Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
The Relationship Between Antipsychotic Treatment and Plasma β-Endorphin Concentration in Patients with Schizophrenia
Authors Urban-Kowalczyk M ![]() , Kotlicka-Antczak M, Strzelecki D
, Kotlicka-Antczak M, Strzelecki D ![]() , Rudecka E, Śmigielski J
, Rudecka E, Śmigielski J
Received 1 November 2020
Accepted for publication 12 January 2021
Published 16 February 2021 Volume 2021:17 Pages 503—512
DOI https://doi.org/10.2147/NDT.S289821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Małgorzata Urban-Kowalczyk,1 Magdalena Kotlicka-Antczak,1 Dominik Strzelecki,1 Ewa Rudecka,2 Janusz Śmigielski3
1Department of Affective and Psychotic Disorders, Medical University of Lodz, Lodz, Poland; 2Faculty of Human Nutrition and Consumer Sciences, Warsaw University of Life Sciences SGGW, Warsaw, Poland; 3State High Vocational School in Konin, Konin, Poland
Correspondence: Małgorzata Urban-Kowalczyk
Department of Affective and Psychotic Disorders, Medical University of Lodz, Czechosłowacka 8/10, 92-216, Lodz, Poland
Tel +48 42 675 73 71
Fax +48 42 675 74 03
Email [email protected]
Objective: Some studies indicate the presence of elevated opioid levels in cases of schizophrenia and their relationship with negative symptoms. The pathogenesis of schizophrenia may be associated with an imbalance in the modulatory effect of opioids on the dopaminergic system. The aim of the study was to identify the association between β-endorphin (BE) concentration and the outcome of short-term schizophrenia treatment.
Methods: We examined 49 patients hospitalized due to exacerbation of schizophrenia symptoms and 47 controls without schizophrenia. The severity of psychopathological symptoms was evaluated using Positive and Negative Syndrome Scale (PANSS) at the onset of hospitalization, and after four, six and ten weeks of treatment. Patients were classified into negative (NEG) and mixed (M) psychopathological subtypes according to the PANSS composite index. &Bgr;-endorphin (BE) plasma concentrations were assessed in all participants; in patients on inclusion to the study and after six weeks of treatment.
Results: The patients with schizophrenia demonstrated higher BE levels than controls. During six-week antipsychotic treatment, BE concentration significantly increased in both NEG (p=0.000) and M (p=0.007), and positive symptoms were effectively reduced. In the NEG group, the prevalence of negative symptoms decreased only transiently and returned to approximately baseline values after 10 weeks (p=0.268). In the M patients, the prevalence of negative symptoms increased gradually (p=0.001), with more severe positive and, notably, negative symptoms correlating with higher BE2 concentrations at the 10-week assessment (R= 0.47, p= 0.0135 vs R= 0.74, p=0.0000). In both NEG and M, a greater rise in BE2 level correlated with a lower composite index during treatment.
Conclusion: Patients with schizophrenia demonstrate higher BE levels compared to controls. These changes in BE concentration during antipsychotic treatment could reflect the interaction between dopaminergic transmission and endogenous opioids. A rise in BE level following effective antipsychotic therapy could be a potential predictor of persisting negative symptoms.
Keywords: negative symptoms, endogenous opioids, schizophrenia, psychosis
Introduction
Despite intensive study, schizophrenia remains one of the most challenging and disabling mental illnesses. A significant percentage of patients develop a chronic course of disease, with predominant negative symptoms and cognitive impairment resulting in poor social drive. Although about 10–15% of individuals recover after first episode psychosis (FEP), the disease commonly fluctuates in severity, and acute psychotic symptoms overlap with negative and neurocognitive symptoms during exacerbation.1 In general, pharmacological treatment is more effective in controlling the positive symptoms of schizophrenia. However, some of the available medication especially amisulpride, and clozapine and olanzapine were found to be significantly effective in controlling the negative symptoms.2,3 Negative symptoms like blunted affect, avolition, alogia, asociality and anhedonia account for a large part of the long-term morbidity and poor functional outcome in patients with schizophrenia. It should be noted that about 60% of patients reveal prominent and clinically relevant negative symptoms.4
Beta-endorphin (BE), which acts as a ligand mainly for μ opioid receptors, is synthesized in the anterior pituitary as a product of proopiomelanocortin degradation. It is regarded as the most powerful endogenous opioid and is found in both the central and peripheral nervous systems, where it exerts a range of biological functions ranging from the cellular to behavioral level.5 The experimental studies revealed the modulatory impact of opioids on dopaminergic system.6 Opioid agonists alter DA release, DA reuptake, and DA metabolism in some brain regions like the striatum and substantia nigra. Neurons containing endorphins modulate the regulation of dopaminergic neuronal activity.7 Moreover, it was reported that chronic antipsychotic medication increases synthesis and release of BE in pituitary gland through DA receptor blockade.6,8 The endogenous opioids modulate the activity of the mesolimbic–mesocortical DA reward–motivation circuitries. Morphine immunoreactivity was also found in the hippocampus. These brain regions are crucial for the pathogenesis of schizophrenia.9–11
The role of endogenous opioids in the pathogenesis of schizophrenia was extensively investigated in the late 1970s and 1980s, but with ambiguous results.12,13 Unfortunately, although many studies indicate the presence of excess endogenous opioids in schizophrenia, this interesting area of research has been abandoned, and the significance of these opioids in the neurobiology of disease remains unclear.14 In previous researches, various biological materials, ie, plasma,15 cerebrospinal fluid (CSF),16 and peripheral blood mononuclear cells,17 were used to assess endogenous opioids level in patient samples. It has been postulated that the etiology of schizophrenia may be based on a hypermorphinergic route related to the dopamine (DA) dysregulation hypothesis. It was found that increased expression of μ opioid receptors is cortical opioid markers in patients with schizophrenia.18 Opioid antagonist administration has been reported to have an effect on psychopathological symptoms. The increased CSF levels of BE in patients with acute psychosis and decreased levels in those with chronic symptoms were found.16 Moreover, some reports indicated that naloxone (opioid antagonist) can decrease symptoms of schizophrenia – it reduces the auditory hallucinations.19,20 Although initial results were promising, and suggested that naloxone supports neuroleptic treatment, their findings were not replicated by subsequent studies.21 Trials using opioids with longer half-lives, such as naltrexone and nalmefene, yielded conflicting results.22 Early studies found naltrexone to be ineffective as monotherapy in schizophrenia.23 Rapaport et al24 reported a decrease in positive symptom severity during nalmefene augmentation of antipsychotic treatment. Interestingly, another randomized trial found naltrexone treatment to result in greater improvement in negative symptoms when added to antipsychotic medication.25 Furthermore, a 12-week double-blind randomized placebo controlled trial found that patients treated with risperidone and naltrexone achieved greater reductions in negative symptoms compared to those receiving risperidone and placebo.26
The aims of the study were to assess if BE plasma concentration varies in the acute phase of psychosis and after neuroleptic treatment in patients with schizophrenia. Furthermore, another purpose was to determine whether negative symptoms predominance is related to specific pattern of BE secretion.
