")
Back to Journals » Journal of Inflammation Research » Volume 15
The Rising Era of “Immunoporosis”: Role of Immune System in the Pathophysiology of Osteoporosis
Authors Srivastava RK , Sapra L
Received 30 November 2021
Accepted for publication 10 February 2022
Published 5 March 2022 Volume 2022:15 Pages 1667—1698
DOI https://doi.org/10.2147/JIR.S351918
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Rupesh K Srivastava, Leena Sapra
Immunoporosis Lab, Department of Biotechnology, All India Institute of Medical Sciences (AIIMS), New Delhi, 110029, India
Correspondence: Rupesh K Srivastava, Tel +91 11-26593548, Email [email protected]; [email protected]
Abstract: Discoveries in the last few years have emphasized the existence of an enormous breadth of communication between bone and the immune system in maintaining skeletal homeostasis. Originally, the discovery of various factors was assigned to the immune system viz. interleukin (IL)-6, IL-10, IL-17, tumor necrosis factor (TNF)-α, receptor activator of nuclear factor kappa B ligand (RANKL), nuclear factor of activated T cells (NFATc1), etc., but now these factors have also been shown to have a significant impact on osteoblasts (OBs) and osteoclasts (OCs) biology. These discoveries led to an alteration in the approach for the treatment of several bone pathologies including osteoporosis. Osteoporosis is an inflammatory bone anomaly affecting more than 500 million people globally. In 2018, to highlight the importance of the immune system in the pathophysiology of osteoporosis, our group coined the term “immunoporosis”. In the present review, we exhaustively revisit the characteristics, mechanism of action, and function of both innate and adaptive immune cells with the goal of understanding the potential of immune cells in osteoporosis. We also highlight the Immunoporotic role of gut microbiota (GM) for the treatment and management of osteoporosis. Importantly, we further discuss whether an immune cell-based strategy to treat and manage osteoporosis is feasible and relevant in clinical settings.
Keywords: immunoporosis, innate immune cells, adaptive immune cells, bone cells, gut microbiota, osteoporosis
Graphical Abstract:
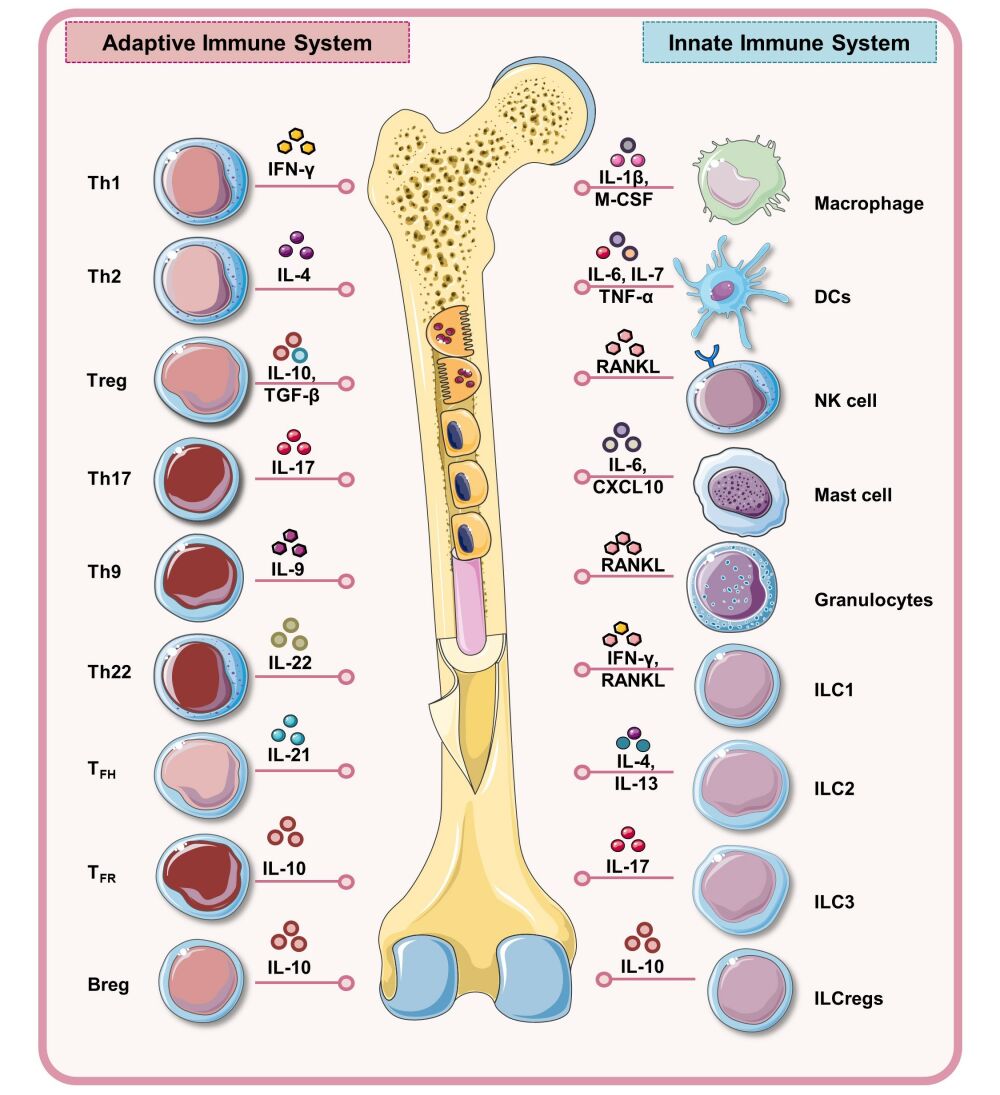
Introduction
Osteoporosis is a systemic skeletal disease that is primarily characterized by the loss of bone mineral density (BMD) and bone mechanical strength (BMS), leading to enhanced rates of fragility-related fractures in the wrist, hip, and spine. It is a growing health issue, and is the fourth most burdensome chronic disease after ischemic heart disease, dementia, and lung cancer (IOF 2021). Globally, a higher prevalence of osteoporosis has been observed in postmenopausal women. According to the International Osteoporosis Foundation (IOF), one-third of females and one-fifth of males will suffer from fragility-related fractures once in their lifetime.1 Common risk factors that are associated with the progression of osteoporosis are age, dietary habits, menopause, long-term glucocorticoid therapy, inherited osteoporosis (osteogenesis-imperfecta), etc.1 On the basis of known causes, osteoporosis can be classified generally into two categories: 1) primary osteoporosis that occurs typically in postmenopausal women, and 2) secondary osteoporosis, with defined etiological mechanisms. Primary osteoporosis is the most common category of osteoporosis, that is further comprised of types 1 and 2. In Type 1 (postmenopausal osteoporosis), bone loss is observed after estrogen deficiency, whereas type 2 (senile osteoporosis) results from systemic senescence.2 Recent advancement in the field has revealed that the currently employed therapies for the treatment of osteoporosis viz. bisphosphonates, denosumab (anti-RANKL monoclonal antibody), teriparatide (parathyroid hormone-PTH), and vitamin D supplementation exhibit the ability to modulate immune mediators. Also, these anti-resorptive and bone anabolic drugs maintain bone health in osteoporotic patients by modulating the pivotal balance of both inflammatory and immunosuppressive immune cells, thus highlighting the interconnection between the bone and immune system in skeletal health.
Twenty years ago, the term “osteoimmunology” was coined which encompasses all the aspects of cross-regulation of the skeletal and immune system.3 In the past two decades, critical work in the field of osteoimmunology has led to the discovery of a receptor activator of the nuclear factor κB ligand (RANKL)-osteoprotegerin (OPG) axis.4 RANKL is a key regulator of osteoclastogenesis. Expression of RANKL is not only observed on bone-forming osteoblasts (OBs) but also by various immune cells such as activated T cells, and B cells which play an indispensable role in the pathophysiology of various bone-related diseases including osteoporosis. Experimental evidence suggests that pro-inflammatory cytokines viz. IL-1β, IL-6, and TNF-α are found to be the causative agents in inducing bone loss in several bone-related diseases, including estrogen-deficient conditions.5
These recent findings pave the path to the birth (coined by our group in 2018) of a novel field, now globally recognized as “Immunoporosis” ie, immunology of osteoporosis.6,7 Immunological insight is now more than ever important for the treatment and management of osteoporosis in clinical applications. In the present review, we discuss and highlight the relevance of the immune microenvironment in osteoporotic conditions with respect to both innate and adaptive immune cells. Also, we discuss the role of various immunotherapeutic targets that inhibit the production of inflammatory cytokines and hold promise in inducing the regeneration of bone. Undoubtedly, this body of work will form the basis of future research and therapeutics exploiting the potential of immune cells as a novel therapeutic tool in osteoporotic conditions.
Importance of Immune–Skeleton Interface (ISI)
Bone is a dynamic organ that undergoes continuous cycles of modeling and remodeling. These processes are governed by the combinatorial action of three bone cells viz. osteoclasts (OCs-bone-resorbing cells), osteoblasts (OBs-bone-forming cells), and osteocytes (OYs), which are OBs that are embedded in the matrix during mineralization.7 During bone remodeling, damaged or old bone is removed by the OCs and replaced by the new bone sculpted by the OBs; thus, the tight coupling between these bone cells is important for skeletal homeostasis (Figure 1). Bone remodeling is a process that is initiated before birth and continues throughout life to manage the routine micro-damage and thus bone integrity. The remodeling process is characterized by a sequential series of the following four phases: 1) Activation phase, which is governed by the systemic factors that induce the differentiation of hematopoietic stem cells (HSCs) derived OC precursors into mature multinucleated OCs. Differentiation of OCs is regulated by the permissive concentrations of two crucial factors viz. macrophage colony-stimulating factor (M-CSF), a survival factor for OCs progenitors, and RANKL, a differentiation factor for OCs. 2) Resorption phase, in which OCs resorb the bone and subsequently detach from the bone surface and undergo apoptosis. 3) Reversal phase, in which mesenchymal derived OBs are recruited to the resorbed site. Differentiation of OBs is regulated by the activation of transcription factors (Runt related transcription factor-2-Runx-2, osterix-Osx); growth factors (bone morphogenetic proteins-BMPs), Wnt, tumor growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), and matrix proteins (collagen type I, fibronectin, laminin). 4) Formation phase, in which OBs lay down the new organic bone matrix which ultimately undergoes mineralization, this phase lasts for several months. At the end of the formation phase, OBs are incorporated either into the bone as OYs or remain on the surface as bone lining cells. OYs are the predominant source of sclerostin that serves to reduce bone formation by OBs via inhibiting the Wnt signaling pathway. In 1997, the potent inhibitor of OCs differentiation OPG was discovered that suppresses bone resorption by OCs via interacting with RANKL. OPG is a well-known decoy receptor of RANKL and the ratio of RANKL and OPG is a crucial determinant of OCs differentiation and bone resorption. Under physiological conditions, both B cells and OBs are important sources of OPG.8 Emerging evidence has reported that inflammatory conditions promote the development of osteoporosis.9 Inflammation is exemplified by the activation of both innate and adaptive immune cells via producing several soluble cytokines. Inflammatory molecules viz. IL-6, TNF-α stimulates the NF-κB signaling pathway, thereby activating the downstream inflammatory cytokine levels. Thus, this vicious cycle of NF-κB signaling facilitates OCs survival and differentiation, thereby becoming detrimental to bone health.10 A study demonstrated that homing of T cells in bone marrow (BM) with the involvement of the C-X-C motif chemokine ligand 10 (CXCL10) and C-X-C motif chemokine receptor-3 (CXCR3), ie, CXCL10-CXCR3 axis, promotes the differentiation of OCs in a RANKL dependent manner.11 Recently, a study proposed that pyroptosis (a programmed cell death mechanism) correlates with inflammation and contributes to the pathogenesis of osteoporosis via the NLR family pyrin domain containing three (NLRP3) inflammasomes mediated production of IL-1β and IL-18 cytokines which ultimately upregulate OCs differentiation.12 Of note, along with the major producers of pro-inflammatory cytokines, monocytes, macrophages (Mϕs), and dendritic cells (DCs) also acts as OCs precursors. Altogether, these studies suggest that immune cells and their secretory cytokines are important players in skeletal homeostasis (Table 1). Recently, a study reported by Frech et al13 examined the role of one particular immunomodulator, butyrophilin (Btn2a2), in skeletal health and reported that Btn2a2 stimulates the differentiation of OCs by enhancing the expression of OCs specific genes viz. RANK, NFATc1, TRAP, and cathepsin K. Butyrophilins (BTN) are comprised of transmembrane proteins that belong to the B7 family and have shown a varied range of functions on different immune cells either as co-stimulatory or co-inhibitory molecules. Taken together, these studies strongly reveal and establish the contribution of immunomodulatory molecules in bone homeostasis. In further sections, we elaborately discuss the role of both innate and adaptive immune cells along with their effector cytokines in the pathophysiology of osteoporosis.
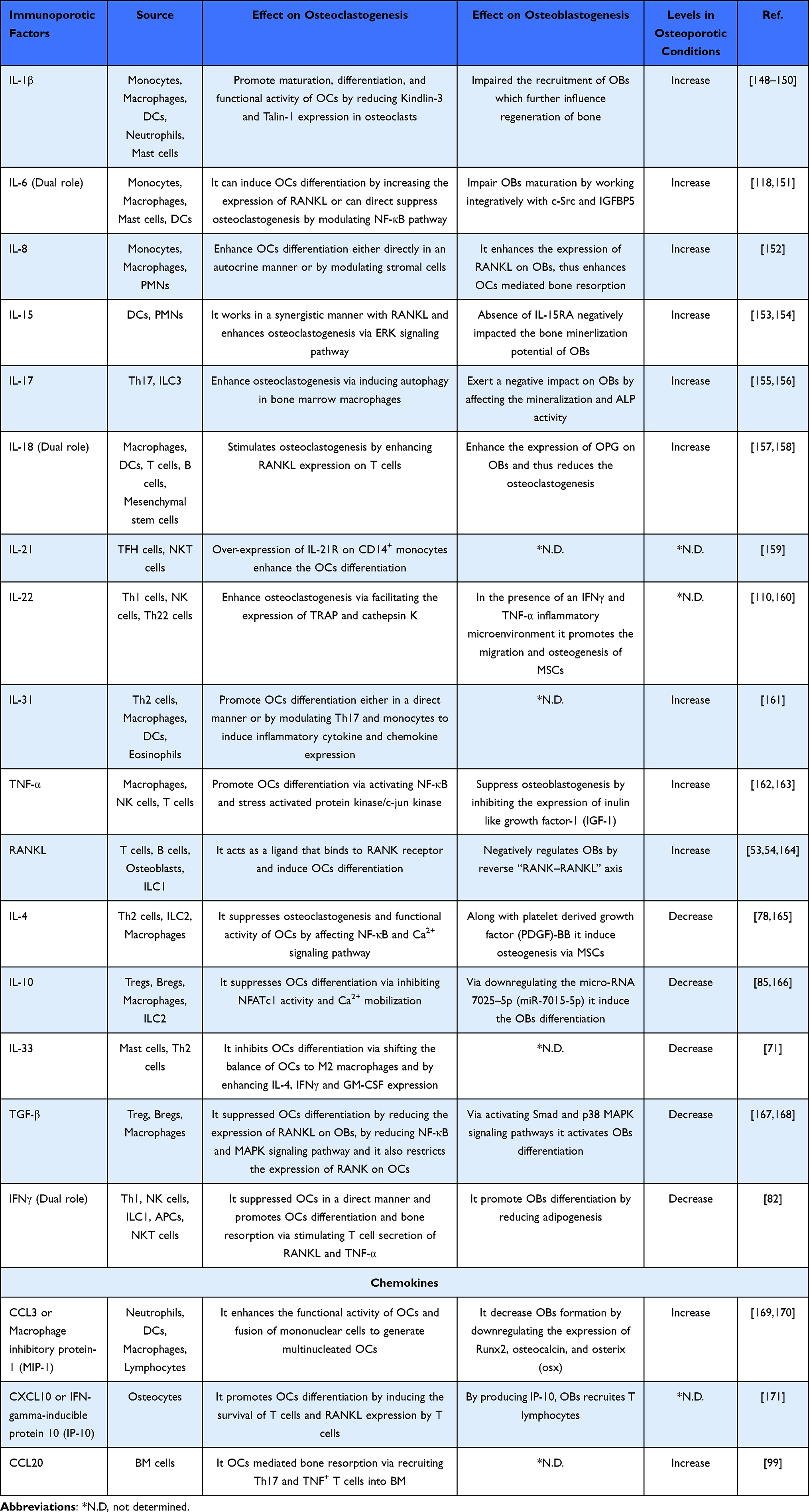 |
Table 1 Role of Immunoporotic Cytokines and Chemokines in Modulating the Bone Remodeling Process |
 |
Figure 1 Bone Remodeling Cycle under physiological and pathological condition: The remodeling process is characterized by a sequential series of four phases: 1) Activation and Resorption phases, which is governed by the systemic concentration of RANKL and M-CSF that induce the differentiation of hematopoietic stem cells (HSCs) derived OC precursors into mature multinucleated OCs. In resorption phase, mature osteoclasts with unique ruffled border induce resorption of bone by secreting cathepsin K, H+, and Cl− in sealing zone and after resorption detaches from the bone surface and undergoes apoptosis (programmed cell death mechanism). 2) Reversal phase: mesenchymal derived OBs differentiate in the presence of Wnt, BMPs, and TGF-β and are recruited to the resorbed site. 3) Formation phase: OBs lay down the new organic bone matrix which ultimately undergoes 4) Mineralization phase. Under estrogen deficient conditions or inflammatory conditions, RANKL is also provided by other immune cells that lead to enhancement in osteoclastogenesis and thus more bone resorption which ultimately leads to osteoporosis. Notes: Figure created with the help of https://smart.servier.com. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/legalcode). |
Immunoporosis: Immunology of Osteoporosis
Osteoporosis has traditionally being considered as a manifestation of a dysregulated bone remodeling process mainly governed by estrogen levels. Nevertheless, recent advancements in the field have now clearly established the pivotal role of the immune system in modulating inflammatory bone loss in osteoporotic conditions, ie, “Immunoporosis”. Immunomodulatory checkpoints and molecules mediating the autocrine, paracrine, and endocrine crosstalk among the bone cells in osteoporosis forms one of the most attractive areas of research in immunoporosis. In further sections, we highlight the specific roles of various innate and adaptive immune cells and their plasticity, and unravel novel therapeutic opportunities for clinical interventions along with their applications in different bone pathological conditions including osteoporosis (Figure 2).
 |
Figure 2 Immunopathogenesis of Osteoporosis: Schematic diagram representing the role of immune cells and their signature cytokines in the pathophysiology of osteoporosis. Solid red arrows represent established roles whereas dotted red arrows represent the proposed mechanism of action. Notes: Figure created with the help of https://smart.servier.com. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/legalcode). |
Innate Immune Cells and Osteoporosis
Innate immune cells are the prime responders towards any invading pathogens within the host and are the major producers of pro-inflammatory mediators which also act as trigger points for the development of various bone pathologies including osteoporosis. A study reported that Mϕs, OCs, and DCs are cells of clinical significance as they all share a common progenitor. Apart from these cells, granulocytes, innate lymphoid cells (ILCs), and natural killer (NK) cells further stimulate the development of osteoporosis via producing various inflammatory mediators.
