")
Back to Journals » OncoTargets and Therapy » Volume 12
The progression of HMGB1-induced autophagy in cancer biology
Authors Xu T, Jiang L , Wang Z
Received 30 August 2018
Accepted for publication 18 November 2018
Published 31 December 2018 Volume 2019:12 Pages 365—377
DOI https://doi.org/10.2147/OTT.S185876
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Tianwei Xu,* Lihua Jiang,* Zhaoxia Wang
Department of Oncology, The Second Affiliated Hospital of Nanjing Medical University, Nanjing, China
*These authors contributed equally to this work
Abstract: Autophagy is an important process of cellular degradation and has been proven to contribute to tumorigenesis. High-mobility group box 1 (HMGB1) is an abundant nonhistone protein that has been widely reported to play a central role in the induction of autophagy. In nucleus, HMGB1 upregulates the expression of HSP27 to induce autophagy. In cytoplasm, the Beclin-1/PI3K-III complex can be activated by HMGB1 to promote autophagy. Extracellular HMGB1 binds to the receptor for advanced glycation end products to induce autophagy. Recent studies have shown that HMGB1-induced autophagy exerts multiple functions in various cancers like proliferation. Moreover, inhibition of HMGB1-induced autophagy can reverse chemoresistance, which is regulated by noncoding RNAs such as microRNAs and lncRNAs. Here, we provide a brief introduction to HMGB1 and HMGB1-induced autophagy in cancer. We also discuss the challenges associated with performing further investigations on this issue. HMGB1-induced autophagy exerts significant functions in cancer and has potential utility for new strategy to reverse drug resistance.
Keywords: HMGB1, autophagy, cancer, noncoding RNA, drug resistance
Introduction
Autophagy is a mechanism of degradation that alters the flow of materials to regulate cellular homeostasis. Following the establishment of the concept of autophagy in 1963, it attracted increasing attention1 and its importance in health and disease was highlighted by the Nobel Prize being awarded for work in this field in 2016. Although autophagy is known to be strongly involved in different aspects of cancer,2 such as metabolism,3,4 metastasis,5,6 and therapy,7,8 the details of this remain obscure. Conflicting evidence on the association of autophagy with cancer has been presented, with some findings showing that an increase of autophagy promoted the survival of tumor cells, whereas in other cases, it prevented cancer development.9 The induction of autophagy is also a double-edged sword during the treatment of cancer due to its ability to participate in both cell death and cell survival.10 Therefore, it is extremely important to determine the specific cellular elements and mechanisms behind the role of autophagy in cancer.
Autophagy is an intricate process that requires the involvement of autophagy-related genes (ATGs) to accomplish the formation of an autophagosome, a double-membraned vesicle.11 It is regulated by a series of factors and signaling pathways.12 Among these elements, Sun et al13 proposed that high-mobility group box 1 (HMGB1) plays a key role in autophagy and believed in the potential for exploiting HMGB1-induced autophagy in cancer therapy. HMGB1 is an evolutionarily conserved nonhistone DNA-binding protein present in all vertebrate cells.14 Substantial evidence has revealed the abnormal expression of HMGB1 in different tumors including non-small cell lung cancer (NSCLC),15 glioma,16 pancreatic cancer,17 and epithelial ovarian tumors.18 Here, we elaborate how HMGB1 induces and influences autophagy in cancer and discuss its significance in clinical applications.
General overview of HMGB1
As an abundant nonhistone chromosomal protein encoded by a single gene located on chromosome 13q12, HMGB1 contains 215 amino acid residues, which are organized into three individual domains: two tandem DNA-binding HMG box domains (A box [aa 9–79] and B box [aa 89–162]) and an acidic C-terminal tail (aa 186–215), which contains numerous glutamic and aspartic acid residues.19 Sanchez et al20 found that HMG box A binds to an AT-rich DNA fragment. The Phe37 residues of two HMG box A domains stack together and intercalate the same CG base pair, generating highly kinked DNA. HMG-B box has also been identified as a functional zone identifiable by toll-like receptor 4 (TLR-4).21 In addition, the A box could compete with the B box since it contains a site antagonistic to the B box.22 The C-terminal tail contains a number of glutamic and aspartic acid residues, whereas the N-terminal is composed of lysine. In other words, the C-terminal tail is negatively charged and the N-terminal is highly positively charged, giving the protein a bipolar nature23 (Figure 1).