Methods
Participants
Forty-nine adult patients with schizophrenia, diagnosed according to ICD-10 criteria, and 47 healthy controls were included to the study. All patients were hospitalized in the Department of Affective and Psychotic Disorders of the Medical University of Lodz due to severe psychotic symptoms. They were divided into negative (NEG; n = 22) and mixed (M; n= 27) psychopathological subtypes according to the PANSS (Positive and Negative Syndrome Scale) composite index (CI),27 calculated by subtracting the PANSS negative symptoms subscale score from the positive subscale score.
The psychopathological subtypes were classified into positive and negative subgroups according to the 25th and 75th percentile limits (score of −8 and +3) of the original PANSS validation data. The routine clinical state examination and psychopathological symptom evaluation were performed by senior psychiatrists.
PANSS examination with CI estimation was performed at the onset of hospitalization, and after two, six and ten weeks of treatment. Blood BE concentration was assessed in controls (BE) and in patient samples: on inclusion to the study (BE1) and after six weeks (BE2). Data about the duration of illness (DOI) and hospitalization number (Hn) were also collected. The study patient subgroups did not differ in terms of gender, age, duration of illness (DOI), in hospitalization frequency (Table 1).
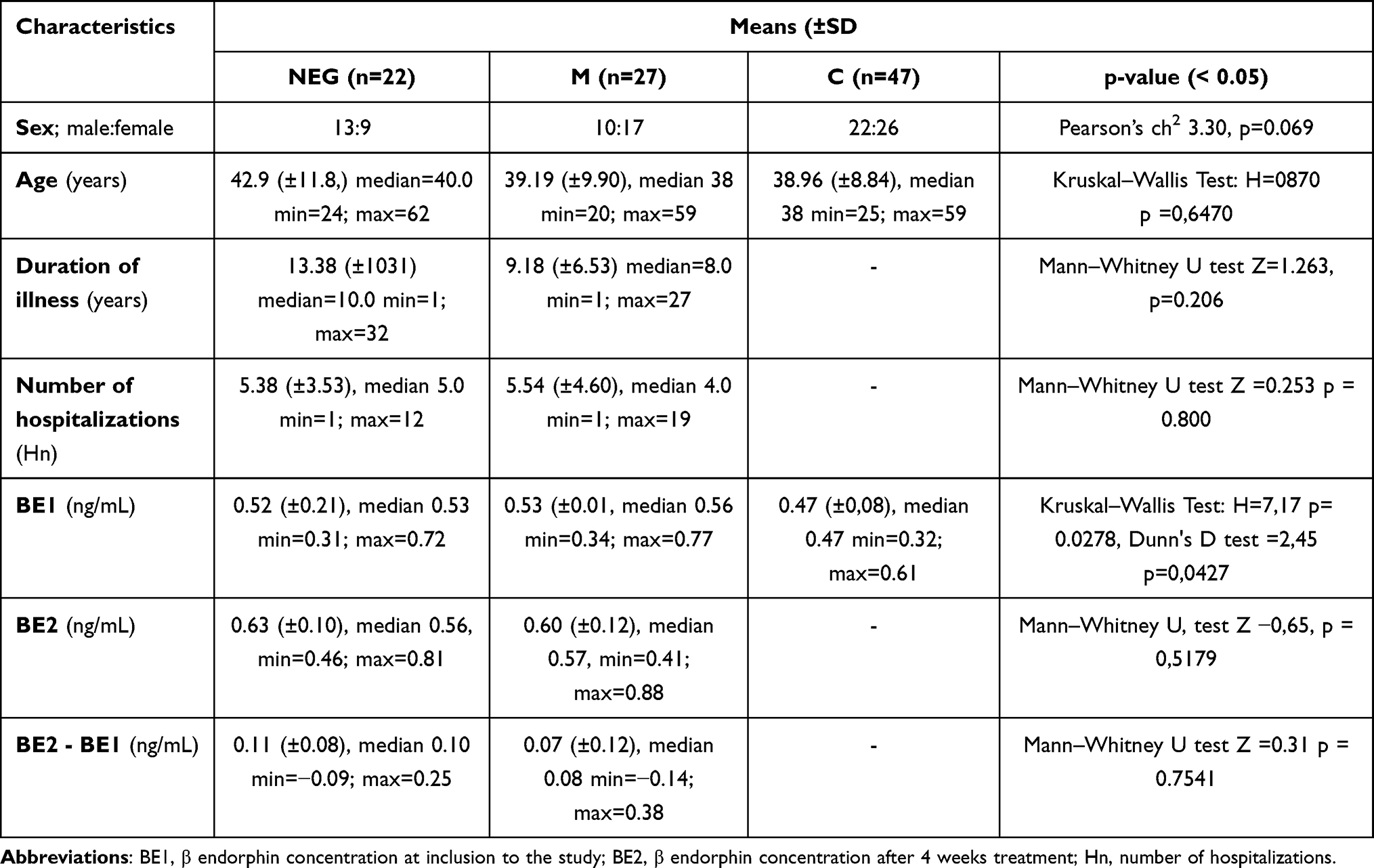 |
Table 1 Sample Characteristics for Schizophrenia with Negative Symptoms (NEG), Mixed Type (M) and Healthy Comparison (C) Groups |
The exclusion criteria were as follows: diagnosis other than schizophrenia, psychoactive substance abuse, chronic somatic disease, age below 18 years. All participants included to the study were somatically healthy and were receiving pharmacological treatment: second-generation antipsychotic and combined therapy with first-generation antipsychotics.