Dendritic Cells (DCs)
In addition to acting as professional antigen-presenting cells (APCs), DCs also play an important role in linking both the innate and adaptive immune system. New emerging evidence suggests that DCs promote inflammation-induced osteoclastogenesis and enhance bone loss by acting as an OCs precursor. Whole-genome sequencing data analysis suggested that OCs and DCs express several overlapping genes such as dendritic cell-specific transmembrane protein (DC-STAMP), indicating the existence of a genetic relationship between these two cells that results from their vertical differentiation relationship.14 Both OCs and DCs express RANK receptor on their surface that leads to the activation of a NF-κB developmental signaling pathway culminating into osteoclastogenesis. Inconsistent with this, a study demonstrated that, upon interaction with CD4+ T cells, CD11c+ DCs can develop into functional multinucleated OCs.15 Under in vitro conditions, DCs transdifferentiate into OCs in the presence of M-CSF and RANKL. It has been observed that, in comparison to monocyte-derived OCs, DCs derived OCs generation and fusion was faster.16 Moreover, in the presence of an inflammatory environment (IL-1β and IL-6) provided by rheumatoid arthritis synovial fluid (RASF) DCs-derived OCs showed higher resorption on dentine slices in comparison to monocyte-derived OCs in RA patients.16,17 In contrast to this, recently a study reported that DCs derived interferon-λ1 (IFN)-λ1 inhibited OCs differentiation by preventing the NF-κB signaling pathway and NLRP3 inflammasome formation.18 Furthermore, this effect was reversed by the addition of the IFN-λ1 monoclonal antibody.18 Thus, this novel finding paves the path towards utilization of exogenous IFN-λ1 as a therapeutic treatment for several OCs related diseases such as inflammatory osteolysis. Recently, a study reported a novel mechanism via which estrogen modulates bone health. It has been observed that, under estrogen-deficient conditions, DCs become long-lived and express higher levels of IL-7 and IL-15 cytokines. These cytokines together induce antigen-independent production of IL-17A and TNF-α cytokines by memory T cells and thus induce bone loss in osteoporotic conditions,9 thereby indicating towards the indirect role of DCs in modulating bone health under estrogen deficient conditions. In summary, these studies suggest that DCs play a dual role in regulating osteoclastogenesis but no study to date has reported the direct role of DCs in the pathophysiology of osteoporosis.
Monocytes and Macrophages (Mϕs)
Monocytes and Mϕs are the innate immune cells derived from myeloid lineage and are known to be the early progenitors of OCs. On the basis of surface antigens, human monocytes are classified under three categories such as classical (CD14+CD16−), intermediate (CD14+CD16+), and non-classical monocytes (CD14−CD16+). Depending on the circumstances, different monocyte subsets can differentiate into OCs. It has been observed that, under physiological conditions, classical monocytes act as OCs precursors, whereas under inflammatory conditions intermediate monocytes differentiate into OCs.19 In blood, the major monocyte population (classical monocytes) acts as OC precursors, whereas, in BM, intermediate monocytes are the predominant OCs precursors. Proteomic analysis revealed that peripheral blood monocytes (PBM) express a novel protein ANXA2 with predominance in subjects with low BMDs.20 One reason for this could be that higher expression of ANXA2 leads to migration of PBM to bone resorptive sites, thereby leading to enhanced bone resorption and thus lower BMD in osteoporotic patients. OCs are formed by the fusion of monocyte/macrophage precursors, and a recent study reported that knockdown of receptor interacting protein 140 (RIP140) in OCs precursors resulted in its enhanced differentiation potential. Together these studies strongly suggest that therapeutic targeting of these novel proteins could be employed for the treatment of osteolytic inflammatory bone loss diseases such as osteoporosis.
Once monocytes leave the bloodstream and enter tissue sites they undergo a series of modifications and finally differentiate into Mϕs. Mϕs play an important role in immune surveillance and eradication of pathogens and are divided into circulating and tissue-resident macrophages (TRMs). Based on origin, TRMs are further categorized into two different subsets. One subset is derived from the yolk sac and the other subset derives from monocytes and hematopoietic progenitors. In the course of embryonic development, Mϕs are also found to be the first emerged immune cell of the immune system that infiltrate into developing organs where they ultimately differentiate into TRMs such as bone resident macrophages. Accumulating evidence suggests that, along with the immune-related functions, Mϕs also play a vital role in bone metabolism and bone tissue engineering. Bone macrophages include bone marrow macrophages (BMMs), OCs, and osteal macrophages. Osteomacs are osteal macrophages that are a distinctive subset of bone macrophages and are found to be f4/80 positive and tartrate-resistant acid phosphatase (TRAP) negative. Osteomacs are seen to be localized near OBs, thus hinting towards its bone-forming potential. Osteomacs differ from the OCs by expressing Siglec-1 (CD169). Various studies demonstrated that depletion of CD169+ osteomacs compromised the bone repair mechanism by causing a striking reduction in OBs cells.21 These studies clearly suggest that, independent of OCs, osteomacs provide anabolic support to OBs during bone repair and in the maintenance of bone homeostasis. In response to a well-defined panel of stimuli, immature Mϕs can be cataloged into two predominant classes: M1 Mϕs (pro-inflammatory) and M2 Mϕs (anti-inflammatory). Even after so many years, the role of Mϕs in modulating the bone remodeling process is a matter of debate. A study demonstrated that lipopolysaccharide (LPS) and IFN-γ induced M1 Mϕs in RAW264.7 cells or bone marrow cells significantly suppressed RANKL induced osteoclastogenesis via secreting IFN-γ and IL-12 cytokines in comparison to M2 Mϕs.22 It was further confirmed in a ligature induced periodontitis mouse model where the adoptive transfer of M1 Mϕs significantly lowered bone loss and reduced the TRAP-positive multinucleated cells in comparison to adoptively transferred M2 Mϕs.22 In-line with this, a study reported that upon RANKL stimulation, CD206+ M2 Mϕs differentiated into OCs that further resulted in enhancement of M1/M2 ratio in an ovariectomized mice model.23 Of note, it has been observed that 17β-estradiol (E2) [via estrogen receptor (ER)-α signaling mediated downstream blockade of NF-κB nuclear translocation] safeguards M2 Mϕs from RANKL stimulation, thereby suppressing its differentiation into OCs. Furthermore, it has been observed that M2 Mϕs showed higher efficiency of differentiating into OCs due to the attenuated expression of interferon regulatory factor-5 (IRF-5).24 Thus, modulating the M1/M2 ratio could be a potential therapeutic target in treating postmenopausal osteoporosis. Moreover, it has been observed that RANKL exhibits the potential of inducing differentiation of bone marrow-derived macrophages (BMDM) into M1 Mϕs. In comparison to LPS and IFN-γ induced M1 Mϕs, RANKL induced M1 Mϕs are actively involved in bone healing.25
Intercellular communication is a biological process that promotes cells to communicate and coordinate spatially and temporally their responses to any physiological alterations. One such member that aids cellular communication is extracellular vesicles (EVs). EVs are membrane-derived phospholipid bilayers with a diameter which lies between 30 and 2,000 nm.26 A predominant class of EVs is exosomes (30–150 nm) that are released by exocytosis. Exosomes derived from the Mϕs upon stimulation with bone morphogenetic protein-2 (BMP-2) showed osteogenic potential in bone marrow-derived mesenchymal stromal cells (BM-MSCs).27 Furthermore, exosomes integration with titanium nanotubes enhanced the bio-functionality of titanium implants that favor osteogenesis by enhancing the expression of early osteoblastic differentiation markers such as BMP-2 and alkaline phosphatase (ALP).27 A study reported that exosomes derived from M1 and M2 Mϕs contain differentially expressed micro-RNAs (miRNAs). Among various miRNAs, miR-5106 was observed to be significantly overexpressed in M2 Mϕs-derived exosomes (M2D-Exos) leading to enhanced osteogenic potential in M2D-Exos in comparison to M1 Mϕs-derived exosomes (M1D-Exos).28 miR-5106 enriched in M2D-Exos induced the osteoblasts differentiation in bone mesenchymal stem cells (BMSCs) by targeting salt inducible kinase 2 and 3 (SIK2 and SIK3) genes.28 Also, a study reported that exosomes derived from the MSCs stimulated the M2 phenotype in Mϕs that further resulted in the exosome-educated macrophages (EEMs). These EEMs showed an acceleration in ligament ligation and reduced inflammation in the rat medial collateral ligament (MCL) injury model.29
These findings indicate the mechanisms of exosomes derived from macrophages that further need investigation in the case of osteoporosis. One of the important alterations observed in macrophage to osteoclasts differentiation is the change in the energy metabolism. A study reported that lysine promotes the polarization of BMDMs to M1 and M2 phenotypes, whereas tyrosine and phenylalanine have different effects.30 Furthermore, differentiated OCs showed enhanced biosynthesis of phenylalanine and tyrosine and are rich in lysine degrading enzymes. These findings altogether suggest that inhibition of macrophage polarization enhances OCs differentiation. Moreover, a study demonstrated that both glycolysis and oxidative phosphorylation promote osteoclastogenesis. Upon RANKL stimulation, expression of glucose transporter viz. GLUT 1 enhances OCs.31 These reports clearly suggest that energy metabolism is enhanced during macrophage to OCs differentiation. Altogether, these studies indicated that plasticity and heterogeneity of Mϕs make them a crucial player in maintaining bone homeostasis. Nevertheless, a more in-depth analysis is essential to understand the potential of Mϕs in dealing with osteoporosis.
Innate Lymphoid Cells (ILCs)
In the past decade, an explosion of research resulted in the identification of an emerging arm of the nnate immune system which is collectively now known as the innate lymphoid cells (ILCs). In 2013, the nomenclature of ILCs was proposed and were classified into three groups: Group 1, Group 2, and Group 3 ILCs. Group 1 includes NK cells and ILC1. These cells are reliant on T-box transcription factor (T-bet) and IFN-γ cytokine for their development and function. Group 2 includes ILC2, which are dependent on transcription factors GATA binding protein-3 (GATA-3) and retinoic acid receptor-related orphan receptor alpha (RORα) for their development and secrete IL-4, IL-5, IL-9, and IL-13 cytokines. Group 3 includes ILC3 and lymphoid tissue inducer cells (LTi) which are dependent on transcription factor RORγt for their development and produce IL-17 and IL-22 cytokines. ILC3s are further divided into subsets based on the expression of either CC chemokine receptor-6 (CCR-6) or natural cytotoxicity receptor (NCR), which consists of NKp30, NKp44, and NKp46.32 ILCs are the growing family of lymphocytes that mirror the phenotypes and functions of T helper cells. Also, ILCs are known to be innate counterparts of T helper cells that share a developmental origin and phenotypes similar to that of T helper cells. In terms of origin and function, ILC1, ILC2, and ILC3 mirror the Th1, Th2, and Th17 CD4+ T helper cells, respectively, of the adaptive immune system. However, in contrast to T cells, ILCs neither express recombination activating gene (RAG) dependent diversified antigenic receptors nor the lymphoid lineage differentiation markers (Lin−). Also, in contrast to T cells, ILCs do not endure clonal selection and expansion that leads to ILCs, getting activated promptly in response to the signals or the inducer cytokines articulated by the tissue-resident cells. As ILCs get activated earlier due to injury or infection, they are expected to regulate the development of the adaptive immune system. Several studies highlighted that ILCs activate the adaptive immune response in two ways either directly or indirectly; by expressing major histocompatibility complex-II (MHC-II), ILCs directly activate antigen-specific T cells or indirectly via regulating DCs.33 In turn, T cells via producing IL-2 cytokine promotes the activities of ILCs, thus, spawning the positive-feedback loop and further amplifying their responses. A study revealed that arthritogenic Th17 cells via secreting IL-17 cytokine orchestrated the expansion of granulocyte monocyte colony-stimulating factor (GM-CSF) producing ILCs that further augments the progression of arthritis in the RA mice model.34 Notably, GM-CSF production by ILCs is regulated by alarmin, IL-33, IL-2, and toll like receptor (TLR)-9 ligands which are released by the damaged cells in inflamed tissues.34
ILCs are largely localized at barrier surfaces and are found to be involved in tissue remodeling, protection against pathogens, and maintenance of tissue homeostasis.32 Various studies revealed that dysregulation in the activation of ILCs has been associated with the pathogenesis of inflammatory and autoimmune diseases such as multiple sclerosis (MS), inflammatory bowel disease (IBD), RA and anti-neutrophil cytoplasmic antibody (ANCA) linked vasculitis.35–37 Abnormalities in the skeleton system are the common comorbidities associated with IBD, Crohn’s disease (CD), and ulcerative colitis (UC). To date, very few studies have underpinned the molecular basis of IBD-associated inflammatory bone loss conditions. A study demonstrated that heightened inflammatory immune response at intestinal tissues in IBD promoted the NF-κB activation in intestinal epithelial cells (IECs) and enhanced frequencies of ILC1, ILC3, and myeloid OCs precursors. In the DSS-induced colitis mice model, it has been observed that pharmacological suppression of inhibitor kappa B kinase 2 (IKK2) effectively reduced the frequencies of ILC1 and ILC3 and thus halted the colitis induced bone loss. These reports thus, suggest towards the bone health regulating potential of ILC1s and ILC3s.38 It has been observed that IL-23 cytokine exhibits paradoxical activities in controlling the conversion of ILC1 to ILC3 and vice versa. In the presence of IL-23 cytokine, ILC3s were found to convert into ILC1, and this was controlled by the higher expression of the signal transducer and activator of transcription (STAT)-4.39 Another study also showed that the presence of IL-23 cytokine converted ILC1 to ILC3.40 But the molecular basis behind the conversion of ILCs remains mysterious. In further sections, we will discuss the different subsets of ILCs in detail and their role in the pathogenesis of several bone pathologies including osteoporosis.
ILC1
Like Th1, ILC1 depends on T-bet for development and produces high amounts of IFN-γ signature cytokine to protect from intracellular pathogens and inflammatory pathologies. ILC1s respond to the intracellular pathogen by activating classical macrophages through IFN-γ. In 2000, Takanayagi et al41 reported that, via production of IFN-γ, Th1 suppressed osteoclastogenesis and since ILC1 are a mirror image of Th1-cell type, it is plausible to suggest that, via secreting IFN-γ, ILC1 may also attenuate bone loss in an osteoporotic mice model. On the other hand, recent studies reported that ILCs also produce RANKL and thus could further lead to enhancement in OCs differentiation.
