Back to Journals » OncoTargets and Therapy » Volume 13
The Pseudogene DUXAP8 Promotes Colorectal Cancer Cell Proliferation, Invasion, and Migration by Inducing Epithelial-Mesenchymal Transition Through Interacting with EZH2 and H3K27me3
Authors He W, Yu Y, Huang W, Feng G, Li J
Received 22 October 2019
Accepted for publication 9 September 2020
Published 29 October 2020 Volume 2020:13 Pages 11059—11070
DOI https://doi.org/10.2147/OTT.S235643
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Wenjing He,1,* Yi Yu,2,* Wei Huang,2 Guoliang Feng,2 Junhe Li2
1Institute of Urology, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 2Department of Oncology, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Junhe Li
Department of Oncology, The First Affiliated Hospital of Nanchang University, No. 17 Yongwai Street, Nanchang 330006, Jiangxi, People’s Republic of China
Email [email protected]
Background: Colorectal cancer (CRC) is the third leading cause of cancer death worldwide. The long noncoding RNA (lncRNA) DUXAP8 has been reported to play an important role in CRC. This study investigated the mechanism by which this lncRNA regulates CRC progression.
Methods: The levels of lncRNA DUXAP8 in CRC tissues and cell lines were detected by qRT-PCR. We then knocked down or forced overexpression of DUXAP8, and the resultant effect on cell proliferation was determined by the Edu assay and a cell cycle analysis, and the effect on cell apoptosis was determined by flow cytometry. The cell invasion/migration ability and the epithelial-to-mesenchymal transition (EMT) markers were determined by Transwell/wound healing assays and Western blotting. CHIP and RNA pull-down assays were performed to determine the binding of Zeste gene enhancer 2 (EZH2) and trimethylated histone H3 to Lys27 (H3K27me3) in the E-cadherin promoter regions, or to DUXAP8.
Results: The levels of lncRNA DUXAP8 were significantly increased in CRC tissues and CRC cell lines. Knockdown of lncRNA DUXAP8 inhibited cell proliferation and the EMT process, and increased cell apoptosis, and overexpression of lncRNA DUXAP8 had an opposite effect. Both ChIP and RNA pull-down assays showed that the E-cadherin promoter region was bound by H3K27me3 and EZH2, which restrained E-cadherin expression. However, that binding was suppressed and E-cadherin expression was markedly induced by lncRNA DUXAP8 knockdown. Furthermore, lncRNA DUXAP8 could interact with EZH2 and H3K27me3.
Conclusion: Our data indicated that lncRNA DUXAP8 could induce the progression of CRC by negatively regulating E-cadherin via interaction with EZH2 and H3K27me3. These findings suggest lncRNA DUXAP8 as target for treating CRC.
Keywords: Long intergenic non-coding RNA, DUXAP8, epithelial-to-mesenchymal transition, EZH2, H3K27me3
Introduction
Colorectal cancer (CRC) is one of the deadliest cancers of the digestive system.1 The incidence rate of CRC is the third highest of all malignant tumors in men and the second highest in women. Furthermore, CRC has the third highest overall mortality rate of all malignant tumors.2 The onset of CRC is insidious, and there are no obvious symptoms during its early stage.3 Most patients already have advanced stage CRR when treatment is initiated, and 50%~60% of those patients have distant metastases.4 Although the overall mortality rate of CRC has decreased significantly with the development of new diagnostic and therapeutic techniques, the prognosis for CRC patients with regional lymph node involvement or distant metastases remains poor.5 The high mortality rate of CRC is due to the invasion and metastasis of cancer cells;6 therefore, new clinical methods must be developed to inhibit the invasion and metastasis of CRC cells. Future strategies for treating CRC must be based on a more comprehensive and in-depth understanding of the molecular mechanisms that regulate cancer cell invasion and metastasis.
Results from the Human Genome Project and the post-genome project revealed that only 2% of the entire human genome has a protein-coding ability, and the remaining 90% of sequences do not encode for proteins.7,8 A pseudogene is a DNA sequence similar to a functional gene, but that has a non-transcribed nucleotide sequence. Pseudogenes were first discovered by Jacq et al in a study of Xenopus laevis genes. In the past, pseudogenes were considered to be “garbage” generated during the evolution of genomes.9 However, with the rapid development of molecular biology, more recent studies have confirmed that pseudogenes play important roles in biological processes.10 Some pseudogenes can transcribe non-protein-encoding RNA molecules in excess of 200 nt.11 Recent studies have shown that dysregulation of lncRNAs might be associated with tumorigenic processes.12
At present, most research examining the development of colorectal cancer has focused on mutations in genes that code for proteins, and mechanisms that regulate the expression of those genes.13–15 However, increasing numbers of studies have reported the dysregulation of many lncRNAs in CRC cells, and those findings might be applicable for the early diagnosis and targeted therapy of colorectal cancer.16–19 Svoboda et al found that HOTAIR was highly expressed in primary colorectal tumors, and its high expression was correlated with a poor prognosis.18 HOTAIR regulates gene expression by affecting chromosome structure, which acts as a “scaffold” for the histone modification complex and interacts with histone complexes PRC2 and LSD1. Reducing HOTAIR levels can inhibit tumor growth in vivo, which suggests HOTAIR as a target for intervention.20 Han et al found that UCA1 is also highly expressed in colorectal cancers, and its expression level is positively correlated with the malignancy of the tumor and negatively correlated with a favorable prognosis.21 Yang et al22 found that MALAT1 can promote the development of colorectal cancer via the AKAP-9 protein. In recent years, additional lncRNAs including SNHG12, PANDAR, SPRY4-IT1, and SBDSP1 have been confirmed to be highly expressed in colorectal cancer tumor cells, promote the proliferation of colorectal cancer tumor cells, and be associated with a poor prognosis.17,23–25 Due to the high tissue and spatiotemporal specific expression characteristics of lncRNAs, there is an urgent need to further study the lncRNA expression profile of CRC, and identify new lncRNAs that are closely associated with CRC progression.
The pseudogene DUXAP8 is located at 22q11, is 2107 bp in length, and can be transcribed to a lncRNA.26 This pseudogene has been shown to be dysregulated in several types of cancer, including gastric, bladder, and lung cancers, and is closely associated with their progression.26–28 DUXAP8 expression is significantly upregulated in CRC, and indicative of a poor prognosis.29 However, the mechanism by which DUXAP8 contributes to colorectal cancer is largely unknown.
Our study confirmed an induction of DUXAP8 expression in CRC, and identified its prognostic role in that disease. Furthermore, the underlying pathway by which DUXAP8 contributes to the progression of CRC was investigated. To the best of our knowledge, our study is the first to reveal that DUXAP8 regulates E-cadherin expression by regulating the binding of EZH2 and H3K27me3 to DUXAP8.
Materials and Methods
Clinical Samples
Tumor specimens were obtained from 30CRC patients who were treated at the First Affiliated Hospital of Nanchang University (Nanchang, Jiangxi, China) from January 2017 to December 2017. A sample of paracancer tissue (located > 3 cm from the tumor margin) was concurrently obtained from each patient for use as a control specimen. No patient had received any radio-, chemo- or interventional therapy prior to their operation, and a postoperative pathological diagnosis was performed for each patient. Each patient provided their written informed consent for study participation, and the study protocol was approved by the Ethics Committee of the First Affiliated Hospital of Nanchang University.