All participants gave their written informed consent prior to their inclusion in the study. The study was approved by the Ethics Committee of the Medical University of Lodz (Nr RNN/53/18/KE). The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2013.
Biochemical Assay
To measure plasma BE concentration, 2.6 mL peripheral blood samples were collected from participants into EDTA tubes. Immediately afterwards, the trypsin inhibitor aprotinin was added to each tube (0.6 TUI, Trypsin Inhibitor Unit per 100 ul of blood) and the samples were placed in storage. The tubes were then centrifuged and the obtained plasma samples were stored at −70°C. Following this, the final BE concentration was assayed using an EK-022-14CE β-endorphin ELISA Kit (Phoenix Pharmaceuticals).
Statistical Analysis
The data was verified for normal distribution using the Shapiro–Wilk test. For parameters with normal distributions, the Student’s t-test was used to compare the means between the groups, in the case of parameters with non-normal distributions, the Mann–Whitney U-test and the Kruskal–Wallis test and Dunn's test were used to compare unpaired samples, and the Wilcoxon test to compare paired samples. The correlations were analyzed with the Spearman rank correlation coefficient. All statistical analyses were performed with Statistica CSS software (version 13). The results of quantitative variables are presented as mean ± SD (standard deviation), median, minimum, maximum. For all analyzes, the limit of statistical significance was set at p <0.05.
Results
Beta-Endorphin Concentration
Both subsamples of individuals with schizophrenia had higher BE concentrations than controls at both the first assay (BE1) and the second (BE2) (p = 0.015 vs p = 0.000); however, no differences were observed among patient groups. All patients revealed significantly higher mean BE concentrations after six-week treatment (BE2) in comparison to first assay (BE1) (NEG: Z = 3.75, p = 0.000; M: Z= 2.65, p=0.007). The two subgroups, NEG and M, demonstrated similar differences range between BE 2 and BE1 (t = 1.263, df =49, p=0.212) Figure 1.
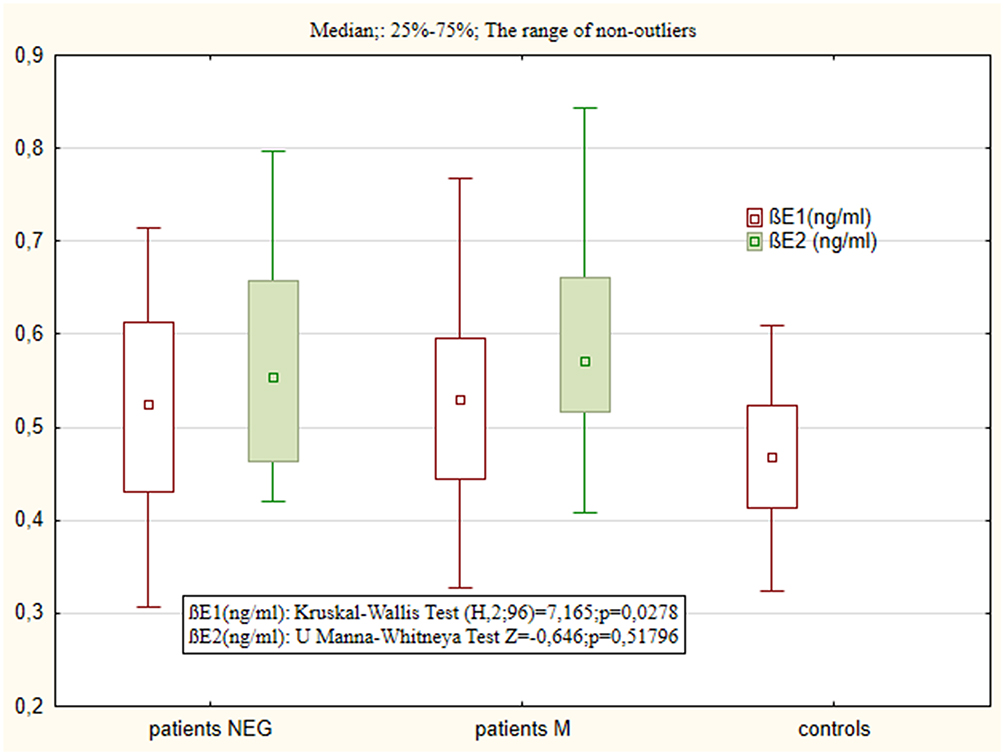 |
Figure 1 Blood concentration of BE1 and BE2 in patient samples (NEG – negative subtype, M – mixed subtype) and BE in controls (C – controls). |
Changes in Psychopathological Symptom Severity
All patients (both NEG and M) demonstrated a significant decrease of PANSS scores in all subscales over the course of pharmacological treatment (Table 2). The M group achieved significantly greater score reduction in PANSS P than those in NEG group (mean 39.46% vs mean 32.74%; Z = 2.462, p=0.0138). A significant increase was observed in the CI value after six-week treatment in NEG individuals (mean CI 1= - 11.36, SD ± 3.23 vs mean CI 3= - 8.73, SD ± 3.51; Z = 2.607, p=0.011). However, no difference was observed between CI 1 and CI 4 at 10 weeks (mean CI 4= −12.41, SD± 4.28; Z = 1.107, p=0.268). Among the M subsample, the prevalence of negative symptoms increased both after six weeks (mean CI 1 = −2.44, SD±5.36 vs mean CI 3 = −5.52, SD±4.63; Z = 3.310, p = 0.001) and after 10 weeks (mean CI 4 = −7.19, SD±5.76; Z = 3.353, p=0.001).