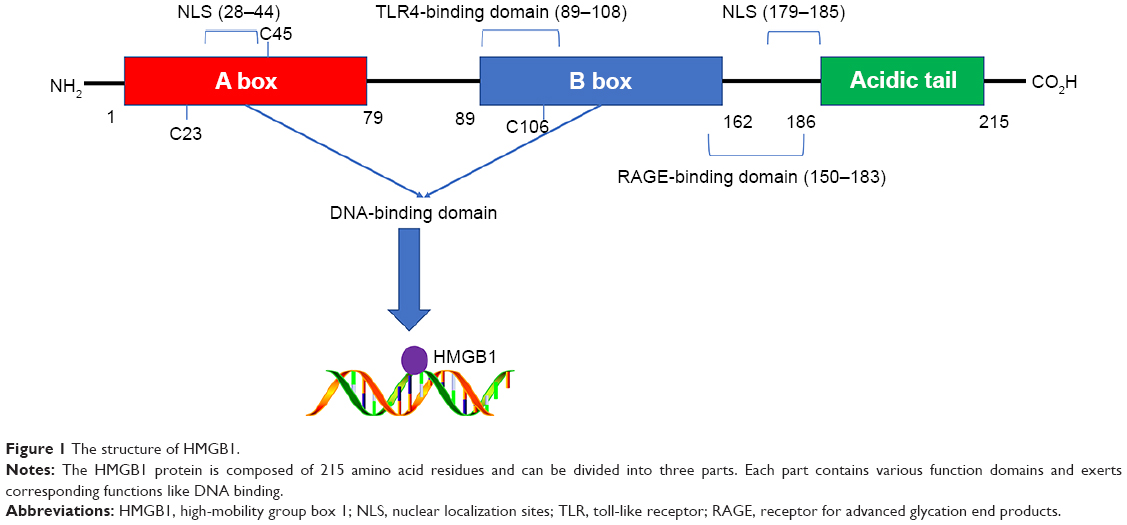 | Figure 1 The structure of HMGB1. |
HMGB1 was considered to be a housekeeping gene because it is widely detected in most cells as the most highly expressed protein in the HMG family.24 However, Muller et al25 disagreed with this and summarized the data on HMGB1 expression in different tissues. They described that the expression of HMGB1 is high in lymphoid tissues and testis, but low in brain and liver. The difference between these sites was ~100-fold, suggesting the correlation between the expression of HMGB1 and the differentiation of tissues. Similar results have been obtained in skeletal myogenic cell lines26 and blood cells.27 In addition, the level of HMGB1 is apparently higher in most tumor samples than in their normal counterparts.25 Furthermore, HMGB1 expression has been reported to be higher in metastasizing tumors.28
HMGB1 is a nuclear protein that alters DNA transcriptional regulation.29 It can also act as a cytokine outside of the nucleus.25 The lysine residues located in the A box (aa 28–44) and the B box (aa 179–185) can form nuclear localization sites (NLSs). HMGB1 can shuttle from the nucleus into the cytoplasm under the control of NLSs. The monomethylation of lysine 42 can inhibit the DNA binding ability of HMGB130 and set HMGB1 free. Bonaldi et al31 also reported that the nuclear-to-cytoplasmic shuttling of HMGB1 depends on the acetylation of NLSs. Immunofluorescence analysis showed that a large amount of fluorescent protein concentrated in the cytoplasm after the acetylation of HMGB1–GFP. At the same time, a mutant form of HMGB1 (whose lysines in NLSs cannot be acetylated) maintained its localization in the nucleus. Further investigation revealed that the JAK/STAT132 and PARP-133 signal pathways mediate the acetylation process. Apart from acetylation, Zhang et al34 demonstrated that the phosphorylation of HMGB1 by calcium/calmodulin-dependent protein kinase (CaMK) IV also plays a role in its nuclear-to-cytoplasmic translocation. Although HMGB1 lacks a leader sequence, which is essential in the classical secretion pathway, it can be released into extracellular space both actively and passively. The HMGB1 in the cytoplasm can be concentrated by secretory lysosomes and released into the extracellular environment.31 HMGB1 can also be passively released by necrotic cells, but this phenomenon does not occur in apoptotic cells. Moreover, immune cells such as macrophages can also actively secrete HMGB1 after the engulfment of dying cells.35 This indicates the potential involvement of a link between dying cells and immune cells in the induction of HMGB1 release.