ILC2
ILC2s are the innate counterparts of Th2 cells that depend on GATA-3 for their development and are characterized by the production of IL-4, IL-5, IL-13, and GM-CSF signature cytokines. Recently, a study reported, via the production of IL-4 and IL-13 cytokines, ILC2s suppressed osteoclastogenesis under both in vitro and in vivo conditions.42 It has been observed that the adoptive transfer of ILC2 abrogated bone loss in a post-menopausal osteoporotic mice model.42 The anti-osteoclastogenic and anti-osteoporotic potential of ILC2 was associated with the production of IL-4/IL-13 cytokine and STAT6 activation in OCs progenitors. These findings suggest the potential role of ILC2s in maintaining bone homeostasis. A study demonstrated that ERα signaling via promoting secretion of IL-33 cytokine augments the proliferation and activation of ILC2.43 Moreover, it has been observed that the adoptive transfer of IL-4+/IL-13+ ILC2 in the initiation phase of arthritis significantly attenuated inflammatory bone loss in both serum-induced arthritis (SIA) and collagen-induced arthritis (CIA) mice models, on the contrary IL-4−/IL-13− ILC2 failed to do the same.44 Importantly, at later time points, even adoptive transfer of IL-4+/IL-13+ ILC2 failed to suppress arthritis in SIA and CIA mice models.44 Thus, these findings suggest that enrichment of ILC2 numbers may efficiently block the progression of inflammatory joint diseases. Also, in RA patients, enhanced numbers of ILC2 in peripheral blood and synovial fluid (SF) of RA patients are correlated with lesser disease severity.44 Recently, a seminal study discovered a unique subset of ILC2 named as ILC210 population that produces a IL-10 cytokine along with downregulated expression of inflammatory cytokines.45 Transcription factors cMaf and Blimp-1 regulate IL-10 production from ILC210 population.46 Recently, a study demonstrated that BM-ILC2s express a higher level of RANKL in the resting stage which in the presence of IL-7 cytokine induces the differentiation of BMMs into TRAP-positive osteoclasts. On the other hand, IL-33 activated ILC2 lowers RANKL expression, which further promotes the differentiation of macrophages into the M2 phenotype.47 This novel finding clearly indicates that ILC2s regulate activation of OCs in both steady state as well as in IL-33 induced inflammation, hence, contributing towards the maintenance of bone metabolism in various inflammatory conditions.
ILC3s
ILC3s are the innate counterparts of Th17 cells that depend on RORγt for their development and contribute towards protection against pathogens via producing IL-17 and IL-22 cytokines. Metabolic profiling studies indicate that proliferation, activation, and effector functions of Th17 cells depend on high-rate glycolysis, whereas ILC3s rely on the metabolic program which is integrative of glycolysis and mitochondrial lipid oxidation and on the generation of reactive oxygen species (mROS).48 ILC3s are the crucial regulators of host-commensal mutualism and are vital sentinels of tissue homeostasis, predominantly of the gastrointestinal tract (GUT), that rapidly respond to damage, infection, and inflammation to restore tissue health. A study revealed the accumulation of CCR6+ ILC3s and its secretory cytokines IL-17 and IL-22 in the inflamed joints of CIA mice model and in SF of active RA patients,49 thus suggesting the arthritogenic role of ILC3s. Moreover, flow cytometric analysis of SF showed higher enrichment of IL-17A producing CCR6+ and NKp46+ ILC3s than the ILC3s in the periphery of psoriatic arthritis (PsA) patients.50 This indicates the existence of an inverse correlation between ILC3s in peripheral blood and PsA disease severity. During inflammatory conditions, it has been observed that GUT-derived IL-17+IL-22+ ILC3s migrate towards inflamed BM, SF of ankylosing spondylitis (AS) patients,51 thereby suggesting the existence of an active homing axis between GUT and various inflamed spondylitis sites. Several studies demonstrated that Th17 cells via secreting IL-17 cytokine enhanced the differentiation of OCs precursors into matured OCs.52 The osteoclastogenic role of IL-17 cytokine is further proven by augmented levels of IL-17 cytokine in an osteoporotic mice model under estrogen-deficient conditions.53,54 Altogether, these studies point towards a crucial involvement of ILC3s in regulating the bone remodeling process. But to date, the role of IL-17 producing ILC3s in regulating osteoclastogenesis and in osteoporotic conditions has not been explored.
ILCregs
In 2017, Wang et al55 discovered a new member in the class of the ILCs family, named regulatory innate lymphoid cells (ILCregs). This subpopulation of ILCs reside in the GUT of mouse and humans and possess a unique gene identity that differs from other subpopulations of ILCs and regulatory T cells (Tregs). Gene expression profile studies demonstrated that ILCregs did not express Tregs specific transcription factor Foxp3, rather ID3 (inhibitor DNA binding 3) was found to be the critical transcription factor that controls the development of ILCregs.55 During intestinal inflammation, TGF-β1 sustains the expansion of ILCregs in an autocrine manner. Moreover, via secretion of IL-10 cytokine, ILCregs suppressed the activation of ILC1 and ILC3 and, thus, favors the resolution of intestinal inflammation. This finding clearly suggests that ILCregs mediate their immunosuppressive functions via secreting IL-10, a known anti-osteoclastogenic cytokine, thereby further signifying that ILCregs can have a vital role in regulating osteoporosis, thereby opening novel avenues for future research.
Mast Cells (MCs)
MCs are tissue-resident effector cells and crucial sensors of the immune system well known for promoting allergic reactions.56 MCs are localized throughout the body in various tissues including the skeletal system. MCs regulate bone health and bone metabolism because these cells store and synthesize several mediators such as enzymes and cytokines which are found to be involved in regulating bone homeostasis and in the pathogenesis of bone disorders. It has been shown that synovial fluid of RA and osteoarthritis (OA) patients contain increased numbers of MCs and their mediators including histamine and tryptase. A study revealed that histamine directly acts on OC precursors, mature OCs and OBs and, thus, promotes osteoclastogenesis in an autocrine and paracrine manner and suggests that anti-histaminic compounds can be employed to suppress osteoclastic activity.57 Increased numbers of MCs have also been observed in the case of postmenopausal osteoporotic patients. After fracture, MCs trigger local and systemic inflammation via inducing the release of various inflammatory mediators. Accumulation of MCs has been shown to regulate the activity of OCs and remodel the bone fracture callus in later stages of healing.58 Systemic mastocytosis (SM) is a rare disorder that is diagnosed with infiltration and accumulation of MCs at different sites.59 SM was also found to be the cause of secondary osteoporosis. MCs infiltration leads to the release of mediators that further develop osteoporosis in 37% of SM patients. Another study demonstrated that female mice lacking mast cell chymase Mcpt4 showed higher serum bone anabolic markers and a higher periosteal bone formation rate60 suggesting the functional effect of mast cell chymase on bone health. Recently, a study was reported by Fischer et al61 in a MCs deficient Mcpt5-Cre-R-DTA mice model where deficiency of MCs protected mice from Ovx-induced osteoporosis and perturbed fracture healing, indicating a crucial role of MCs in the pathophysiology of osteoporosis under estrogen-deficient conditions. In wild-type conditions, MCs triggered fracture provoked an inflammatory response by promoting release of inflammatory molecules IL-6, CXCL10, and midkine (Mdk) along with inducing the infiltration of neutrophils at the fracture site in ovariectomized mice.61 IL-31, secreted by MCs, are also being employed as biomarker to monitor disease severity in various allergic diseases. Interestingly, a study reported that increased levels of IL-31 in post-menopausal osteoporotic patients are correlated with reduced BMD.62 Collectively, these findings suggest that MCs can also be employed as a potential target for the treatment of inflammatory bone disorders including osteoporosis along with suppressing inflammatory bone loss associated with allergic diseases.
Granulocytes
Granulocytes are the polymorphonuclear (PMN) cells that involve neutrophils, basophils, and eosinophils. Among these three immune cells, neutrophils are the predominant type of granulocyte observed in the blood that plays a vital role in protection against bacterial infection. In the circulation PMNs derived from bone marrow (BM) are referred to as circulatory PMNs (cPMNs). In response to various inflammatory conditions such as periodontitis, diabetes mellitus (DM), RA, and osteoporosis the percentage of cPMNs was found to be increased.63 In 2009, Chakravarti et al64 demonstrated that LPS activated human or murine neutrophils stimulated osteoclastogenesis and bone resorption in human monocyte-derived OCs and RAW 264.7 cells, respectively. Moreover, flow cytometric and confocal laser microscopy data suggested that neutrophil aids osteoclastogenesis by expressing membrane-bound RANKL. In contrast to this, recently a study reported limited RANKL expression on neutrophils after stimulation for 24 and 48 hours with LPS63 with limited potential for inducing osteoclastogenesis. Thus, it still remains unclear whether neutrophils play any pivotal role in providing signals to promote differentiation of monocytes to OCs. Furthermore, it is observed that neutrophils enhanced OCs differentiation induced by vitamin D3 in the co-cultures of BM cells and OBs.65 Moreover, it was found that via secreting elastase as a component of neutrophil extracellular trap (NETs), neutrophils promote the degradation of OPG, enhancing osteoclastogenesis in BM cells, thereby suggesting an important role of neutrophils in inflammatory bone loss.65 These findings pave the path towards exploring the role of neutrophils in regulating osteoclastogenesis, particularly in the field of immunoporosis. Under inflammatory conditions, the neutrophils-to-lymphocyte ratio (NLR) is employed as an effective inflammatory marker that has been linked with several inflammatory disease conditions. A study revealed the existence of a negative correlation between NLR and BMD and indicated its utilization as a predictor for the occurrence of osteoporosis.66 Moreover, a study showed the correlation between percentages of neutrophils and osteonecrosis in the femoral head and thus provides a new perspective for future study.67 In contrast to these, a study recently reported that, in the case of fracture healing, neutrophils contribute to bone regeneration by synthesizing a fibronectin positive emergency extracellular matrix (ECM).68 In addition, recruitment of neutrophils post-fracture is important for promoting the activation of downstream processes, thereby leading to regeneration of bone.69 Moreover, treatment with anti-Ly-6G antibody showed reduction in the numbers of neutrophils recruited to the fracture site, thereby further leading to impaired fracture healing.69
Earlier osteoporosis and allergic diseases were considered to be unrelated, however, growing knowledge in the field of osteoimmunology provided support to the existence of a correlation between bone and allergy, thereby providing a fascinating area of research. Eosinophils and basophils are leading cells involved in the pathogenesis of airway allergic inflammation. Recently, IL-31 has been indicated as a biomarker for allergy and several other immunologic diseases, and is produced by CD4+ T cells and in lower amounts by MCs and DCs.70 Levels of IL-31 cytokine are correlated with the disease severity in allergic diseases as it’s receptors are expressed on eosinophils. Interestingly, a study reported that increased levels of IL-31 cytokine in post-menopausal osteoporotic patients are correlated with reduced BMD.62 In addition, IL-33 that induces eosinophil adhesion in allergic diseases is also known to protect from inflammatory bone loss via inhibiting RANKL mediated OCs differentiation, along with inducing secretion of anti-osteoclastogenic molecules (IL-4, IL-10, IFN-γ, and GM-CSF).71 However, further research is still needed for correlating allergic inflammation and osteoporosis.
Natural Killer (NK) Cells
NK cells are innate immune cells that play a crucial role in the elimination of transformed, stressed, or virus-infected cells. A study demonstrated that NK cells express M-CSF and RANKL at higher levels and were found to be linked with CD14+ monocytes in RA synovium and induced OCs differentiation in a M-CSF and RANKL dependent manner.72 The importance of NK cells was further suggested in bone loss when depletion of NK cells in the CIA model reduces the severity and completely prevented bone destruction. Interestingly, a study by the same group in 2015 demonstrated that IL-15 activated NK cells suppress osteoclastogenesis by triggering apoptosis of OCs in a dose-dependent manner via expressing leukocyte function-associated antigen-1 (LFA-1) and TNF related apoptosis-inducing ligand (TRAIL).73 In contrast to these studies, a study revealed the regulatory role of NK cells in systemic juvenile arthritis (sJIA), where NK cells were observed to impede disease development in CFA-challenged mice in an NGK2D dependent manner.74 INF-γ is an anti-osteoclastogenic cytokine that suppresses osteoclastogenesis. NK cells are known producers of INF-γ cytokine, but in the case of RA patients it has been observed that NK cells exhibit poor capacity to produce INF-γ, thereby leading to enhanced bone deterioration.72 Moreover, apart from enhancing osteoclastogenesis it has also been observed that NK cells via IL-15 (bone destructive) promote apoptosis in OBs in a caspase-3 dependent manner.75 Altogether, these studies give a glimpse into the osteoclastogenic role of NK cells in osteoporosis, but it needs further investigation.
Adaptive Immune Cells and Osteoporosis
Innate immune cells provide a first line defense against various pathogens and also play a vital role in activating adaptive immunity. The adaptive immune system provides a versatile means of defense and also plays a vital role in providing protection against reinfection with the same pathogens via its memory response. Lymphocytes are major representatives of the adaptive immune system, comprised mainly of B and T lymphocytes. In further sections, we will discuss the potential of these lymphocytes in modulating bone health in the case of osteoporosis.
T Cells
T cells are the crucial mediators of the adaptive immune system that play an important role in the regulation of bone health. In accordance to the subunits of T cell receptor (TCR), T cells are further subdivided into distinct classes. TCR are either comprised of αβ or γδ subunits through which T cells recognize a wide range of antigens. T cells play a dual role in regulating bone health, in the resting state it can prevent bone resorption by OCs, whereas activated T cells via expressing RANKL mediate the generation of OCs and thus enhance bone resorption. Takanayagi et al41 demonstrated that interferon-gamma (IFN-γ) provides a negative correlation between T cell activation and the bone resorption process where IFN-γ interferes with RANKL-RANK signaling and induces the degradation of RANK adaptor protein, ie, tumor necrosis receptor-associated factor 6 (TRAF6), thereby leading to inhibition of osteoclastogenesis. TRAF6 is an adaptor protein that induces survival and differentiation of OCs by activating the downstream signaling pathways upon RANKL–RANK interaction. In further sections, we explain the role of various T helper cells concerning osteoporosis.
