")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
The protective effect of PRMT6 overexpression on cigarette smoke extract-induced murine emphysema model
Authors He X, Li T, Kang N, Zeng H, Ren S, Zong D, Li J, Cai S, Chen P , Chen Y
Received 26 June 2017
Accepted for publication 26 September 2017
Published 3 November 2017 Volume 2017:12 Pages 3245—3254
DOI https://doi.org/10.2147/COPD.S144881
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Xue He,1–3 Tiao Li,1–3 Naixin Kang,1–3 Huihui Zeng,1–3 Siying Ren,1–3 Dandan Zong,1–3 Jinhua Li,1–3 Shan Cai,1–3 Ping Chen,1–3 Yan Chen1–3
1Department of Respiratory Medicine, The Second Xiangya Hospital, 2Research Unit of Respiratory Disease, 3Diagnosis and Treatment Center of Respiratory Disease, Central South University, Changsha, Hunan, China
Background: Cigarette smoke exposure is the most common risk factor for emphysema, which is one of the major pathologies of COPD. Protein arginine methyltransferase 6 (PRMT6) is a nuclear enzyme that specially catalyzes dimethylation of R2 in histone H3 (H3R2me2a). H3R2me2a prevents trimethylation of H3K4 (H3K4me3), which is located in the transcription start sites of genes in mammalian genomes. We attempted to determine the expression of PRMT6 in human samples, and investigate whether the upregulation of PRMT6 expression can attenuate the development of cigarette smoke extract (CSE)-induced emphysema. Further experiments were performed to elucidate the molecular mechanisms involved.
Materials and methods: Human lung tissues were obtained from patients undergoing pneumonectomy for benign pulmonary lesions. BALB/c mice were treated with lentiviral vectors intratracheally and injected with CSE three times. The protein expression of PRMT6, H3R2me2a, and H3K4me3 in human and mouse samples, as well as B-cell lymphoma-2 (Bcl-2), Bcl-2-associated X protein (Bax), and endothelial nitric oxide synthase (eNOS) in mice were detected in lung homogenates by Western blotting. The mRNA expression of cyclooxygenase-2, interleukin-6, Bcl-2, Bax, and eNOS in mice was measured by quantitative real-time polymerase chain reaction.
Results: The expression of PRMT6 was significantly downregulated in the pulmonary parenchyma in smokers with COPD as well as in mice treated with CSE. Overexpression of PRMT6 was detected in the CSE + Lenti-PRMT6 group of mice, which reversed the expression of H3R2me2a and H3K4me3. Inflammation, apoptosis, and oxidative stress levels were severe in the CSE-treated emphysema mice compared with the control group, which was inhibited by the overexpression of PRMT6.
Conclusion: The overexpression of PRMT6 might inhibit inflammation, apoptosis, and oxidative stress in CSE-induced emphysema mediated by H3R2me2a.
Keywords: COPD, cigarette smoke extract, inflammation, apoptosis, oxidative stress, PRMT6
Introduction
COPD, according to the definition by the Global initiative for chronic Obstructive Lung Disease (GOLD), is a complex chronic airway disease characterized by persistent airflow limitation that is usually progressive.1 This disease is now the third leading cause of death in the world.2 The worldwide morbidity and mortality are expected to increase in the coming decades; as such, COPD poses a huge burden on the economy and society.3 The strongest and most common risk factor for COPD is exposure to cigarette smoke (CS),4 which leads to alveolar wall destruction with airspace enlargement (emphysema), one of the major pathologies of COPD.1,5 It is thought that CS-induced inflammation,6 apoptosis,7 and oxidative stress8,9 are the major drivers of emphysema.
Chen et al reported that interleukins (ILs)-1β play an important role in the inflammatory pathogenesis of emphysema in an animal model.10 Apoptosis of alveolar wall cells has been observed in COPD, and has been related to disease progression.7,11,12 The B-cell lymphoma-2 (Bcl-2) family of proteins is an essential factor that regulates apoptosis.13,14 Another major pathogenic factor contributing to emphysema/COPD is CS-associated oxidative stress, related to the formation of reactive oxygen species (ROS)/reactive nitrogen species.15,16 Endothelial cells produce nitric oxide synthase,17 which has been correlated with alveolar repair in a rat model of pulmonary emphysema.18
Protein arginine methyltransferase 6 (PRMT6) is a nuclear enzyme that modifies histone tails.19 It catalyzes the asymmetric dimethylation of histone H3 arginine 2 (H3R2me2a) specially. The trimethylation of H3 lysine 4 (H3K4me3) appears to counter-correlate with H3R2me2a in E-box-containing gene promoters.20,21 Interestingly, overexpression and knockdown analysis identify PRMT6 as a critical negative regulator of H3K4me3, which is related to global transcriptional activation in mammalian genomes.22–24 In addition, the level of PRMT6 is associated with apoptosis in vivo.25,26 In our previous study,27 human umbilical vein endothelial cells (HUVECs) transfected with PRMT6 expressing plasmid inhibited the cigarette smoke extract (CSE)-induced upregulation of Bcl-2-associated X protein (Bax) and cyclooxygenase-2 (COX-2). The inhibition of PRMT6 promoted the apoptosis and inflammation level in HUVECs induced by CSE. Meanwhile, CS-induced oxidative stress also has a close relationship with histone methylation, which could promote or depress gene activation.28
We hypothesized that the attenuation of PRMT6 may play a role in the pathogenesis of COPD and that upregulation of PRMT6 might possibly protect against CSE-induced emphysema in a mouse model. To the best of our knowledge, this study is the first investigation on the overexpression of PRMT6 with the aim of preventing CSE-induced emphysema in a mouse model by inhibiting inflammation, apoptosis, and oxidative stress at the mRNA and protein levels.
Materials and methods
Ethics statement
This study was approved by the Institutional Human and Animal Care Ethics Committee at the Second Xiangya Hospital of Central South University. Written informed consent was obtained from all human participants before their enrollment into the study.