Due to its extensive distribution, HMGB1 exerts a great influence on cellular activity both inside and outside of cells. As mentioned above, nuclear HMGB1 can bind to and bend DNA to regulate gene expression.36 Different posttranslational modifications (PTMs) such as acetylation, methylation, and phosphorylation are involved in the distribution of HMGB1 and endow it with various functions. Ugrinova et al37 revealed that the acetylation of HMGB1 at lysine 2 can promote its affinity to distorted DNA structures. Moreover, Yu et al38 believed that oxidative stress plays a central role in regulating HMGB1 translocation, release, and functions. The redox state of three cysteines (C23, C45, and C106) in extracellular HMGB1 is of great importance in the regulation of cell activities. Entirely reduced HMGB1, which has three reduced cysteines, shows chemotactic activity, but this can be abolished by partial or full oxidation.37 The redox state is also a checkpoint between autophagy and apoptosis after the binding of receptor for advanced glycation end products (RAGE).39,40 Although the mechanisms by which PTMs exert effects on the cell have not been comprehensively revealed, the evidence obtained thus far suggests that they play an important role in the functions of HMGB1.
Typical receptors such as RAGE, toll-like receptors (TLRs), and T cell immunoglobulin domain and mucin domain-3 (TIM3), which directly or indirectly bind with HMGB1, also play roles in the pathological and physiological functions of HMGB136 (Table 1). As the first reported receptor of HMGB1, RAGE is a transmembrane receptor that has high affinity for HMGB1.41 Numerous recent reports23,35,36 have asserted that RAGE plays an indispensable role in HMGB1-induced cell activities such as autophagy. In addition, RAGE can interact with other receptors such as TLR942 and Mac-143 to impact immune responses, adhesion, and migration of cells. The mechanisms underlying this could involve the activation of downstream factors such as MAPK,44 nuclear factor (NF)-κB,45 and mTOR.46 TLRs expressed on innate immune cells can induce and regulate adaptive immune responses. TLRs are also expressed on tumor cells. HMGB1 released by dying tumor cells activates the TLRs–MyD88 signaling pathway to confer anti- or protumor effects.47 TIM3 is a biomarker for activation and maturation of natural killer (NK) cells. The interaction between HMGB1 and TIM3 interferes with the recruitment of nucleic acids into endosomes. This reduces the immunogenicity of nucleic acids from dying tumor cells and weakens the efficacy of antitumor treatment such as chemotherapy.48 C-X-C chemokine receptor type 4 (CXCR4) has been proved to be related to tumor aggressiveness, metastasis and recurrence.49 Evidence has shown that HMGB1 can promote the migration of inflammatory cells via CXCR4.50 The NF-κB signaling pathway plays a critical role in inflammatory cell recruitment.51 TREM-1 belongs to the immunoglobulin superfamily and induces the inflammatory response. HMGB1 can activate TREM-1 in various cell types.52 HMGB1 upregulates the production of cytokines via the NF-κB signaling pathway after binding with TREM-1, which participates in the inflammatory response.53 At present, few partners of HMGB1 have been identified, but they are assumed to be involved in many novel signaling pathways to participate in the functions of HMGB1.36 Further investigation is needed to obtain a comprehensive understanding of these receptors.