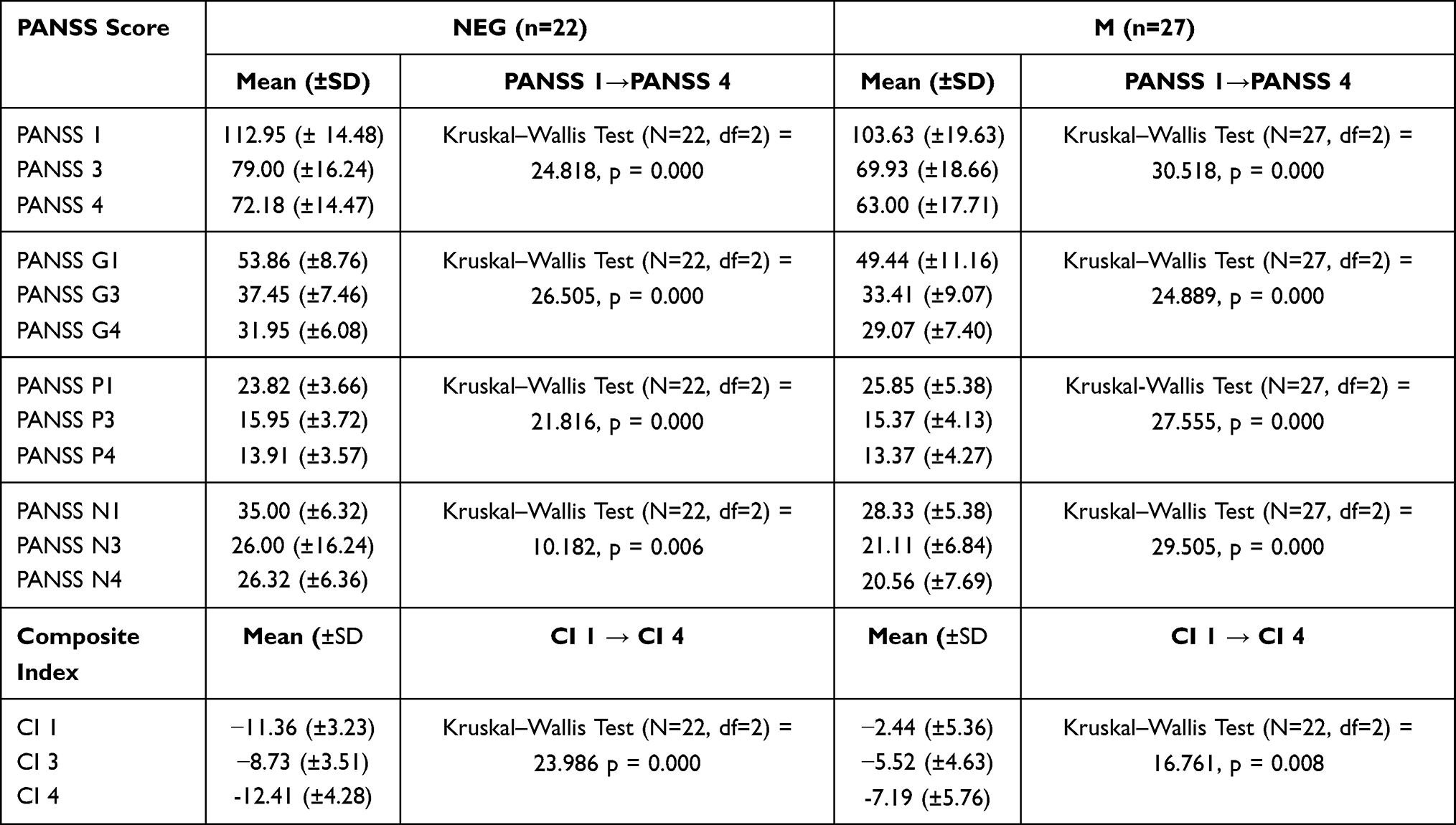 |
Table 2 PANSS Score and Composite Index (CI) in Patient Subsamples (NEG – Negative Subtype; M – Mixed Subtype) at Inclusion to the Study (1), After 4 Weeks Treatment (3) and After 10 Weeks Treatment (4) |
Relationship Between BE Concentration and Changes in the Severity of Psychopathological Symptoms
No relationship was observed between BE1 concentrations and PANSS subscale score at any mental state assessment among both groups of patients. Additionally, BE 2 concentration was not related to changes in PANSS score in the NEG sample. In the M group, it was found that patients with higher BE2 concentration demonstrated more severe psychopathological symptoms after six-week treatment, according to total PANSS score (R=0.498; p=0.008). A similar correlation was observed for negative symptoms according to the PANSS N score (R=0.676; p=0.000) and for positive symptoms, according to the PANSS P (R=0.423; p= 0.0281). Moreover, increased BE2 levels were correlated with persistent greater symptom severity in PANSS (R=0.77, p=0.000), PANSS N (R= 0.74, p=0.000) and PANSS P (R= 0.47, p= 0.013) after 10 weeks compared to baseline. The greater the difference between BE2 and BE1 concentration (BE2-BE1), the greater the severity of negative symptoms persisting after 10 weeks of treatment in the M group (R= 0.69, p=0.000).