Cell Culture
NCM460 human colorectal epithelial cells and SW480, SW620, LoVo, HCT116, HT29, and RKO CRC cells were obtained from the American Type Culture Collection (Manassas, VA, USA). All cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) (GE Healthcare, Chicago, IL, USA) in a humidified atmosphere containing 5% CO2 at 37°C.
Subcellular Fractionation
The nuclear and cytosolic fractions of SW260 cells were isolated by using a NE-PER Nuclear and Cytoplasmic Extraction Reagents kit according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA, USA). In brief, the cultured cells were harvested, washed with pre-cooled PBS, and then suspended in 200 μL of cytoplasmic extraction reagent I. Next, 11 μL of cytoplasmic extraction reagent II was added, and the cell suspension was vortexed for 5 s; after which, it was incubated on ice for 1 min and centrifuged at 4°C for 5 min at 16,000 g. The supernatant (cytoplasmic extract) was carefully transferred into a pre-chilled EP tube. The pellet contained crude nuclei.
Protein Extraction and Western Blotting
Harvested cells were transferred into a 1.5 mL EP tubes and centrifuged at 800 rpm for 5 min at 4°C; the pelleted cells were retained. Next, 100 μL of RIPA buffer (Beyotime, Shanghai, China) containing protease inhibitor cocktail II (Beyotime) was added to the lysed the cells, which were then stored on ice for 30 min with shaking every 5 min. The lysed cells were then centrifuged at 14,000 rpm for 20 min, and the supernatant fractions were collected in new EP tubes. The protein concentration in each supernatant was measured using a Coomassie Plus Assay Kit (BioRad Laboratories, Hercules, CA, USA). Next, an equal amount of protein from each sample was loaded onto a sodium dodecyl sulfate-polyacrylamide gel and separated by electrophoresis (SDS-PAGE). The separated protein bands were then transferred onto nitrocellulose filter membranes (GE Healthcare), which were subsequently blocked by incubation with 5% skimmed milk at 37°C for 5 minutes. Next, the membranes were incubated with primary antibodies against E-cadherin, N-cadherin, and GAPDH (1:500, Cell Signaling Technology, Danvers, MA, USA), vimentin and snail (1:200, Abcam, Cambridge, MA, USA), EZH2 (Abcam, ab186006, 1:2000), and H2K27me3 (Abcam, ab272155, 1:2000). After being washed with TBST buffer, the membranes were incubated with a goat anti-rabbit secondary antibody (1:3000, BioRad, USA) labeled with horseradish peroxidase (HRP) at 37°C for 1 h, and the immunostained protein bands were visualized with ECL chemiluminescent reagent (eBioscience, San Diego, CA, USA). The ratio of the staining intensity of the target protein to that of GAPDH (the internal reference protein) was used as a measure of protein expression.
Immunofluorescence
SW620 cells and LoVo cells were fixed with 4% paraformaldehyde for 15 min and then permeabilized with saponin (Sigma-Aldrich, St. Louis, USA) for 5 min. Next, the cells were blocked with 5% BSA for 30 min, and then incubated for 1 h at room temperature with primary antibodies against E-cadherin (Abcam, ab15148, 1: 500), snail (Abcam, ab216347, 1: 200), vimentin (Abcam, ab92547, 1: 1000), and N-cadherin (Abcam, ab18203, 1: 500). Next, the cells were washed 3 times with PBST for (5 min per wash); after which, a fluorescent secondary antibody (CST, 2985 or 2975, 1:1000) was added, and the cells were incubated at room temperature for 1 h in the dark. After being rinsed 3 times with PBST, followed by ultimate rinse with distilled water, the cells were sealed for examination by fluorescence microscopy.
RNA Extraction and qRT-PCR
Specimens of CRC tissue (~ 0.1 g each) were washed twice with pre-cooled PBS and ground into a homogenate with an autoclaved tissue grinder in an ice bath. Each homogenate was transferred into an EP tube. Next, 1 mL of Trizol Reagent (Takara, Otsu, Japan) was added to each tube and the tubes were gently shaken. For harvested cells, Trizol Reagent was added to the nuclear and cytoplasmic fractions that had been separated by centrifugation. After 5 min, each mixture was transferred into a 1.5 mL Eppendorf tube, and total RNA was extracted. Next, cDNA was obtained by using a High Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA). An ABI PRISM 7700 System and TaqMan reagents (Applied Biosystems) were used to perform qRT-PCR with the following conditions: initial denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 94°C for 20 s, annealing at 58°C for 20 s, and elongation at 72°C for 20 s. GAPDH and U6 were used as internal reference genes. The relative expression levels of various genes and DUXAP8 were calculated by the 2−ΔΔCt method. The primers used for qRT-PCR were as follows: DUXAP8 forward primer: 5ʹ-ACCCAAACACTAATTGTAGACT-3ʹ and reverse primer 5ʹ- TGTCTGGGAGACTGCTTACA-3ʹ; EZH2 forward primer: 5ʹ-GTACACGGGGATAGAGAATGTGG-3ʹ and reverse primer 5ʹ- GGTGGGCGGCTTTCTTTATCA-3ʹ; E-cadherin forward primer: 5ʹ- ATTTTTCCCTCGACACCCGAT-3ʹ and reverse primer 5ʹ-TCCCAGGCGTAGACCAAGA-3ʹ; U6 forward primer: 5ʹ-CTCGCTTCGGCAGCACA-3ʹ and reverse primer 5ʹ- AACGCTTCACGAATTTGCGT-3ʹ; GAPDH forward primer: 5ʹ-TGTTCGTCATGGGTGTGAAC-3ʹ and reverse primer 5ʹ- ATGGCATGGACTGTGGTCAT −3ʹ.
Cell Transfection
Short hairpin RNAs (shRNAs) targeting DUXAP8 (sh-DUXPAP8) and EZH2 (sh-EZH2), and the corresponding negative controls (NCs) were purchased from GenePharma Co., Ltd. (Shanghai, China). A recombinant plasmid pcDNA3.0-DUXAP8 was purchased from VIPOTION (Guangzhou, China). SW620 cells were transfected with sh-DUXAP8, sh-EZH2 or the NC for 48 h at 37°C using Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA). An empty vector (NC) and a lncRNA DUXAP8 vector were also transfected into LoVo cells by using Lipofectamine® 2000 according to the manufacturer’s instructions.
Cell Proliferation Assay
A Cell Counting Kit-8 (CCK8; Dojindo Lab, Japan) was used to determine cell viability. Briefly, SW620 or LoVo cells were cultured in 96-well plates (3000 cells/well) at 37°C for 24 h, 48 h or 72 h, respectively. Next, 10 μL of CCK-8 solution was added to each well and the plate was incubated for an additional 24 h. Following incubation, the absorbance of each well at 450 nm was determined with a Varioskan Flash Plate Reader (Thermo Fisher Scientific). The mean value was calculated based on results obtained from 3 separate experiments.