Lung tissues of patients with COPD
Pulmonary tissues were obtained from patients undergoing pneumonectomy at the Second Xiangya Hospital of Central South University between August 2012 and January 2013. Final diagnosis was benign lesions (such as lung abscesses and bullae), excluding cancer patients. Regarding smoking history and pulmonary function, the patients were divided into three groups: nonsmokers without COPD group (n=10), smokers without COPD group (n=10), and smokers with COPD group (stable stage, n=10). According to GOLD, the diagnosis of COPD was a post-bronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ≤0.7.29
Preparation of CSE
CSE was prepared following a modification of the method proposed by Zhang et al.30 Briefly, five cigarettes (China Tobacco Hunan Industrial Co, Ltd, Hunan, China; tar: 12 mg, nicotine: 1.1 mg, carbon monoxide: 14 mg) were burned; the smoke was collected in a vessel containing phosphate-buffered saline (PBS; 10 mL). The pH of the solution was 7.2–7.4. CSE was prepared fresh before use.
Lentivirus plasmid and transduction
Lentivirus containing the PRMT6 gene was purchased from Thermo Fisher Scientific (Waltham, MA, USA). The lentivirus vectors were constructed by the company. The final concentration of the lentivirus was 1.0×109 ifu/mL. The lentivirus was stored at −80°C and repetitive freeze–thaw occurred no more than three times.
Animal protocols
Six-week-old, male, specific pathogen-free, BALB/c mice (21–23 g each) were purchased from the Shanghai Animal Husbandry Advisory Services (China). The Second Xiangya Hospital Experimental Animal Center of Central South University was responsible for feeding. All animal care and experimental protocols were approved by the Animal Care and Use Committee of the Second Xiangya Hospital, Central South University, and were conducted in accordance with the committee’s animal care and use guidelines. Thirty-two mice were randomly divided into four groups: the control group, CSE group, CSE + Lenti-NULL group, and CSE + Lenti-PRMT6 group, with eight mice in each group. The control group was injected intraperitoneally with 0.3 mL PBS at days 0, 11, and 22.30 The other three groups were injected intraperitoneally with 0.3 mL CSE at days 0, 11, and 22. The control group and CSE group received intratracheal instillation of PBS at day 14. CSE + Lenti-NULL group and CSE + Lenti-PRMT6 group received an intratracheal instillation of 108 ifu/mL (100 μL) Lenti-NULL and Lenti-PRMT6 correspondingly at day 14. All mice were sacrificed at day 28.
Lung tissues preparation
The left lungs were inflated with 10% formalin at a constant pressure of 25 cm H2O and fixed for 24 hours. Paraffin-embedded sections were prepared using standard techniques.31 Total protein and RNA were extracted from the right middle lobe and lower lobe separately as previously described.30
Morphometry
Lung sections were stained with hematoxylin and eosin (HE), and the morphology of lung tissues of mice was observed by light microscopy. The mean linear intercept (MLI) and destructive index (DI) were measured at a magnification of 100× as previously described.32,33
Western blotting
In lung homogenates, the expression of PRMT6, H3R2me2a, and H3K4me3 in mouse and human samples, as well as Bcl-2, Bax, and endothelial nitric oxide synthase (eNOS) in mouse samples were detected by standard Western blotting techniques. Homogenate supernatants were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Beyotime Institute of Biotechnology, Shanghai, China), transferred onto polyvinylidene fluoride membranes, blocked for 1 hour, then washed, incubated with rabbit monoclonal antibodies against PRMT6, H3R2me2a, H3K4me3, Bcl-2, Bax, and eNOS (Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C. The next day, the membrane was washed and incubated for 2 hours at 37°C with HRP-labeled goat anti-rabbit secondary antibody (Santa Cruz Biotechnology Inc, Dallas, TX, USA), and then washed again. Labeled proteins were visualized by the electrochemiluminescence plus Western blotting detection system (Immobilon-P; EMD Millipore, Billerica, MA, USA). Band densities were quantified using Quantity One Analysis Software (Bio-Rad Laboratories Inc, Hercules, CA, USA).
Real-time quantitative polymerase chain reaction
Total RNA was isolated from lung tissues of mice using Trizol reagent (Thermo Fisher Scientific) and then treated with DNase I (Thermo Fisher Scientific) according to the manufacturer’s instructions. The primers used in this study were either synthesized according to previous protocols27 or designed with Primer Premier 5.0 (Premier Biosoft international, Palo Alto, CA, USA). The sequences of the primers used in the present study were:
- actin-F: 5′-CATCCTGCGTCTGGACCTGG-3′;
- actin-R: 5′-TAATGTCACGCACGATTTCC-3′;
- IL-6-F: 5′-AGAACTCGAACGGGAACGCG-3′;
- IL-6-R: 5′-CGGCGACGGCAGACGGGC-3′;
- COX-2-F: 5′-TGCAGAATGTTGTTGTTTCTG-3′;
- COX-2-R: 5′-AATTAGAAACTTCAATCCGATTA-3′;
- Bcl-2-F: 5′-TGCAGAATGTTGTTGTTTCTG-3′;
- Bcl-2-R: 5′-AATTAGAAACTTCAATCCGATTA-3′;
- Bax-F: 5′-AGAACTCGAACGGGAACGCG-3′;
- Bax-R: 5′-CGGCGACGGCAGACGGGC-3′;
- eNOS-F: 5′-AGAACTCGAACGGGAACGCG-3′;
- eNOS-R: 5′-CGGCGACGGCAGACGGGC-3′.
Relative changes in transcript abundance were expressed as ΔCT values (ΔCT = ΔCT reference − ΔCT target), where higher ΔCT values indicate higher transcript abundance.
Statistical analysis
A software package (SPSS 19.0; IBM Corporation, Armonk, NY, USA) was used to perform all statistical analyses. The data distribution was tested by the Shapiro–Wilk test. Continuous data were expressed as mean ± standard error of the mean. Differences were evaluated by one-way analysis of variance followed by the unpaired Student’s t-test for multiple comparisons. Pearson’s correlation was used to analyze the correlations between MLI and DI and the expression of PRMT6, H3R2me2a, and H3K4me3 in mice. A p-value <0.05 was considered statistically significant.