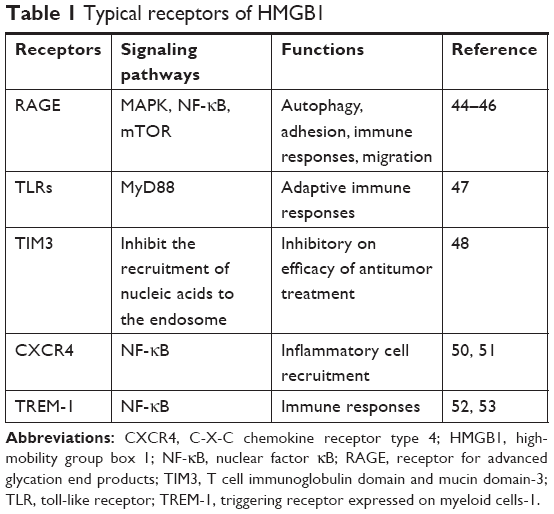 | Table 1 Typical receptors of HMGB1 |
The mechanism of HMGB1-induced autophagy
As mentioned above, HMGB1 is widely distributed inside and outside the cell. Numerous studies have reported that HMGB1 can induce autophagy through various pathways in different intra- and extracellular locations.13 We also reviewed the mechanism of HMGB1-induced autophagy based on the distribution of this molecule (Figure 2).
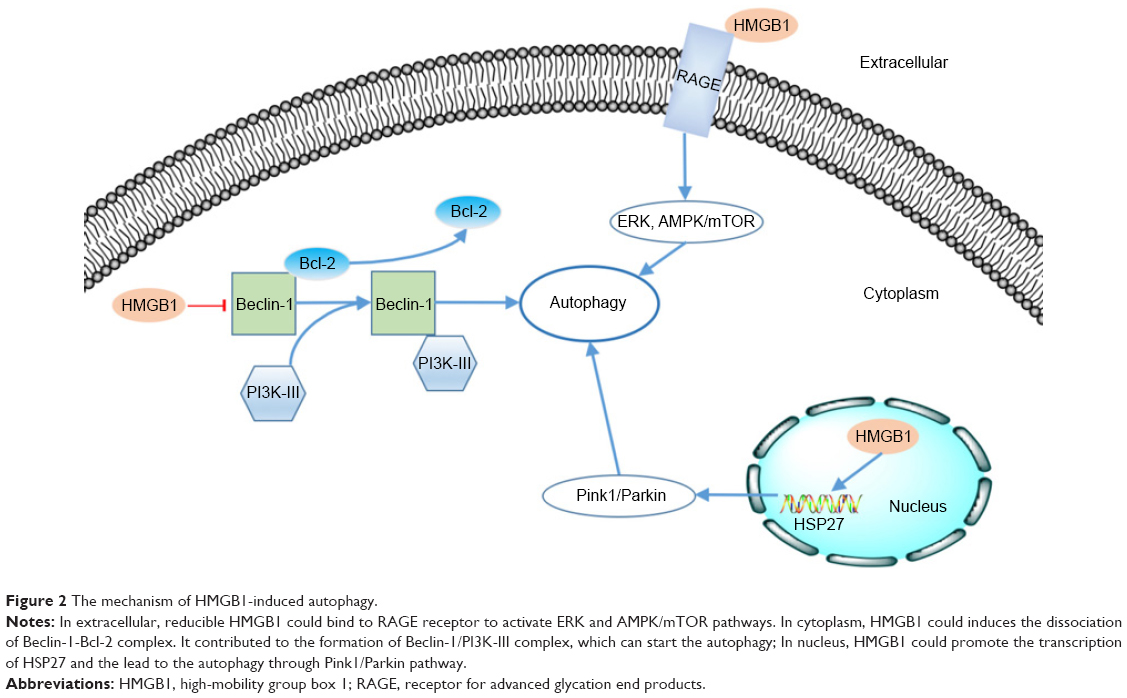 | Figure 2 The mechanism of HMGB1-induced autophagy. |
As a kind of cargo-specific autophagy, mitophagy is a selective form of autophagy of mitochondria54,55 and is strongly correlated with tumorigenesis.56 Kuramitsu et al57 confirmed that the expression of HMGB1 and MAPKAPK2 was upregulated in gemcitabine-resistant pancreatic cancer cells. HMGB1 can act as a transcription factor in the nucleus. Tang et al58 proved that HMGB1 can upregulate the transcription of the heat-shock protein 27 (HSP27) gene. Consequently, nuclear HMGB1 can activate the HSP27 pathway in autophagy. HSP27 phosphorylated by MAPKAPK2 has the ability to modulate actin polymerization and reorganization and then participates in mitophagy.59 The Pink1/Parkin pathway plays a key role in mitochondrial health and quality control.60 One of its major roles is in the mediation of mitophagy.54 As a kinase of outer mitochondrial membranes, PINK can mediate the mitochondrial translocation of Parkin, which leads to ubiquitination of the voltage-dependent anion channel 1 (VACD1). This information can be identified by performing analyses using p62, the autophagic adaptor. After the abovementioned translocation, p62 binds directly to LC3 to accelerate the formation of the autophagosome. Kang et al59 observed that the HMGB1/HSP27 signal pathway participates in the translocation of Parkin and the ubiquitination of VACD1. Furthermore, knockdown of Pink1 or Parkin prevents the HSP27-induced functional recovery of mitochondria in HMGB1-deficient cells, indicating that the Pink1/Parkin pathway is required for HMGB1/HSP27-dependent mitophagy. We know that the HSP27 pathway is a potential mechanism by which nuclear HMGB1 regulates autophagy.