EdU Analysis
A 5-ethynyl-2-deoxyuridine (EdU) labeling/detection kit (Ribobio, Guangzhou, China) was used to detect cell proliferation. SW620 and LoVo cells transfected with sh-DUXAP8, DUXAP vector or the corresponding NCs were cultured in 96-well plates at the density of 1 × 103 cells per well. Next 50 μM EdU labeling medium was added to each well, and the cells were cultured for an additional 2 h. The cells were then fixed with 4% paraformaldehyde, stained with anti-EdU working solution, and treated with 0.5% Triton X-100. EdU-positive cells were photographed with a fluorescence microscope and their numbers were recorded.
Flow Cytometry Analysis
SW620 and LoVo cells were harvested, and the stained with FITC-Annexin V and propidium iodide (PI) double dyes contained in a FITC Annexin V Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA). After double staining, the cells were analyzed by flow cytometry (FACScan®; BD Biosciences). For cell cycle investigations, PI was used to stain the LoVo and SW620 cells, and the data were analyzed by a FACScan flow cytometer.
Cell Migration and Invasion Assays
For wound healing assays, cells were seeded into 6-well plates and cultured until confluence. Next, a horizontal line was drawn on the bottom of the plate, and wounds were made by moving a sterile 200 μL pipet tip through the cultured cells along the drawn line. The plates were observed and photographed at 0 h and again at 48 h after wounding. For Transwell assays, 1 × 105 cells in 100 μL of serum-free RPMI-1640 medium were plated into the upper chamber of a Transwell plate, and 500 μL of RPMI-1640 medium containing 20% FBS was added to the lower chamber. After 48 h, cells in the lower chamber were fixed with methanol and then stained with 0.1% crystal violet. Finally, the number of invaded cells in five randomly selected microscopic fields was recorded and used to calculate the final results.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were conducted by using an EZ-CHIP kit according to the manufacturer’s protocol (Millipore, Burlington, MA, USA). SW620 cells were incubated with formaldehyde for 10 min, and then sonicated to obtain cell lysates. The cell lysates were immunoprecipitated with EZH2- and H3K27me3-specific antibodies (Millipore); IgG was used as a control. The precipitated chromatin DNA was analyzed by qPCR.
RNA Pull-Down Assay
A biotin-labeled DUXAP8 probe and RNA complementary to the specific region of the E-cadherin promoter or corresponding negative control were obtained from GenePharma. After incubation with the cell lysates for 1 h at room temperature, the complexes were extracted with streptavidin agarose beads (Invitrogen). The beads were washed three times with PBS, and then boiled in sodium dodecyl sulfate buffer to elute proteins. The eluents were subjected to Western blot analysis.
Statistical Analysis
All statistical analyses were performed using Graphpad software, and results are reported as a mean value ± standard deviation. The student’s t-test was used to make comparisons between groups. The patients were divided into a high DUXAP8 group and a low DUXAP8 group based on the median level of DUXAP8 (2.812) expression. Survival curves were created using the Kaplan–Meier method and Log rank tests were used to analyze differences between patients with high or low levels of DUXAP8 expression and also differences in overall survival. All P-values are bilateral, and a P-value < 0.05 was considered to be statistically significant.
