")
Back to Journals » OncoTargets and Therapy » Volume 12
The Role Of Circulating Tumor DNA In Therapeutic Resistance
Authors Xu C, Cao H, Shi C, Feng J
Received 6 August 2019
Accepted for publication 9 October 2019
Published 8 November 2019 Volume 2019:12 Pages 9459—9471
DOI https://doi.org/10.2147/OTT.S226202
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Chenxin Xu,1,* Haixia Cao,2,* Chen Shi,1 Jifeng Feng1
1The Affiliated Cancer Hospital of Nanjing Medical University, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research, Nanjing, Jiangsu Province, People’s Republic of China; 2Research Center for Clinical Oncology, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, Jiangsu Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jifeng Feng
The Affiliated Cancer Hospital of Nanjing Medical University, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research, 42 Baiziting, Nanjing, Jiangsu Province, People’s Republic of China
Tel +86 02583283584
Fax +86 02583283588
Email [email protected]
Abstract: The application of precision medicine in cancer treatment has partly succeeded in reducing the side effects of unnecessary chemotherapeutics and in improving the survival rate of patients. However, with the long-term use of therapy, the dynamically changing intratumoral and intertumoral heterogeneity eventually gives rise to therapeutic resistance. In recent years, a novel testing technology (termed liquid biopsy) using circulating tumor DNAs (ctDNAs) extracted from peripheral blood samples from patients with cancer has brought about new expectations to the medical community. Using ctDNAs, clinicians can trace the heterogeneity pattern to duly adjust individual therapy and prolong overall survival for patients with cancer. Technological advances in detecting and characterizing ctDNAs (eg, development of next-generation sequencing) have provided clinicians with a valuable tool for genotyping tumors individually and identifying genetic and epigenetic alterations of the entire tumor to capture mutations associated with therapeutic resistance.
Keywords: cancer, heterogeneity, drug resistance, targeted therapies, sequencing technologies
Introduction
Cancer is a leading cause of death worldwide in both developed and developing countries. Based on GLOBOCAN estimates, in 2012, approximately 14.1 million new cancer cases and 8.2 million cancer-induced deaths occurred globally.1 Cancer is a genetic disease characterized by years of progressive accumulation of genomic aberrations that are occasionally augmented by predisposing germline mutations.2 In 1976, Nowell proposed that this accumulation of mutations is caused by a process of diversification and selection of beneficial mutations promoting tumor cell proliferation and survival.3 This process also occurs during long-term targeted therapy, causing changing mutations within tumor cells and eventually inducing intratumoral and intertumoral heterogeneity. This reduces the efficacy of therapeutic regimens, a phenomenon referred to as development of therapeutic resistance. The identification of selected genomic aberrations that target clinically relevant signaling pathways in patients, especially those giving rise to drug tolerance, remains a fundamental challenge.4 Malignant tumors occasionally release fragments of nucleic acids into peripheral blood. Therefore, a noninvasive blood test detecting circulating tumor DNAs (ctDNAs) in the bloodstream (termed liquid biopsy) can inform clinicians regarding changes in the clonal composition of tumor tissues over time, thus allowing dynamic stratification of treatment.5 Recent technological advances in isolating and analyzing ctDNAs bring about hope that liquid biopsies performed at different time points may be useful in tracing heterogeneous changes during treatment. Based on this concept, more precise therapeutic schedules can be formulated for patients with cancer of different stages.
This review focuses on the biological and technical aspects of ctDNAs and their valuable clinical applications in monitoring therapeutic resistance and improving outcomes in patients with cancer, as well as perspectives and challenges for future use.
Underlying Reasons For Therapeutic Resistance: Intratumoral And Intertumoral Heterogeneity
Clonal mutations are detected in the majority of neoplastic cells in tumors. Beyond that, subclonal mutations resulting in heterogeneity within tumors also play an important role in tumor evolution, which must be considered when making individualized treatment decisions. The newly arisen concept of precision medicine aims to stratify patients into novel subgroups based on their individual information (eg, genetic, biomarker, phenotypic, or psychosocial characteristics) and offer personalized treatment strategies to each patient.6 The application of targeted medicine offers an opportunity to improve patient care without the use of unnecessary chemotherapy or radiotherapy and reduce the side effects caused by these therapeutic modalities. An example which has shown remarkable clinical success to some extent is the administration of breakpoint cluster region-Abelson murine leukemia tyrosine kinase inhibitor (TKI) imatinib for the treatment of chronic myeloid leukemia.7 However, tumor heterogeneity may give rise to differences in treatment responses and lead to the development of treatment resistance in patients with cancer. Tumor heterogeneity has two meanings: 1) intratumor heterogeneity, occurring within an individual tumor and 2) intertumor heterogeneity, appearing across several different tumors.8 Explanations for the origin of intratumor heterogeneity fall into two categories. The cancer stem cell hypothesis proposes that a subset of cells with stem cell properties of indefinite self-renewal drive tumor initiation and progression. The differentiation of these cells may generate intratumor heterogeneity prior to or after treatment. In contrast, the clonal evolution model suggests that premalignant or malignant cells accumulate various hereditary changes over time, and those acquired advantageous genetic changes are selected according to natural selection. The accumulation of continuous mutations in cancer cells ultimately contributes to the diversification of the tumor and emergence of further genetic and epigenetic alterations. These changes confer more aggressive, invasive, and drug-resistant phenotypes, thus resulting in intratumor heterogeneity.9,10 Although surgical resection can remove the primary tumor along with a large proportion of the trunk mutations in them, a fraction of the residual branch heterogeneous mutation may cause tumor metastasis or relapse. In addition, the changing heterogeneity may reduce the initial efficacy of treatment. Mechanisms of drug resistance that have been identified thus far include the following: prevention of drug entry into the cells; extrusion of drugs out of cells; induction of enzymatic inactivation of drugs; prevention of drug activity by mutation or altered expression of the target and defects in apoptosis, senescence and repair mechanisms, etc.11 Clinicians must consider this aspect in diagnosis and treatment planning to achieve optimal therapeutic efficacy. When selecting optimized therapy approaches, it is necessary to obtain an in-depth characterization of the clonal composition of each tumor.12
Andor et al found a link between individuals with intermediate copy number variation burdens detected in their primary tumor prior to receiving treatment and their worst overall survival (OS) across several types of tumors.13 However, the strength of this association varies depending on the different types of therapies the patients receive. Islam et al conducted a study involving patients with high-grade diffuse large B-cell lymphomas treated with the aurora kinase inhibitor alisertib. They found that the generation of drug-induced aneuploid/polyploid cells, which are thought to be responsible for treatment resistance owing to their capability of re-entering the cell cycle during off-therapy periods, are commonly induced by anti-diffuse large B-cell lymphoma therapies.14 The factors underlying the contribution of drug-induced aneuploid/polyploid cells to therapeutic resistance are the following: amplifying receptor tyrosine kinase or T-cell receptor signaling, or MYC-mediated dysregulation of three spindle assembly checkpoints Ran GTPase-activating protein 1 (RanGAP1), TPX2, and karyopherin alpha 2 (KPNA2). These factors can be targeted in combination with the administration of aurora kinase inhibitors to reduce the occurrence of therapeutic resistance. Goodall et al recently reported an analysis of targeted and whole-exome sequencing of serial circulating-free DNA samples collected during a Phase II clinical trial (TOPARP-A) of the poly(ADP)-ribose polymerase inhibitor olaparib in metastatic prostate cancer. As disease progressed following response to olaparib, they detected multiple sub-clonal aberrations reverting the reading frame of mutated homologous recombination genes in tumors harboring germline and somatic loss of DNA repair mutations (eg, BRCA2, PALB2). These aberrations emerged as mechanisms of therapeutic resistance.15 Collectively, these discoveries suggest that drug resistance may be induced by the long-term use of a single chemotherapeutic drug. Hence, it is of great importance to monitor the alterations occurring in tumor cells during the course of treatment and duly adjust the therapeutic strategies.