No relationship was found between composite index values and BE1 in the patient samples. A negative correlation was found between BE2 concentration and CI 4 value in the NEG group (R= - 0.557, p=0.0071) Figure 2. Individuals from the M group with higher BE2 concentration demonstrated lower CI 3 and CI 4 values (greater severity of negative symptoms) (R= - 0.415, p=0.0314 and R= - 0.585, p= 0.0013) Figure 3.
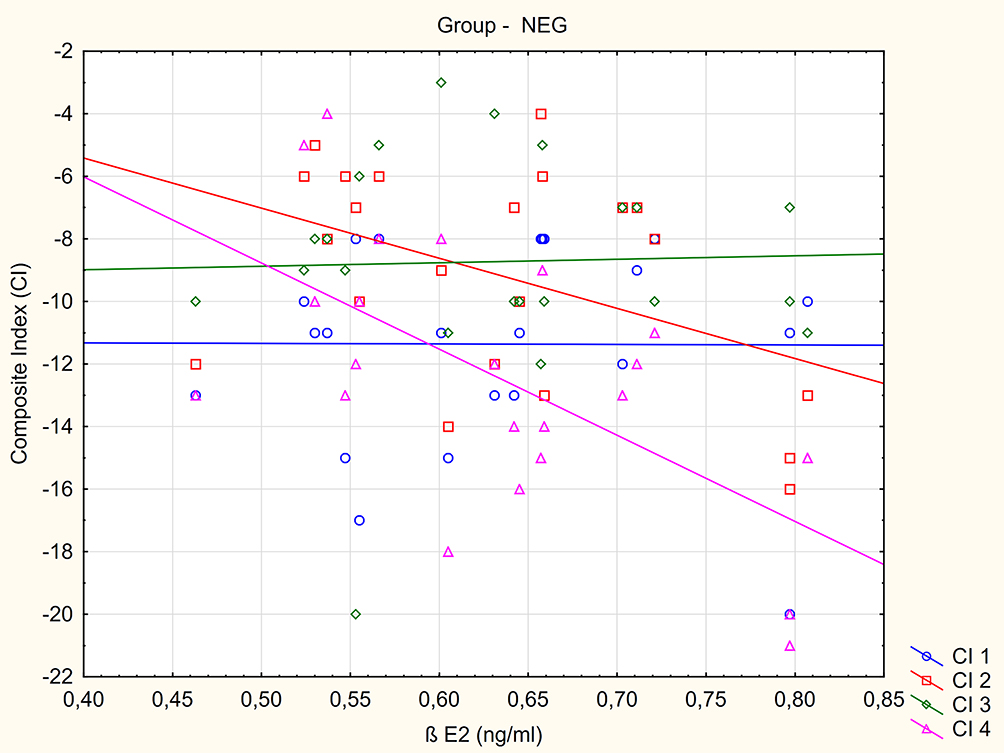 |
Figure 2 The relationship between composite index (CI) value measured at inclusion to the study (CI 1), after 2 weeks (CI 2), 4 weeks (CI 3) and 10 weeks (CI 4) treatment and BE2 blood concentration in negative subtype (NEG) group. |
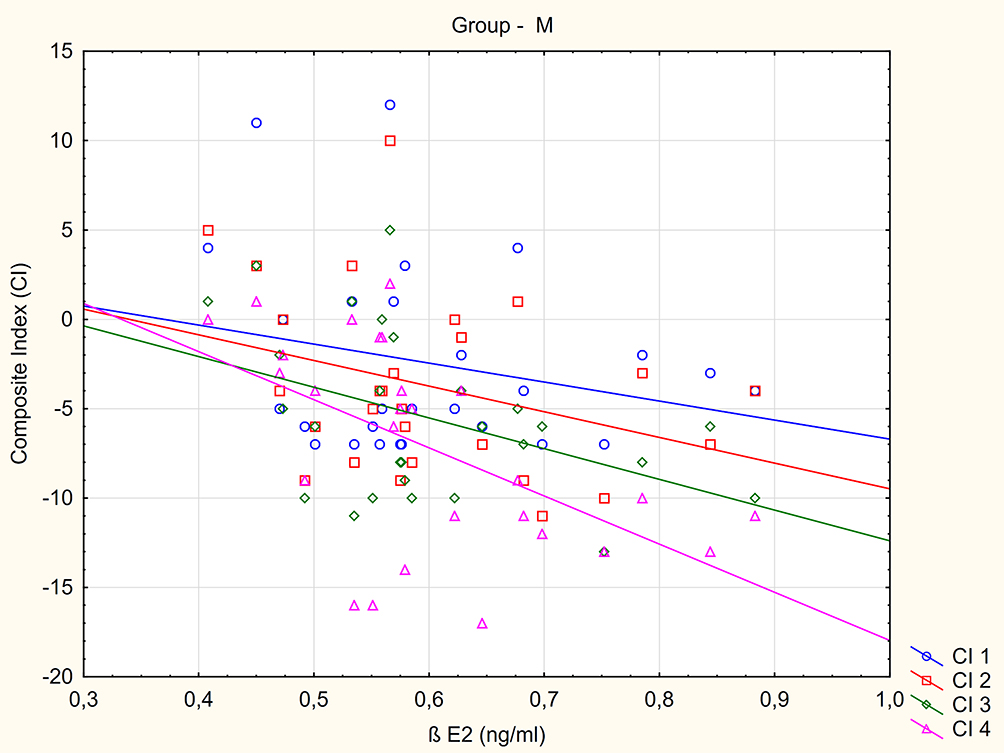 |
Figure 3 The relationship between composite index (CI) value measured at inclusion to the study (CI 1), after 2 weeks (CI 2), 4 weeks (CI 3) and 10 weeks (CI 4) treatment and BE2 blood concentration in mixed subtype (M) group. |
Discussion
Increased BE concentrations have been reported among patients with schizophrenia, with the highest BE being observed among patients with predominant negative symptoms.28,29 However, no such significant difference in BE levels was observed among patient samples in the current study. This difference could be accounted for by the selection of study groups: the previous study compared samples with both negative and positive symptoms, the present study compared the NEG group with a group of patients with mixed psychopathology but without predominant positive symptoms. Interestingly, BE concentration significantly increased during antipsychotic treatment in all patients. Inpatients have previously been found to demonstrate higher BE concentrations than those with exacerbations of positive symptoms and healthy controls during relapse with predominant negative symptoms.28
Various clinical stages or manifestations may display different BE secretion patterns. Brambilla et al31 report higher BE plasma concentration in 37 neuroleptic-free patients with chronic schizophrenia than in healthy ones. In line with these observations increased basal BE concentrations were observed among pharmacologically treated individuals with chronic schizophrenia.32 Endorphin level was found to increase only in patients under long-term antipsychotic treatment. Mauri et al,17 assessed neuropeptide concentrations in peripheral blood mononuclear cells at baseline in 30 drug-naïve schizophrenic patients; however, second assessment was performed four weeks later, among only nine haloperidol-treated individuals. They reported that basal BE tended to be lower in patients with percent amelioration higher than 40% according to the Brief Psychiatric Rating Scale (BPRS) and Scale for the Assessment of Positive Symptoms (SAPS), and in patients with less than 40% amelioration on the Scale for the Assessment of Negative Symptoms (SANS). After four-week treatment, BE concentration did not significantly change in nine evaluated patients. Previously, Panza et al33 described that during treatment with haloperidol, BE increased significantly after two and 15 days in comparison to baseline levels and to controls. Authors proposed that reduction of inhibitory tone of dopamine on BE secretion via haloperidol antidopaminergic properties. In contrast, most of the patients participating in our study were treated with second-generation antipsychotics which have also other mechanisms of action. Moreover, we evaluated BE concentration at baseline but also after a relatively longer period of pharmacological treatment, providing a greater overview of BE level over more stable symptomatic improvement.