Beclin-1 was identified as an ortholog of the Atg6/vacuolar protein sorting (Vps)–30 protein and was proven to be important in the initiation of autophagy in the cytoplasm.61,62 Beclin-1 can form a complex with class III phosphatidylinositol-3 kinase (PI3K-III).63 This Beclin-1/PI3K-III complex initiates the recruitment of Atg proteins to the phagophore.64 The Beclin-1/PI3K-III complex can be reduced by the binding between Beclin-1 and Bcl-2.65 HMGB1 can induce dissociation of the Beclin-1–Bcl-2 complex through the ERK/MAPK pathway.66 Kang et al67 found that HMGB1 may promote the phosphorylation of Bcl-2 by ERK/MAPK to inhibit formation of the Beclin-1–Bcl-2 complex. Formation of the Beclin-1/PI3K-III complex could be promoted by HMGB1-induced dissociation of the Beclin-1–Bcl-2 complex. Consequently, autophagy would be promoted.
As mentioned above, extracellular HMGB1 released by cancer cells could bind to several receptors such as RAGE and induce autophagy activity in other cells.68 Tang et al39 reported that Beclin-1-dependent autophagy can be activated by the binding of RAGE and extracellular HMGB1. They also demonstrated that HMGB1 may play a role as a checkpoint between autophagy and apoptosis. Reducible HMGB1 leads to the activation of autophagy, while oxidized HMGB1 increases the apoptosis in cancer cells. Su et al69 found that the induction of autophagy by the HMGB1/RAGE axis can be reduced after treatment with Erk1/2 inhibitor in Lewis cells. Moreover, Zhang et al70 also proved that the HMGB1/RAGE axis promoted the phosphorylation of ERK1/2 and consequently induced autophagy in gastric cancer cells. Substantial evidence has implicated the AMPK/mTOR signaling pathway in the process of autophagy in cancer.71,72 Furthermore, Li et al73 also reported that the AMPK/mTOR signaling pathway may be the downstream mechanism through which the HMGB1/RAGE axis affects hepatocellular carcinoma (HCC).
The role of HMGB1-induced autophagy in tumors
Autophagy has been widely acknowledged to be an important biological process in cancer.74 It contributes to various aspects of cancer,9 especially drug resistance.75,76 HMGB1-induced autophagy is part of this phenomenon, as discussed below.