The Definition And Characterization Of ctDNA
The tumor molecular landscape is continuously altered during treatment with targeted drugs. Thus, the genomic profiles of patients with cancer ought to be repeatedly evaluated during the course of therapy.16 The repeated use of tissue biopsies is hindered by the inherent risk of complications and limited availability of samples. As a result, this approach cannot reveal all subclone oncogenic mutations caused by tumor heterogeneity, which are concealed in primary or metastasized tumor tissue. In response to this limitation, a new technology for the analysis of ctDNAs (termed liquid biopsy) has emerged.
ctDNA is a small fraction of cell-free DNA, which refers to DNA fragments released from cells, presenting in body fluids (eg, plasma, cerebrospinal fluid, and urine). In plasma, most of the circulating-free DNA originates from leukocytes. A small proportion of this DNA, termed ctDNA, is derived from tumor cells.17 However, the quantity of ctDNA ranges from 3% to 93% of all circulating DNA. The source of ctDNA in the peripheral blood of patients with cancer is disintegrating cancer cells due to apoptosis and/or necrosis, like other circulating DNA which is released from normal dying cells.18 This brings difficulty in distinguishing ctDNAs from other cell-free DNAs through the detection of apoptotic or necrosis-related markers. Therefore, the development of accurate and specific technologies for isolating ctDNAs is warranted. The detection of ctDNA can identify somatic genomic alterations missed in biopsy studies on account of tumor heterogeneity or lesions in distant sites.
Apart from ctDNAs, liquid biopsies include other circulating molecular indices, such as circulating tumor cells (CTCs), exosomes, tumor-specific proteins, and several kinds of non-coding RNAs. The clinical applications of these molecular indicators rely on advantages that distinguish them from other factors (Table 1). Among them, CTCs are most frequently investigated and compared with ctDNAs.19 CTCs are a type of tumor cells derived from the multicellular groupings of a primary tumor, entering the hematogenous or lymphatic circulation to initiate metastasis.20 The vast majority of CTCs floating in the bloodstream die during circulation. However, a small cluster of surviving CTCs are capable of migrating to a distant site and initiate the invasion of another organ. Among all molecules containing genetic information in the blood, CTCs are the first to be approved for use as a biomarker of metastatic cancer by the Food and Drug Administration (USA).21 Owing to intratumor heterogeneity, CTCs offer whole tumor cell-derived materials to noninvasively provide a better representation of the invasive clones for serial analysis during treatment compared with traditional surgical biopsies.
 |
Table 1 Comparison Of Strengths And Limitations Between ctDNAs And Other Circulating Biomarkers |
Nevertheless, a few limitations hinder the investigation of CTCs. In recent years, advances in detection technologies and devices have optimized the methods of CTC enrichment and isolation, rendering the analysis of this measurement index achievable. Both ctDNAs and CTCs show great value in temporally and spatially tracing mutation profiles. However, ctDNAs have the preponderance of superior sensitivity to CTCs, larger quantities in the bloodstream, and greater dynamic range that are in accord with changes in tumor burden.22 Hence, ctDNA is the preferred source for molecular diagnosis, especially in patients with early stage cancer.
Current Sequencing Techniques Applied To The Analysis Of ctDNA In Clinical Cancer Research
Recent advances in sequencing techniques provide new opportunities for analyzing ctDNAs (Table 2). These developments provide clinicians with a more valuable tool for monitoring tumor burden and treatment response based on molecular profiles. Here, we present a portion of these newly developed techniques.
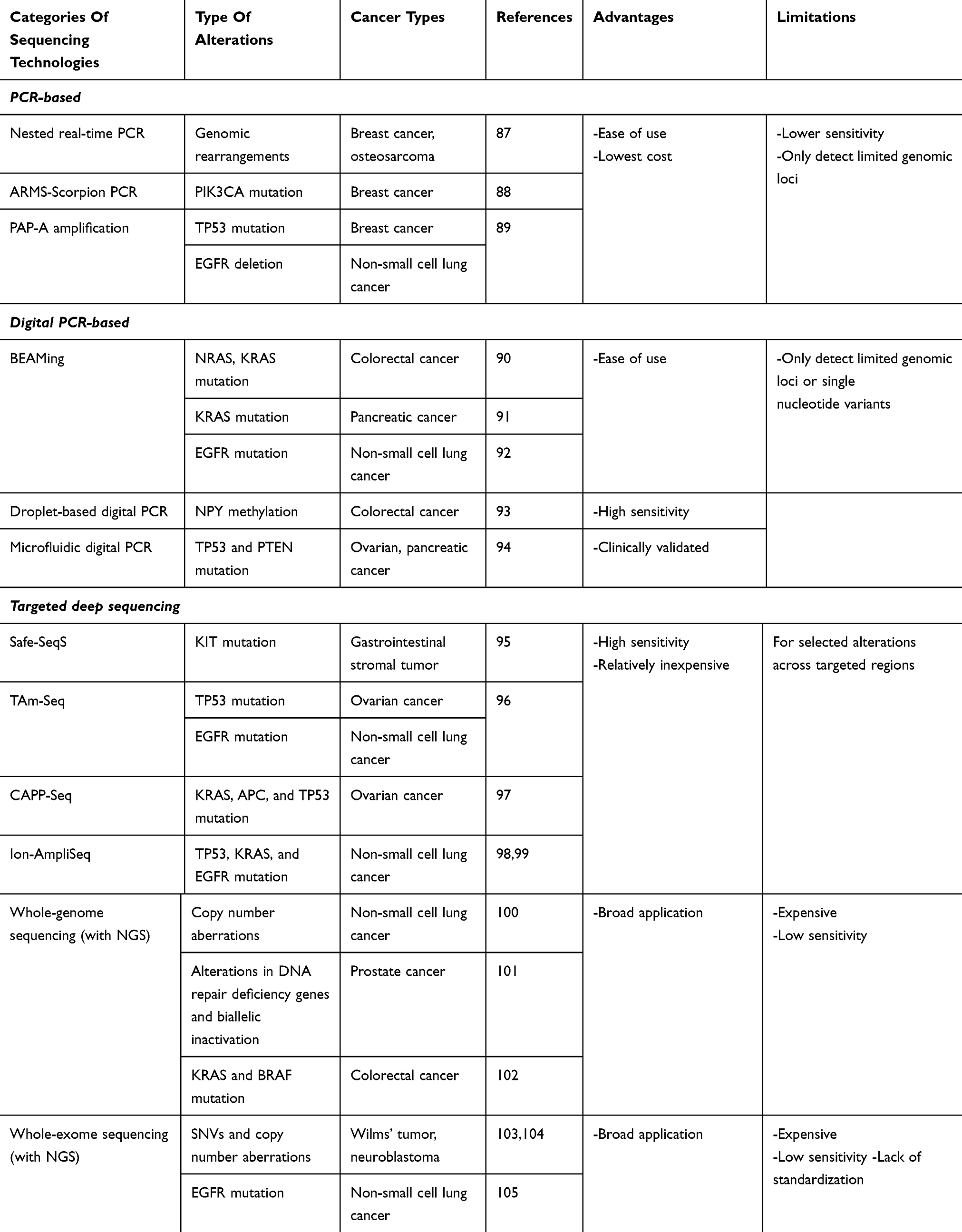 |
Table 2 Sequencing Technologies For ctDNA Analysis |
Digital Droplet Polymerase Chain Reaction (ddPCR) And Next-Generation Sequencing
Genotyping of ctDNA provides a chance to probe into heterogeneity during the evolution of cancer, monitor cancer development, and potentially detect the emergence of therapeutic resistance. However, the development of new technologies, such as ddPCR and next-generation sequencing (NGS), is required for the identification of valuable variants to be targeted when making clinical decisions. Plasma ddPCR is capable of rapidly detecting mutations, such as epidermal growth factor receptor (EGFR) and Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations, with a low rate of false-positive test results and the required robustness for real-world testing. Sacher AG et al conducted a plasma ddPCR assay on 180 patients with non-small cell lung cancer (NSCLC) and showed a sensitivity of 82% for EGFR 19 del, 77% for T790M, 74% for L858R, and 64% for KRAS. It also exhibited a positive predictive value of 100% for EGFR 19 del, L858R, and KRAS; of note, this value was lower for T790M (79%).23 This evidence indicated the ability of ddPCR to rapidly and sensitively