous versus oral iron supplementation for the treatment of anemia in CKD: systematic review and meta-analysis. Am J Kidney Dis. 2008;52(5):897–906. | |
Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005;293(1):90–95. | |
Arcasoy MO. The non-haematopoietic biological effects of erythropoietin. Br J Haematol. 2008;141(1):14–31. | |
Rastogi S, Imai M, Sharov VG, Mishra S, Sabbah HN. Darbepoetin-alpha prevents progressive left ventricular dysfunction and remodeling in nonanemic dogs with heart failure. Am J Physiol Heart Circ Physiol. 2008;295(6):H2475–H2482. | |
van der Meer P, Lipsic E, Henning RH, et al. Erythropoietin induces neovascularization and improves cardiac function in rats with heart failure after myocardial infarction. J Am Coll Cardiol. 2005;46(1):125–133. | |
Amann K, Tyralla K. Cardiovascular changes in chronic renal failure – pathogenesis and therapy. Clin Nephrol. 2002;58(Suppl 1):S62–S72. | |
US Food and Drug Administration. Information for Healthcare Professionals. Erythropoiesis Stimulating Agents (ESA). FDA Alert [webpage on the Internet]. Silver Spring, MD: US Food and Drug Administration; 2007. Available from: http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm126481.htm. Accessed July 10, 2014. | |
KDOQI; National Kidney Foundation. KDOQI Clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis. 2006;47(Suppl 5):S11–S145. | |
McMurray JJ, Adamopoulos S, Anker SD, et al; Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology; ESC Committee for Practice Guidelines. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14(8):803–869. | |
Qaseem A, Humphrey LL, Fitterman N, Starkey M, Shekelle P; Clinical Guidelines Committee of the American College of Physicians. Treatment of anemia in patients with heart disease: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2013;159(11):770–779. | |
Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med. 2010;363(12):1146–1155. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.