Th1/Th2 Paradigm in Osteoporosis
In 1986, Mosmann et al76 discovered that CD4+ T cells can be characterized by specific cytokines that led to the emergence of the “division of labor” Th1/Th2 paradigm. Th1 and Th2 cells could be differentiated from each other on the basis of signature cytokines and transcription factors. Concerning the cytokine expression, Th1 cells express IFN-γ and IL-2 cytokines and lineage commitment depends on the T-bet transcription factor. On the other hand, Th2 cells express IL-4 cytokine, and lineage commitment depends on the GATA-3 transcription factor. Several studies reported that, via secreting these IFN-γ and IL-4 classical cytokines Th1 and Th2 cells inhibit osteoclastogenesis and, thus, are now considered to be anti-osteoclastogenic in nature (Figure 3). Lower serum levels of IFN-γ and IL-4 cytokines (low levels would enhance bone loss) in postmenopausal osteoporotic patients further suggest its osteoprotective role and thus targeting these cytokines could be beneficial in the treatment of postmenopausal osteoporosis.77 A study reported that IL-4 cytokine inhibits osteoclastogenesis and bone resorption by suppressing NF-κB and Ca2+ signaling in mature OCs.78 Also, it has been observed that the IL-4 cytokine suppresses NFATc1, a transcription factor that is found to be involved in OCs differentiation. Also, together with IL-13 cytokine, IL-4 strongly inhibited the OCs formation by enhancing the OPG production by OBs.79 Moreover, a study demonstrated that IL-4 cytokine directly suppressed osteoclastogenesis by inducing the peroxisome proliferator-activated receptor γ1 (PPARγ1) ligands that further target the PPARγ1 receptor and thus suppressed osteoclastogenesis.80 Also, a study suggested that the IL-4 cytokine exhibits the potential to suppress osteoclastogenesis that was induced by TNF-α via STAT6.81
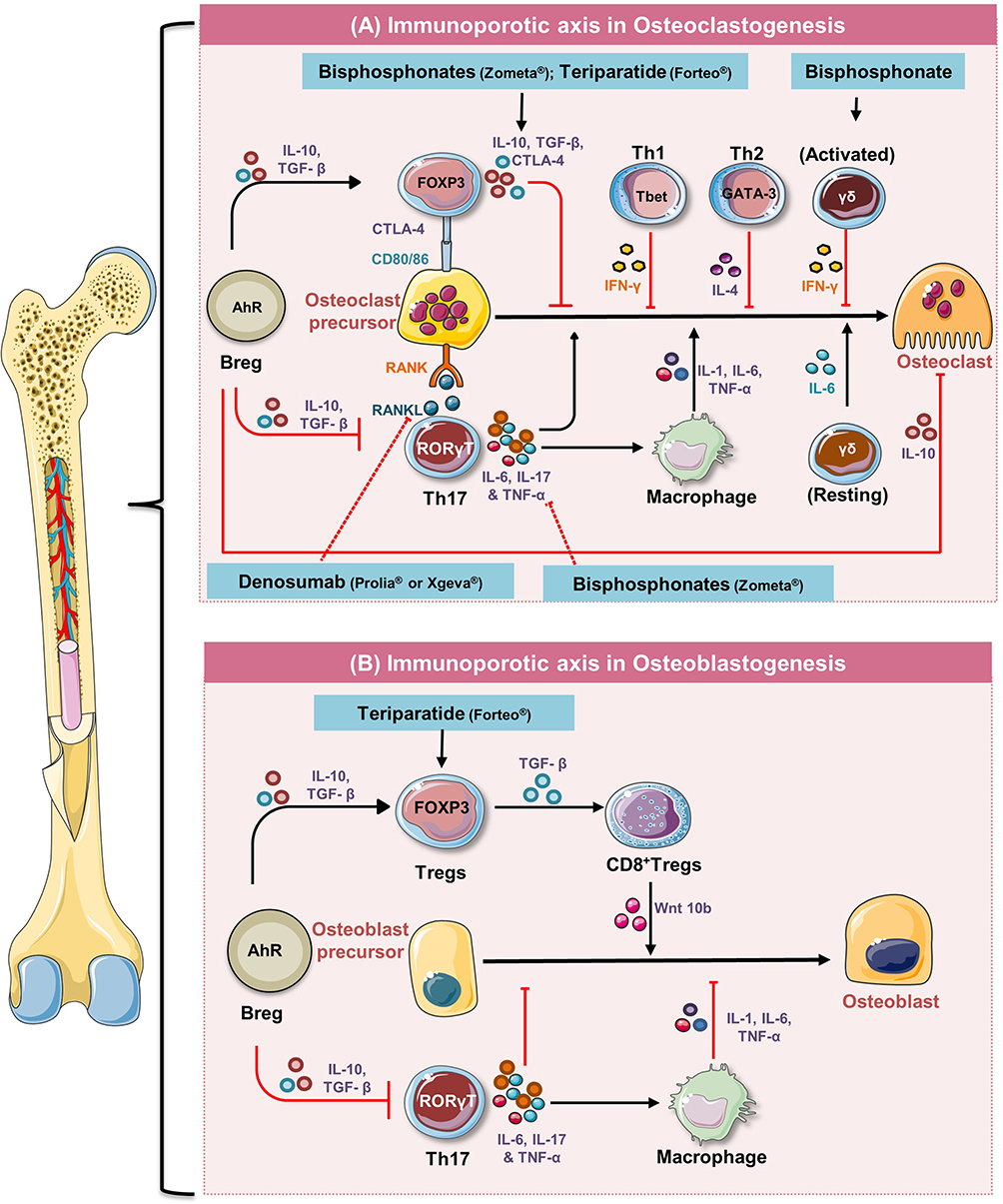 |
Figure 3 Immunomodulation of bone remodelling and potential therapeutic checkpoints: (A) Differentiation of osteoclasts is a multi-step process where various immune cells viz. Breg, Treg, Th1, and Th2 by producing anti-inflammatory cytokines such as IL-10, TGF-β, IFNγ, and IL-4 suppress the differentiation of osteoclasts precursors to mature osteoclasts and thus suppress osteoclastogenesis. On the contrary, by producing inflammatory cytokines Th17 enhance osteoclastogenesis either in a direct or indirect manner. (B) Tregs enhance osteoblastogenesis via CD8+ Tregs and Th17 suppress osteoblastogenesis via IL-17 cytokine or by enhancing macrophage mediated reduction of osteoblasts differentiation. Notes: Figure created with the help of https://smart.servier.com. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/legalcode). |
In 2007, a study revealed that IFN-γ plays a dual role in regulating the bone resorption process. It has been observed that IFN-γ exhibits the potential of directly blunting osteoclastogenesis, nevertheless it can also promote OCs generation by inducing expression of RANKL on activated T cells.82 Moreover, its direct effect on osteoclastogenesis is dependent on the stage of OCs differentiation. In the early stage, it suppresses osteoclastogenesis by targeting the RANK–RANKL signaling pathway wherein at later stages it promotes differentiation by encouraging fusion of mononuclear cells to form multinucleated OCs. In summary, these shreds of evidence point towards pro and anti-osteoclastogenic functions of IFN-γ. Few studies indicated that under estrogen-deficient conditions the net balance between these two opposing forces is biased towards bone resorption.82 But few studies suggested that this balance is biased more towards the anti-osteoclastogenic potential of IFN-γ in an ovariectomy-induced post-menopausal osteoporotic mice model.53 In addition, IFN-γ also cooperates with TLRs to inhibit osteoclastogenesis via suppressing the expression of RANK receptor and CSF1R gene that encodes the c-fms receptor for M-CSF.83 Recently, a study demonstrated that IFN inhibits OCs formation via regulating guanylate binding proteins (GBPs) such as GBP1, GBP2, GBP3, GBP5, and GBP7.84 Among these GBPs, GBP2 and GBP5 inhibit OCs differentiation and out of these two, GBP5 restricts both age-associated and inflammation-induced bone loss.84 The most relevant finding is that IFN-γ also acts as an anabolic agent whose absence leads to a lack of bone formation. Thus, altogether these studies shed light on the osteo-protective role of Th1 and Th2 cells in bone homeostasis that could act as a therapeutic target in the treatment and management of osteoporosis.
Tregs/Th17 Axis in Osteoporosis
Over the years, the regulatory T cells (Tregs) and inflammatory T cells (Th17) research field has undergone an incredible boom as the balance between these cells is important for sustaining immune homeostasis. Recently, our group along with others has reported that the imbalance between these immune cells plays a key role in the pathogenesis of bone-related diseases including osteoporosis6,54,85 (Figure 3). Th17 cells belong to T effector cell lineage, whereas both peripheral Tregs (pTregs) and thymus derived Tregs (tTregs) belong to regulatory lineage. Recent advancements in the research led to the discovery of markers viz. Helios and semaphorin receptor neuropilin-1 (Nrp-1) that can distinguish pTregs (CD4+Foxp3+Nrp-1−Helios−) from tTregs (CD4+Foxp3+Nrp-1+Helios+). A study reported that tTregs are involved in the maintenance of immune homeostasis and broad-spectrum autoimmune diseases, whereas pTregs are locally involved in controlling effector functions of T cells at the site of inflammation.86 A study demonstrated that tTregs induce tolerance against antigens that are produced by intestinal microbiota.87 It has been observed that estrogen signaling plays a vital role in inducing pTregs in the gut. Absence of an ERβ receptor in Tregs from Crohn’s disease patients induces aberrant expression of Tsc22d3 (GILZ) protein, a glucocorticoid responsive transcription factor not expressed in Tregs,88 suggesting that estrogen signaling suppresses the expression of GILZ protein, thereby inducing Tregs generation in the gut. Recently, based on proteomics and transcriptomics, Rianne et al89 discovered a marker GPA33 (Ig superfamily protein) expressed on tTregs that can be employed as a marker to distinguish human tTregs and pTregs. Generally, Tregs express the Foxp3 transcription factor and are characterized by the production of anti-inflammatory cytokines such as IL-10, TGF-β, etc. A study reported that CD4+CD25+Foxp3+ Tregs suppress osteoclastogenesis and bone resorptive functions of OCs via TGF-β1 and IL-10 cytokines in human PBMCs.90 Moreover, E2 at a concentration of 1 nM to 100 nM further augmented the anti-osteoclastogenic potential of Tregs via promoting the secretion of TGF-β1 and IL-10 cytokines,90 representing a potential therapeutic target for the treatment of postmenopausal osteoporosis. TGF-β1 is an abundant isoform of the TGF-β superfamily present in bone and is found to be a major modulator of OCs. OC precursors express both type I and type II TGF-β receptors and it has been observed that TGF-β1 (1 ng/mL) promotes OCs apoptosis by activating the intrinsic apoptosis pathway that includes caspase 9 and Bim.91 This also corresponds to the regulatory mechanism by which TGF-β1 modulates OCs activity. A study by Fischer et al92 demonstrated that Foxp3+ Tregs via expressing cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) interacts with costimulatory molecules CD80/86 expressed on OCs precursors and suppressed its differentiation. Upon this interaction, OCs precursors express a programmed death ligand-1 (PD-L1) that interacts with PD-1 on T cells, thus representing a crucial immune checkpoint. Moreover, osteoanabolic compound viz. teriparatide (parathyroid fragment) is reported to augment bone health in mice and humans by promoting 2–3-fold enhancement of the Tregs population.93 Thus, pharmacological enhancement of Tregs represents a novel bone anabolic therapy. Remarkably, via Foxp3 fate mapping, a study revealed that one particular type of Th17 cells (exFoxp3+ Th17 cells), derived from the Foxp3+ T cells showed much stronger osteoclastogenic potential as compared to the conventional Th17 cells.94 Moreover, it has been observed that IL-6 further aids in the conversion of Tregs to Th17 cells.
After the discovery of Th17 cells, Ivanov et al95 identified a Rorγt transcription factor that promotes differentiation of Th17 cells. Rorγt expression in Th17 cells is induced by the combinatorial action of TGF-β and IL-6 cytokines via STAT3. Th17 cells are osteoclastogenic subsets of T cells that are personified by the production of inflammatory cytokines such as IL-6, IL-17, TNF-α, RANKL, etc.5 In the case of autoimmune arthritis, bone loss is associated with the IL-23/IL-17A cytokine axis that links the activation of T cells with bone destruction.96 A study demonstrated that IL-17A is a crucial modulator of cortical bone loss rather than trabecular bone loss after ovariectomy. It has been observed that loss of trabecular bone in IL-17A knockout mice is associated with enhanced adipogenesis shown by the increased leptin levels.97 One of the severe complications associated with hyperparathyroidism is enhanced bone loss. By employing antibiotic-treated mice and germ-free mice, Yu et al98 demonstrated that PTH induced bone loss in the study having enriched microbiota with Th17 inducing segmented filamentous bacteria (SFB). SFB+ microbiota enabled the PTH to promote egress of TNF+ T cells and Th17 cells from the intestine to BM via a sphingosine-1-phosphate (S1P) receptor-1 mediated mechanism.98 The significance of Gut in modulating Th17 and TNF+ T cells in BM under ovariectomy conditions is further documented by Pacifici et al. They showed that the absence of estrogen hormone led to egress of intestinal Th17 and TNF+ T cells from the intestine via a S1P mediated mechanism and subsequently increased their migration to BM via CXCR3 and CCL-20.99 This study suggests that migratory blockade of intestinal Th17 cells could also be employed as a potent therapeutic strategy for the treatment of osteoporosis. Recently, a study reported that high frequencies of Th17 cells and IL-17 cytokine levels are correlated with lower BMDs in post-menopausal osteoporotic patients.100 Altogether, these studies clearly indicate that the homeostatic balance of the “Tregs-Th17 cells” axis is crucial for the maintenance of bone health in the case of osteoporosis.
Gamma-Delta (γδ) T Cells
A small subpopulation of T cells bears the T-cell receptor (TCR) composed of gamma (γ) and delta (δ) subunits. γδ T cells are innate-like lymphocytes that bridge innate and adaptive immunity. Unlike the conventional αβ T cells, γδ T cells are minor subpopulation of T cells present in low number in the peripheral circulation (1–5%).101 γδ T cells are abundant at barrier sites such as the gut, skin, reproductive tract, and lungs: almost 20% of human colonic intraepithelial lymphocytes (IELs) express γδ TCR on its surface.101 The role of γδ T cells is well studied in inflammatory and autoimmune disease conditions but its role in bone biology needs investigation. Amino bisphosphonates which are being used as an anti-osteoporotic drug are found to be potent activators of γδ T cells. Interestingly, a study showed that freshly isolated γδ T cells enhanced osteoclastogenesis, whereas activated γδ T cells [α-CD3/CD28+rhIL-2 or BrHPP (phosphoantigen) + rhIL-2] suppressed osteoclastogenesis.102 Furthermore, cytokine analysis and neutralization assays revealed that IL-6 and INF-γ cytokines dictate both pro and anti-osteoclastogenic roles of γδ T cells, respectively102 (Figure 3). In 2009, Colburn et al103 demonstrated that the development of fracture in γδ T cells knock out mice improved the biomechanical strength and stability at repair site due to a rise in the expression of bone sialoprotein and BMP. In contrast to this, a study reported that IL-17A producing γδ T cells contributes to the healing process after tissue injury associated with bone fracture and muscle damage.104 γδ17+ T cells are the versatile subset of cells that react quickly to innate stimuli and further strengthen the inflammatory functions of lymphoid and myeloid cells in various inflammatory and autoimmune diseases. In mice and humans, under inflammatory conditions, strong IL-23 signals induce de-novo differentiation of γδ17+ T cells even from uncommitted precursors. Based on these studies, it is probable to suggest the bone protective effect of γδ17+ T cells in osteoporosis, but unfortunately no study to date has ever investigated the potential of γδ17+ T cells in postmenopausal osteoporosis.
Th9 Cells
A recent identified subset of CD4+ T cells is Th9 cells that play a pivotal role in various inflammatory diseases, parasitic infection, anti-tumor immunity, etc. In the presence of IL-4 and TGF-β cytokines, naïve T cells differentiate into Th9 cells. In mice, Th9 cells secrete IL-9 and IL-10 cytokines whereas human Th9 only secretes the IL-9 cytokine. Differentiation of Th9 cells requires the involvement of transcription factors such as STAT6, interferon response factor 4 (IRF4), PU.1, and GATA-3.105 A study demonstrated that Th9 cells are found to be positively correlated with RA. Higher levels of IL-9 cytokine in synovial fluid of RA patients promote the survival of neutrophils and induce the differentiation of Th17 cells which are evidenced by the increase in RORγt and STAT3 phosphorylation. Th9 cells are known to harbor both inflammatory and anti-inflammatory potential, thus indicating functional heterogeneity. Of note, a single-cell gene expression analysis revealed the existence of two subsets of Th9 cell lineage: CD96high (low inflammatory) and CD96low (high inflammatory). It has been observed that adoptive transfer of CD96low Th9 cells into Rag−/− mice led to chronic inflammation, whereas CD96high did not show colitis development, suggesting the inhibitory role of CD96 in Th9 cells.106 Recently, a study reported by Kar et al showed higher levels of IL-9 cytokine in serum and SF of RA patients, pointing towards an inflammatory role of IL-9 in the pathophysiology of bone related diseases. In addition, they observed that IL-9 cytokines modulates the expression of several genes involved in osteoclastogenesis such as matrix metalloproteinases (MMPs) and significantly enhanced the OCs differentiation.107 This study strongly suggests that blocking of IL-9 cytokine could be an attractive target to prevent bone deterioration in several bone pathologies including osteoporosis. However, the role of Th9 cells in the case of osteoporosis is still lacking and warrants further research.
Th22 Cells
Another novel subset of T cells is Th22 cells whose differentiation is positively regulated by RORγt and negatively regulated by T-bet transcription factors.108 Recently, the aryl hydrocarbon receptor (AhR) is found to be associated with its differentiation and functional activity. One study showed that the IL-21 cytokine induces IL-22 expression in CD4+ T cells by activating STAT3 which in turn regulates the epigenetic status of il22 promoter and its interaction with the AhR.109 Since the IL-22 cytokine is produced by ILCs and CD4+ T cells and plays a crucial role in host defense and mucosal immunity, highlighting its possible contribution to bone-related disease. A study demonstrated that Th22 cells by secreting IL-22 cytokine promote OCs differentiation by inducing NFATc1 expression in CD14+ monocytes and enhanced bone deterioration in RA patients. The data indicated that, via expressing chemokine receptors viz. CCR4, CCR6, and CCR10, Th22 migrates to the inflamed synovial site where the ligands of these chemokine receptor CCL28 are highly expressed, suggesting a pivotal role of Th22 in bone destruction observed in RA patients.110 Th22 cells are also known to induce Th17 cell differentiation via DCs.111 Further, a study found that the IL-22 cytokine via enhancing the RANKL expression induced the OCs differentiation and caused bone resorption in sub-chondral bone in temporomandibular joint osteoarthritis (TMJ-OA) and in periodontitis.112 Altogether, these studies indicate that blockade of the IL-22 cytokine may be employed as a therapeutic strategy for the treatment of bone-related diseases including osteoporosis.
T Follicular Helper Cells (TFH)–T Follicular Regulatory Cells (TFR) Axis in Osteoporosis
TFH differentiation is a multi-step procedure that involves B cell lymphoma-6 (Bcl-6) transcription factor. TFH is a special T cell help provider to B cells that are important for germinal center reaction, affinity maturation, and high-affinity antibody production along with the generation of memory B cells. In RA patients, it has been observed that frequencies of CD4+CXCR5+ICOShigh TFH cells are positively correlated with the anti-cyclic citrullinated peptide (CCP) antibodies. TFH cells mediate its inflammatory function by producing IL-21 cytokines, thus pointing towards the possible involvement of TFH in RA progression.113 In auto-immune diseases viz. RA, TFH promotes the auto-antibody generation by B cells and leads to the progression of disease whereas recently discovered TFR suppress the generation of autoantibodies in RA. One study revealed that enhanced IL-6/pSTAT3 signaling was found to be responsible for the elevated TFH/ TFR cell ratio in inflammatory bone loss conditions.114 Surprisingly, in parasitic infections, TFH was found to be the source of RANKL cytokine that further enhance bone loss in parasite-infected patients.115 ERα is a receptor for female sexual estrogen hormone that inhibits the TFH response by suppressing the expression of Bcl-6 and IL-21 cytokine, thereby inhibiting autoantibody production in autoimmune diseases.116 ERα is found to be crucial for bone homeostasis that modulates the RANKL expression and thus suppresses RANKL mediated osteoclastogenesis. Deficiency of ERα leads to enhanced bone loss in ERα knock-out mice.117 Altogether, these studies indicate the probable contribution of TFH in bone destruction associated with osteoporosis. To date, only one study reported the role of TFR in the prevention of inflammation-induced bone loss in RA, however, studies in osteoporosis still need further investigation.