detect EGFR and KRAS mutations. This approach offers the advantage of absolute quantification and sample differentiation with tiny concentration distinction, highlighting its potential for application in the clinical setting. Nevertheless, ddPCR is limited by its inability to detect unknown genotypes or rearrangements and difficulty in multiplexing to determine the amounts of genetic variants. The breakthrough concept for the detection of circulating genomic subclones through ctDNA‐NGS has recently been investigated as a formidable platform to develop noninvasive blood tests. The purpose of this approach is to complement and further enhance the currently available screening and diagnostic strategies. With similar sensitivity to ddPCR, plasma NGS can compensate for the disadvantage of ddPCR by detecting a wide range of driver and resistance mutations in NSCLC, such as anaplastic lymphoma kinase (ALK), ROS1, and RET rearrangements, mesenchymal–epithelial transition (MET) amplification, and human epidermal growth factor receptor 2 (HER2) insertions, with 100% specificity.24 NGS mainly consists of three specific techniques, namely whole-genome sequencing (WGS), whole-exome sequencing, and targeted region sequencing. Single ctDNA samples are inadequate to reveal subclonal populations or their responses to therapy. Therefore, the combined application of whole-exome sequencing and targeted longitudinal monitoring of the ctDNA can reveal both the presence of individual subclones with their genetic constitution and distinct responses to therapy.25 Compared with WGS, exomic analyses exhibit markedly higher sequence coverage, with generally more than 100-fold versus just approximately 30-fold of WGS. Nevertheless, whole-genome plasma DNA sequencing enables the identification of novel mutant clones and potentially facilitates early adjustments of clinical therapies that may delay or prevent the progression of cancer.26
Deep Sequencing Of ctDNA
Diehn et al developed a new strategy based on NGS, termed CAncer Personalized Profiling by deep Sequencing (CAPP-seq), for the analysis of ctDNA. This strategy involves the design of a “selector” consisting of labeled DNA oligonucleotides that target recurrently mutated regions in ctDNAs of interest for their quantification. This methodology is the first to achieve ctDNA analysis with an ultralow detection limit and broad patient coverage (ie, early or advanced stages of human malignancy with available recurrent mutation data) at a reasonable cost.27 Integrated digital error suppression (iDES) was developed to further improve the performance of CAPP-seq by eliminating most artifacts observed in ctDNA sequencing data and simultaneously maximizing the recovery of ctDNA molecules. As a result, the iDES-enhanced CAPP-seq can sensitively and precisely facilitate the noninvasive detection of variants across hundreds of kilobases. For example, when applied to ctDNAs obtained from a cohort of patients with NSCLC, this approach enabled the profiling of mutations in the EGFR kinase domain. At the variant level, the sensitivity and specificity were 92% and nearly 100%, respectively. At the patient level, these values were almost 90% and 96%, respectively.28
Safe-Sequencing System (Safe-SeqS) And Simple, Multiplexed, PCR-Based Barcoding Of DNA
Compared with conventional digital PCR methods, massively parallel sequencing can sequentially and easily analyze hundreds of millions of template molecules and multiple bases in an automated fashion. However, this sequencing technique cannot be used for the detection of rare variants (ie, variant alleles with fractions ≤0.1% in ctDNA), owing to the high error rate associated with the sequencing process. Vogelstein described that Safe-SeqS can substantially increase the sensitivity of massively parallel sequencing by assigning a unique identifier (UID) to each amplified template molecule to create UID families, followed by implementation of redundant sequencing of the amplification products. In this method, the presence of an identical mutation in ≥95% of the PCR fragments with the same UID indicates mutation (“supermutants”).29 Although Safe-SeqS has considerably improved massively parallel sequencing, this technology has not been widely used probably due to its complex protocol and the requirement of a gel purification step. In a laboratory in Boston, Tony E. Godfrey et al developed a method based on Safe-SeqS termed Simple, Multiplexed, PCR-based barcoding of DNA for Sensitive mutation detection using Sequencing (SiMSen-Seq). This approach employed a molecular hairpin serving as the UID to protect the barcode sequences from polymerase-induced errors for a more efficient identification of true mutations during the preparation of a PCR-based NGS library. Moreover, it involves a standard multiplex preamplification approach using lower primer concentrations as well as an elongated PCR extension time.30 The strengths of the SiMSen-Seq methodology are as follows: 1) facilitates the detection of sequence variants at an allele frequency ≤0.1%; 2) requires <50 ng of DNA input; and 3) can be applied to interrogate multiple genome loci covering >1000 kb of a target sequence.