According to the dopamine hypothesis of schizophrenia, the presence of excess dopamine in mesolimbic structures is a necessary and sufficient condition to develop psychotic symptoms. Nevertheless, the biology of negative symptoms and cognitive impairment in schizophrenia is a more complex phenomenon. Therefore, we hypothesize that changes in BE concentration related with treatment results may reflect a predisposition to the development of a specific pattern of symptoms and course of the disease. In a 15-year follow-up MUFUSSAD study,34 a chronic course was observed in 57% of patients with schizophrenia and an episodic-remitted course in 39%. The chronic course was predicted by high positive symptoms at admission to the hospital and negative symptom severity at discharge. Interestingly, a significant increase in negative symptoms almost similar to that observed during first assessment was also found at 15-year follow-up.
Another follow-up study found 33.6% of patients with schizophrenia to be continuously ill over two years, with negative symptoms being prominent in half of individuals with this pattern of schizophrenia (Harrison 2001).35 Significant positive correlations have also been found between BE concentration and negative symptoms severity among patients with first episode psychosis; of these, patients with higher BE concentration revealed higher PANSS N but not PANSS P.30 Significantly higher levels of BE were also observed in chronic patients with predominant negative symptoms in comparison to those with predominant positive symptoms of schizophrenia.29 During the present study, in the acute phase of treatment, the prevalence of negative symptoms initially decreased (C1 <C3) but returned to initial levels after 10 weeks (C1=C4) in the NEG sample; in contrast, the negative symptoms were maintained throughout the observation period in the M group. In the NEG sample, BE concentration increased after treatment independent of PANSS score changes; the participants of the M group with higher BE2 levels tended to demonstrate a poorer treatment effect, particularly in negative symptoms, after six and 10 weeks. Moreover, the patients with higher rise of BE during pharmacotherapy revealed more severe negative symptoms at the final mental state evaluation. Additionally, higher BE2 concentrations among both patient samples were related to the significant predominance of negative symptoms according to composite index value. Interaction between dopaminergic transmission and endogenous opioids might play a crucial role. Endorphins alter DA release, DA reuptake, and DA metabolism in the striatum and substantia nigra. Moreover, in contrast to DA, opioids stimulate pituitary prolactin release. Therefore, neuroleptic (antidopaminergic) treatment stimulate BE and prolactin secretion.8
The BE assessment was performed using peripheral blood samples. In somatically healthy humans endogenous opioids are detectable in plasma only in low levels ranging from pM to nM. However, BE concentrations are higher in pathological states, particularly inflammation.14 It is known that a lot of factors which we did not consider in this study can alter the brain-blood-barrier permeability.36 Obviously, this biological material does not directly reflect CNS endogenous opioid status and a measurement of cerebrospinal fluid BE would be more informative. However, CSF concentration is a methodologically better choice but it requires much more invasive procedures and is less acceptable to participants, especially for samples with acute psychotic symptoms. Both patient samples in our study were drawn from an inpatient unit, which could be considered both a limitation and an advantage of this study. The patient samples are more homogenic in terms of symptoms severity however it does not reflect BE status in schizophrenia as general. Another limitation was that no direct analysis was made of the relationship between pharmacological treatment and measured variables. Antipsychotic medication could affect BE concentration. The patients' participants included to our study revealed relapse of psychosis, which at least in part was precipitated because of treatment withdrawal. Nevertheless, the drug-naïve patient sample was not evaluated. However, there is not enough evidence that long-term neuroleptic administration is the sole cause of BE concentration excess in schizophrenia. Brambilla et al15 found higher BE plasma concentrations in 37 neuroleptic-free patients with chronic schizophrenia than in healthy controls.