Results
Pulmonary function of patients
The pulmonary function test results of the patients are given in Table 1. Compared to the nonsmokers without COPD and smokers without COPD, FEV1 as a percentage of the normal predicted value (FEV1/pre[%]), FEV1/FVC (%), and peak expiratory flow (PEF) as a percentage of the normal predicted value ([PEF]/pre%) were lower in the smokers with COPD (p<0.01).
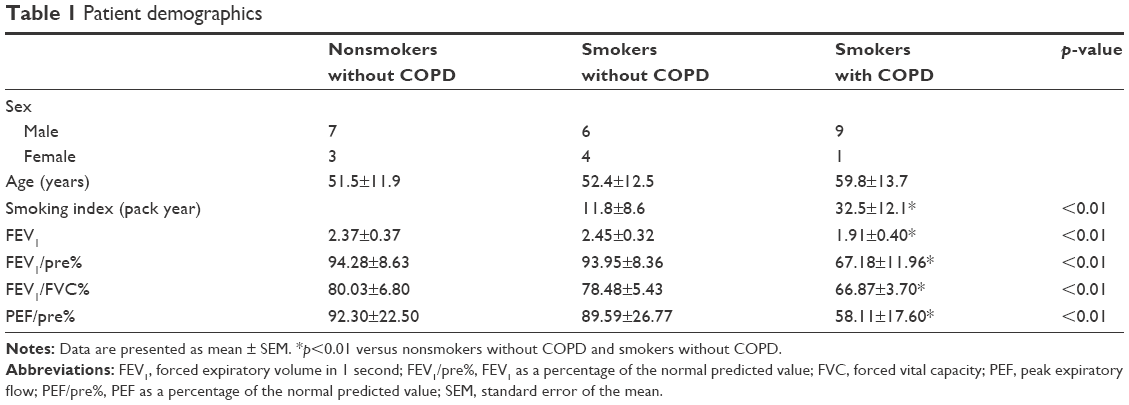 | Table 1 Patient demographics |
Expression of PRMT6, H3R2me2a, and H3K4me3 in human lung tissues
As shown in Figure 1, the protein abundance of PRMT6 was significantly decreased (p<0.05) in lung tissues of patients with COPD compared with those subjects without COPD. Meanwhile, the protein abundance of H3R2me2 was significantly decreased and the protein abundance of H3K4me3 was significantly increased in the lung tissues of smokers with COPD compared with subjects without COPD, indicating a change in the expression of these proteins in COPD patients.
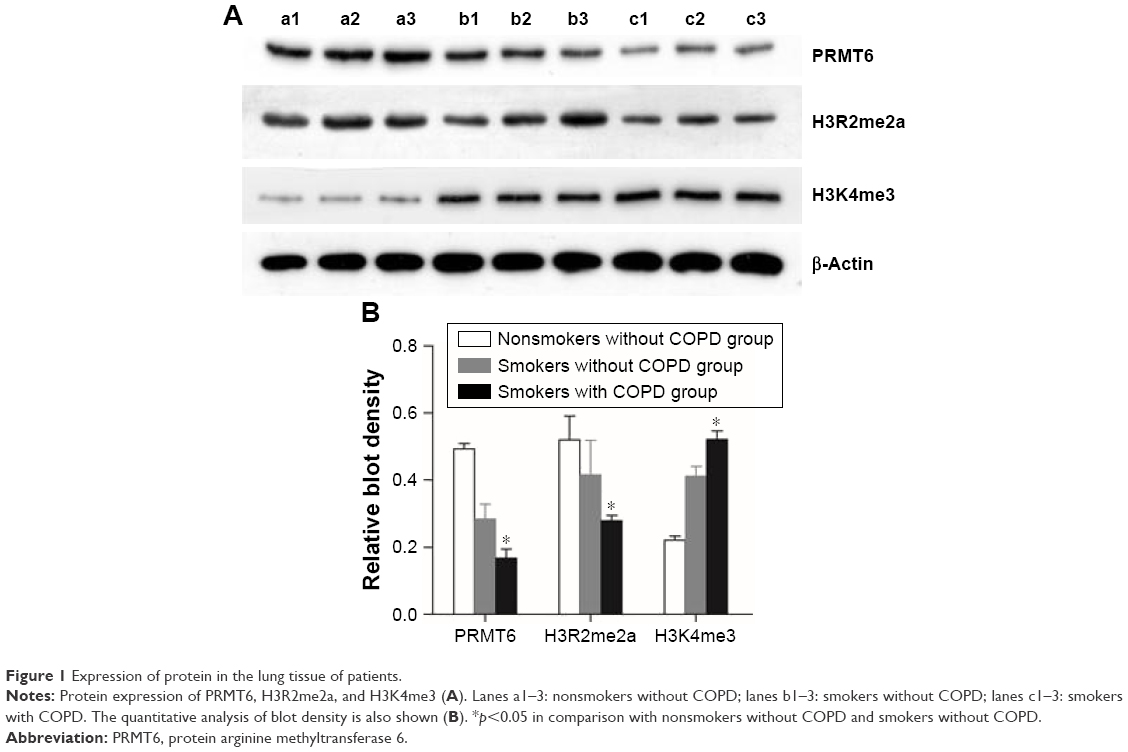 | Figure 1 Expression of protein in the lung tissue of patients. |
Histological examination of mice
All HE stained lung tissue slices were observed at low magnification (100×). The CSE group showed CSE-induced alveolar airspace enlargement, lung parenchymal destruction, and inflammatory infiltration (p<0.05, Figure 2Ab). However, the CSE + Lenti-PRMT6 group showed significant improvements (Figure 2Bd) in alveolar architecture in contrast to the CSE group and the CSE + Lenti-NULL group, but was still worse than the control group (p<0.05, Figure 2A).