Assessment Of Epigenetic Heterogeneity Using DREAMing (Discrimination Of Rare EpiAlleles By Melt)
The ever-growing number of genes that show epigenetic alterations in cell biology and tissue physiology in the course of human disease emphasizes the crucial role of these epigenetic alterations, especially DNA methylation, in future diagnosis, prognosis, and prediction of response to therapies. Epigenetic drugs which target mutated modifying enzymes, such as DNA methyltransferase, improve to some extent the outcome of various patients with cancer whose methylation profiles had been analyzed. Thus, comprehensive DNA methylation profiling presents a valid tool for the analysis of regions offering a possibility of therapeutic intervention.31 High-resolution data can serve as a source of cancer subtype-specific profiles at various stages of disease. This is achieved by performing analyses of alterations in methylation profiles using convenient and noninvasive methods with powerful technologies, such as ctDNAs. Wang at Johns Hopkins University (Baltimore, MD, USA) discussed a method termed DREAMing for ultrasensitive assessment of locus-specific epigenetic heterogeneity in specimens obtained from liquid biopsies. This approach applies semi-limiting dilution and precise melt curve analysis to discriminate and enumerate individual copies of epiallelic variants at single-CpG-site resolution, with fractions as low as 0.005%, while using only a single 96-well microtiter plate.32 This technique is particularly suitable for the ultrasensitive assessment of epigenetic heterogeneity in specimens with low abundance of epialleles and may be utilized for the evaluation of methylation dynamics in ctDNAs during progression of cancer.
Clinical Implication Of ctDNAs As A Monitoring Tool Of Drug Resistance
According to the time of development, drug resistance can be divided as follows: present at the time of initial treatment (ie, primary resistance) or arising during the course of treatment (ie, acquired resistance).33 Thus far, four main strategies employed by cancer cells to induce drug resistance have been found: 1) direct reactivation of small-molecule targets; 2) activation of signaling nodes upstream or downstream of oncogenes; 3) engagement of parallel oncogenic signaling pathways; and 4) adaptive survival mechanisms.34 An important commonality of all four strategies is the persistent activation of the drug target itself or its critical signaling pathway.35 ctDNAs can reveal the tremendous heterogeneity and genetic composition of primary or metastatic tumors; thus, they can also reflect mutations associated with changes in the activation state.36 Based on this, the isolation, purification, and analysis of ctDNAs can expound the reasons for the generation and development of primary or acquired resistance after chemotherapy and guide the administration of drugs in the clinical setting.37 Thus far, numerous mutations resulting in resistance to different kinds of targeted medicines have been found in ctDNAs. Here, we discuss some of these mutations according to the classification of cancer types and targeted medicine.
Resistance To EGFR-Targeted Therapies
Anti-EGFR Therapy With The Monoclonal Antibodies In Colorectal Cancer (CRC)
Overexpression of EGFR has been associated with more aggressive clinical progression and acts as a factor of poor prognosis in a wide variety of cancers. In CRC, the EGFR gene is frequently amplified and overexpressed at the RNA and protein levels.38 Thus, EGFR-targeted therapies may improve the OS and progression-free survival (PFS) of patients with CRC. Cetuximab and panitumumab are monoclonal antibodies binding to EGFR, which are routinely used in the treatment of metastatic CRC (mCRC). However, these drugs show clinical benefits in only a portion of patients with mCRC owing to molecular alterations in EGFR pathway effectors.39 The EGFR effector KRAS is frequently mutated in CRC, resulting in activation of the downstream RAS-RAF-mitogen-activated protein kinase (RAS-RAF-MAPK) signaling pathway, regardless of EGFR activation or blockade. Therefore, KRAS mutations may predict the development of resistance to anti-EGFR antibodies and are consistently correlated with reduced OS and PFS.40
RAS testing has become mandatory for patients with mCRC to identify the specific tumor genotype for treatment decision. Diaz et al traced KRAS mutations in ctDNAs obtained from patients with CRC who received monotherapy with anti-EGFR drugs.41 They found that 38% of the patients had a detectable conversion from KRAS wild type to KRAS mutant type in their ctDNAs under EGFR blockade. According to the KRAS type tested through allele-specific real-time PCR, clinicians decide to further analyze all RAS through Sanger sequencing (if KRAS ex2 wild-type is mutated) or treat patients with standard chemotherapy (if KRAS ex2 is mutated). Standard chemotherapy is required for patients with identified RAS mutations. Only all RAS wild-type patients receive targeted treatment with an anti-EGFR monoclonal antibody.42
Apart from RAS mutations, amplification of the MET protooncogene is also associated with acquired resistance in patients treated with anti-EGFR therapy. Amplification of the MET locus can be present in ctDNA before relapse becomes clinically evident, and timely administration of MET kinase inhibitors can overcome the induced resistance to EGFR blockade.43 Low-abundance somatic mutations, such as phosphoinositide-3-kinase, catalytic, alpha polypeptide (PIK3CA), can also be detected through a targeted amplicon ultra-deep sequencing method in ctDNAs collected from blood of patients with mCRC.44 As the PIK3CA mutation may result in acquired resistance to cetuximab in patients with mCRC, ctDNA sequencing can provide a reference for adjusting the clinical therapeutic approach.
EGFR-TKI In NSCLC
EGFR-TKI have been introduced into the first-line treatment of patients with NSCLC harboring an EGFR mutation, improving PFS and OS to various degrees.45 Nevertheless, almost all patients with EGFR-mutant NSCLC ultimately develop clinically progressive disease, as a result of resistance to treatment with an EGFR-TKI (ie, gefitinib or erlotinib). Thus far, two main mechanisms underlying acquired resistance to TKI have been identified. Secondary EGFR T790M mutation is a major mechanism, accounting for half of all reported resistance cases. Using the ddPCR method, T790M ctDNA can be detected in plasma as a poor prognostic factor and an indicator for modifying therapeutic strategies.46 MET amplification is another cause of resistance to gefitinib, which can be detected in approximately 20% of the patients. Amplification of MET can drive ERBB3 (HER3) sustained phosphorylation in the presence of gefitinib. In addition, upregulation of the ERBB3/PI3K/Akt signaling pathway impedes the induction of apoptosis by gefitinib in EGFR-mutant cells.47 In a CAPP-Seq ctDNA analysis, researchers found that MET amplification was the most frequent mechanism of resistance in patients with NSCLC receiving the third-generation EGFR inhibitor rociletinib.48 Those results indicated that ctDNA analysis is capable of noninvasively presenting resistance mechanisms and defining patterns of resistance to targeted therapies.
According to the T790M-mutation status shown in ctDNA analysis through multiplexed deep sequencing, 52% of the patients with NSCLC who were T790M-mutation positive and received treatment with osimertinib ultimately achieved a 62.5% response rate and a 12-month PFS.49 Another trial showed that even in patients with NSCLC with unknown tumor mutation status, but merely harboring T790M mutation detected through ctDNA analysis, treatment with osimertinib resulted in favorable efficacy.50 Similarly, crizotinib is a common targeted medicine used to counter MET amplification, and ctDNA can reveal a number of genetic mutations which contribute to resistance to crizotinib and progression of cancer (eg, multiple secondary MET mutations, increase of fibroblast growth factor receptor 2 gene relative copy number, etc).51 Thus, ctDNA analysis may illustrate the complex and heterogeneous molecular mechanisms of drug resistance, and allow personalized selection of therapies in response to resistance.