Results
Human CRC Tissues Had Elevated Levels of DUXAP8
A qRT-PCR analysis showed that DUXAP8 was obviously upregulated in CRC tissues when compared with paracarcinoma tissues (P < 0.0001, Figure 1A). A Kaplan-Meier analysis revealed that patients with high levels of DUXA8 had a lower overall survival (OS) survival rate than patients with low levels of DUXAP8 (P = 0.0475, Figure 1B). We also analyzed the levels of DUXAP8 mRNA in normal human NCM460 cells and six human CRC cell lines. Those results showed that DUXAP8 mRNA production was also up-regulated in the CRC cell lines when compared with the normal NCM460 cells, with SW620 cells expressing the highest levels of DUXAP8 mRNA, and LoVo cells expressing much lower levels of DUXAP8 mRNA (P < 0.01, Figure 1C). Furthermore, a statistical analysis showed that DUXAP8 expression in tumor tissues from patients with severe or metastatic CRC was significantly increased (Table 1). These results indicated that DUXAP8 might play a vital role in CRC progression.
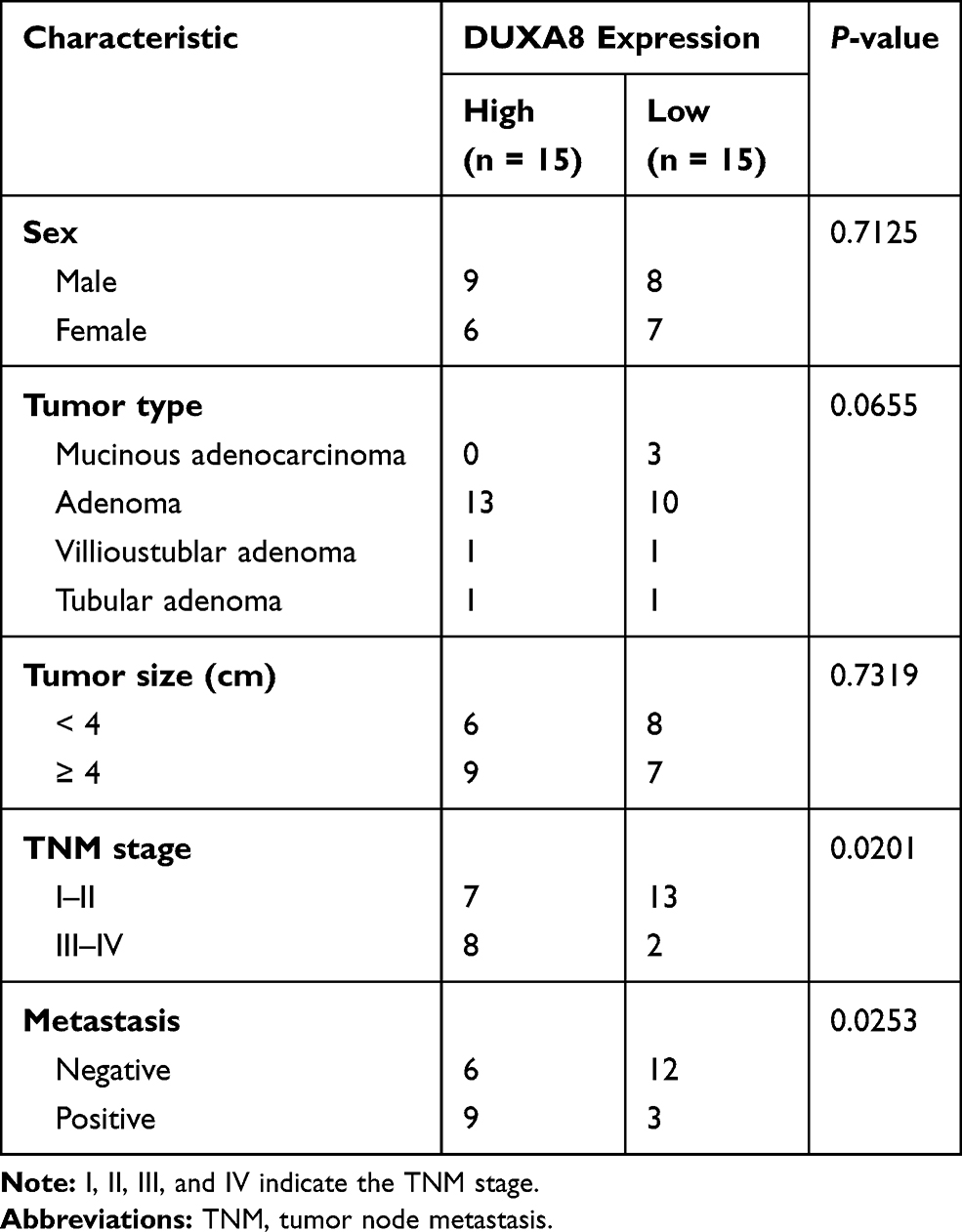 |
Table 1 Correlations Between DUXAP8 Expression Levels and the Pathological Characteristics of CRC Specimens |
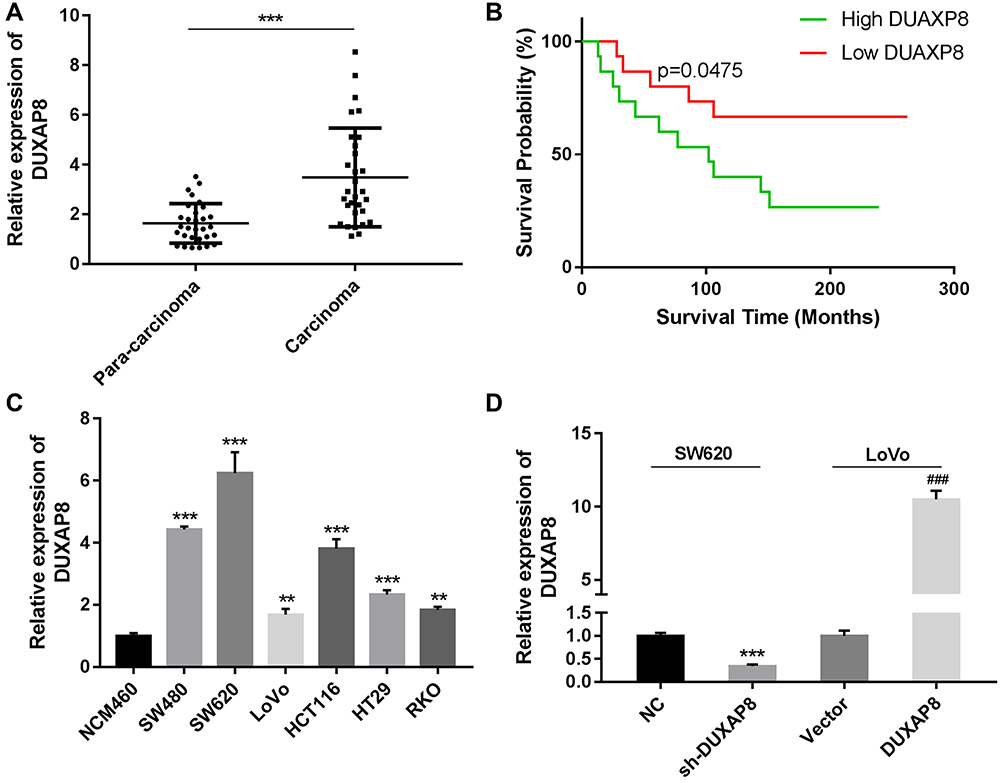 |
Figure 1 Relative levels of DUXAP8 expression in colorectal cancer (CRC) tissues and their clinical significance. (A) DUAXP8 expression was measured in CRC tissues and their adjacent tissues by qRT-PCR (n = 30/group). (B) Overall survival analysis of CRC patients with high or low levels of DUAXP8 expression (n = 15 patients/group). P = 0.0457. (C) qRT-PCR analysis of DUXAP8 expression in human normal colonic epithelial cells (NCM460) and CRC cells (SW480, SW620, L0V0, HCT116, HT29, and RKO). (D) qRT-PCR analysis of DUXAP8 expression in SW620 cells transfected with the NC and sh-DUXAP8 and in LoVo cells transfected with the control vector and pcDNA-DUXAP8 expression vector. **P < 0.01, ***P < 0.001, ###P < 0.001. |
To further study the function of DUXAP8 in CRC tumorigenesis and progression, SW620 cells were transfected with DUXAP8 shRNA or the NC. A subsequent qRT-PCR analysis confirmed the transfection efficiency of DUXAP8 shRNA in those cells (Figure 1D). Moreover, LoVo cells were also transfected with the control vector or pcDNA-DUXAP8, and a subsequent analysis confirmed the up-regulation of DUXAP8 in LoVo cells (Figure 1D).
DUXAP8 Enhanced CRC Cell Proliferation, Inhibited Apoptosis, and Accelerated the Cell Cycle
To investigate the functional role of DUXAP8 in CRC cells, we performed ethynyl deoxyuridine (EdU) staining with SW620 cells and LoVo cells. The results suggested that downregulation of DUXAP8 suppressed the proliferation of SW620 cells (Figure 2A), while its overexpression promoted the proliferation of those cells (Figure 2B), suggesting that DUXAP8 functions as an oncogene to accelerate the proliferation of CRC cells. These results were also confirmed by CCK8 assays (Figure 2C and D).
We further explored the effects of DUXAP8 on the cell cycle and apoptosis. Flow cytometry results indicated that knockdown of DUXAP8 induced cell apoptosis and blocked the cell cycle at the G0/G1 phase in SW620 cells (Figure 2E and F), whereas overexpression of DUXAP8 in LoVo cells produced the opposite effects on cell apoptosis and cycle progression (Figure 2E and F). The apoptosis ratio (Figure 2G) and cell cycle distribution (Figure 2H) also confirmed that DUXAP8 enhanced the proliferation and suppressed the apoptosis of CRC cells.