Patient samples were small which caused lower statistic power of presented findings. A sample group with clearly dominant positive symptoms and mild negative symptoms could reveal more pronounced differences between patients. Moreover, BE levels were not measured after 10 weeks treatment, when the concentration could be even higher. Furthermore, the follow-up period was quite short and did not allow the long-term outcome to be predicted.
Conclusions
Negative symptoms are a heterogeneous clinical construct. Primary negative symptoms are treatment resistant and persist in spite of pharmacological efficacy in other psychopathological areas. Secondary negative symptoms might be related to other factors like isolation, positive symptoms and affective symptoms, and could be reduced following improvement of concomitant symptoms. Our findings suggest that increased BE levels could be a characteristic feature in patient with predominant negative symptoms. In addition, in patients with “mixed” psychopathology, the presence of elevated BE levels despite effective antipsychotic treatment might bring on persistent negative symptoms, even after the resolution of positive symptoms. Thus, the various psychopathological manifestations of schizophrenia might be related to specific patterns of BE secretion and increases in BE level during pharmacotherapy may be potential predictors of negative symptom persistence.
Acknowledgments
The project was funded by Medical University of Lodz (grant number: 502-03/1-155-02/502-14-269).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Millan MJ, Andrieux A, Bartzokis G, et al. Altering the course of schizophrenia: progress and perspectives. Nature Rev. 2016;15:485–515. doi:10.1038/nrd.2016.28
2. Krause M, Zhu Y, Huhn M, et al. Antipsychotic drugs for patients with schizophrenia and predominant or prominent negative symptoms: a systematic review and meta-analysis. Eur Arch Psychiatry Clin Neurosci. 2018;268(7):625–639. doi:10.1007/s00406-018-0869-3
3. Huhn M, Nikolakopoulou A, Schneider-Thoma J, et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: a systematic review and network meta-analysis. Lancet. 2019;394:939–951. doi:10.1016/S0140-6736(19)31135-3
4. Correll C, Schooler NR. Negative symptoms in schizophrenia: A review and clinical guide for recognition, assessment, and treatment. Neuropsychiatr Dis Treat. 2020;16:519–534. doi:10.2147/NDT.S225643
5. Veening JG, Barendregt HP. The effects of beta-endorphin: state change modification. Fluids Barriers CNS. 2015;12:3. doi:10.1186/2045-8118-12-3
6. Schmauss C, Emrich HM. Dopamine and the action of opiates: A reevaluation of the dopamine hypothesis of schizophrenia with special consideration of the role of endogenous opioids in the pathogenesis of schizophrenia. Biol Psychiatry. 1985;20(11):1211–1231. doi:10.1016/0006-3223(85)90179-9
7. Stefano GB, Goumon Y, Casares F, et al. Endogenous morphine. Trends Neurosci. 2000;23:436–442.
8. Kream RM, Stefano GB, Ptacek R. Psychiatric implications of endogenous morphine: up-to-date review. Folia Biologica. 2010;56:231–241.
9. Lipska BK, Weinberger DR. Delayed effects of neonatal hippocampal damage on haloperidol-induced catalepsy and apomorphine-induced stereotypic behaviors in the rat. Brain Res Dev Brain Res. 1993;15(75):213–222. doi:10.1016/0165-3806(93)90026-7
10. Neri C, Ghelardini C, Sotak B, et al. Dopamine is necessary to endogenous morphine formation in mammalian brain in vivo. J Neurochem. 2008;106:2337–2344. doi:10.1111/j.1471-4159.2008.05572.x
11. Stefano GB, Kream R. Endogenous opiates, opioids, and immune function: evolutionary brokerage of defensive behaviors. Semin Cancer Biol. 2008;18:190–198. doi:10.1016/j.semcancer.2007.12.001
12. Volavka J, Davis LG, Ehrlich YH. Endorphins, dopamine, and schizophrenia. Schizophr Bull. 1970;5(2):227–239. doi:10.1093/schbul/5.2.227
13. De Wied D, Sigling HO. Neuropeptides involved in psychopathology of schizophrenia and major depression. Neurotoxicity Res. 2000;4:453–468.
14. Laux-Biehlmann A, Mouheiche J, Veriepe J, Goumon Y. Endogenous morphine and its metabolites in mammals: history, synthesis, localization and perspectives. Neuroscience. 2013;233:95–117.