Memory T Cells
Memory T cells are generated after first antigenic exposure and are crucial to sustain long lasting immunological responses against these antigens. Memory T cells normally reside in BM as it provides an appropriate niche for their self renewal. Recently, a study reported a novel mechanism via which estrogen modulates bone health. It has been observed that in estrogen-deficient conditions, DCs become long-lived and express higher levels of IL-7 and IL-15 cytokines. These cytokines together induce antigen-independent production of IL-17A and TNF-α cytokines by memory T cells.9 Recently, Ohmachi et al118 demonstrated that Th2 dominant OVA-specific T cells induced bone loss in a food allergic enteropathy mice model via increasing effector memory T cells (CD4+CD44hiCD62Llo) in mesenteric lymph nodes and BM. This novel report thus provides the crucial role of effector memory T cells in allergy and bone loss. These studies indicate that there could be the possibility of an existence of an antigenic entity against which memory T cells are specifically generated, but further studies are required for identification of this antigenic entity which would thereby prime osteoporosis to be included under the category of autoimmune diseases.
Invariant Natural Killer T (iNKT) Cells
iNKT cells or classical NKT cells are a distinct population of T cells that express invariant TCRVα14Jα18 chain on cell surface. iNKT cells are activated by glycolipids which are presented by MHC-class I like molecules (ie, CD1d). iNKT cells exhibit the potential to modulate both innate and adaptive immunity via activating myeloid DCs and Mϕs. In the Innate Immune Cells section, we discussed that both DCs and Mϕs can give rise to OCs which orchestrate the bone remodeling process. Interestingly, a study demonstrated that activation of iNKT cells with α-galactosylceramide enhances the egress of myeloid cells, OCs progenitor, and enhances the bone resorptive activity of OCs.119 These data thus demonstrate the novel mechanism of iNKT cells where it couples the osteoclastogenesis process with the egress of myeloid cells during immune activation conditions. In consistent to this, recently it has also been observed that iNKT cells showed higher expression of RANKL in osteoporotic patients having a T score of −2.5 in comparison to control groups,120 suggesting the critical contribution of iNKT cells in inducing bone loss even under osteoporotic conditions.
B Cells
In addition to the immune-related functions, B cells share a multifaceted relationship with the bone cells. Emerging evidence highlights the potential of B cells in osteo-immunological regulation that is further supported by the physiological and anatomical coexistence of B cells and bone cells in BM.121 Recently, several findings suggested that B cells are the active modulator of the RANKL/OPG axis and the production of RANKL by B cells further augmented bone loss in ovariectomy induced bone loss.122 Interestingly, a report suggested that along with enhancing the OCs differentiation RANKL also increases B cell number that further acts as an OCs supporter cell.123 Along with affecting the OCs differentiation, B cells also negatively affect the differentiation of OBs. It has also been observed that by producing higher levels of OBs inhibitors such as CCL3 and TNF, B cells suppress OBs differentiation by activating extracellular signal-regulated kinase (ERK) and NF-κB signaling pathway that leads to dysfunctioning of OBs which in turn causes the inhibition of bone formation in a RA mice model.124 Bone deterioration potential of B cells is further augumented in an osteoporotic mice model where treatment with E2 enhanced bone health by reducing the percentages of B cells throughout its developmental stages in an ovariectomized mice model.125 Extensive research revealed the ability of B cells to mediate their immunomodulatory properties independent of secreting antibodies, ie, “non-humoral” functions of B cells. In further sections, we discuss the regulatory effect of B cells in inflammatory bone loss conditions. Among B cell lineage, regulatory B cells (Bregs) are the immunosuppressive subset of B cells that are known to suppress inflammation in various autoimmune and inflammatory conditions.
Regulatory B Cells (Bregs)
The discovery of suppressor B cells dates to the 1970s and later after 30 years, these suppressor B cells were termed as Bregs. In the last few years, the number of different subsets of Bregs has increased steadily which confirms the emerging interest in understanding the significance of Bregs in modulating immune responses in various inflammatory diseases including osteoporosis. Bregs are potent regulators of immune response which prevents unwarranted inflammation after infection or injury. Various studies in murine models and clinical settings have reported that Bregs exhibits distinct phenotypic markers which could be grouped into distinct subsets and the action of these Bregs depends on specific pathological conditions. In the arthritis model, it has been observed that, via producing IL-10 cytokine, Bregs suppressed the development of disease.126 In 2008, Tedder et al127 identified a subpopulation of Bregs, ie, CD19+CD1dhiCD5+ B cells, which is greatly enriched for IL-10 cytokine. Recently, our group demonstrated for the first time that IL-10 producing CD19+CD1dhiCD5+ Bregs possess anti-osteoclastogenic properties.85 Of note, our group also revealed that reduction in the frequency of CD19+CD1dhiCD5+ Bregs along with their tendency to produce IL-10 cytokine is significantly reduced in an osteoporotic mice model. This pioneer study thereby opens novel avenues in the field of cellular immunotherapy for the treatment and management of osteoporosis. Inconsistent to our study, Wang et al128 further demonstrated that adoptive transfer of B10 cells in ovariectomized mice decelerated the development of osteoporosis. During the normal healing process, it has been observed that frequencies of IL-10 producing Bregs (IgM+CD27+) were observed to be significantly enhanced during the early healing state (6 weeks post-surgery) in bone fracture patients, highlighting the ability of Bregs in mediating the fracture healing process. The inhibitory mechanism of Bregs is primarily but not exclusively associated with the IL-10 cytokine. The IL-35 cytokine is a new emerging immunomodulator that belongs to the IL-12 cytokine family.129 One of the mechanisms by which IL-35 mediates its immunosuppressive functions is by inducing Breg cell development. It also suppresses bone resorption by inducing OPG production along with simultaneously reducing RANKL expression. Wang et al130 reported that IL-35 converts human B cells into IL-35 producing Bregs (IL-35+ Bregs) by activating the STAT1/STAT3 signaling pathway via an IL-35 receptor. To date, multiple subsets of Bregs have been identified in both humans and mice but, unfortunately, only limited studies reported the potential of a particular subset in modulating bone health in various bone-related anomalies (Table 2).
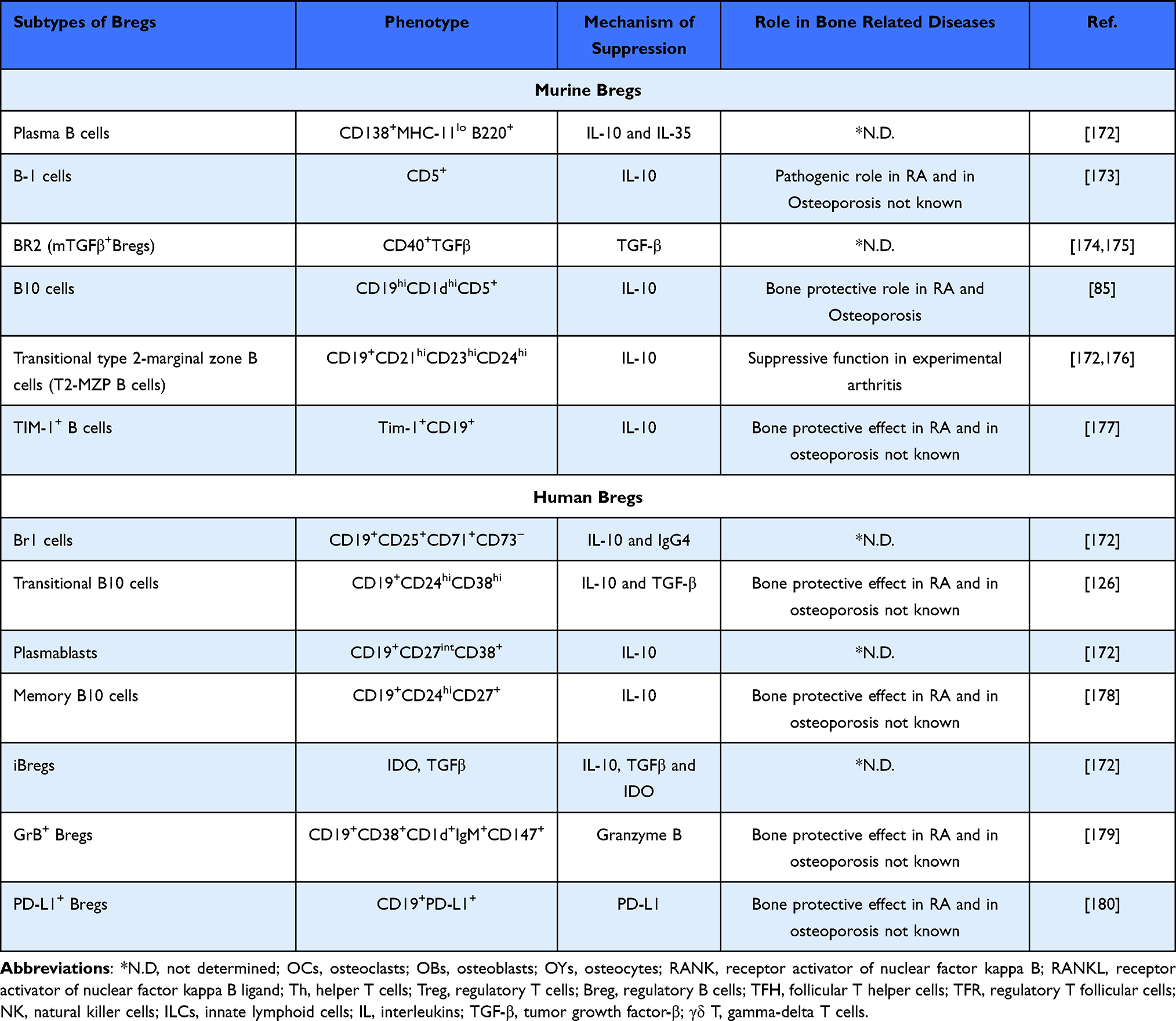 |
Table 2 Subsets of Bregs in Mice and Humans |
Currently Available Treatment Strategies Highlighting Immunoporosis
Currently, various pharmacological therapies are being employed for the treatment of osteoporosis. These therapeutic agents are further categorized under two divisions: Anti-resorptive drugs that reduce bone resorption via suppressing OCs activity and bone anabolic compounds that enhance the formation of bone via stimulating OBs function. Anti-resorptive drugs include denosumab, bisphosphonates, hormonal therapy, calcitonin, vitamin D, and calcium supplementation. Bone anabolic drugs include strontium ranelate, teriparatide, and recently available romosozumab. Bisphosphonates (BPs) are the first-line therapy for the treatment of osteoporosis. BPs include zoledronic acid, alendronate, risedronate, and ibandronate, which reduce the risk of vertebral fractures in postmenopausal osteoporosis. Recently, concern has been raised with the long-term administration of BPs. Apart from inducing well-recognized inhibitory effects on OCs functional activity, it also exhibits immunomodulatory potential where treatment with BPs reduced the levels of IL-6, IL-17, IL-23, and IFNγ cytokines along with simultaneously inducing significant enhancement in the levels of IL-4, IL-10, and TGF-β cytokines in postmenopausal osteoporotic patients.131 Thus, it can be considered that BPs might be affecting the Breg/ Treg /Th17 cell balance via modulating their signature cytokines (Figure 3).
Denosumab, a human monoclonal antibody, prevents the interaction of RANKL with its RANK receptor on OCs precursors, thereby suppressing the maturation and functional activity of OCs. In addition to OCs precursors, RANK receptors are also expressed on monocytes, macrophages, and DCs; and RANKL plays a vital role in the development of the immune system which forms the reason behind the observed enhanced risk of serious infections in denosumab treated patients. Teriparatide, which is a recombinant fragment of PTH, exhibits potent osteoanabolic potential. Along with inducing bone formation by OBs, teriparatide causes 2–3-fold enhancement in the frequencies of Tregs in both mice and humans93 (Figure 3). Moreover, a study revealed that blockade of Tregs enhancement also abolished the bone-forming potential of parathyroid hormone, representing a novel osteo-anabolic therapeutic strategy. Altogether, these studies further highlight the importance of the immune system in maintaining skeletal homeostasis as the majority of the first-line therapies that are currently being employed for the treatment of osteoporosis exhibit immunomodulatory potential via regulating the “Immunoporotic axis”.
Inflammaging: A Driving Force in Osteoporosis?
With advancing age, low grade inflammation creeps in and contributes towards the pathogenesis of several age-related diseases, termed “Inflammaging”. Inflammaging is a low grade chronic inflammation which is characterized by a complex network of inflammatory and anti-inflammatory responses. Age-related diseases such as osteoporosis and ageing share the fundamental mechanistic pillars which largely congregate on inflammation. The major contributor of inflammatory stimuli is represented by the self/endogenous, pathogens (non-self), altered molecules resulting from the damaged or dead cells, and gut microbiota (quasi self)132. These molecules are recognized by the immune cells (viz. macrophages) which are key players in the process of inflammaging.133 Levels of altered molecules increase with age along with declined removal of these molecules by proteasomal degradation pathways (autophagy or mitophagy).133 In old age, this may lead to remarkable enhancement in bone resorption (decline in BMD) which is predominantly due to the transition from osteoblastogenesis to adipogenesis in BM.134 Remarkably, a study reported that aging also led to enhancement in the numbers of BMDMs skewed more towards the M1 phenotype indicating towards enhanced osteoclastogenesis and thus more bone resorption.135 Moreover, inflammaging cytokines such as IL-17 and IFN-γ enhance osteoclastogenesis in BMMs. Altogether inflammaging should be considered as one of the crucial unexplored niches for therapeutic targeting of osteoporosis (Figure 4).
 |
Figure 4 Inflammaging and Osteoporosis: (A) Various factors such as dysfunctioning of mitochondria, autophagy/mitophagy, dysbiosis of gut microbiota (GM), senescence of cells, cell debris, pathogen associated molecular patterns (PAMPs), death associated molecular patterns (DAMPs) and altered metabolites promote the activation of NLR family pyrin domain containing 3 inflammasome in macrophages. Activation of macrophages leads to conversion of the inactive form of IL-1β cytokine to the active form along with IL-18 cytokine. These osteoclastogenic cytokines further skew the balance towards bone resorption, thereby enhancing bone loss. (B) Dogma representing the nexus between ageing, Inflammaging, and age-related diseases including osteoporosis. Notes: Figure created with the help of https://smart.servier.com. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/legalcode). |
Harnessing the Immunoporotic Role of Gut Microbiota (GM)
Cumulative data from recent reports clearly emphasize the role of the immune system in the pathophysiology of osteoporosis. Multiple parameters regulate the development of the immune system, among them Gut-Microbiota (GM) plays a vital role in the development of the host’s innate and adaptive immune system. GM are the colonized indigenous microorganisms that inhabit the bodies of animals and humans and are collectively referred to as microbiota. Recently, our group reported that interplay between GM and the immune system is believed to contribute to the pathogenesis of various immune-mediated diseases including osteoporosis.136 Rosser et al137,138 demonstrated that GM via producing IL-6 and IL-1β cytokines promotes the development of IL-10 producing Bregs, and in RA dysbiosis of GM impaired the immunosuppressive abilities of Bregs. Dysbiosis of the GM and loss of gut permeability were also found to be the profound causative factors for the development of osteoporosis.139 Our group, along with others, reported that modification of the GM, via probiotics (a viable micro-organism), could be a potential therapeutic strategy in regulating bone health, ie, Osteomicrobiology.136 Among various immune cells, the intricate balance between Tregs and Th17 plays a vital role in bone homeostasis. Our group has reported that probiotics administration viz. Lactobacillus acidophilus, Bacillus clausii and Lactobacillus rhamnosus attenuates inflammatory bone loss in an osteoporotic mice model by modulating the Th17/Tregs cell balance.53,54,140
Under physiological conditions, it has been observed that differentiation of naïve T cells into Tregs and Th17 is further regulated by Bregs. Several studies reported that, via IL-10 and TGF-β cytokines, Bregs restrains differentiation of Th17 cells along with simultaneously inducing differentiation of Foxp3+ Tregs. In both mice and human studies, it has been observed that the immunosuppressive phenotype of Bregs is controlled by the expression of the AhR transcription factor. AhR is a ligand stimulated transcription factor that perceive signals from microbial, dietary, and metabolic stimuli that binds to the IL10 locus in Bregs. Relevance of AhR in inducing Bregs is further supported by a study where B cell-specific deletion of AhR aggravated arthritis in a mice model by reducing the differentiation of Tregs along with enhancing the expansion of Th17 cells.141 In search of identifying a suitable stimulus apart from cytokine stimuli that activate AhR in B cells, Rosser et al demonstrated that microbial derived short chain fatty acid (SCFA) viz. butyrate supports Bregs functionality by amplifying the activation of AhR and by reprogramming the differentiation of B cells towards Bregs. Notably, butyrate promotes Bregs differentiation via enhancing the production of a tryptophan derived metabolite viz. 5-hydroxyindole-3-acetic acid (5-HIAA). Markedly, other SCFAs such as acetate and pentanoate also enhanced IL-10 production by B cells. Acetate increased IL-10 production in mice by B1a cells and CD19+CD24hiCD27+ B cells in humans via enhancing the production of acetyl coenzyme A, ultimately fueling the tricarboxylic acid (TCA) cycle for energy metabolism. In addition, a fiber rich diet intervention can also enhance acetate levels and thus B10 Breg differentiation in human PBMCs. Pentanoate potentiate IL-10 production in CpG stimulated mice B cells by enhancing glycolysis and mTOR activation in B cells. Activation of Bregs via these metabolites further enhances the downstream differentiation of Tregs along with inhibition of Th17 cells. These studies along with our own work clearly highlight that modulation of dietary habits could be exploited as a novel, safe, and effective approach in modulating the pivotal “Bregs-Treg-Th17” cell axis in osteoporosis (Figure 5).