Resistance To anti-HER2 Therapies In Breast Cancer
Amplification of the HER2/neu oncogene occurs relatively frequently in breast cancer and correlates with disease relapse and OS.52 With the development of HER2-targeted agents (eg, trastuzumab, pertuzumab, and other small molecule TKI of HER2), addition of these drugs to chemotherapy can prolong the time to disease progression and lengthen patient survival.53 Although the efficacy of anti-HER2 therapy is obvious, therapeutic resistance prevents patients from achieving sustained clinical remission with this treatment strategy. The ctDNA assay can reveal markers of resistance to HER2 blockade, such as mutations in TP53 and genes implicated in the PI3K/mTOR pathway.54 In a longitudinal follow-up study analyzing ctDNAs collected from 42 patients with metastatic breast cancer, it was demonstrated that ctDNA can show more dynamic changes to mutations and gene amplification than cancer antigen 15-3. Thus, ctDNA carries the potential of monitoring response to treatment, analyzing dynamic tumor heterogeneity, and stratifying targeted therapies.55
Resistance To BRAF And MEK Inhibitor Therapies In Melanoma And CRC
BRAF inhibitors (eg, dabrafenib and vemurafenib) in combination with a MEK inhibitor (eg, trametinib) can improve the OS of patients with previously untreated BRAF V600-mutant metastatic melanoma.56 However, drug resistance may cause progression of disease within a year, which usually involves MAPK reactivation, commonly via BRAF amplification and mutations affecting NRAS or MEK2.57 Gray et al analyzed the ctDNAs extracted from melanoma patients with progressive disease who initially responded to treatment with dabrafenib/trametinib.58 They found that NRAS mutations, such as NRASQ61K and NRASQ61R, can be detected in ctDNAs. In addition, the number of these mutations was consistently lower than that of the BRAF mutation, possibly owing to their origin from a subset of tumor cells. Daniele et al reported a case of CRC relapse after combined treatment with BRAF and MEK inhibitors, with a KRAS G12C mutation and an amplification of mutant BRAF V600E shown through ctDNA analysis. They systematically assessed the efficacy of several candidate therapies across a panel of resistant cell lines. The results revealed that most resistant cells remain sensitive to suppression of the MAPK pathway after combination of extracellularly regulated kinase, BRAF, and EGFR inhibitors.59
Resistance To Other Therapies In Various Cancers
Patients with castration-resistant prostate cancer demonstrated improved clinical efficacy and OS following the application of abiraterone acetate and enzalutamide; however, the inevitable occurrence of resistance impedes long-term therapeutic benefit. Genomic profiling of ctDNAs collected from 65 patients with metastatic castration-resistant prostate cancer receiving enzalutamide showed that aberrations associated with primary resistance include androgen receptor (AR) amplification, multiple AR mutations, and RB1 loss. Acquired resistance can also be identified in ctDNAs, such as AR-L702H, AR-T878A, and catenin beta 1 mutations, and PI3K pathway alterations.60 ctDNAs can also be used as an identifier for patients with breast cancer who have become resistant to endocrine therapies. The detection of estrogen receptor 1 mutations in ctDNAs correlates with resistance to aromatase inhibitors (AIs) and may point to the application of more efficient treatments in such patients.61 Through utilization of a targeted ctDNA NGS test, mechanisms of resistance to ALK inhibitors (eg, kinase domain mutations, alternative oncogenic mutations, and amplifications) have been found in patients with NSCLC.62 ctDNAs were detected in all patients with metastatic gastrointestinal stromal tumors, and their levels were associated with tumor size. The mutations resulting in resistance to treatment with imatinib clustered in the region of the adenosine triphosphate-binding pocket or the activation loop; both could be detected in ctDNAs.63 Furthermore, a study of four patients who underwent resection of imatinib-resistant gastrointestinal stromal tumor identified secondary C-KIT exon 13 or 18 mutations in ctDNAs with a mutation fraction range of 0.010–9.385%.64 Consequently, the application of liquid biopsy is a promising option to comprehensively determine the mutational profiles of various heterogeneous tumors and guide therapeutic decisions in drug-resistant cancers.
Clinical Trials Demonstrating The Role Of ctDNAs In Treatment Monitoring
Based on the great potential value of ctDNAs in demonstrating mutations underlying primary or acquired resistance, researchers worldwide have conducted clinical trials to prove the practicability of this approach. The phase I/II multi-cohort eXalt2 trial (NCT01625234) employed hybrid-capture NGS to analyze ctDNAs in plasma collected from 76 patients with ALK+ NSCLC. The results showed serial changes in ALK mutation allelic frequencies while receiving treatment with ensartinib, demonstrating the clinical utility of ctDNAs in serially tracking genetic determinants of resistance.65 A prospective, multicancer, biomarker trial applied Safe-SeqS to analyze plasma ctDNAs in 42 patients with early stage operable pancreatic adenocarcinoma (Australia New Zealand Clinical Trials registry number ACTRN12612000763842). The analysis revealed a KRAS mutation in 23 patient samples pre-operatively. Although 18 patients received standard therapy, 19 of the 23 patients relapsed at a median of 38.4 months, probably due to resistance. The researchers suggested to explore the use of neoadjuvant therapy strategies in these patients rather than immediately proceeding to surgery.66 Using plasma samples collected from 83 patients with advanced breast cancer, a prospective plasma DNA AI study (CCR3297, London-Bromley Research Ethics Committee, REC 10/H0808/50) also demonstrated the potential value of ctDNA analysis in describing the mutant genetics of drug-resistant disease. ddPCR and enhanced tagged-amplicon sequencing of ctDNA samples showed high levels of genetic heterogeneity, such as estrogen receptor 1 mutations and sub-clonal KRAS mutations, with cancers progressing after first-line treatment with AI. The genetic diversity of AI-resistant cancers shown by ctDNAs may limit the effectiveness of subsequent targeted therapy approaches which target only one of the clones.67 Thus, ctDNA analysis is essential for establishing clinical therapeutic strategies, as well as tracing their levels and changes in genetic heterogeneity during treatment to adjust or combine medications.
Conclusions
Surgical techniques and targeted drugs are rapidly developing. However, the prognosis of a proportion of patients with cancer remains poor owing to the development of resistance to chemotherapeutic and targeted agents during long-term use. The primary or mutant selection-induced intratumoral and intertumoral heterogeneity renders the adjustment of the therapeutic schedule essential for improving the outcomes of patients and effectively prolonging their survival. Liquid biopsy, such as ctDNAs analysis, is likely to play a complementary role as a cancer biomarker to show the mutation status in tumor cells and guide clinical practice. As plasma is easy to collect and there is no need to enrich or isolate a rare cluster of cells, ctDNA analysis is more appealing for clinical application. However, it can only provide information regarding genomes and has a limitation in detecting mutations occurring in the RNA or protein levels. Thus, CTC analysis is also useful, as it can compensate for the shortcomings of ctDNAs. Improvements in sequencing techniques, such as NGS and other sequencing techniques, have promoted the study of ctDNAs and CTCs, and offer the possibility of clinical application in the future. Nonetheless, obstacles continue to hinder the extensive use of liquid biopsies. For example, the sensitivity and specificity of ctDNA or CTC detection warrant further verification. It is currently unclear whether all alterations in tumor cells contribute equally to changes in the composition of CTCs or ctDNAs in different stages of cancer. In addition, the concentration of ctDNAs and CTCs in plasma is low. Hence, the development of more specific techniques for their isolation and detection is necessary. Another problem that impedes widespread clinical application of this method is the relatively high cost of sequencing. A number of prospective clinical trials have been conducted to detect genetic heterogeneity in several common cancers. However, whether ctDNAs can detect mutations in rare cancers warrants further clinical investigation. In spite of these difficulties, we firmly believe that liquid biopsies hold great promise for future genetic studies concerned with monitoring mutations in tumor cells. This method has the capacity to conveniently and noninvasively offer a wealth of information over the course of treatment, which is not possible with traditional tissue biopsies. Future advances in technologies will make ctDNA and CTC analysis essential in precision medicine.