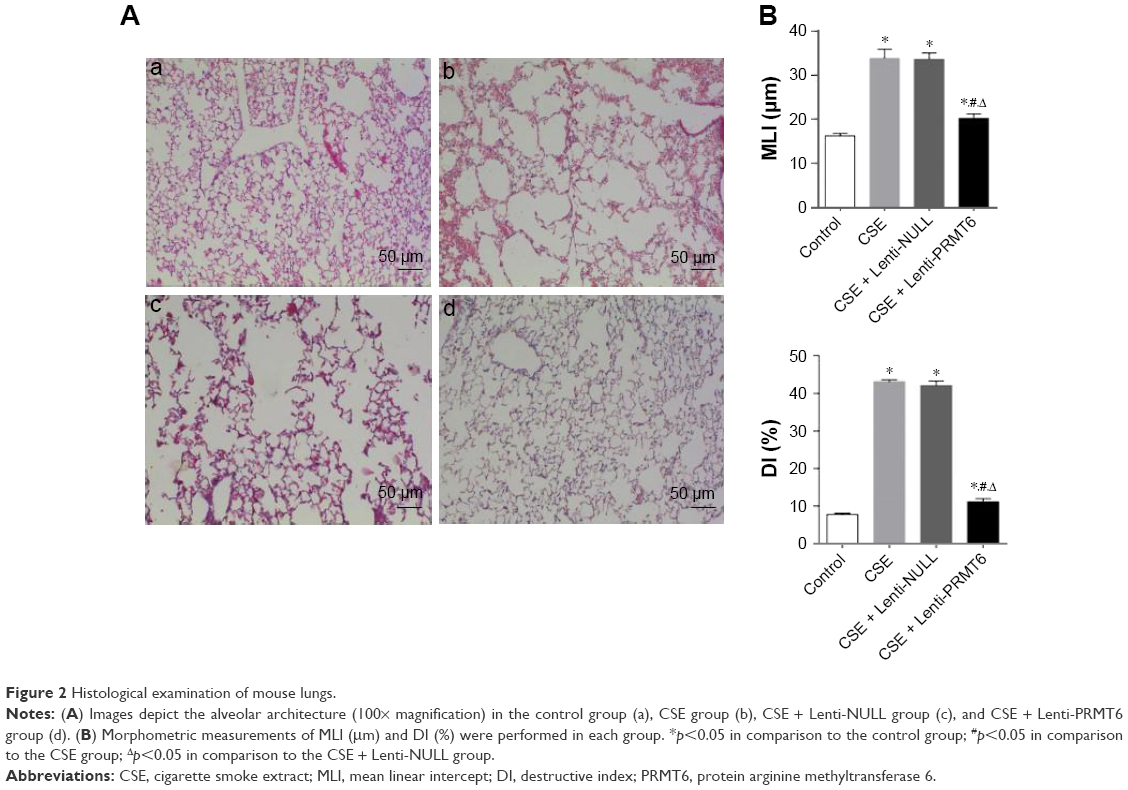 | Figure 2 Histological examination of mouse lungs. |
Expression of PRMT6, H3R2me2a, and H3K4me3 in mice
As shown in Figure 3, the protein abundance of PRMT6 was significantly decreased (p<0.05) in the CSE group compared with the control group. The overexpression of PRMT6 was successful in mice with CSE-induced emphysema as the protein abundance of PRMT6 was significantly increased in the CSE + Lenti-PRMT6 group (p<0.05) compared to that in the CSE group and the CSE + Lenti-NULL group. Moreover, the protein abundance of H3R2me2a was significantly decreased and H3K4me3 was significantly increased (p<0.05) in the CSE group compared with levels in the control group. However, the protein abundance of H3R2me2a was significantly increased and H3K4me3 was significantly decreased in the CSE + Lenti-PRMT6 group with the overexpression of PRMT6 compared with mice not overexpressing PRMT6. As shown in Figure 4, the expression of PRMT6 and H3R2me2a was negatively correlated with MLI and DI (p<0.05). In addition, the expression of H3K4me3 was positively correlated with MLI and DI (p<0.05).
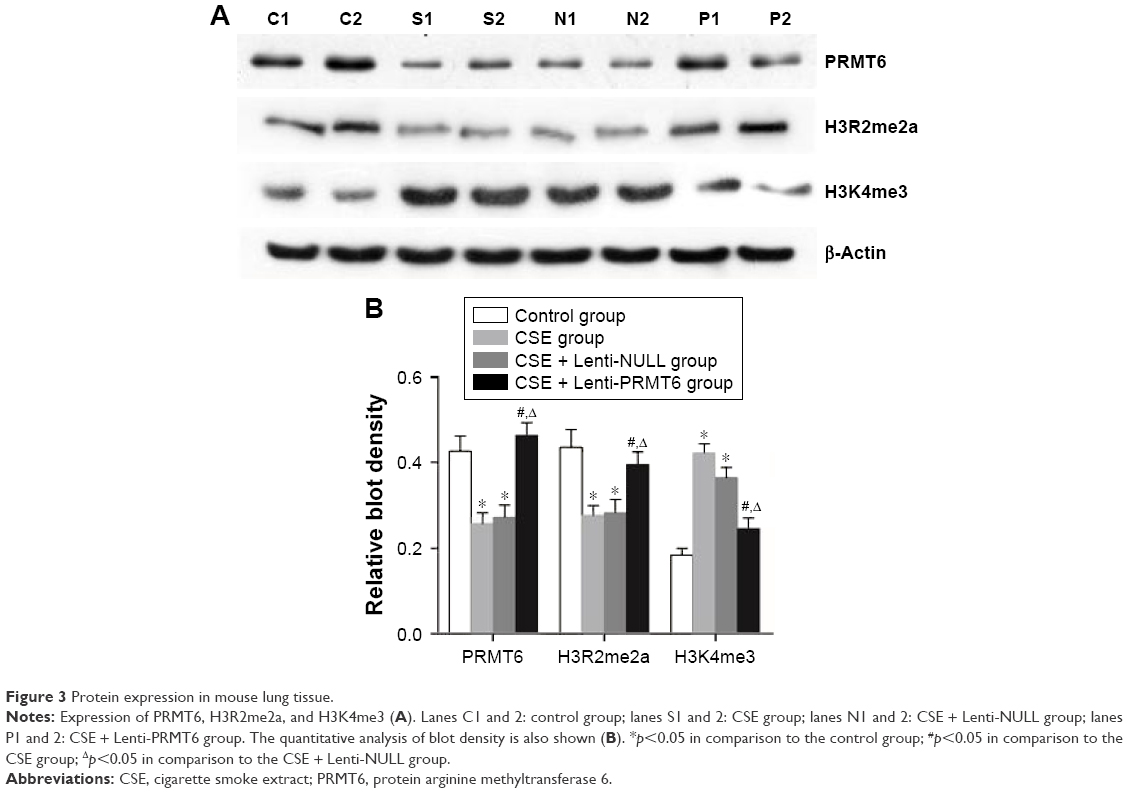 | Figure 3 Protein expression in mouse lung tissue. |