Lung cancer
Non-small cell lung cancer (NSCLC) is a devastating form of cancer with high morbidity and mortality.77 As an approved therapeutic for NSCLC, pemetrexed can interact with sildenafil to enhance the death of NSCLC cells; factors such as Beclin-1, ULK-1, and mTOR, which are involved in the autophagy, may be involved in the mechanism underlying this.78,79 Booth et al80 found that treatment with pemetrexed+ sildenafil in vitro led to the release of HMGB1 and that the subsequent autophagy could participate in regulating the response of NSCLC cells to immunotherapy. Zhang et al81 demonstrated that interference with HMGB1 can increase the sensitivity of NSCLC to chemotherapy via HMGB1-induced autophagy. The expression of HMGB1 in different NSCLC cell lines increased greatly after treatment with chemotherapeutic drugs. The drug-resistant A549 had higher levels of HMGB1 than the drug-sensitive A549. In addition, interference with endogenous HMGB1 by HMGB1-specific shRNA could reverse the drug resistance in drug-resistant A549. At the same time, the autophagy in drug-resistant A549 was inhibited along with the interference of endogenous HMGB1. Further investigation by Chen et al82 showed that the knockdown of HMGB1 could increase PI3K, Akt, and mTOR phosphorylation to restrain the autophagy in NSCLC. In addition, luciferase reporter assay results demonstrated the interaction between miR-142–3 p and HMGB1-induced autophagy. miR-142–3 p can directly suppress HMGB1-induced autophagy by targeting the 3′-UTR of HMGB1 in NSCLC. Thus, the miR-142–3 p/HMGB1 signaling pathway is involved in the chemosensitivity of NSCLC via HMGB1-induced autophagy. Pan et al83 reported a similar result obtained in an analysis of human lung adenocarcinoma (LAD). From both in vivo and in vitro results, HMGB1-induced autophagy contributes to docetaxel resistance in LAD cells. Mechanistic investigations revealed that formation of the Beclin-1/PI3K-III complex could be promoted by HMGB1 through activation of the MEK/ERK signaling pathway, consequently promoting autophagosome formation.
Hematological malignancy
Autophagy is commonly believed to play a pivotal role in hematological malignancy.84 Research focused on HMGB1-induced autophagy has also indicated the importance of HMGB1 in autophagy in hematological malignancy.85,86 Kong et al87 recruited 30 children with B-cell acute lymphoblastic leukemia (ALL) and 8 control subjects to evaluate the correlation between the levels of HMGB1 and clinical stages of ALL. The results showed that HMGB1 was upregulated in ALL patients with primary, relapsed, or recurrent disease, while its level was equal to that at baseline in those with complete remission. Yu et al85 concluded that HMGB1 acts as a proautophagic protein in leukemia and has the potential to be a therapeutic target for leukemia. Liu et al88 found that HMGB1 released from leukemia cell lines in response to chemotherapy could render these cells more resistant to chemotherapy via regulating autophagy. Yang et al89 also proved that endogenous HMGB1 could act as an intrinsic regulator of autophagy to promote drug resistance in leukemia. We know that both exogenous and endogenous HMGB1 can regulate autophagy to promote the drug resistance of leukemia.
HMGB1-induced autophagy is also involved in childhood chronic myeloid leukemia (CML). Zhao et al90 found that autophagy induced by HMGB1 could decrease the sensitivity of CML cells to chemotherapy drugs. In addition, Kong et al87 asserted that targeting the autophagic complexes of HMGB1-like HMGB1–Beclin1 complex may inhibit HMGB1-induced autophagy and overcome drug resistance in leukemia. Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia (AML) characterized by distinctive morphology. All-trans retinoic acid, a specific drug capable of curing APL, can induce the differentiation of leukemia cells. Yang et al91 demonstrated the vital role of HMGB1-induced autophagy in this process. They reported that HMGB1-induced autophagy can be activated by the ROS induced by ATAR in APL. The HMGB1-induced autophagy can directly promote the degradation of PML/RARα, an oncogene in APL, to mediate APL cell differentiation.
MicroRNAs play an important role in the pathogenesis of AML.92 Liu et al93 found that miR-34a acts as a tumor suppressor gene in AML, one of the regulatory mechanisms of which may involve HMGB1-induced autophagy. A dual luciferase reporter system confirmed HMGB1 as a potential target of miR-34a and it was shown that overexpression of HMGB1 significantly reversed the inhibition of autophagy mediated by miR-34a. Zhang et al94 reported that miR-142–3 p negatively regulated HMGB1 expression to repress autophagy in AML and promoted the chemosensitivity of AML. In addition, Gao et al95 claimed that HMGB1-induced autophagy is involved in the proliferation of multiple myeloma (MM) cells. Tumor growth was greatly inhibited in tumor-bearing mice that had reduced expression of HMGB1, LC3B, and Beclin-1 in tumor tissues. In addition, the overexpression of HMGB1 reversed the repression of LC3B and Beclin-1 mediated by LV-si-MALAT-1 and consequently promoted the viability of MM cells. MALAT-1 can prevent degradation of HMGB1 by modification of the ubiquitination of HMGB1. This indicated that HMGB1-induced autophagy can suppress apoptosis and promote the survival of MM tumor cells.