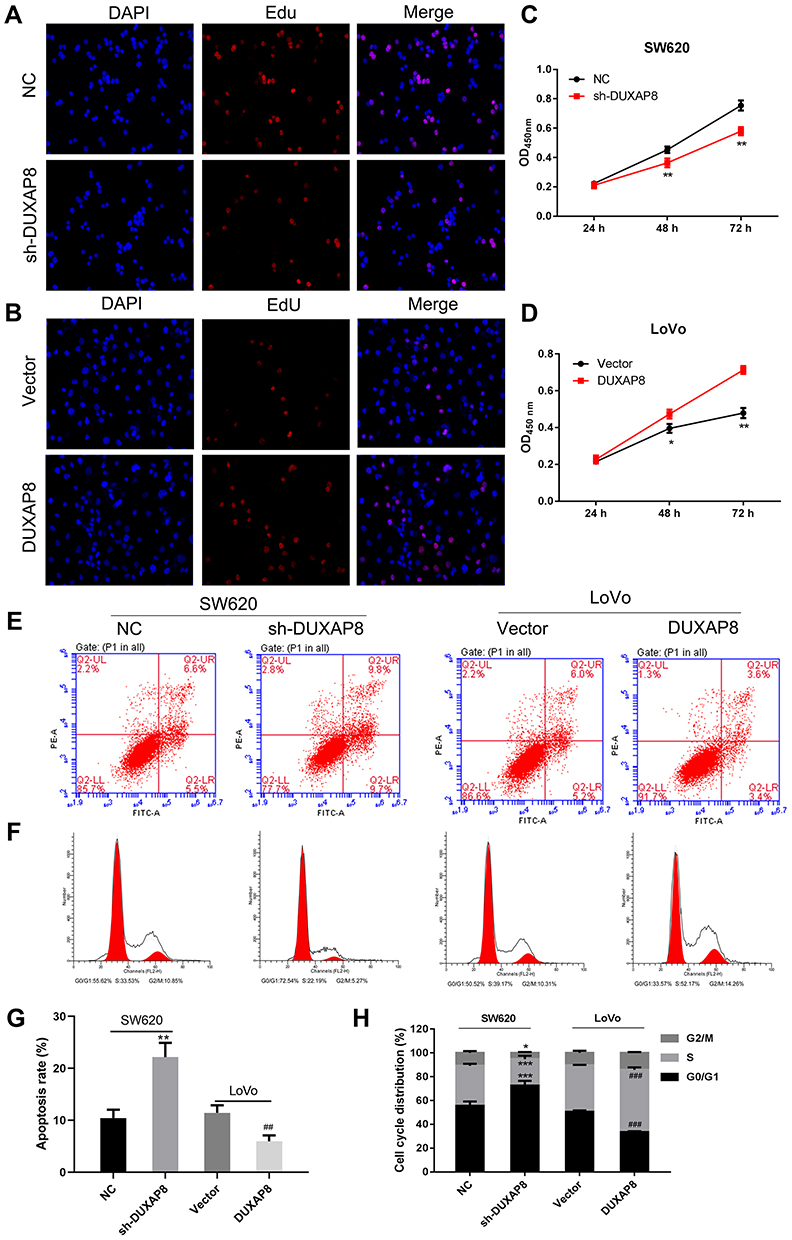 |
Figure 2 DUXAP8 increased CRC cell proliferation and cell cycle progression, and inhibited apoptosis. (A, B) Proliferating SW620 and LoVo cells were labeled with Edu. The click-it reaction revealed Edu staining (red). Cell nuclei were stained with DAPI (blue). (C, D) CCK8 assays revealed that DUXAP8 silencing inhibited SW620 cell proliferation, while overexpression DUXAP8 promoted LoVo cell proliferation. (E) Flow cytometry was used to detect the apoptotic rates of cells. UR, early apoptotic cells; LR, terminal apoptotic cells. (F) Flow cytometry was used to detect the cell cycle distribution of cells. Silencing of DUXAP8 increased the percentage of SW620 cells in G0/G1 phase, while overexpression of DUXAP8 reduced the percentage of LoVo cells in G0/G1 phase. (G) The apoptosis ratio as affected by DUXAP8 silencing and DUXAP8 overexpression. (H) The cell cycle distribution as affected by DUXAP8 silencing and DUXAP8 overexpression. *P < 0.05, **P < 0.01, ***P < 0.001 vs NC; ##P < 0.01, ###P < 0.001 vs Vector. |
DUXAP8 Promoted CRC Cell Invasion, Migration, and Epithelial Mesenchymal Transition
Transwell assays were performed to determine whether DUXAP8 affects the migration and invasion of CRC cells. Those results showed that knockdown of DUXAP8 expression reduced the invasiveness of LoVo cells while overexpression of DUXAP8 promoted the invasion of LoVo cells (Figure 3A). Additionally, downregulation of DUXAP8 inhibited the wound healing ability of SW620 cells, but had the opposite effect on LoVo cells (Figure 3B).
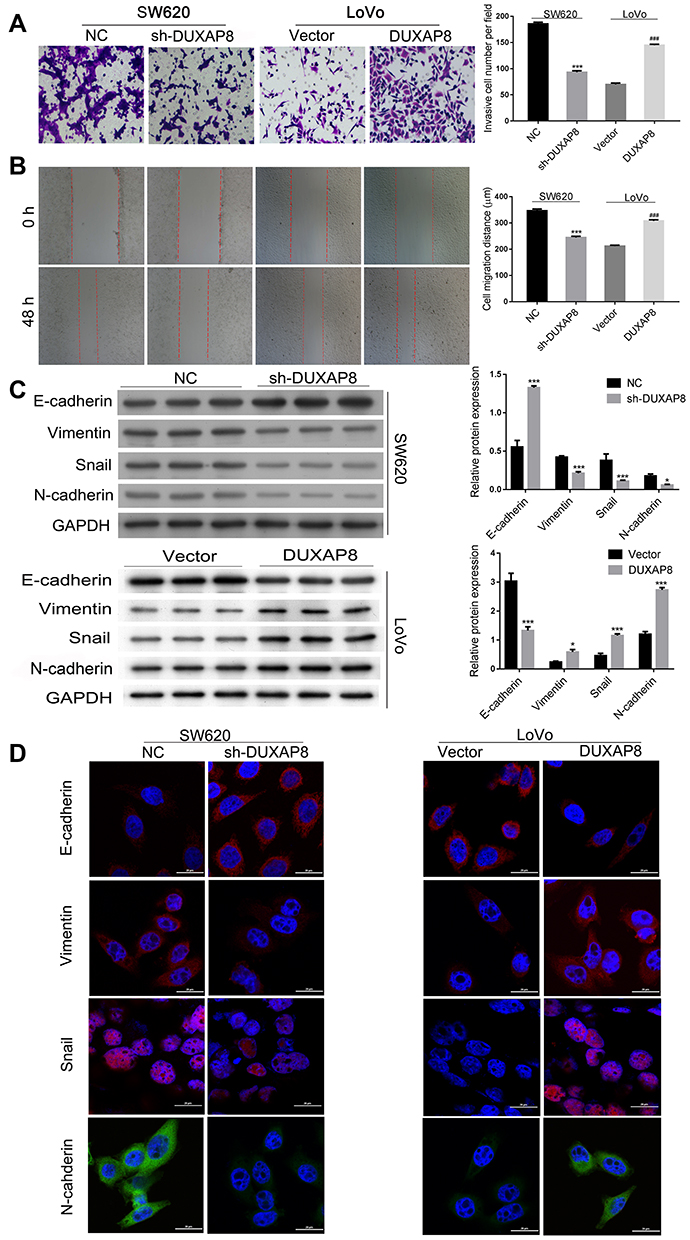 |
Figure 3 DUXAP8 promoted CRC cell invasion, migration, and EMT. (A) Transwell assays revealed that knockdown of DUXAP8 inhibited the migration of SW620 cells, while overexpression DUXAP8 promoted the migration of LoVo cells. (B) Wound healing assays showed that knockdown of DUXAP8 significantly reduced the wound healing ability of SW620 cells, while its overexpression greatly increased the wound healing ability of LoVo cells. ***P < 0.01, ###P < 0.001. (C) Western blotting was performed to evaluate the levels of E-cadherin, N-cadherin, snail, and vimentin expression after knockdown of DUXAP8 in SW620 cells and overexpression of DUXAP8 in LoVo cells. *P < 0.05, ***P < 0.001. (D) Immunofluorescence studies detected the expression of E-cadherin, N-cadherin, Snail, and Vimentin in DUXAP8 knockdown SW620 cells and DUXAP8 overexpressing LoVo cells. |
The EMT process has been shown to be involved in the metastasis of cancers.30–32 Therefore, we then examined the effect of DUXAP8 on EMT. As shown in Figure 3C, knockdown of DUXAP8 induced E-cadherin expression, but decreased vimentin, snail, and N-cadherin expression in SW620 cells. Meanwhile, overexpression of DUXAP8 suppressed E-cadherin expression, but increased vimentin, snail, and N-cadherin levels in LoVo cells. Those results were further confirmed by immunofluorescence experiments (Figure 3D). Taken together, our studies showed that DUXAP8 could promote the migration, invasion, and EMT of CRC cells.