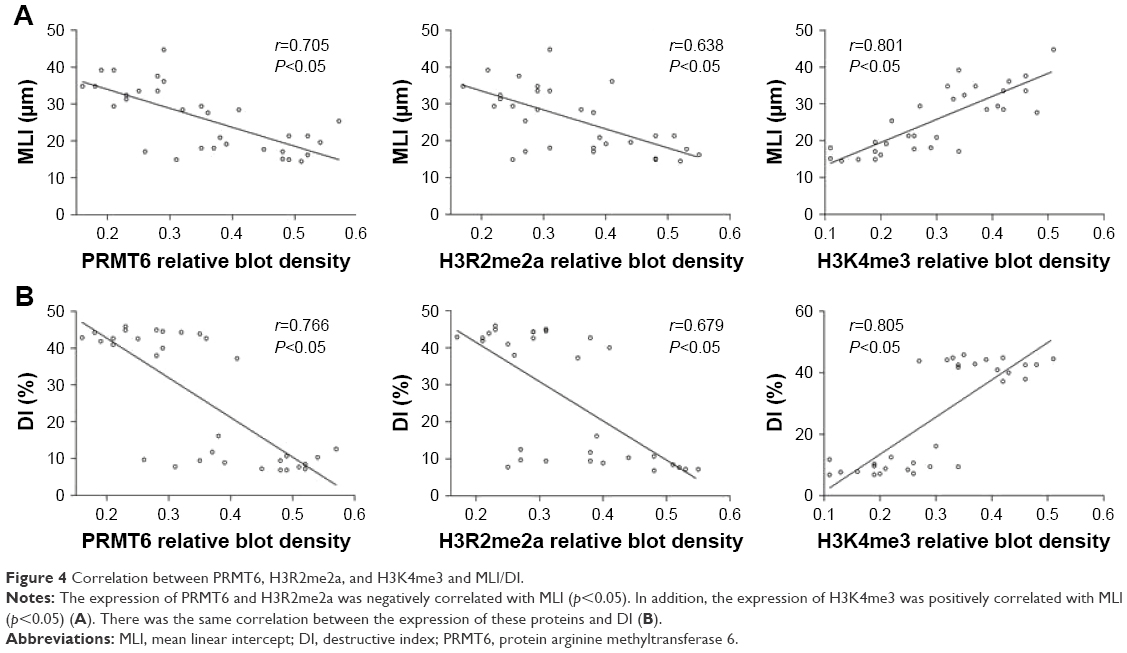 | Figure 4 Correlation between PRMT6, H3R2me2a, and H3K4me3 and MLI/DI. |
Overexpression of PRMT6 inhibits inflammation in the lungs of mice with CSE-induced emphysema
The CSE group showed increased expression of COX-2 and IL-6 in lung tissues compared with the control group and the CSE + Lenti-PRMT6 group (p<0.05, Figure 5). In addition, there were no significant differences between the CSE group and the CSE + Lenti-NULL group (p>0.05, Figure 5). The overexpression of PRMT6 attenuated the expression of inflammatory biomarkers.
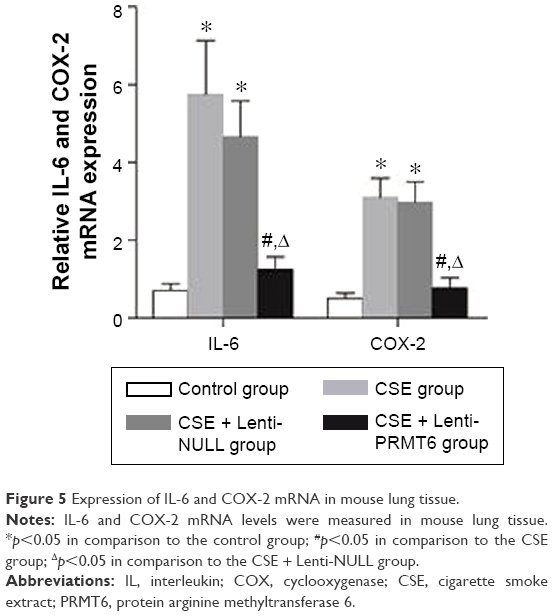 | Figure 5 Expression of IL-6 and COX-2 mRNA in mouse lung tissue. |
Overexpression of PRMT6 inhibits apoptosis in the lungs of mice with CSE-induced emphysema
The Bcl-2 family proteins are essential factors that regulate apoptosis, and include antiapoptotic proteins and proapoptotic proteins. The CSE group showed decreased expression of Bcl-2 and increased expression of Bax in the lung compared with the control group and the CSE + Lenti-PRMT6 group (p<0.05, Figure 6). Lenti-NULL had no effect on the expression of Bcl-2 and Bax. The overexpression of PRMT6 inhibited apoptosis in the lungs of mice.