15. Brambilla F, Genazzani AR, Facchinetti F, et al. Beta-endorphin and beta-lipotropin plasma levels in chronic schizophrenia, primary affective disorders and secondary affective disorders. Psychoneuroendocrinology. 1981;6(4):321–330. doi:10.1016/0306-4530(81)90017-2
16. Domschke W, Dickschas A, Mitznegg PCSF. beta-endorphin in schizophrenia. Lancet. 1979;12:1024. doi:10.1016/S0140-6736(79)92769-7
17. Mauri MC, Rudelli R, Vanni S, et al. Cholecystokinin, beta-endorphin and vasoactive intestinal peptide in peripheral blood mononuclear cells of drug-naive schizophrenic patients treated with haloperidol compared to healthy controls. Psychiatry Res. 1997;78(1–2):45–50. doi:10.1016/S0165-1781(97)00145-5
18. Volk DW, Radchenkova PV, Walker EM, Sengupta EJ, Lewis DA. Cortical opioid markers in schizophrenia and across postnatal development. Cereb Cortex. 2012;22:15–23. doi:10.1093/cercor/bhr202
19. Gunne LM, Lindstrom L, Terenius L. Naloxone-induced reversal of schizophrenic hallucinations. J Neural Transm. 1977;40:13–19. doi:10.1007/BF01250276
20. Pickar D, Vartanian F, Bunney WE, et al. Short-term naloxone administration in schizophrenic and manic patients: a World Health Organization collaborative study. Arch Gen Psychiatry. 1982;39(3):313. doi:10.1001/archpsyc.1982.04290030047009
21. Janowsky DS, Segal DS, Bloom F, Abrams A, Guillemin R. Lack of effect of naloxone on schizophrenic symptoms. Am J Psychiatry. 1977;134:926–927.
22. Welch EB, Thompson BF. Opiate antagonists for the treatment of schizophrenia. J Clin Pharm Therapeutics. 1994;19:279–283. doi:10.1111/j.1365-2710.1994.tb00814.x
23. Gitlin MJ, Gerner RH, Rosenblatt M. Assessment of naltrexone in the treatment of schizophrenia. Psychopharmacology. 1981;74:51–53. doi:10.1007/BF00431756
24. Rapaport MH, Wolkowitz O, Kelsoe JR, Pato C, Konicki PE, Pickar D. Beneficial effects of nalmefene augmentation in neuroleptic-stabilized schizophrenic patients. Neuropsychopharmacology. 1993;9(2):111–115. doi:10.1038/npp.1993.49
25. Marchesi GF, Santone G, Cotani P, Troiani G. Naltrexone integrated antipsychotic treatment in schizophrenia. Biol Psychiatry. 1991;29(suppl):536.
26. Tatari F, Shakeri J, Farnia V, et al. Naltrexone augmentation of risperidone in treatment of schizophrenia symptoms: a randomized placebo-controlled study. Ann Psychiatry Ment Health. 2014;2(3):1016.
27. Kay SR Positive and negative syndromes in schizophrenia. In, Van Praag PH editor. Clinical and Experimental Psychiatric.1991 Monograph No 5, New York: Brunnel/Mazer.
28. Urban-Kowalczyk M, Śmigielski J, Strzelecki D. Comparison of beta-endorphin and CGRP levels before and after treatment for severe schizophrenia. Neuropsychiatr Dis Treat. 15(12):863–868. doi:10.2147/NDT.S101647
29. Urban-Kowalczyk M, Śmigielski J, Kotlicka-Antczak M. Overrated hedonic judgment of odors in patients with schizophrenia. CNS Neurosci Therapeutics. 2018;24(12):1156–1162. doi:10.1111/cns.12849
30. Urban-Kowalczyk M, Strzelecki D, Śmigielski J, Kotlicka-Antczak M. Odor perception and hedonics in chronic schizophrenia and in first episode psychosis. Neuropsychiatr Dis Treat. 2019;5(15):647–654. doi:10.2147/NDT.S192523
31. Brambilla F, Facchinetti F, Petraglia F, Vanzulli L, Genazzani AR. Secretion pattern of endogenous opioids in chronic schizophrenia. Am J Psychiatry. 1984;141(10):1183–1189.
32. Brambilla F, Facchinetti F, Petraglia F, et al. Effects of neuroleptic treatments on peripheral opioid secretion. Neuropsychobiology. 1978;18(2):68–73. doi:10.1159/000118395
33. Panza G, Monzani E, Sacerdote P, Penati G, Panerai AE. Beta-endorphin, vasoactive intestinal peptide and cholecystokinin in peripheral blood mononuclear cells from healthy subjects and from drug-free and haloperidol-treated schizophrenic patients. Acta Psychiatr Scand. 1992;85(3):207–210. doi:10.1111/j.1600-0447.1992.tb08596.x
34. Möller H-J, Jäger M, Riedel M, Obermeier M, Strauss A, Bottlender R. The Munich 15-year follow-up study (MUFUSSAD) on first-hospitalized patients with schizophrenic or affective disorders: comparison of psychopathological and psychosocial course and outcome and prediction of chronicity. Eur Arch Psychiatry Clin Neurosci. 2010;260:367–384. doi:10.1007/s00406-010-0117-y
35. Harrison G, Hopper K, Craig T, et al. Recovery from psychotic illness: a 15- and 25-year international follow-up study. Br J Psychiatry. 2001;178:506–517. doi:10.1192/bjp.178.6.506
36. Witt KA, Davis TP. CNS drug delivery: opioid peptides and the blood-brain- barrier. AAPS J. 2006;8(1):E76–E88. doi:10.1208/aapsj080109
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.