DUXAP8 Epigenetically Silenced E-Cadherin mRNA Transcription by Interacting with EZH2 and H3K27me3
We performed a series of mechanism validation experiments to explore how DUXAP8 affects CRC cell proliferation, invasion, and metastasis. First, we fractioned SW620 cells into their cytoplasmic and nuclear components to determine the location of DUXAP8. A subsequent qRT-PCR analysis indicated that DUXAP8 was mainly expressed in the nucleus, suggesting that it functions at the RNA level (Figure 4A). U6 was used as a control for nuclear expression of DUXAP8.
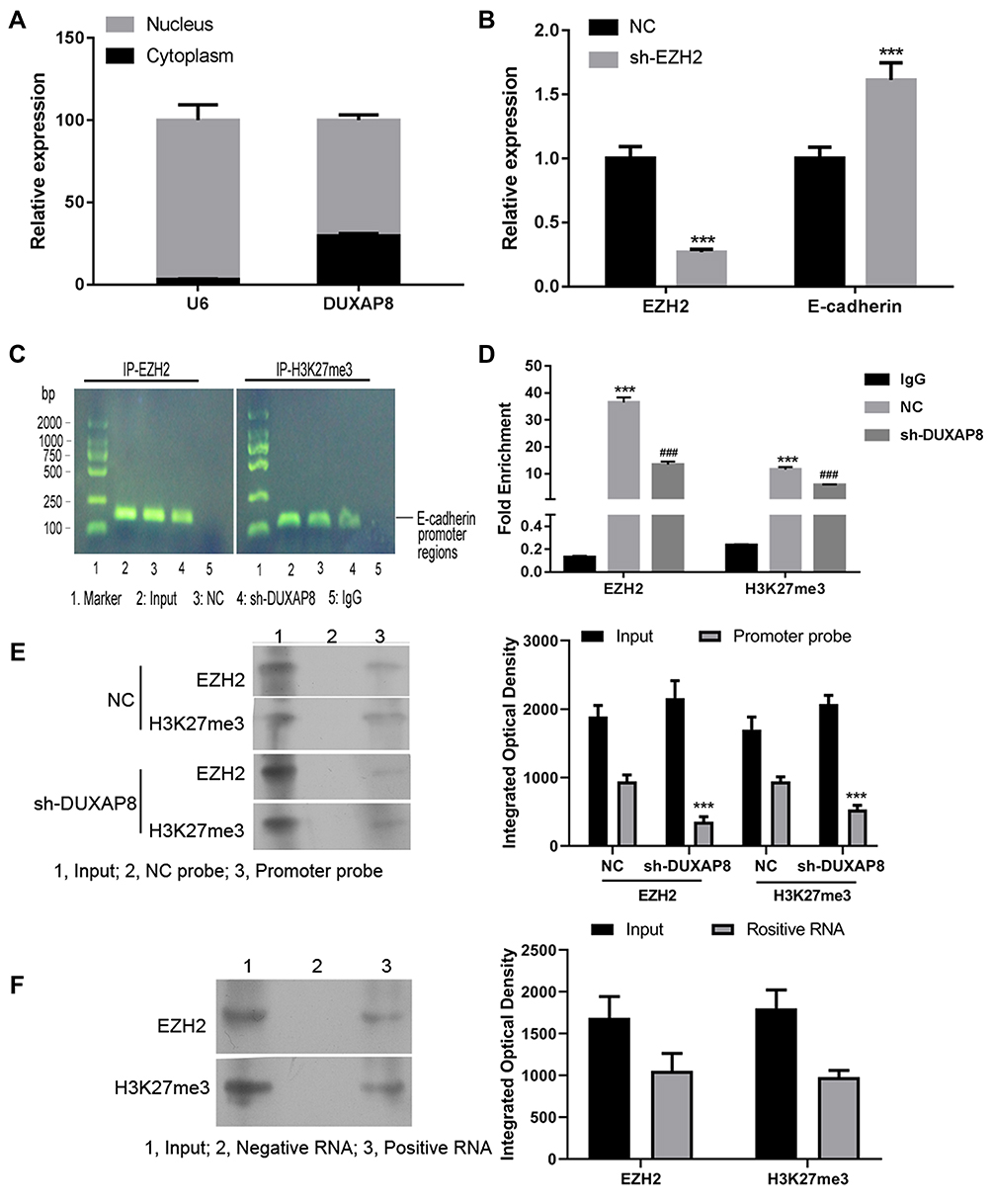 |
Figure 4 DUXAP8 epigenetically silenced E-cadherin transcription by binding with EZH2 and H3K27me3. (A) The levels of DUXAP8 expression in the nucleus or cytoplasm of SW620 cells were detected by qRT-PCR. U6 was used as a nucleus marker. (B) QRT-PCR was performed to analyze the levels of EZH2 and E-cadherin in SW620 cells transfected with shRNA against EZH2 (sh-EZH2) or the NC. ***P < 0.001. (C) Agarose gel assays were performed on the PCR products obtained from immunoprecipitation of the EZH2 antibody and H3K27me3 antibody in the NC and sh-DUXAP8-treated SW620 cells. (D) qPCR analysis of EZH2 binding and H3K27me3 occupancy in the E-cadherin promoter regions of SW620 cells treated with the NC or sh-DUXAP8; IgG served as a negative control. ***P < 0.001 vs IgG, ###P < 0.001 vs NC. (E) RNA pull-down assays were conducted using RNA supplementary to the E-cadherin promoter in SW620 cells transfected with sh-DUXAP8 or the NC; proteins bound to the E-cadherin promoter were detected by western blotting. ***P < 0.001 vs NC group with the same probe. (F) EZH2 binding and H3K27me3 occupancy in the DUXAP8 promoter region were examined using RNA pull down assays performed with a DUXAP8 probe. |
Recent studies have shown that many lncRNAs can bind with PRC2 (polycomb repressive complex 2) to impact the expression of downstream genes by trimethylating H3K27 (histone H3 lysine 27) and thereby suppressing the transcription of target genes. Enhancer of zeste homolog 2 (EZH2) is a major component of PRC2.26 When the NC siRNA and EZH2 siRNA was transfected into SW620 cells, we found that knockdown of EZH2 expression by EZH2 siRNA increased the expression of E-cadherin in SW620 cells (Figure 4B). ChIP assays showed that both EZH2 and H3K27me3 bound to a DNA fragment (Figure 4C) that was located in the E-cadherin gene promoter region (Figure 4D). In the cells with DUXAP8 knockdown, the interaction between EZH2, H3K27me3, and the E-cadherin promoter was significantly suppressed (Figure 4C and D). Pull-down studies performed using RNA supplementary to the E-cadherin promoter showed that binding of EZH2 and H3K27me3 to the promoter was inhibited after DUXAP8 knockdown (Figure 4E). Additionally, pull-down assays performed with the DUXAP8 probe and subsequent western blot examinations revealed the interaction between EZH2, H3K27me3 and DUXAP8 (Figure 4F). These results suggest that DUXAP8 promotes CRC cell proliferation by decreasing E-cadherin levels. This decrease might be partially achieved by DUXAP8 interaction with EZH2 and H3K27me3.