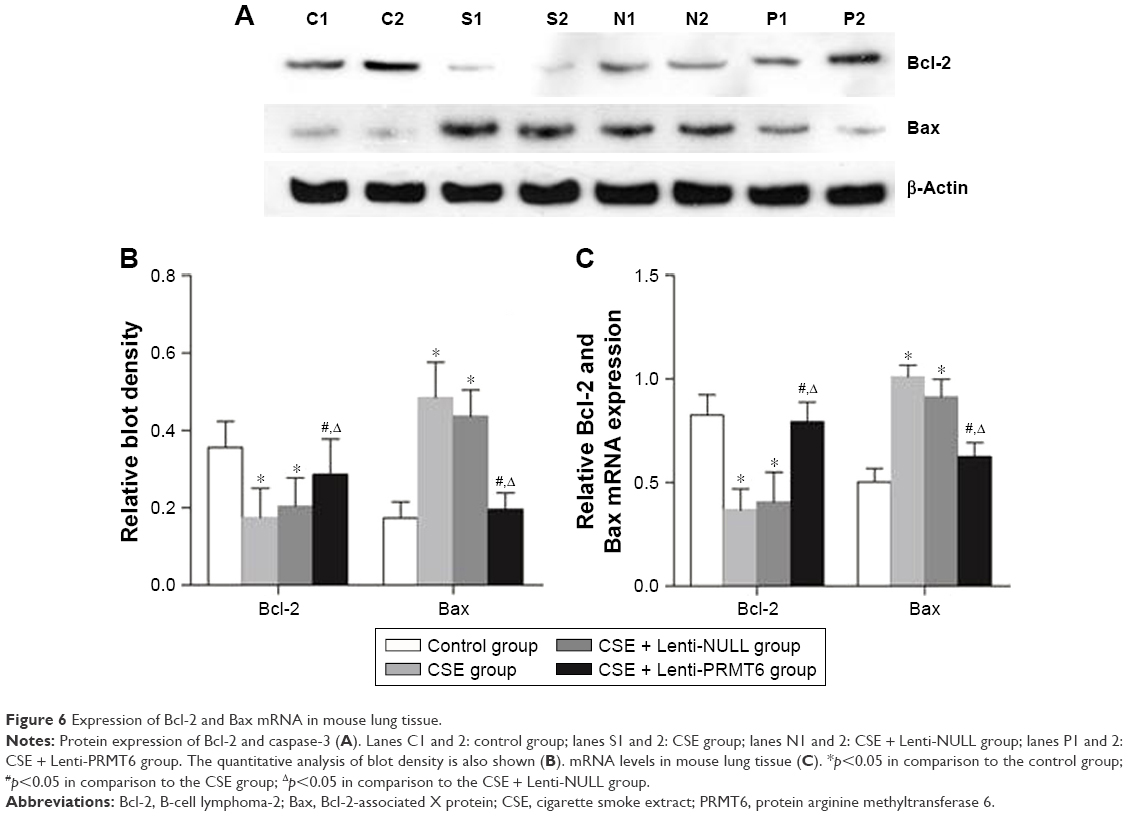 | Figure 6 Expression of Bcl-2 and Bax mRNA in mouse lung tissue. |
Overexpression of PRMT6 inhibits oxidative stress in the lungs of mice with CSE-induced emphysema
eNOS catalyzes the oxidation of NO, and is an important enzyme during oxidative stress. The CSE group showed decreased expression of eNOS in the lung tissues of mice compared with the control group and the CSE + Lenti-PRMT6 group (p<0.05, Figure 7). The expression of eNOS in the CSE + Lenti-PRMT6 group was lower than that in the control group (p<0.05, Figure 7). The overexpression of PRMT6 partly inhibited oxidative stress in the mouse lung.
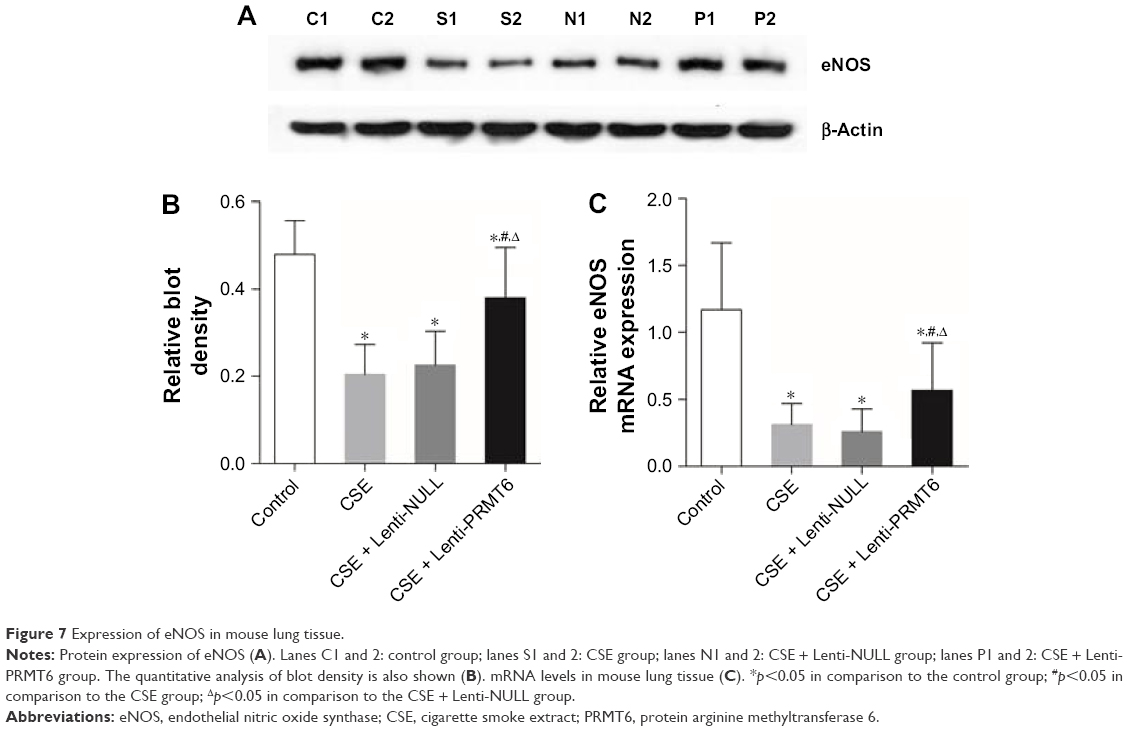 | Figure 7 Expression of eNOS in mouse lung tissue. |
Discussion
This study demonstrated that the expression of PRMT6 was decreased in the lung tissues of mice with CSE-induced emphysema, which was confirmed in the lung tissues of COPD patients. The most remarkable finding of this study was that the overexpression of PRMT6 was able to partly reduce levels of biomarkers associated with inflammation, apoptosis, and oxidative stress in lung tissues of mice with CSE-induced emphysema. These results indicate that the overexpression of PRMT6 might inhibit the inflammatory response by reducing proinflammatory cytokine levels (such as COX-2, IL-6), attenuate apoptosis by affecting the expression of anti/proapoptotic factors (such as Bcl-2 and Bax), and downregulate the expression of eNOS in the oxidative stress pathway in the lung tissues of mice with CSE-induced emphysema.