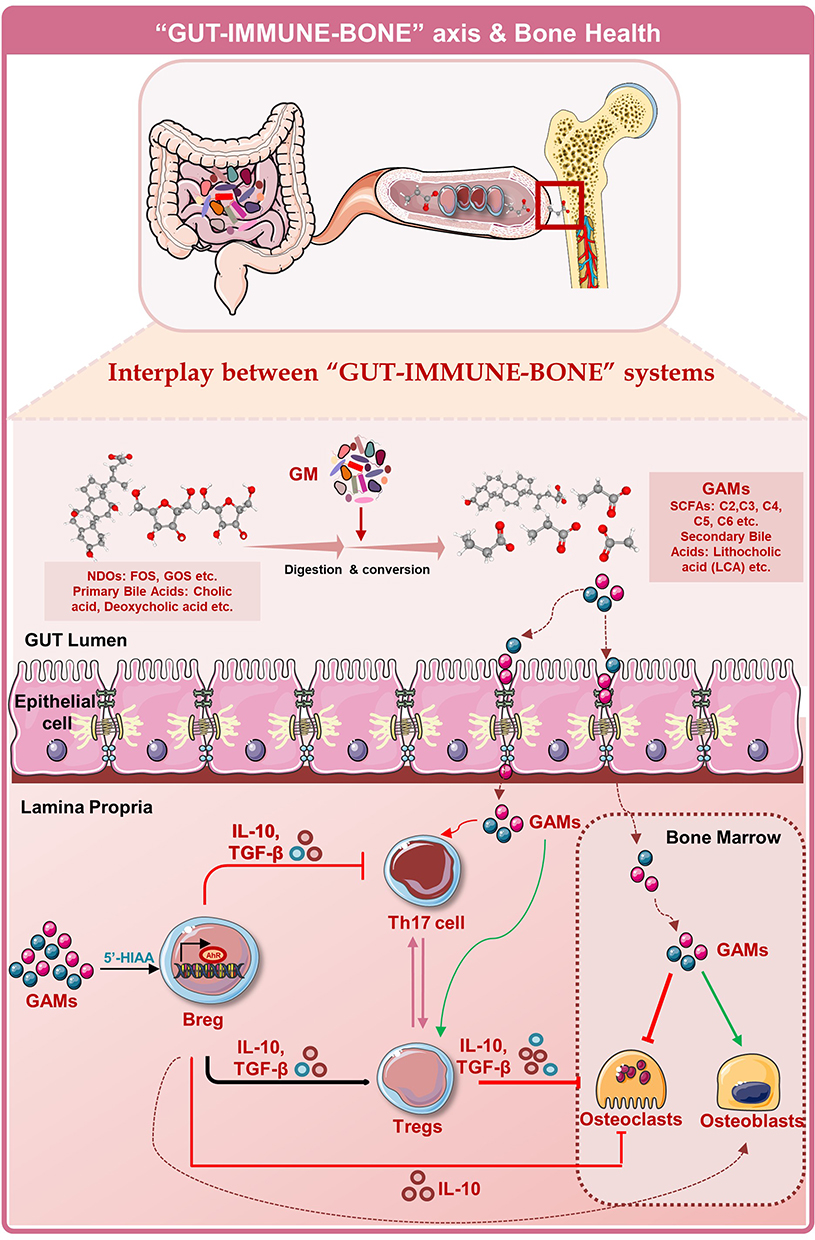 |
Figure 5 Harnessing “GUT-IMMUNE-BONE” axis in Bone Health: GUT microbiota (GM) acts on the non-digestible carbohydrates (NDOs) such as fructo-oligosaccharides (FOS), galacto-oligosaccharides (GOS) etc. and convert them into various Gut Associate Metabolites (GAMs): Short chain fatty acids (SCFAs), ie, acetate (C2), propionate (C3), butyrate (C4), pentanoate (C5), and hexanoate (C6). Moreover, primary bile acids produced by liver such as cholic acid and deoxycholic acid are converted into secondary bile acids (by the GM) such as lithocholic acid (LCA), etc. These GAMs can cross the intestinal lining and upon entry into lamina propria modulate Breg, Tregs, and Th17 cells which further regulate bone remodelling after reaching BM. Also, GAMs can directly regulate bone remodelling via the peripheral circulation, thereby maintaining bone health. Notes: Figure created with the help of https://smart.servier.com. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/legalcode). |
In 1995, Marcel Roberfroid and Glenn Gibson introduced the concept of prebiotics, which is described as non-digestible food that imparts beneficial affects on the host by promoting the growth and activity of gut microbial species in the colon and thus maintains host health. In comparison to probiotics, the use of prebiotics has been observed to be more valuable due to the susceptibility and vulnerability of probiotics to environmental stresses such as heat and the vanishing conditions during storage.142 These novel dietary fibers enhance calcium, inorganic phosphorus (Pi), and magnesium absorption in the intestine of both preclinical and clinical models. These effects are primarily due to the interaction of prebiotics and GM in the large intestine. Prebiotics enhance mineral absorption in the intestine via regulating various modifications in the host including GM composition, SCFAs production, immune cells regulation, altering the intestinal pH, etc. Prebiotics include non-digestible oligosaccharides such as galacto-oligosaccharides, inulin type fructans, fructo-oligosaccharides (FOS), oligofructose, etc., and exhibit bone health modulating potential.143 These prebiotics exhibit the potential to stimulate the growth of bifidobacteria and to a lesser extent lactobacillus. In addition, a study reported that treatment with FOS significantly enhanced bone health in Ovx rats by enhancing the butyrate levels in serum.144
To overcome the survival difficulties of probiotics, the concept of synbiotics was further introduced. Synbiotic is the mixture of prebiotics and probiotics that imparts beneficial effects on the host by enhancing the survival and functional activity of gut microbiota. Bacterial genes are responsible for encoding the enyzmes that produce the metabolites such as SCFAs, branched chain fatty aids, vitamins, and bile acid derivatives. Interestingly, the production of these metabolites is dependent on the accessibility of substrate produced by the prebiotics and GM. A study investigated the effect of synbiotic fermented milk comprising Lactobacillus paracasei and inulin on bone health in post-menopausal osteoporotic women.145 It was observed that consumption of synbiotic fermented milk followed by a single dose of soy significantly increased the bioavailibity of isoflavones that further improved the metabolism of estrogen hormone and bone health in post-menopausal women, thereby alleviating the risk of osteoporosis.145 Altogether, these studies provide potential checkpoints which can be targeted for the treatment and management of osteoporosis.
Conclusion and Future Outlook
In the past two decades, growing advancements in the field of osteoimmunology have enhanced our understanding of the interrelation between immune and bone cells in maintaining skeletal health. Nevertheless, the full realization of various immune cells with immunoporotic potential such as DCs, ILCs, Th9, Th22, TFH, Bregs, etc., will require deeper insight, thereby opening novel avenues for future research. Existing research has primarily focused on the role of both innate and adaptive immunity in bone destruction observed in osteoporosis, but studies reported by Cline-Smith et al9 and Iseme et al146 strongly hint towards the role of memory T cells and auto-antibodies in the pathophysiology of osteoporosis. These studies suggest that osteoporosis could have an autoimmune component, but further research is required to confirm the same.
Current therapies available for the treatment of osteoporosis have long-term adverse health affects. Recently, the Food and Drug Administration (FDA) has approved Evenity® (romosozumab-aqqg) for treatment of osteoporosis. It is a monoclonal antibody which blocks the protein sclerostin (secreted by osteocytes to suppress bone formation) and thus enhances bone formation. However, it has been observed that it may also enhance the risk of cardiovascular diseases and stroke, thereby cautioning its use in patients with cardiac history. Recently, a study also highlighted the novel role of sclerostin in regulation of inflammation and myelopoiesis in BM. It has been observed that short-term depletion of sclerostin alters immune cell development in the BM and spleen.147 In addition, depletion of sclerostin correlates with enhanced levels of inflammatory cytokines such as MCP-1, TNF-α, etc,147 thereby, questioning the employment of sclerostin in postmenopausal osteoporotic patients. Thus, in search for a safe, novel, and cost-effective therapeutic strategy our group along with other’s have reported that probiotics exhibit immunoporotic potential, thereby enhancing bone health in several bone anomalies including osteoporosis. Generally, biotics (prebiotics, probiotics, or synbiotics) work by modulating the GM and the significance of GM in modulating the immune system has already been established. Various evidences suggests that GM plays a vital role in maintaining the homeostatic balance of different subsets of ILCs and vice versa. Loosening of the gut barrier integrity is also one of the major causative factors for the onset and progression of osteoporosis. Thus, it is plausible to suggest that the dysregulated “ILC1-ILC2-ILC3-ILCreg” axis would increase the risk of osteoporosis (Figure 6). In summary, we conclude that alterations in the dietary interventions would be a better strategy to enhance bone health via modulating the “GUT–IMMUNE–BONE” axis.
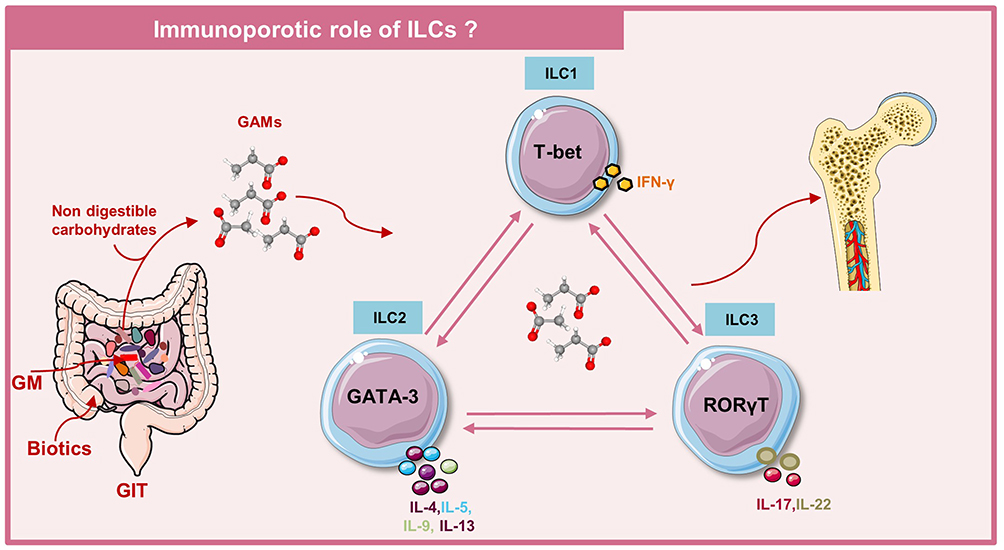 |
Figure 6 Immunoporotic role of ILCs: Schematic representation of proposed mechanism of the role of ILCs in Osteoporosis. GM modulation strategies (prebiotics, probiotics, and synbiotics) can be employed for enhancing bone health via modulating the plasticity of ILCs (ILC1, ILC2, and ILC3). Notes: Figure created with the help of https://smart.servier.com. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/legalcode). |
Acknowledgments
This work was financially supported by projects: DST-SERB (EMR/2016/007158), DBT(BT/PR41958/MED/97/524/2021) Govt. of India, Intramural project from All India Institute of Medical Sciences (AIIMS), New Delhi-India (AI-798 and AC-21) project sanctioned to RKS. LS and RKS acknowledge the Department of Biotechnology, AIIMS, New Delhi-India for providing infrastructural facilities. LS thanks UGC for the research fellowship. Figures are created with the help of https://smart.servier.com.
Author Contributions
Both authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. de Villiers TJ, Goldstein SR. Bone health 2022: an update. Climacteric. 2022;25(1):1–3. doi:10.1080/13697137.2021.1965408
2. Dobbs MB, Buckwalter J, Saltzman C. Osteoporosis: the increasing role of the orthopaedist. Iowa Orthop J. 1999;19:43–52. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10847516
3. Arron JR, Choi Y. Bone versus immune system. Nature. 2000;408(6812):535–536. doi:10.1038/35046196
4. Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473(2):139–146. doi:10.1016/j.abb.2008.03.018
5. Srivastava RK. Osteoimmunology The i Nexus i between bone and immune system. Front Biosci. 2018;23(2):4600. doi:10.2741/4600
6. Srivastava RK, Dar HY, Mishra PK. Immunoporosis: immunology of osteoporosis—role of T cells. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.00657
7. Sapra L, Azam Z, Rani L, et al. “Immunoporosis”: immunology of osteoporosis. Proc Natl Acad Sci India Sect B Biol Sci. 2021;91:511–519. doi:10.1007/s40011-021-01238-x
8. Cawley KM, Bustamante-Gomez NC, Guha AG, et al. Local production of osteoprotegerin by osteoblasts suppresses bone resorption. Cell Rep. 2020;32(10):108052. doi:10.1016/j.celrep.2020.108052
9. Cline‐Smith A, Axelbaum A, Shashkova E, et al. Ovariectomy activates chronic low‐grade inflammation mediated by memory T cells, which promotes osteoporosis in mice. J Bone Miner Res. 2020;35(6):1174–1187. doi:10.1002/jbmr.3966
10. Xing L, Bushnell TP, Carlson L, et al. NF-κB p50 and p52 expression is not required for RANK-expressing osteoclast progenitor formation but is essential for RANK- and cytokine-mediated osteoclastogenesis. J Bone Miner Res. 2002;17(7):1200–1210. doi:10.1359/jbmr.2002.17.7.1200
11. von Gunten S, Simon H-U. Linking glucocorticoid-induced osteoporosis to osteoimmunology. Cell Death Dis. 2020;11(12):1026. doi:10.1038/s41419-020-03250-x
12. Tao Z, Wang J, Wen K, et al. Pyroptosis in osteoblasts: a novel hypothesis underlying the pathogenesis of osteoporosis. Front Endocrinol (Lausanne). 2021;11. doi:10.3389/fendo.2020.548812
13. Frech M, Schuster G, Andes FT, Schett G, Zaiss MM, Sarter K. RANKL-induced Btn2a2 – a T cell immunomodulatory molecule – during osteoclast differentiation fine-tunes bone resorption. Front Endocrinol (Lausanne). 2021;12. doi:10.3389/fendo.2021.685060
14. Gallois A, Lachuer J, Yvert G, et al. Genome-wide expression analyses establish dendritic cells as a new osteoclast precursor able to generate bone-resorbing cells more efficiently than monocytes. J Bone Miner Res. 2010;25(3):661–672. doi:10.1359/jbmr.090829
15. Alnaeeli M, Teng Y-TA. Dendritic cells differentiate into osteoclasts in bone marrow microenvironment in vivo. Blood. 2009;113(1):264–265. doi:10.1182/blood-2008-09-180836
16. Rivollier A, Mazzorana M, Tebib J, et al. Immature dendritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood. 2004;104(13):4029–4037. doi:10.1182/blood-2004-01-0041
17. Maitra R, Follenzi A, Yaghoobian A, et al. Dendritic cell-mediated in vivo bone resorption. J Immunol. 2010;185(3):1485–1491. doi:10.4049/jimmunol.0903560
18. Chen Y, Wang Y, Tang R, et al. Dendritic cells-derived interferon-λ1 ameliorated inflammatory bone destruction through inhibiting osteoclastogenesis. Cell Death Dis. 2020;11(6):414. doi:10.1038/s41419-020-2612-z
19. Kylmäoja E, Nakamura M, Turunen S, et al. Peripheral blood monocytes show increased osteoclast differentiation potential compared to bone marrow monocytes. Heliyon. 2018;4(9):e00780. doi:10.1016/j.heliyon.2018.e00780
20. Deng F-Y, Lei S-F, Zhang Y, et al. Peripheral blood monocyte-expressed ANXA2 gene is involved in pathogenesis of osteoporosis in humans. Mol Cell Proteomics. 2011;10(11):
21. Batoon L, Millard SM, Wullschleger ME, et al. CD169+ macrophages are critical for osteoblast maintenance and promote intramembranous and endochondral ossification during bone repair. Biomaterials. 2019;196:51–66. doi:10.1016/j.biomaterials.2017.10.033
22. Yamaguchi T, Movila A, Kataoka S, et al. Proinflammatory M1 macrophages inhibit RANKL-induced osteoclastogenesis. Infect Immun. 2016;84(10):2802–2812. doi:10.1128/IAI.00461-16
23. Dou C, Ding N, Zhao C, et al. Estrogen deficiency-mediated M2 macrophage osteoclastogenesis contributes to M1/M2 ratio alteration in ovariectomized osteoporotic mice. J Bone Miner Res. 2018;33(5):899–908. doi:10.1002/jbmr.3364
24. Yang J, Park O, Kim J, Kwon Y, Yun C, Han SH. Modulation of macrophage subtypes by IRF5 determines osteoclastogenic potential. J Cell Physiol. 2019;234(12):23033–23042. doi:10.1002/jcp.28863
25. Huang R, Wang X, Zhou Y, Xiao Y. RANKL-induced M1 macrophages are involved in bone formation. Bone Res. 2017;5(1):17019. doi:10.1038/boneres.2017.19
26. Théry C. Exosomes: secreted vesicles and intercellular communications. F1000 Biol Rep. 2011;3. doi:10.3410/B3-15
27. Wei F, Li M, Crawford R, Zhou Y, Xiao Y. Exosome-integrated titanium oxide nanotubes for targeted bone regeneration. Acta Biomater. 2019;86:480–492. doi:10.1016/j.actbio.2019.01.006
28. Xiong Y, Chen L, Yan C, et al. M2 Macrophagy-derived exosomal miRNA-5106 induces bone mesenchymal stem cells towards osteoblastic fate by targeting salt-inducible kinase 2 and 3. J Nanobiotechnol. 2020;18(1):66. doi:10.1186/s12951-020-00622-5
29. Chamberlain CS, Kink JA, Wildenauer LA, et al. Exosome-educated macrophages and exosomes differentially improve ligament healing. Stem Cells. 2021;39(1):55–61. doi:10.1002/stem.3291
30. Bordbar A, Mo ML, Nakayasu ES, et al. Model‐driven multi‐omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol Syst Biol. 2012;8(1):558. doi:10.1038/msb.2012.21
31. Kim J-M, Jeong D, Kang HK, Jung SY, Kang SS, Min B-M. Osteoclast precursors display dynamic metabolic shifts toward accelerated glucose metabolism at an early stage of RANKL-stimulated osteoclast differentiation. Cell Physiol Biochem. 2007;20(6):935–946. doi:10.1159/000110454
32. Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517(7534):293–301. doi:10.1038/nature14189
33. Eberl G, Colonna M, Di Santo JP, McKenzie ANJ. Innate lymphoid cells: a new paradigm in immunology. Science (80-). 2015;348(6237):aaa6566–aaa6566. doi:10.1126/science.aaa6566
34. Hirota K, Hashimoto M, Ito Y, et al. Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity. 2018;48(6):1220–1232.e5. doi:10.1016/j.immuni.2018.04.009
35. Geremia A, Arancibia-Cárcamo CV, Fleming MPP, et al. IL-23–responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. 2011;208(6):1127–1133. doi:10.1084/jem.20101712