Discussion
While pseudogenes have long been considered to be non-functional, many recent studies have shown that pseudogenes play important roles in tumor development.12 In this study, we found that levels of the pseudogene DUXAP8 were significantly up-regulated in CRC tissues. Cell function studies revealed that interference with DUXAP8 expression in CRC cell lines significantly inhibited tumor cell proliferation and metastasis in vitro.
Currently, only three studies have examined the effects of DUXAP8 on tumor proliferation and metastasis. Those studies found that when expressed at high levels, DUXAP8 may function as a proto-oncogene in gastric cancer. After binding to PRC2, DUXAP8 silences the downstream target gene PLEIm01 and promotes the proliferation and metastasis of gastric cancer cells. Our study suggests that DUXAP8 can be used as an independent risk factor for predicting the survival times of gastric cancer patients, and can also aid in diagnosing and assessing the prognosis of gastric cancer.28 In addition, another study found that the pseudogene DUXAP8 promotes the proliferation and invasion of lung cancer cells by epigenetic silencing of EGR1 and RHOB, which is possibly mediated by the binding of DUXAP8 to EZH2 and LSD1. This binding may allow DUXAP8 to act as an oncogene in NSCLC, suggesting its usefulness as a new therapeutic target for that disease.26 LncRNA DUXAP8 was also found to be overexpressed in bladder cancer tissues, where it promoted the progression of bladder cancer by inhibiting PTEN, and its expression was positively correlated with TNM stage and tumor size, but negatively correlated with patient survival times.27 In our study, we found that overexpression of DUXAP8 could not only enhance CRC cell proliferation, invasion, migration, and epithelial mesenchymal transition, but also inhibit apoptosis and accelerate the cell cycle.
Studies have confirmed that the morphological basis for invasion and metastasis of epithelial-derived malignant cells is the EMT process.30 During the EMT of CRC cells, expression of the EMT marker E-cadherin is reduced and the levels of vimentin and neural cadherin (N-cadherin) expression become elevated.31 In this study, the overexpression of DUXAP8 suppressed the expression of E-cadherin, but increased the levels of vimentin, snail, and N-cadherin expression. However, knockdown of lncRNA DUXAP8 up-regulated E-cadherin, but reduced the levels of vimentin, snail, and N-cadherin. Taken together, these results suggest that DUXAP8 promotes the EMT of CRC cells.
In recent years, studies have also been conducted on the epigenetic regulation mechanism of EMT in tumor cells.33,34 Those studies showed that in different types of highly invasive malignancies, histone methyltransferase could catalyze the methylation of H3K27 or H3K9 in the promoter region of the E-cadherin gene, and thereby promote the EMT process in tumor cells.35,36 EZH2 is the catalytic subunit of PRC2, and catalyzes methylation of the 27th (H3K27m3) lysine of histone H3 by a highly conserved SET region of histone methyltransferase, thereby inhibiting the expression of various tumor suppressor genes and regulating the cell cycle.37 The E-cadherin protein is encoded by the CDH1 gene.38 Studies have shown that SNAIL can bind to the CDH1 promoter sequence in tumor cells and then recruit EZH2 and SUZl2 to the promoter region. SNAIL then catalyzes the trimethylation of H3K27 to ultimately inhibit the transcription of CDH1, which subsequently down-regulates E-cadherin expression.39 Here, we demonstrated that DUXAP8 could epigenetically silence E-cadherin transcription by binding with EZH2 and H3K27me3, and thus promote the EMT process in CRC cells.
In conclusion, our study proved that the pseudogene DUXAP8 was induced in CRC tissues and cells. Our findings showed that DUXAP8 could enhance CRC cell proliferation, invasion, migration, and EMT, and inhibit apoptosis and accelerate the cell cycle. These findings greatly extend our knowledge of the role played by pseudogenes in CRC pathogenesis. We also proved that DUXAP8 could epigenetically silence E-cadherin transcription by binding with EZH2 and H3K27me3, and thereby promote the occurrence of EMT during CRC development. Those findings suggest DUXAP8 as a potential therapeutic target for CRC intervention. Nevertheless, other target genes and regulatory mechanisms related to DUXAP8 remain to be studied in the future. At present, the research conducted on lncRNAs remains in a very early stage, and only a few functions of lncRNAs have been clarified. The expression of lncRNAs has not been examined in large-scale studies, and their specific molecular mechanisms in tumorigenesis and development remain unclear. The detection and screening of differentially expressed lncRNAs in clinical tissue specimens may provide results that can aid in the early diagnosis and targeted therapy of CRC, and also in developing a prognosis for CRC patients. At present, the rapid development of next-generation sequencing technology has allowed researchers to gradually establish a gene catalogue of lncRNA characteristics that can be used to further understand the role played by lncRNA in tumor development.
Data Sharing Statement
Any data associated with this study will be made available.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Wenjing He and Yi Yu contributed equally to this study.
Funding
This study was supported by the Natural Science Foundation of Jiangxi Province (No. 20192ACB20029), the Foundation of the Jiangxi Provincial Health Department (No. 20181036), and the Support Program of the Jiangxi Science and Technology Department (No. 20123BBG70243).
Disclosure
The authors declare that they have no competing interests for this work.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107
2. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383(9927):1490–1502. doi:10.1016/S0140-6736(13)61649-9
3. Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin. 2010;59(6):366–378. doi:10.3322/caac.20038
4. Weitz J, Koch M, Debus J, Höhler T, Galle PR, Büchler MW. Colorectal cancer. Lancet. 2004;365(9454):153–165. doi:10.1016/S0140-6736(05)17706-X
5. Chubak J, Boudreau DM, Rulyak SJ, Mandelson MT. Colorectal cancer risk in relation to antidepressant medication use. J China Univ Geosci. 2018;128:227–232.
6. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):104–117. doi:10.3322/caac.21220
7. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291(5507):1304. doi:10.1126/science.1058040
8. Jianxin L, Juecun Z, Sizhong Z. Human genome project and the post genome era. Prog Biotechnol. 2003;23.
9. Jacq C, Miller JR, Brownlee GG. A pseudogene structure in 5S DNA of Xenopus laevis. Cell. 1977;12(1):109–120. doi:10.1016/0092-8674(77)90189-1
10. Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA. 2011;17:792.
11. Gao P, Wei GH. Genomic insight into the role of lncRNA in cancer susceptibility. Int J Mol Sci. 2017;18(6):1239.
12. Shi X, Nie F, Wang Z, Sun M. Pseudogene-expressed RNAs: a new frontier in cancers. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2016;37(2):1471. doi:10.1007/s13277-015-4482-z
13. Oku Y, Shimoji T, Takifuji K, et al. Identification of the molecular mechanisms for dedifferentiation at the invasion front of colorectal cancer by a gene expression analysis. Clin Cancer Res off J Am Assoc Cancer Res. 2008;14(22):7215–7222. doi:10.1158/1078-0432.CCR-08-0370
14. Qi L, Zhang W, Cheng Z, Tang N, Ding Y. Study on molecular mechanism of ANOS1 promoting development of colorectal cancer. PLoS One. 2017;12(8):e0182964. doi:10.1371/journal.pone.0182964
15. Bognár G, Ledniczky G, István G, Ondrejka P. Molecular mechanisms in development of colorectal cancer metastasis. Magy Seb. 2006;59:342–349.
16. Yang Y, Zhao L, Lei L, et al. LncRNAs: the bridge linking RNA and colorectal cancer. Oncotarget. 2017;8(7):12517–12532. doi:10.18632/oncotarget.13573
17. Wang JZ, Xu CL, Wu H, Shen SJ. LncRNASNHG12promotes cell growth and inhibits cell apoptosis in colorectal cancer cells. Braz J Med Biol Res. 2017;50(3):e6079. doi:10.1590/1414-431x20176079
18. Svoboda M, Slyskova J, Schneiderova M, et al. HOTAIR long non-coding RNA is a negative prognostic factor not only in primary tumors, but also in the blood of colorectal cancer patients. Carcinogenesis. 2014;35(7):1510–1515. doi:10.1093/carcin/bgu055
19. Ling H, Spizzo R, Atlasi Y, et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013;23(9):1446–1461. doi:10.1101/gr.152942.112
20. Ryunosuke K, Teppei S, Koshi M, et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71(20):6320–6326. doi:10.1158/0008-5472.CAN-11-1021
21. Yu H, Ying-Nan Y, Heng-Heng Y, et al. UCA1, a long non-coding RNA up-regulated in colorectal cancer influences cell proliferation, apoptosis and cell cycle distribution. Pathology. 2014;46:396–401.
22. Yang MH, Hu ZY, Xu C, et al. MALAT1 promotes colorectal cancer cell proliferation/migration/invasion via PRKA kinase anchor protein 9. Biochim Biophys Acta. 2015;1852(1):166–174. doi:10.1016/j.bbadis.2014.11.013
23. Lu M, Liu Z, Li B, Wang G, Li D, Zhu Y. The high expression of long non-coding RNA PANDAR indicates a poor prognosis for colorectal cancer and promotes metastasis by EMT pathway. J Cancer Res Clin Oncol. 2017;143(1):1–11. doi:10.1007/s00432-016-2252-y
24. Jin J, Chu Z, Ma P, Meng Y, Yang Y. Long non-coding RNA SPRY4-IT1 promotes proliferation and invasion by acting as a ceRNA of miR-101-3p in colorectal cancer cells. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2017;39(7):101042831771625. doi:10.1177/1010428317716250
25. Shi D, Liang L, Zheng H, et al. Silencing of long non-coding RNA SBDSP1 suppresses tumor growth and invasion in colorectal cancer. Biomed Pharmacother. 2017;85:355–361. doi:10.1016/j.biopha.2016.11.036
26. Sun M, Nie FQ, Zang C, et al. The pseudogene DUXAP8 promotes non-small-cell lung cancer cell proliferation and invasion by epigenetically silencing EGR1 and RHOB. Mol Ther J Am Soc Gene Ther. 2017;25(3):739. doi:10.1016/j.ymthe.2016.12.018
27. Lin MG, Hong YK, Zhang Y, Lin BB, He XJ. Mechanism of lncRNA DUXAP8 in promoting proliferation of bladder cancer cells by regulating PTEN. Eur Rev Med Pharmacol Sci. 2018;22:3370–3377.
28. Ma HW, Xie M, Sun M, et al. The pseudogene derived long noncoding RNA DUXAP8 promotes gastric cancer cell proliferation and migration via epigenetically silencing PLEKHO1 expression. Oncotarget. 2016;8:52211.
29. Du C, Wang HX, Chen P, Chen CH. STAT3-induced upregulation of lncRNA DUXAP8 functions as ceRNA for miR-577 to promote the migration and invasion in colorectal cancer through the regulation of RAB14. Eur Rev Med Pharmacol Sci. 2019;23:6105–6118.
30. De CB, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110.
31. Bates RC, Mercurio AM. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther. 2005;4(4):371–376. doi:10.4161/cbt.4.4.1655
32. Santamaria PG, Moreno‐Bueno G, Portillo F, Cano A. EMT: present and future in clinical oncology. Mol Oncol. 2017;11(7):718–738. doi:10.1002/1878-0261.12091
33. Lin Y, Dong C, Zhou BP. Epigenetic regulation of EMT: the snail story. Curr Pharm Des. 2014;20(11):1698–1705. doi:10.2174/13816128113199990512
34. Zha L, Cao Q, Cui X, et al. Epigenetic regulation of E-cadherin expression by the histone demethylase UTX in colon cancer cells. Med Oncol. 2016;33(3):1–11. doi:10.1007/s12032-016-0734-z
35. Hsiao SM, Chen MW, Chen CA, et al. The H3K9 methyltransferase G9a represses E-cadherin and is associated with myometrial invasion in endometrial cancer. Ann Surg Oncol. 2015;22(S3):1556–1565. doi:10.1245/s10434-015-4379-5
36. Zhou Z, Zhang HS, Liu Y, et al. Loss of TET1 facilitates DLD1 colon cancer cell migration via H3K27me3-mediated down-regulation of E-cadherin. J Cell Physiol. 2018;233(2):1359–1369. doi:10.1002/jcp.26012
37. Liu F, Gu L, Cao Y, Fan X, Zhang F, Sang M. Aberrant overexpression of EZH2 and H3K27me3 serves as poor prognostic biomarker for esophageal squamous cell carcinoma patients. Biomarkers. 2016;21(1):80–90. doi:10.3109/1354750X.2015.1118537
38. Richards FM, Mckee SA, Rajpar MH, et al. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 1999;8(4):607. doi:10.1093/hmg/8.4.607
39. Peña C, García JM, Silva J, et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet. 2005;14(22):3361–3370. doi:10.1093/hmg/ddi366
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.