The present mouse model of emphysema was established by an intraperitoneal injection of CSE every 11 days, that is, three times within 28 days. Airspace enlargement and alveolar destruction were evidenced by increases in MLI and DI in mice injected with CSE. The increases in MLI and DI were partly ameliorated by the intratracheal instillation of PRMT6 in mice with CSE-induced emphysema.
We found that PRMT6, a type of arginine methyltransferase, was downregulated in the lung tissues of COPD patients as well as in mice with emphysema. A previous study showed that CSE treatment decreased the protein abundance of PRMT6 in vitro,19 but, the mechanism of the reduction was not described.27 One possible mechanism might be related to activation of the ubiquitin–proteasome pathway, an ATP-dependent proteolytic pathway, in CSE-induced emphysema. Ottenheijm et al demonstrated that the reduced contractile protein content was attributed to increased activity of the ubiquitin–proteasome pathway in patients with COPD stage I/II.34 Interestingly, the ubiquitin–proteasome system is also involved in the response to hypoxemia,35 which is important in the pathogenesis of COPD. A growing body of evidence has shown that the ubiquitin–proteasome system plays a crucial role in the degradation of contractile proteins in patients with COPD.36 Moreover, Kim et al demonstrated that apoptosis and cell cycle arrest induced by CS occurred partly through Akt degradation via the ubiquitin–proteasome system.37 It is worth to further investigate the role of the ubiquitin–proteasome system in PRMT6 degradation, resulting in the attenuation of PRMT6 levels in mice with CSE-induced emphysema.
Chronic inflammation, apoptosis, and oxidative stress and the related networks are the most well-known and important mechanisms in cigarette smoking-induced emphysema. The present investigation undoubtedly demonstrates that the protective role of PRMT6 overexpression regarding inflammation, apoptosis, and oxidative stress was associated with alterations in pro-inflammatory cytokines (COX-2, IL-6), apoptosis biological markers (Bcl-2 family), and oxidative stress indicators (eNOS) in the mouse emphysematous lung. Moreover, the protective effects of PRMT6 overexpression on CSE-induced inflammation, apoptosis, and oxidative stress appeared to be mediated through H3R2me2a in emphysematous mice, because inhibition of PRMT6 in the CSE-induced emphysematous lung was associated with decreased expression of H3R2me2a and increased expression of H4K3me3 in the present study. In agreement with this, previous studies have indicated that PRMT6 catalyzes H3R2 dimethylation and controls global levels of H3R2me2a in vivo, which conversely prevents the di- and trimethylation of H3K4.20,21 Moreover, alterations to H3R2me2a correspond with CSE-induced inflammation and apoptosis.27 All through, PRMT6 plays a potential oncogenic role in specific cancer tissue and tumor apoptosis.38,39 The molecular mechanism between PRMT6 and lung cancer is still unknown. Further investigation concerning PRMT6 and apoptosis in lung cancer should gain more concern. Furthermore, H3K4me3 has been found in the 5′ region of the transcribed genes and correlates with most of the genes expression.22–24 Oxidative stress induced by CS leads to chromatin modifications (histone acetylation/deacetylation and histone methylation/demethylation), which result in gene activation (such as H3K4).28 However, further investigation is necessary to investigate the detailed function of H3R2me2a and H3K4me3 in apoptosis, inflammation, and oxidative stress.
There are also some limitations in this study. We did not evaluate the level of ROS or antioxidant capacity in CSE-induced emphysema mice. The current results may not be sufficient to conclude that oxidative stress is implicated. Further studies are necessary to confirm this pathway.
In summary, the remarkable results of this study suggest that the overexpression of PRMT6 might partially protect against CSE-induced emphysema in mice through pathways relating to inflammation, apoptosis, and oxidative stress, mediated by H3R2me2a. This compelling finding is helpful for understanding the pathogenesis of COPD, and thus has important implications on the clinical treatment of COPD.