Abbreviations
ADP, adenosine diphosphate; AF, allelic frequency; AI, aromatase inhibitor; ARMS, amplification refractory mutation system; CAPP-seq: Cancer Personalized Profiling by deep Sequencing; CRC, colorectal cancer; CRPC, castration-resistant prostate cancer; CTC, circulating tumor cell; ctDNA, circulating tumor DNA; DREAMing, Discrimination of Rare EpiAlleles by Melt; ddPCR, digital droplet PCR; EGFR, epidermal growth factor receptor; HER2, human epidermal receptor 2; iDES, integrated digital error suppression; mCRC, metastatic colorectal cancer; MET, mesenchymal–epithelial transition; NGS, next-generation sequencing; NSCLC, non-small cell lung cancer; OS, overall survival; PAP-A, pyrophosphorolysis-activated polymerization allele-specific-amplification; PCR, polymerase chain reaction; PFS, progression-free survival; Safe-SeqS, Safe-Sequencing System; SiMSen-Seq, Simple, Multiplexed, PCR-based barcoding of DNA for Sensitive mutation detection using Sequencing; SNV, single-nucleotide variants; TAm-Seq, tagged amplicon deep sequencing; TKI, tyrosine kinase inhibitors; UID, unique identifier; WGS, whole-genome sequencing.
Acknowledgements
This study was supported by Jiangsu Provincial Key Medical Discipline (grant No. ZDXKA2016009) and the Program of Jiangsu Commission of Health (grant No. Z2018047).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Schwartz R, Schaffer AA. The evolution of tumour phylogenetics: principles and practice. Nat Rev Genet. 2017;18(4):213–229. doi:10.1038/nrg.2016.170
3. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. doi:10.1126/science.959840
4. Barrett MT, Lenkiewicz E, Evers L, et al. Clonal evolution and therapeutic resistance in solid tumors. Front Pharmacol. 2013;4:2. doi:10.3389/fphar.2013.00002
5. von Bubnoff N. Liquid biopsy: approaches to dynamic genotyping in cancer. Oncol Res Treat. 2017;40(7–8):409–416. doi:10.1159/000478864
6. Konig IR, Fuchs O, Hansen G, von Mutius E, Kopp MV. What is precision medicine? Eur Respir J. 2017;50(4):1700391. doi:10.1183/13993003.00391-2017
7. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344(14):1038–1042. doi:10.1056/NEJM200104053441402
8. Lovly CM, Salama AKS, Salgia R, Tumor heterogeneity and therapeutic resistance. Am Soc Clin Oncol Educ Book. 2016;(36):e585–e593. doi:10.1200/EDBK_158808
9. Michor F, Polyak K. The origins and implications of intratumor heterogeneity. Cancer Prev Res. 2010;3(11):1361–1364. doi:10.1158/1940-6207.CAPR-10-0234
10. Welch DR. Tumor heterogeneity–a ‘contemporary concept’ founded on historical insights and predictions. Cancer Res. 2016;76(1):4–6. doi:10.1158/0008-5472.CAN-15-3024
11. Kruh GD. Introduction to resistance to anticancer agents. Oncogene. 2003;22(47):7262–7264. doi:10.1038/sj.onc.1206932
12. Grzywa TM, Paskal W, Wlodarski PK. Intratumor and intertumor heterogeneity in Melanoma. Transl Oncol. 2017;10(6):956–975. doi:10.1016/j.tranon.2017.09.007
13. Andor N, Graham TA. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22(1):105–113.
14. Islam S, Paek AL, Hammer M, et al. Drug-induced aneuploidy and polyploidy is a mechanism of disease relapse in MYC/BCL2-addicted diffuse large B-cell lymphoma. Oncotarget. 2018;9(89):35875–35890. doi:10.18632/oncotarget.v9i89
15. Goodall J, Mateo J, Yuan W, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017;7(9):1006–1017. doi:10.1158/2159-8290.CD-17-0261
16. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA
17. Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C, Beljanski M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology. 1989;46(5):318–322. doi:10.1159/000226740
18. Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61(4):1659–1665.
19. Ye Q, Ling S, Zheng S, Xu X. Liquid biopsy in hepatocellular carcinoma: circulating tumor cells and circulating tumor DNA. Mol Cancer. 2019;18(1):114. doi:10.1186/s12943-019-1043-x
20. Aceto N, Bardia A, Miyamoto DT, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–1122. doi:10.1016/j.cell.2014.07.013
21. Cai X, Janku F, Zhan Q, Fan JB. Accessing genetic information with liquid biopsies. Trends Genet. 2015;31(10):564–575. doi:10.1016/j.tig.2015.06.001
22. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368(13):1199–1209. doi:10.1056/NEJMoa1213261
23. Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2016;2(8):1014–1022. doi:10.1001/jamaoncol.2016.0173
24. Paweletz CP, Sacher AG, Raymond CK, et al. Bias-corrected targeted next-generation sequencing for rapid, multiplexed detection of actionable alterations in cell-free DNA from advanced lung cancer patients. Clin Cancer Res. 2016;22(4):915–922. doi:10.1158/1078-0432.CCR-15-1627-T
25. Gremel G, Lee RJ, Girotti MR, et al. Distinct subclonal tumour responses to therapy revealed by circulating cell-free DNA. Ann Oncol. 2016;27(10):1959–1965. doi:10.1093/annonc/mdw278
26. Mohan S, Heitzer E, Ulz P, et al. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet. 2014;10(3):e1004271. doi:10.1371/journal.pgen.1004271
27. Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–554. doi:10.1038/nm.3519
28. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547–555. doi:10.1038/nbt.3520
29. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108(23):9530–9535. doi:10.1073/pnas.1105422108
30. Stahlberg A, Krzyzanowski PM, Jackson JB, Egyud M, Stein L, Godfrey TE. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Res. 2016;44(11):e105. doi:10.1093/nar/gkw224
31. Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13(10):679–692. doi:10.1038/nrg3270
32. Pisanic TR
33. Ramirez M, Rajaram S, Steininger RJ, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun. 2016;7:10690. doi:10.1038/ncomms10690
34. Sabnis AJ, Bivona TG. Principles of resistance to targeted cancer therapy: lessons from basic and translational cancer biology. Trends Mol Med. 2019;25(3):185–197. doi:10.1016/j.molmed.2018.12.009
35. Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2(3):214–226. doi:10.1158/2159-8290.CD-12-0012
36. Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol Ther. 2016;157:120–124. doi:10.1016/j.pharmthera.2015.11.007
37. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;61(1):112–123. doi:10.1373/clinchem.2014.222679
38. Spano JP, Lagorce C, Atlan D, et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol. 2005;16(1):102–108. doi:10.1093/annonc/mdi006
39. Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. doi:10.1200/JCO.2007.14.7116
40. Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: anti-epidermal growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154(1):37–49. doi:10.7326/0003-4819-154-1-201101040-00006
41. Diaz Jr LA
42. Bronte G, Silvestris N, Castiglia M, et al. New findings on primary and acquired resistance to anti-EGFR therapy in metastatic colorectal cancer: do all roads lead to RAS? Oncotarget. 2015;6(28):24780–24796.
43. Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–673. doi:10.1158/2159-8290.CD-12-0558
44. Xu JM, Wang Y, Wang YL, et al. PIK3CA mutations contribute to acquired cetuximab resistance in patients with metastatic colorectal cancer. Clin Cancer Res. 2017;23(16):4602–4616. doi:10.1158/1078-0432.CCR-16-2738
45. Nan X, Xie C, Yu X, Liu J. EGFR TKI as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer. Oncotarget. 2017;8(43):75712–75726. doi:10.18632/oncotarget.v8i43
46. Zheng D, Ye X, Zhang MZ, et al. Plasma EGFR T790M ctDNA status is associated with clinical outcome in advanced NSCLC patients with acquired EGFR-TKI resistance. Sci Rep. 2016;6:20913. doi:10.1038/srep20913
47. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi:10.1126/science.1141478
48. Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi:10.1038/ncomms11815
49. Remon J, Caramella C, Jovelet C, et al. Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Ann Oncol. 2017;28(4):784–790. doi:10.1093/annonc/mdx017
50. Park CK, Cho HJ, Choi YD, Oh IJ, Kim YC. A Phase II trial of osimertinib in the second-line treatment of non-small cell lung cancer with the EGFR T790M mutation, detected from circulating tumor DNA: liquidLung-O-cohort 2. Cancer Res Treat. 2018;51:777.
51. Du J, Wu X, Tong X, et al. Circulating tumor DNA profiling by next generation sequencing reveals heterogeneity of crizotinib resistance mechanisms in a gastric cancer patient with MET amplification. Oncotarget. 2017;8(16):26281–26287. doi:10.18632/oncotarget.15457
52. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi:10.1126/science.3798106
53. de Melo Gagliato D, Jardim DL, Marchesi MS, Hortobagyi GN. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget. 2016;7(39):64431–64446. doi:10.18632/oncotarget.7043
54. Ma F, Zhu W, Guan Y, et al. ctDNA dynamics: a novel indicator to track resistance in metastatic breast cancer treated with anti-HER2 therapy. Oncotarget. 2016;7(40):66020–66031. doi:10.18632/oncotarget.v7i40
55. Page K, Guttery DS, Fernandez-Garcia D, et al. Next generation sequencing of circulating cell-free DNA for evaluating mutations and gene amplification in metastatic breast cancer. Clin Chem. 2017;63(2):532–541. doi:10.1373/clinchem.2016.261834
56. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. doi:10.1056/NEJMoa1412690
57. Long GV, Fung C, Menzies AM, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi:10.1038/ncomms6694
58. Gray ES, Rizos H, Reid AL, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget. 2015;6(39):42008–42018. doi:10.18632/oncotarget.v6i39
59. Oddo D, Sennott EM, Barault L, et al. Molecular landscape of acquired resistance to targeted therapy combinations in BRAF-mutant colorectal cancer. Cancer Res. 2016;76(15):4504–4515. doi:10.1158/0008-5472.CAN-16-0396
60. Wyatt AW, Azad AA, Volik SV, et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol. 2016;2(12):1598–1606. doi:10.1001/jamaoncol.2016.0494
61. Beddowes E, Sammut SJ, Gao M, Caldas C. Predicting treatment resistance and relapse through circulating DNA. Breast. 2017;34 Suppl 1:S31–S35. doi:10.1016/j.breast.2017.06.024
62. McCoach CE, Blakely CM, Banks KC, et al. Clinical utility of cell-free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non-small cell lung cancer. Clin Cancer Res. 2018;24(12):2758–2770. doi:10.1158/1078-0432.CCR-17-2588
63. Namlos HM, Boye K, Mishkin SJ, et al. Noninvasive detection of ctDNA reveals intratumor heterogeneity and is associated with tumor burden in gastrointestinal stromal tumor. Mol Cancer Ther. 2018;17(11):2473–2480. doi:10.1158/1535-7163.MCT-18-0174
64. Wada N, Kurokawa Y, Takahashi T, et al. Detecting secondary C-KIT mutations in the peripheral blood of patients with imatinib-resistant gastrointestinal stromal tumor. Oncology. 2016;90(2):112–117. doi:10.1159/000442948
65. Horn L, Whisenant JG, Wakelee H, et al. Monitoring therapeutic response and resistance: analysis of circulating tumor DNA in patients with ALK+ lung cancer. J Thoracic Oncol. 2019. doi:10.1016/j.jtho.2019.08.003
66. Lee B, Lipton L, Cohen J, et al. Circulating tumor DNA as a potential marker of adjuvant chemotherapy benefit following surgery for localized pancreatic cancer. Ann Oncol. 2019;30(9):1472–1478. doi:10.1093/annonc/mdz200
67. Fribbens C, Garcia Murillas I, Beaney M, et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann Oncol. 2018;29(1):145–153. doi:10.1093/annonc/mdx483
68. Lin J, Ma L, Zhang D, et al. Tumour biomarkers-Tracing the molecular function and clinical implication. Cell Prolif. 2019;52(3):e12589. doi:10.1111/cpr.2019.52.issue-3
69. Wu Z, Yang Z, Dai Y, Zhu Q, Chen LA. Update on liquid biopsy in clinical management of non-small cell lung cancer. Onco Targets Ther. 2019;12:5097–5109. doi:10.2147/OTT.S203070
70. Leers MPG. Circulating tumor DNA and their added value in molecular oncology. Clin Chem Lab Med. 2019. doi:10.1515/cclm-2019-0436
71. Wu TH, Hsiue EH, Yang JC. Opportunities of circulating tumor DNA in lung cancer. Cancer Treat Rev. 2019;78:31–41. doi:10.1016/j.ctrv.2019.07.002
72. Ignatiadis M, Lee M, Jeffrey SS. Circulating tumor cells and circulating tumor DNA: challenges and opportunities on the path to clinical utility. Clin Cancer Res. 2015;21(21):4786–4800. doi:10.1158/1078-0432.CCR-14-1190
73. Scilla KA, Rolfo C. The role of circulating tumor DNA in lung cancer: mutational analysis, diagnosis, and surveillance now and into the future. Curr Treat Options Oncol. 2019;20(7):61. doi:10.1007/s11864-019-0653-2
74. Moon DH, Lindsay DP, Hong S, Wang AZ. Clinical indications for, and the future of, circulating tumor cells. Adv Drug Deliv Rev. 2018;125:143–150. doi:10.1016/j.addr.2018.04.002
75. Micalizzi DS, Maheswaran S, Haber DA. A conduit to metastasis: circulating tumor cell biology. Genes Dev. 2017;31:1827–1840. doi:10.1101/gad.305805.117
76. Ignatiadis M, Dawson SJ. Circulating tumor cells and circulating tumor DNA for precision medicine: dream or reality? Ann Oncol. 2014;25(12):2304–2313. doi:10.1093/annonc/mdu480
77. Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–1215. doi:10.1172/JCI81135
78. Ruivo CF, Adem B, Silva M, Melo SA. The biology of cancer exosomes: insights and new perspectives. Cancer Res. 2017;77(23):6480–6488. doi:10.1158/0008-5472.CAN-17-0994
79. Bjornetro T, Redalen KR, Meltzer S, et al. An experimental strategy unveiling exosomal microRNAs 486-5p, 181a-5p and 30d-5p from hypoxic tumour cells as circulating indicators of high-risk rectal cancer. J Extracell Vesicles. 2019;8(1):1567219. doi:10.1080/20013078.2019.1567219
80. Zheng M, Hou L, Ma Y, et al. Exosomal let-7d-3p and miR-30d-5p as diagnostic biomarkers for non-invasive screening of cervical cancer and its precursors. Mol Cancer. 2019;18(1):76. doi:10.1186/s12943-019-0999-x
81. Marcuello M, Vymetalkova V, Neves RPL, et al. Circulating biomarkers for early detection and clinical management of colorectal cancer. Mol Aspects Med. 2019;69:107–122. doi:10.1016/j.mam.2019.06.002
82. Cui M, Wang H, Yao X, et al. Circulating MicroRNAs in Cancer: potential and Challenge. Front Genet. 2019;10:626. doi:10.3389/fgene.2019.00626
83. Min L, Zhu S, Chen L, et al. Evaluation of circulating small extracellular vesicles derived miRNAs as biomarkers of early colon cancer: a comparison with plasma total miRNAs. J Extracell Vesicles. 2019;8(1):1643670. doi:10.1080/20013078.2019.1643670
84. Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77(15):3965–3981. doi:10.1158/0008-5472.CAN-16-2634
85. Ramnarine VR, Kobelev M, Gibb EA, et al. The evolution of long noncoding RNA acceptance in prostate cancer initiation, progression, and its clinical utility in disease management. Eur Urol. 2019;76:546–559. doi:10.1016/j.eururo.2019.07.040
86. Sole C, Arnaiz E, Manterola L, Otaegui D, Lawrie CH. The circulating transcriptome as a source of cancer liquid biopsy biomarkers. Semin Cancer Biol. 2019;58:100–108. doi:10.1016/j.semcancer.2019.01.003
87. McBride DJ, Orpana AK, Sotiriou C, et al. Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors. Genes Chromosomes Cancer. 2010;49(11):1062–1069. doi:10.1002/gcc.20815
88. Board RE, Wardley AM, Dixon JM, et al. Detection of PIK3CA mutations in circulating free DNA in patients with breast cancer. Breast Cancer Res Treat. 2010;120(2):461–467. doi:10.1007/s10549-010-0747-9
89. Chen Z, Feng J, Buzin CH, et al. Analysis of cancer mutation signatures in blood by a novel ultra-sensitive assay: monitoring of therapy or recurrence in non-metastatic breast cancer. PLoS One. 2009;4(9):e7220. doi:10.1371/journal.pone.0007220
90. Garcia-Foncillas J, Alba E, Aranda E, et al. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: an expert taskforce review. Ann Oncol. 2017;28(12):2943–2949. doi:10.1093/annonc/mdx501
91. Kruger S, Heinemann V, Ross C, et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann Oncol. 2018;29(12):2348–2355. doi:10.1093/annonc/mdy417
92. Krug AK, Enderle D, Karlovich C, et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann Oncol. 2018;29(3):700–706. doi:10.1093/annonc/mdx765
93. Boeckx N, Op de Beeck K, Beyens M, et al. Mutation and methylation analysis of circulating tumor DNA can be used for follow-up of metastatic colorectal cancer patients. Clin Colorectal Cancer. 2018;17(2):e369–e379. doi:10.1016/j.clcc.2018.02.006
94. Guan Y, Mayba O, Sandmann T, et al. High-throughput and sensitive quantification of circulating tumor DNA by microfluidic-based multiplex PCR and next-generation sequencing. J Mol Diagn. 2017;19(6):921–932. doi:10.1016/j.jmoldx.2017.08.001
95. Fredebohm J, Mehnert DH, Lober AK, et al. Detection and quantification of KIT mutations in ctDNA by plasma safe-SeqS. Adv Exp Med Biol. 2016;924:187–189.
96. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra168. doi:10.1126/scitranslmed.3003726
97. Iwahashi N, Sakai K, Noguchi T, et al. A comprehensive gene mutation analysis of liquid biopsy samples from patients with metastatic colorectal cancer to the ovary: a case report. Oncol Lett. 2018;16(5):6431–6436. doi:10.3892/ol.2018.9467
98. Demuth C, Winther-Larsen A, Madsen AT, Meldgaard P, Sorensen BS. A method for treatment monitoring using circulating tumour DNA in cancer patients without targetable mutations. Oncotarget. 2018;9(57):31066–31076. doi:10.18632/oncotarget.v9i57
99. Sung JS, Chong HY, Kwon NJ, et al. Detection of somatic variants and EGFR mutations in cell-free DNA from non-small cell lung cancer patients by ultra-deep sequencing using the ion ampliseq cancer hotspot panel and droplet digital polymerase chain reaction. Oncotarget. 2017;8(63):106901–106912. doi:10.18632/oncotarget.22456
100. Chen X, Chang CW, Spoerke JM, et al. Low-pass whole-genome sequencing of circulating cell-free DNA demonstrates dynamic changes in genomic copy number in a squamous lung cancer clinical cohort. Clin Cancer Res. 2019.
101. Mayrhofer M, De Laere B, Whitington T, et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018;10(1):85. doi:10.1186/s13073-018-0595-5
102. Jin CE, Koo B, Lee TY, et al. Simple and low-cost sampling of cell-free nucleic acids from blood plasma for rapid and sensitive detection of circulating tumor DNA. Adv Sci. 2018;5(10):1800614. doi:10.1002/advs.v5.10
103. Jimenez I, Chicard M, Colmet-Daage L, et al. Circulating tumor DNA analysis enables molecular characterization of pediatric renal tumors at diagnosis. Int J Cancer. 2019;144(1):68–79. doi:10.1002/ijc.v144.1
104. Chicard M, Colmet-Daage L, Clement N, et al. Whole-exome sequencing of cell-free DNA reveals temporo-spatial heterogeneity and identifies treatment-resistant clones in neuroblastoma. Clin Cancer Res. 2018;24(4):939–949. doi:10.1158/1078-0432.CCR-17-1586
105. Li C, Liu H, Zhang B, et al. Whole-exome sequencing identifies key mutated genes in T790M wildtype/cMET-unamplified lung adenocarcinoma with acquired resistance to first-generation EGFR tyrosine kinase inhibitors. J Cancer Res Clin Oncol. 2018;144(6):1079–1086. doi:10.1007/s00432-018-2634-4
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.