Breast cancer
Estradiol (E2) is believed to play a crucial role in mediating autophagy in breast cancer.96 Wang et al97 reported that the knockdown of HMGB1 can attenuate E2-induced autophagosome formation in BT474 breast cancer cells. Western blot showed that the knockdown of HMGB1 inhibited the expression of LC3-II and Beclin-1 mediated by E2. Ladoire et al98,99 investigated the expression of nuclear HMGB1 and the occurrence of cytoplasmic LC3B (microtubule-associated protein 1 light chain 3B)-positive puncta, which is an immunohistochemical sign of autophagy, in tumor tissues from breast cancer patients who were treated with adjuvant chemotherapy. The results showed that the combination of nuclear HMGB1 and positivity for LC3B(+) puncta was an independent prognostic marker significantly associated with prolonged metastasis-free survival and improved breast cancer-specific survival. HMGB1(+) LC3B(+) double-positive tumors exhibited better prognosis than breast cancer that lacked one or both of these markers. In other words, the combined analysis of HMGB1 and the level of autophagy is of great value in determining the prognosis of breast cancer. Further investigation demonstrated that the mechanism behind these results may involve the loss of HMGB1 and the blockage of autophagy having a negative impact on anticancer immunosurveillance.100 Based on this we know that HMGB1 is related to autophagy in breast cancer and that HMGB1-induced autophagy plays a vital role in the progression of breast cancer. Moreover, HMGB1-induced autophagy participates in the chemoresistance and radioresistance of breast cancer.
Amornsupak et al101 found that the promotion of HMGB1 induced a degree of resistance to doxorubicin in the human breast cancer cell line MDA-MB-231, whereas anti-HMGB1 neutralizing antibody reduced this effect. MDA-MB-231 showed an increase in doxorubicin resistance and the induction of LC3B after the treatment of recombinant HMGB1. HMGB1-specific antibody also reversed the effect of rHMGB1 on autophagy and doxorubicin resistance. This indicates that HMGB1 can lead to autophagy in breast cancer and that HMGB1-induced autophagy contributes to doxorubicin resistance. Radiotherapy is an important adjuvant treatment after breast-conserving surgery. Luo et al102 focused on the impact of autophagy induced by the miR-129–5 p/HMGB1 axis on the radioresistance of breast cancer. The knockdown of HMGB1 suppressed irradiation-induced autophagy and substantially enhanced radiosensitivity in breast cancers. Online prediction revealed potential binding sites between miR-129–5 p and HMGB1. Further study showed that miR-129–5 p could be an upstream regulator of HMGB1-induced autophagy and consequently participate in the radioresistance of breast cancer.