Acknowledgments
The authors would like to thank all the participants involved in this study. This study was supported by the National Science Foundation of China (no 81270100, 81400032, 81600031), the Project of Hunan Science and Technology Department (no 2015SK20403), the National Science Foundation of Hunan Province (no 13JJ3024), the Fundamental Research Funds for the Central Universities of Central South University (no 2016zzts551), and the National Key Clinical Specialty Construction Projects of China.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
The Global Burden of Disease 2010 Study Consortium. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2013;380(9859):2095–2128. | ||
Lopez AD, Shibuya K, Rao C, et al. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27(2):397–412. | ||
Hooper R, Burney P, Vollmer WM, et al. Risk factors for COPD spirometrically defined from the lower limit of normal in the BOLD project. Eur Respir J. 2012;39(6):1343–1353. | ||
Shergis JL, Di YM, Zhang AL, et al. Therapeutic potential of Panax ginseng and ginsenosides in the treatment of chronic obstructive pulmonary disease. Complement Ther Med. 2014;22(5):944–953. | ||
Barnes PJ. The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2009;41(6):631–638. | ||
Yokohori N, Aoshiba K, Nagai A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest. 2004;125(2):626–632. | ||
Barreiro E, Peinado VI, Galdiz JB, et al; ENIGMA in COPD Project. Cigarette smoke-induced oxidative stress: a role in chronic obstructive pulmonary disease skeletal muscle dysfunction. Am J Respir Crit Care Med. 2010;82(4):477–488. | ||
Repine JE, Bast A, Lankhorst IDA. Oxidative stress in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1997;156(2):341–357. | ||
Chen Y, Hanaoka M, Droma Y, Chen P, Voelkel NF, Kubo K. Endothelin-1 receptor antagonists prevent the development of pulmonary emphysema in rats. Eur Respir J. 2010;35(4):904–912. | ||
Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25(2):250–258. | ||
Chen ZH, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3(10):e3316. | ||
Zeng H, Kong X, Peng H, et al. Apoptosis and Bcl-2 family proteins, taken to chronic obstructive pulmonary disease. Eur Rev Med Pharmacol Sci. 2012;16(6):711–727. | ||
Hu W, Xie J, Zhao J, Xu Y, Yang S, Ni W. Involvement of Bcl-2 family in apoptosis and signal pathways induced by cigarette smoke extract in the human airway smooth muscle cells. DNA Cell Biol. 2009;28(1):13–22. | ||
Fischer BM, Voynow JA, Ghio AJ. COPD: balancing oxidants and antioxidants. Int J Chron Obstruct Pulmon Dis. 2015;10:261. | ||
Postma DS, Kerkhof M, Boezen HM, Koppelman GH. Asthma and chronic obstructive pulmonary disease: common genes, common environments? Am J Respir Crit Care Med. 2011;183(12):1588–1594. | ||
Babu BR, Griffith OW. Design of isoform-selective inhibitors of nitric oxide synthase. Curr Opin Chem Biol. 1998;2(4):491–500. | ||
Ito H, Matsushita S, Ishikawa S, et al. Significant correlation between endothelial nitric oxide synthase (eNOS) expression and alveolar repair in elastase-induced rat pulmonary emphysema. Surgery Today. 2013;43(3):293–299. | ||
Frankel A, Yadav N, Lee J, Branscombe TL, Clarke S, Bedford MT. The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J Biol Chem. 2002;277(5):3537–3543. | ||
Guccione E, Bassi C, Casadio F, et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449(7164):933–937. | ||
Hyllus D, Stein C, Schnabel K, et al. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007;21(24):3369–3380. | ||
Schübeler D, MacAlpine DM, Scalzo D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18(11):1263–1271. | ||
Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. | ||
Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120(2):169–181. | ||
Sgarra R, Lee J, Tessari MA, et al. The AT-hook of the chromatin architectural transcription factor high mobility group A1a is arginine-methylated by protein arginine methyltransferase 6. J Biol Chem. 2006;281(7):3764–3772. | ||
Wang X, Huang Y, Zhao J, Zhang Y, Lu J, Huang B. Suppression of PRMT6-mediated arginine methylation of p16 protein potentiates its ability to arrest A549 cell proliferation. Int J Biochem Cell Biol. 2012;44(12):2333–2341. | ||
Kang N, Chen P, Chen Y, Zeng H, He X, Zhu Y. PRMT6 mediates CSE induced inflammation and apoptosis. Int Immunopharmacol. 2015;24(1):95–101. | ||
Sundar IK, Yao H, Rahman I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid Redox Signal. 2013;18(15):1956–1971. | ||
Celli BR, MacNee W; ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. | ||
Zhang Y, Cao J, Chen Y, et al. Intraperitoneal injection of cigarette smoke extract induced emphysema, and injury of cardiac and skeletal muscles in BALB/C mice. Exp Lung Res. 2013;39(1):18–31. | ||
Halbower AC, Mason RJ, Abman SH, Tuder RM. Agarose infiltration improves morphology of cryostat sections of lung. Lab Invest. 1994;71(1):149–153. | ||
Choe KH, Taraseviciene-Stewart L, Scerbavicius R, Gera L, Tuder RM, Voelkel NF. Methylprednisolone causes matrix metalloproteinase-dependent emphysema in adult rats. Am J Respir Crit Care Med. 2003;67(11):1516–1521. | ||
Saetta M, Shiner RJ, Angus GE, et al. Destructive index: a measurement of lung parenchymal destruction in smokers. Am Rev Respir Dis. 1985;131(5):764–769. | ||
Ottenheijm CA, Heunks LM, Li YP, et al. Activation of the ubiquitin–proteasome pathway in the diaphragm in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174(9):997–1002. | ||
Chaudhary P, Suryakumar G, Prasad R, Singh SN, Ali S, Ilavazhagan G. Chronic hypobaric hypoxia mediated skeletal muscle atrophy: role of ubiquitin–proteasome pathway and calpains. Mol Cell Biochem. 2012;364(1–2):101–113. | ||
Debigaré R, Côté CH, Maltais F. Ubiquitination and proteolysis in limb and respiratory muscles of patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2010;7(1):84–90. | ||
Kim SY, Lee JH, Huh JW, et al. Cigarette smoke induces Akt protein degradation by the ubiquitin–proteasome system. J Biol Chem. 2011;286(37):31932–31943. | ||
Yoshimatsu M, Toyokawa G, Hayami S, et al. Dysregulation of PRMT1 and PRMT6, type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer. 2011;128(3):562–573. | ||
Almeida-Rios D, Graça I, Vieira FQ, et al. Histone methyltransferase PRMT6 plays an oncogenic role in prostate cancer. Oncotarget. 2016;7(33):53018. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.