36. Kwong B, Rua R, Gao Y, et al. T-bet-dependent NKp46+ innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat Immunol. 2017;18(10):1117–1127. doi:10.1038/ni.3816
37. Fazekas B, Moreno-Olivera A, Kelly Y, et al. Alterations in circulating lymphoid cell populations in systemic small vessel vasculitis are non-specific manifestations of renal injury. Clin Exp Immunol. 2018;191(2):180–188. doi:10.1111/cei.13058
38. Ke K, Chen T, Arra M, Mbalaviele G, Swarnkar G, Abu‐Amer Y. Attenuation of NF‐κB in intestinal epithelial cells is sufficient to mitigate the bone loss comorbidity of experimental mouse colitis. J Bone Miner Res. 2019;34(10):1880–1893. doi:10.1002/jbmr.3759
39. Mikami Y, Scarno G, Zitti B, et al. NCR + ILC3 maintain larger STAT4 reservoir via T‐BET to regulate type 1 features upon IL‐23 stimulation in mice. Eur J Immunol. 2018;48(7):1174–1180. doi:10.1002/eji.201847480
40. Bernink JH, Krabbendam L, Germar K, et al. Interleukin-12 and −23 control plasticity of CD127+ group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity. 2015;43(1):146–160. doi:10.1016/j.immuni.2015.06.019
41. Takayanagi H, Ogasawara K, Hida S, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature. 2000;408(6812):600–605. doi:10.1038/35046102
42. Omata Y, Frech M, Lucas S, et al. Type 2 innate lymphoid cells inhibit the differentiation of osteoclasts and protect from ovariectomy-induced bone loss. Bone. 2020;136:115335. doi:10.1016/j.bone.2020.115335
43. Cephus J, Fuseini H, Davis JB, Yung JA, Sucharski H, Newcomb DC. Estrogen receptor alpha signaling increases ILC2-mediated allergic airway inflammation. J Allergy Clin Immunol. 2019;143(2):AB2. doi:10.1016/j.jaci.2018.12.006
44. Omata Y, Frech M, Primbs T, et al. Group 2 innate lymphoid cells attenuate inflammatory arthritis and protect from bone destruction in mice. Cell Rep. 2018;24(1):169–180. doi:10.1016/j.celrep.2018.06.005
45. Seehus CR, Kadavallore A, de la Torre B, et al. Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat Commun. 2017;8(1):1900. doi:10.1038/s41467-017-02023-z
46. Howard E, Lewis G, Galle-Treger L, et al. IL-10 production by ILC2s requires Blimp-1 and cMaf, modulates cellular metabolism, and ameliorates airway hyperreactivity. J Allergy Clin Immunol. 2021;147(4):1281–1295.e5. doi:10.1016/j.jaci.2020.08.024
47. Momiuchi Y, Motomura Y, Suga E, et al. Group 2 innate lymphoid cells in bone marrow regulate osteoclastogenesis in a reciprocal manner via RANKL, GM-CSF and IL-13. Int Immunol. 2021;33(11):573–585. doi:10.1093/intimm/dxab062
48. Di Luccia B, Gilfillan S, Cella M, Colonna M, Huang S-C-C. ILC3s integrate glycolysis and mitochondrial production of reactive oxygen species to fulfill activation demands. J Exp Med. 2019;216(10):2231–2241. doi:10.1084/jem.20180549
49. Takaki-Kuwahara A, Arinobu Y, Miyawaki K, et al. CCR6+ group 3 innate lymphoid cells accumulate in inflamed joints in rheumatoid arthritis and produce Th17 cytokines. Arthritis Res Ther. 2019;21(1):198. doi:10.1186/s13075-019-1984-x
50. Leijten EFA, Van Kempen TS, Boes M, et al. Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol. 2015;67(10):2673–2678. doi:10.1002/art.39261
51. Ciccia F, Guggino G, Rizzo A, et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis. 2015;74(9):1739–1747. doi:10.1136/annrheumdis-2014-206323
52. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7(9):e44552. doi:10.1371/journal.pone.0044552
53. Dar HY, Shukla P, Mishra PK, et al. Lactobacillus acidophilus inhibits bone loss and increases bone heterogeneity in osteoporotic mice via modulating Treg-Th17 cell balance. Bone Reports. 2018;8:46–56. doi:10.1016/j.bonr.2018.02.001
54. Sapra L, Dar HY, Bhardwaj A, et al. Lactobacillus rhamnosus attenuates bone loss and maintains bone health by skewing Treg-Th17 cell balance in Ovx mice. Sci Rep. 2021;11(1):1807. doi:10.1038/s41598-020-80536-2
55. Wang S, Xia P, Chen Y, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell. 2017;171(1):201–216.e18. doi:10.1016/j.cell.2017.07.027
56. Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18(5):693–704. doi:10.1038/nm.2755
57. Biosse-Duplan M, Baroukh B, Dy M, de Vernejoul M-C, Saffar J-L. Histamine promotes osteoclastogenesis through the differential expression of histamine receptors on osteoclasts and osteoblasts. Am J Pathol. 2009;174(4):1426–1434. doi:10.2353/ajpath.2009.080871
58. Kroner J, Kovtun A, Kemmler J, et al. Mast cells are critical regulators of bone fracture-induced inflammation and osteoclast formation and activity. J Bone Miner Res. 2017;32(12):2431–2444. doi:10.1002/jbmr.3234
59. Zechner C, Gruntmanis U. Systemic mastocytosis with decreased bone density and fractures. Mayo Clin Proc. 2015;90(6):843–844. doi:10.1016/j.mayocp.2014.10.014
60. Lind T, Gustafson A-M, Calounova G, et al. Increased bone mass in female mice lacking mast cell chymase. PLoS One. 2016;11(12):e0167964. doi:10.1371/journal.pone.0167964
61. Fischer V, Ragipoglu D, Diedrich J, et al. Mast cells trigger disturbed bone healing in osteoporotic mice. J Bone Miner Res. 2021. doi:10.1002/jbmr.4455
62. Ginaldi L, De Martinis M, Ciccarelli F, et al. Increased levels of interleukin 31 (IL-31) in osteoporosis. BMC Immunol. 2015;16(1):60. doi:10.1186/s12865-015-0125-9
63. Moonen CGJ, de Vries TJ, Rijkschroeff P, Poubelle PE, Nicu EA, Loos BG. The possible role of neutrophils in the induction of osteoclastogenesis. J Immunol Res. 2019;2019:1–14. doi:10.1155/2019/8672604
64. Chakravarti A, Raquil M-A, Tessier P, Poubelle PE. Surface RANKL of Toll-like receptor 4–stimulated human neutrophils activates osteoclastic bone resorption. Blood. 2009;114(8):1633–1644. doi:10.1182/blood-2008-09-178301
65. Sugisaki R, Miyamoto Y, Yoshimura K, et al. Possible involvement of elastase in enhanced osteoclast differentiation by neutrophils through degradation of osteoprotegerin. Bone. 2020;132:115216. doi:10.1016/j.bone.2019.115216
66. Yilmaz H, Uyfun M, Yilmaz TS, et al. Neutrophil-lymphocyte ratio may be superior to C-reactive protein for predicting the occurrence of postmenopausal osteoporosis. Endocr Regul. 2014;48(01):25–33. doi:10.4149/endo_2014_01_25
67. Jiang J, Liu X, Lai B, et al. Correlational analysis between neutrophil granulocyte levels and osteonecrosis of the femoral head. BMC Musculoskelet Disord. 2019;20(1):393. doi:10.1186/s12891-019-2778-7
68. Bastian OW, Koenderman L, Alblas J, Leenen LPH, Blokhuis TJ. Neutrophils contribute to fracture healing by synthesizing fibronectin + extracellular matrix rapidly after injury. Clin Immunol. 2016;164:78–84. doi:10.1016/j.clim.2016.02.001
69. Kovtun A, Bergdolt S, Wiegner R, Radermacher P, Huber-Lang M, Ignatius A. The crucial role of neutrophil granulocytes in bone fracture healing. Eur Cells Mater. 2016;32:152–162. doi:10.22203/eCM.v032a10
70. Fischer KD, Agrawal DK. Hematopoietic stem and progenitor cells in inflammation and allergy. Front Immunol. 2013;4. doi:10.3389/fimmu.2013.00428
71. Ginaldi L, De Martinis M, Saitta S, et al. Interleukin-33 serum levels in postmenopausal women with osteoporosis. Sci Rep. 2019;9(1):3786. doi:10.1038/s41598-019-40212-6
72. Soderstrom K, Stein E, Colmenero P, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci. 2010;107(29):13028–13033. doi:10.1073/pnas.1000546107
73. Feng S, Madsen SH, Viller NN, et al. Interleukin-15-activated natural killer cells kill autologous osteoclasts via LFA-1, DNAM-1 and TRAIL, and inhibit osteoclast-mediated bone erosion in vitro. Immunology. 2015;145(3):367–379. doi:10.1111/imm.12449
74. Vandenhaute J, Avau A, Filtjens J, et al. Regulatory role for NK cells in a mouse model of systemic juvenile idiopathic arthritis. J Immunol. 2019;203(12):3339–3348. doi:10.4049/jimmunol.1900510
75. Takeda H, Kikuchi T, Soboku K, et al. Effect of IL-15 and natural killer cells on osteoclasts and osteoblasts in a mouse coculture. Inflammation. 2014;37(3):657–669. doi:10.1007/s10753-013-9782-0
76. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136(7):2348–2357.
77. Zhang J, Fu Q, Ren Z, et al. Changes of serum cytokines-related Th1/Th2/Th17 concentration in patients with postmenopausal osteoporosis. Gynecol Endocrinol. 2015;31(3):183–190. doi:10.3109/09513590.2014.975683
78. Mangashetti LS, Khapli SM, Wani MR. IL-4 inhibits bone-resorbing activity of mature osteoclasts by affecting NF-κB and Ca 2+ signaling. J Immunol. 2005;175(2):917–925. doi:10.4049/jimmunol.175.2.917
79. Yamada A, Takami M, Kawawa T, et al. Interleukin-4 inhibition of osteoclast differentiation is stronger than that of interleukin-13 and they are equivalent for induction of osteoprotegerin production from osteoblasts. Immunology. 2007;120(4):573–579. doi:10.1111/j.1365-2567.2006.02538.x
80. Bendixen AC, Shevde NK, Dienger KM, Willson TM, Funk CD, Pike JW. IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator-activated receptor 1. Proc Natl Acad Sci. 2001;98(5):2443–2448. doi:10.1073/pnas.041493198
81. Moreno JL, Kaczmarek M, Keegan AD, Tondravi M. IL-4 suppresses osteoclast development and mature osteoclast function by a STAT6-dependent mechanism: irreversible inhibition of the differentiation program activated by RANKL. Blood. 2003;102(3):1078–1086. doi:10.1182/blood-2002-11-3437
82. Gao Y, Grassi F, Ryan MR, et al. IFN-γ stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117(1):122–132. doi:10.1172/JCI30074
83. Ji J-D, Park-Min K-H, Shen Z, et al. Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-γ in human osteoclast precursors. J Immunol. 2009;183(11):7223–7233. doi:10.4049/jimmunol.0900072
84. Place DE, Malireddi RKS, Kim J, Vogel P, Yamamoto M, Kanneganti T-D. Osteoclast fusion and bone loss are restricted by interferon inducible guanylate binding proteins. Nat Commun. 2021;12(1):496. doi:10.1038/s41467-020-20807-8
85. Sapra L, Bhardwaj A, Mishra PK, et al. Regulatory B cells (Bregs) inhibit osteoclastogenesis and play a potential role in ameliorating ovariectomy-induced bone loss. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.691081
86. Yadav M, Stephan S, Bluestone JA. Peripherally induced tregs – role in immune homeostasis and autoimmunity. Front Immunol. 2013;4. doi:10.3389/fimmu.2013.00232
87. Cebula A, Seweryn M, Rempala GA, et al. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature. 2013;497(7448):258–262. doi:10.1038/nature12079
88. Goodman WA, Bedoyan SM, Havran HL, Richardson B, Cameron MJ, Pizarro TT. Impaired estrogen signaling underlies regulatory T cell loss-of-function in the chronically inflamed intestine. Proc Natl Acad Sci U S A. 2020;117(29):17166–17176. doi:10.1073/pnas.2002266117
89. Opstelten R, de Kivit S, Slot MC, et al. GPA33: a marker to identify stable human regulatory T cells. J Immunol. 2020;204(12):3139–3148. doi:10.4049/jimmunol.1901250
90. Luo C, Wang L, Sun C, Li D. Estrogen enhances the functions of CD4+CD25+Foxp3+ regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol. 2011;8(1):50–58. doi:10.1038/cmi.2010.54
91. Houde N, Chamoux E, Bisson M, Roux S. Transforming growth factor-β1 (TGF-β1) induces human osteoclast apoptosis by up-regulating bim. J Biol Chem. 2009;284(35):23397–23404. doi:10.1074/jbc.M109.019372
92. Fischer L, Herkner C, Kitte R, et al. Foxp3+ regulatory T cells in bone and hematopoietic homeostasis. Front Endocrinol (Lausanne). 2019;10. doi:10.3389/fendo.2019.00578
93. Yu M, D’Amelio P, Tyagi AM, et al. Regulatory T cells are expanded by Teriparatide treatment in humans and mediate intermittent PTH ‐induced bone anabolism in mice. EMBO Rep. 2018;19(1):156–171. doi:10.15252/embr.201744421
94. Tsukasaki M, Takayanagi H. Osteoimmunology: evolving concepts in bone–immune interactions in health and disease. Nat Rev Immunol. 2019;19(10):626–642. doi:10.1038/s41577-019-0178-8
95. Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor rorγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–1133. doi:10.1016/j.cell.2006.07.035
96. Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–2682. doi:10.1084/jem.20061775
97. Scheffler JM, Grahnemo L, Engdahl C, et al. Interleukin 17A: a Janus-faced regulator of osteoporosis. Sci Rep. 2020;10(1):5692. doi:10.1038/s41598-020-62562-2
98. Yu M, Malik Tyagi A, Li J-Y, et al. PTH induces bone loss via microbial-dependent expansion of intestinal TNF+ T cells and Th17 cells. Nat Commun. 2020;11(1):468. doi:10.1038/s41467-019-14148-4
99. Yu M, Pal S, Paterson CW, et al. Ovariectomy induces bone loss via microbial-dependent trafficking of intestinal TNF+ T cells and Th17 cells. J Clin Invest. 2021;131(4). doi:10.1172/JCI143137
100. Bhadricha H, Patel V, Singh AK, et al. Increased frequency of Th17 cells and IL-17 levels are associated with low bone mineral density in postmenopausal women. Sci Rep. 2021;11(1):16155. doi:10.1038/s41598-021-95640-0
101. Wo J, Zhang F, Li Z, Sun C, Zhang W, Sun G. The role of gamma-delta T cells in diseases of the central nervous system. Front Immunol. 2020;11. doi:10.3389/fimmu.2020.580304
102. Phalke SP, Chiplunkar SV. Activation status of γδ T cells dictates their effect on osteoclast generation and bone resorption. Bone Reports. 2015;3:95–103. doi:10.1016/j.bonr.2015.10.004
103. Colburn NT, Zaal KJM, Wang F, Tuan RS. A role for γ/δ T cells in a mouse model of fracture healing. Arthritis Rheum. 2009;60(6):1694–1703. doi:10.1002/art.24520
104. Ono T, Okamoto K, Nakashima T, et al. IL-17-producing γδ T cells enhance bone regeneration. Nat Commun. 2016;7(1):10928. doi:10.1038/ncomms10928
105. Kaplan MH. Th9 cells: differentiation and disease. Immunol Rev. 2013;252(1):104–115. doi:10.1111/imr.12028
106. Stanko K, Iwert C, Appelt C, et al. CD96 expression determines the inflammatory potential of IL-9–producing Th9 cells. Proc Natl Acad Sci. 2018;115(13):E2940–E2949. doi:10.1073/pnas.1708329115
107. Kar S, Gupta R, Malhotra R, et al. Interleukin-9 facilitates osteoclastogenesis in rheumatoid arthritis. Int J Mol Sci. 2021;22(19):10397. doi:10.3390/ijms221910397
108. Plank MW, Kaiko GE, Maltby S, et al. Th22 cells form a distinct Th lineage from Th17 cells in vitro with unique transcriptional properties and tbet-dependent Th1 plasticity. J Immunol. 2017;198(5):2182–2190. doi:10.4049/jimmunol.1601480
109. Yeste A, Mascanfroni ID, Nadeau M, et al. IL-21 induces IL-22 production in CD4+ T cells. Nat Commun. 2014;5(1):3753. doi:10.1038/ncomms4753
110. Miyazaki Y, Nakayamada S, Kubo S, et al. Th22 cells promote osteoclast differentiation via production of IL-22 in rheumatoid arthritis. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.02901
111. Shi L, Ji Q, Liu L, et al. IL‐22 produced by Th22 cells aggravates atherosclerosis development in ApoE −/− mice by enhancing DC‐induced Th17 cell proliferation. J Cell Mol Med. 2020;24(5):3064–3078. doi:10.1111/jcmm.14967
112. Monasterio G, Budini V, Fernández B, et al. IL-22-expressing CD4 + AhR + T lymphocytes are associated with RANKL-mediated alveolar bone resorption during experimental periodontitis. J Periodontal Res. 2019;54(5):513–524. doi:10.1111/jre.12654
113. Ma J, Zhu C, Ma B, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol. 2012;2012:1–7. doi:10.1155/2012/827480
114. Niu Q, Huang Z, Wu X, et al. Enhanced IL-6/phosphorylated STAT3 signaling is related to the imbalance of circulating T follicular helper/T follicular regulatory cells in patients with rheumatoid arthritis. Arthritis Res Ther. 2018;20(1):200. doi:10.1186/s13075-018-1690-0
115. Li W, Wei C, Xu L, et al. Schistosome infection promotes osteoclast-mediated bone loss. PLOS Pathog. 2021;17(3):e1009462. doi:10.1371/journal.ppat.1009462
116. Kim D-H, Park H-J, Park H-S, et al. Estrogen receptor α in T cells suppresses follicular helper T cell responses and prevents autoimmunity. Exp Mol Med. 2019;51(4):1–9. doi:10.1038/s12276-019-0237-z
117. Streicher C, Heyny A, Andrukhova O, et al. Estrogen regulates bone turnover by targeting RANKL expression in bone lining cells. Sci Rep. 2017;7(1):6460. doi:10.1038/s41598-017-06614-0
118. Ono-Ohmachi A, Yamada S, Uno S, et al. Effector memory CD4+T cells in mesenteric lymph nodes mediate bone loss in food-allergic enteropathy model mice, creating IL-4 dominance. Mucosal Immunol. 2021;14(6):1335–1346. doi:10.1038/s41385-021-00434-2
119. Hu M, Bassett JHD, Danks L, et al. Activated invariant NKT cells regulate osteoclast development and function. J Immunol. 2011;186(5):2910–2917. doi:10.4049/jimmunol.1002353
120. Tilkeridis K, Kiziridis G, Ververidis A, et al. Immunoporosis: a new role for invariant natural killer T (NKT) cells through overexpression of nuclear factor-κB ligand (RANKL). Med Sci Monit. 2019;25:2151–2158. doi:10.12659/MSM.912119
121. Li S, Liu Q, Wu D, et al. PKC-δ deficiency in B cells displays osteopenia accompanied with upregulation of RANKL expression and osteoclast–osteoblast uncoupling. Cell Death Dis. 2020;11(9):762. doi:10.1038/s41419-020-02947-3
122. Onal M, Xiong J, Chen X, et al. Receptor activator of nuclear factor κB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem. 2012;287(35):29851–29860. doi:10.1074/jbc.M112.377945
123. Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O’Brien CA. RANKL (receptor activator of nfκb ligand) produced by osteocytes is required for the increase in B cells and bone loss caused by estrogen deficiency in mice. J Biol Chem. 2016;291(48):24838–24850. doi:10.1074/jbc.M116.742452
124. Sun W, Meednu N, Rosenberg A, et al. B cells inhibit bone formation in rheumatoid arthritis by suppressing osteoblast differentiation. Nat Commun. 2018;9(1):5127. doi:10.1038/s41467-018-07626-8
125. Bernardi AI, Andersson A, Grahnemo L, et al. Effects of lasofoxifene and bazedoxifene on B cell development and function. Immun Inflamm Dis. 2014;2(4):214–225. doi:10.1002/iid3.37
126. Flores-Borja F, Bosma A, Ng D, et al. CD19 + CD24 hi CD38 hi B cells maintain regulatory T cells while limiting T H 1 and T H 17 differentiation. Sci Transl Med. 2013;5:173. doi:10.1126/scitranslmed.3005407
127. Yanaba K, Bouaziz J-D, Haas KM, Poe JC, Fujimoto M, Tedder TF. A rB cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28(5):639–650. doi:10.1016/j.immuni.2008.03.017
128. Sun G, Wang Y, Ti Y, Wang J, Zhao J, Qian H. Regulatory B cell is critical in bone union process through suppressing proinflammatory cytokines and stimulating Foxp3 in Treg cells. Clin Exp Pharmacol Physiol. 2017;44(4):455–462. doi:10.1111/1440-1681.12719
129. Sakkas LI, Mavropoulos A, Perricone C, Bogdanos DP. IL-35: a new immunomodulator in autoimmune rheumatic diseases. Immunol Res. 2018;66(3):305–312. doi:10.1007/s12026-018-8998-3
130. Wang R-X, Yu C-R, Dambuza IM, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. 2014;20(6):633–641. doi:10.1038/nm.3554
131. Talaat RM, Sidek A, Mosalem A, Kholief A. Effect of bisphosphonates treatment on cytokine imbalance between TH17 and Treg in osteoporosis. Inflammopharmacology. 2015;23(2–3):119–125. doi:10.1007/s10787-015-0233-4
132. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14(10):576–590. doi:10.1038/s41574-018-0059-4
133. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab. 2017;28(3):199–212. doi:10.1016/j.tem.2016.09.005
134. Demontiero O, Vidal C, Duque G. Aging and bone loss: new insights for the clinician. Ther Adv Musculoskelet Dis. 2012;4(2):61–76. doi:10.1177/1759720X11430858
135. Kim O-H, Kim H, Kang J, et al. Impaired phagocytosis of apoptotic cells causes accumulation of bone marrow-derived macrophages in aged mice. BMB Rep. 2017;50(1):43–48. doi:10.5483/BMBRep.2017.50.1.167
136. Bhardwaj A, Sapra L, Tiwari A, Mishra PK, Sharma S, Srivastava RK. “Osteomicrobiology”: the nexus between bone and bugs. Front Microbiol. 2022;12. doi:10.3389/fmicb.2021.812466
137. Rosser EC, Oleinika K, Tonon S, et al. Regulatory B cells are induced by gut microbiota–driven interleukin-1β and interleukin-6 production. Nat Med. 2014;20(11):1334–1339. doi:10.1038/nm.3680
138. Rosser EC, Piper CJM, Matei DE, et al. Microbiota-derived metabolites suppress arthritis by amplifying Aryl-hydrocarbon receptor activation in regulatory B cells. Cell Metab. 2020;31(4):837–851.e10. doi:10.1016/j.cmet.2020.03.003
139. Tyagi AM, Darby TM, Hsu E, et al. The gut microbiota is a transmissible determinant of skeletal maturation. Elife. 2021;10. doi:10.7554/eLife.64237
140. Dar HY, Pal S, Shukla P, et al. Bacillus clausii inhibits bone loss by skewing Treg-Th17 cell equilibrium in postmenopausal osteoporotic mice model. Nutrition. 2018;54:118–128. doi:10.1016/j.nut.2018.02.013
141. Piper CJM, Rosser EC, Oleinika K, et al. Aryl hydrocarbon receptor contributes to the transcriptional program of IL-10-producing regulatory B cells. Cell Rep. 2019;29(7):1878–1892.e7. doi:10.1016/j.celrep.2019.10.018
142. Lilian Ilesanmi-Oyelere B, Cathorina Kruger M. Prebiotics, probiotics and synbiotic for bone health. In: Prebiotics and Probiotics - from Food to Health [Working Title]. IntechOpen; 2021. doi10.5772/intechopen.100525
143. Whisner CM, Castillo LF. Prebiotics, bone and mineral metabolism. Calcif Tissue Int. 2018;102(4):443–479. doi:10.1007/s00223-017-0339-3
144. Porwal K, Pal S, Kulkarni C, et al. A prebiotic, short-chain fructo-oligosaccharides promotes peak bone mass and maintains bone mass in ovariectomized rats by an osteogenic mechanism. Biomed Pharmacother. 2020;129:110448. doi:10.1016/j.biopha.2020.110448
145. Timan P, Rojanasthien N, Manorot M, Sangdee C, Teekachunhatean S. Effect of synbiotic fermented milk on oral bioavailability of isoflavones in postmenopausal women. Int J Food Sci Nutr. 2014;65(6):761–767. doi:10.3109/09637486.2014.908169
146. Iseme RA, Mcevoy M, Kelly B, Agnew L, Walker FR, Attia J. Is osteoporosis an autoimmune mediated disorder? Bone Reports. 2017;7:121–131. doi:10.1016/j.bonr.2017.10.003
147. Donham C, Chicana B, Robling AG, et al. Sclerostin depletion induces inflammation in the bone marrow of mice. Int J Mol Sci. 2021;22(17):9111. doi:10.3390/ijms22179111
148. Mbalaviele G, Novack DV, Schett G, Teitelbaum SL. Inflammatory osteolysis: a conspiracy against bone. J Clin Invest. 2017;127(6):2030–2039. doi:10.1172/JCI93356
149. Shiratori T, Kyumoto-Nakamura Y, Kukita A, et al. IL-1β induces pathologically activated osteoclasts bearing extremely high levels of resorbing activity: a possible pathological subpopulation of osteoclasts, accompanied by suppressed expression of kindlin-3 and talin-1. J Immunol. 2018;200(1):218–228. doi:10.4049/jimmunol.1602035
150. Hengartner N-E, Fiedler J, Ignatius A, Brenner RE. IL-1β inhibits human osteoblast migration. Mol Med. 2013;19(1):36–42. doi:10.2119/molmed.2012.00058
151. Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-κB signaling pathways. J Biol Chem. 2008;283(17):11535–11540. doi:10.1074/jbc.M607999200
152. Bendre MS, Montague DC, Peery T, Akel NS, Gaddy D, Suva LJ. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone. 2003;33(1):28–37. doi:10.1016/S8756-3282(03)00086-3
153. Okabe I, Kikuchi T, Mogi M, et al. IL-15 and RANKL play a synergistically important role in osteoclastogenesis. J Cell Biochem. 2017;118(4):739–747. doi:10.1002/jcb.25726
154. Loro E, Ramaswamy G, Chandra A, et al. IL15RA is required for osteoblast function and bone mineralization. Bone. 2017;103:20–30. doi:10.1016/j.bone.2017.06.003
155. Song L, Tan J, Wang Z, et al. Interleukin‑17A facilitates osteoclast differentiation and bone resorption via activation of autophagy in mouse bone marrow macrophages. Mol Med Rep. 2019. doi:10.3892/mmr.2019.10155
156. Kim Y-G, Park J-W, Lee J-M, et al. IL-17 inhibits osteoblast differentiation and bone regeneration in rat. Arch Oral Biol. 2014;59(9):897–905. doi:10.1016/j.archoralbio.2014.05.009
157. Mansoori MN, Shukla P, Kakaji M, et al. IL-18BP is decreased in osteoporotic women: prevents Inflammasome mediated IL-18 activation and reduces Th17 differentiation. Sci Rep. 2016;6(1):33680. doi:10.1038/srep33680
158. Makiishi-Shimobayashi C, Tsujimura T, Iwasaki T, et al. Interleukin-18 up-regulates osteoprotegerin expression in stromal/osteoblastic cells. Biochem Biophys Res Commun. 2001;281(2):361–366. doi:10.1006/bbrc.2001.4380
159. Bolzoni M, Ronchetti D, Storti P, et al. IL21R expressing CD14 + CD16 + monocytes expand in multiple myeloma patients leading to increased osteoclasts. Haematologica. 2017;102(4):773–784. doi:10.3324/haematol.2016.153841
160. El-Zayadi AA, Jones EA, Churchman SM, et al. Interleukin-22 drives the proliferation, migration and osteogenic differentiation of mesenchymal stem cells: a novel cytokine that could contribute to new bone formation in spondyloarthropathies. Rheumatology. 2016;kew384. doi:10.1093/rheumatology/kew384
161. De Martinis M, Sirufo MM, Suppa M, Ginaldi L. IL-33/IL-31 axis in osteoporosis. Int J Mol Sci. 2020;21(4):1239. doi:10.3390/ijms21041239
162. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106(12):1481–1488. doi:10.1172/JCI11176
163. Gilbert L, He X, Farmer P, et al. Inhibition of osteoblast differentiation by tumor necrosis factor-α. Endocrinology. 2000;141(11):3956–3964. doi:10.1210/endo.141.11.7739
164. Cao X. RANKL-RANK signaling regulates osteoblast differentiation and bone formation. Bone Res. 2018;6(1):35. doi:10.1038/s41413-018-0040-9
165. Zhang N, Lo C-W, Utsunomiya T, et al. PDGF-BB and IL-4 co-overexpression is a potential strategy to enhance mesenchymal stem cell-based bone regeneration. Stem Cell Res Ther. 2021;12(1):40. doi:10.1186/s13287-020-02086-8
166. Xiong Y, Yan C, Chen L, et al. IL‐10 induces MC3T3‐E1 cells differentiation towards osteoblastic fate in murine model. J Cell Mol Med. 2020;24(1):1076–1086. doi:10.1111/jcmm.14832
167. Lee B, Oh Y, Jo S, Kim T-H, Ji JD. A dual role of TGF-β in human osteoclast differentiation mediated by Smad1 versus Smad3 signaling. Immunol Lett. 2019;206:33–40. doi:10.1016/j.imlet.2018.12.003
168. Chen G, Deng C, Li Y-P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci. 2012;8(2):272–288. doi:10.7150/ijbs.2929
169. Wan H, Qian T-Y, Hu X-J, Huang C-Y, Yao W-F. Correlation of serum CCL3/MIP-1α levels with disease severity in postmenopausal osteoporotic females. Balkan Med J. 2018;35(4):320–325. doi:10.4274/balkanmedj.2017.1165
170. Fu R, Liu H, Zhao S, et al. Osteoblast inhibition by chemokine cytokine ligand3 in myeloma-induced bone disease. Cancer Cell Int. 2014;14(1):132. doi:10.1186/s12935-014-0132-6
171. Brylka LJ, Schinke T. Chemokines in physiological and pathological bone remodeling. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.02182
172. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42(4):607–612. doi:10.1016/j.immuni.2015.04.005
173. Deng J, Wang X, Chen Q, et al. B1a cells play a pathogenic role in the development of autoimmune arthritis. Oncotarget. 2016;7(15):19299–19311. doi:10.18632/oncotarget.8244
174. Parekh VV, Prasad DVR, Banerjee PP, Joshi BN, Kumar A, Mishra GC. B cells activated by lipopolysaccharide, but not by anti-Ig and anti-CD40 antibody, induce anergy in CD8 + T cells: role of TGF-β1. J Immunol. 2003;170(12):5897–5911. doi:10.4049/jimmunol.170.12.5897
175. Dar HY, Rani L, Sapra L, et al. A chronological journey of breg subsets: implications in health and disease. In Systems and Synthetic Immunology. Springer Singapore; 2020:125–152. doi:10.1007/978-981-15-3350-1_5
176. Evans KE, Fox SW. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007;8(1):4. doi:10.1186/1471-2121-8-4
177. Ding Q, Yeung M, Camirand G, et al. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. J Clin Invest. 2011;121(9):3645–3656. doi:10.1172/JCI46274
178. Daien CI, Gailhac S, Mura T, et al. Regulatory B10 cells are decreased in patients with rheumatoid arthritis and are inversely correlated with disease activity. Arthritis Rheumatol. 2014;66(8):2037–2046. doi:10.1002/art.38666
179. Xu L, Liu X, Liu H, et al. Impairment of gB-producing regulatory B cells correlates with exacerbated rheumatoid arthritis. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.00768
180. Zacca ER, Onofrio LI, Acosta CDV, et al. PD-L1+ regulatory B cells are significantly decreased in rheumatoid arthritis patients and increase after successful treatment. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.02241
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.