Urogenital cancer
Bladder cancer is the most common malignancy of the genitourinary system.103 Radiation therapy can preserve urinary and sexual function and predict a high quality of life in patients.104 Liao et al105 showed that a decreased level of HMGB1-induced autophagy in bladder urothelial carcinoma (BUC) cells significantly inhibited the viability of BUC cells. Shrivastava et al106 reported the important role of HMGB1 as a predictive marker in bladder cancers subjected to radiation therapy and its correlation with autophagy. Higher expression of HMGB1 had a positive correlation with radiation resistance, while HMGB1 knockdown led to the inhibition of autophagy by more than three-fold (P<0.01) and a better response to radiation treatment in in vitro and in vivo studies. These findings highlight the value of HMGB1 for radiation therapy in bladder cancer and that HMGB1-induced autophagy plays a vital role in this process. As another alternative treatment of bladder cancer, chemotherapy also occupies a position in bladder cancer treatment.107 Yin et al108 focused on the function of HMGB1 in bladder cancer cells treated with gemcitabine. The results showed that gemcitabine induced an increase of HMGB1 in a dose- and time-dependent manner and an increase of LC3-II, which is a marker of autophagy, at the same time in bladder cancer cells. Moreover, Western blotting showed the activation of JNK and ERK in this process, while knockdown of HMGB1 attenuated the JNK and ERK activation. It has been asserted that the MAPK signal is involved in autophagy induction. It has also been demonstrated that HMGB1-induced autophagy induced the chemoresistance of bladder cancer and that targeting this process could improve the anticancer efficacy of chemotherapeutics. Moreover, Lin et al109 proved that cytolethal distending toxin (CDT) could enhance radiosensitivity in prostate cancer via HMGB1-induced autophagy. CDT could reduce the expression of HMGB1, thereby inhibiting the formation of the Beclin-1/PI3K-III complex and restraining autophagy. Moreover, HMGB1-induced autophagy leads to chemoresistance to gemcitabine in hormone-independent prostate cancer.110
Xu et al111 investigated the role of LC3 and HMGB1 in squamous cervical cancer (SCC). Their results showed that both HMGB1 and LC3 are associated with the prognosis of SCC. In addition, they found a negative correlation between nuclear HMGB1 and LC3, which means that HMGB1-induced autophagy plays a role in SCC. Xia et al112 successfully reversed the cisplatin resistance in HeLa cells by inhibiting the nuclear-to-cytoplasmic translocation of HMGB1. Studies also showed that cisplatin resistance was reestablished by the administration of recombinant HMGB1. Further research revealed that the autophagy induced by HMGB1 via the phosphorylation of ERK contributed to cisplatin resistance in HeLa cells. Therefore, HMGB1-induced autophagy can represent a therapeutic target for chemotherapy-resistant cervical cancer. Ran et al113 reported similar results in endometrial carcinoma (EC) cells. In paclitaxel-resistant EC cells, HMGB1 was upregulated and mediated the autophagy contributing to chemoresistance. The inhibition of HMGB1-induced autophagy by miR-218 could protect against chemotherapy resistance in EC cells.
Digestive system carcinoma
Hsieh et al114 reported that the autophagy-related release of HMGB1 plays a key role in the protection of oral cancer cells treated with vincristine. Their results showed that the blockade of autophagy abolished the release of HMGB1-induced by vincristine in SCC9 and OECM-1 cells. At the same time, they found that the knockdown of HMGB1 substantially increased apoptosis. Further investigation demonstrated that the HMGB1 released by autophagy inhibited the expression of Mcl-1 by binding to RAGE. Consequently, the apoptosis was inhibited by HMGB1-induced autophagy. Notably, Luo et al68 revealed a novel relationship in energy production between cancer and its host tissues. Colorectal cancer could lead to the increase of HMGB1, along with the increase of autophagy, in mouse muscle tissues. The autophagy induced by HMGB1 provided glutamine to the cancer cells by the degradation of host muscle tissues. Liu et al115 also reported the vital role of HMGB1-induced autophagy in the chemoresistance of CRC. HMGB1 could induce autophagy to reduce the sensitivity of CRC to oxaliplatin via the MEK/ERK pathway.
Research has suggested the importance of autophagy in the process of development of pancreatic tumors.116 For example, Søreide et al117 asserted that autophagy plays an important role in tumor growth and the suppression of pancreatic tumors; their findings also suggested that the HMGB1–RAGE pathway, which can induce autophagy, is also involved in the process of pancreatic tumor development. Moreover, Xiong et al118 demonstrated that miR-410–3 p could alleviate the resistance to gemcitabine in pancreatic ductal adenocarcinoma by reducing HMGB1-induced autophagy. As a member of the TNF family, TNFSF10/TRAIL is considered to be of great value in cancer therapy. For example, Yang et al119 discovered that the suppression of HMGB1-induced autophagy reduced the resistance of the pancreatic cancer cell line PANC-1 to the drug TNFSF10/TRAIL.
Chemically modified tetracycline 3 (CMT-3) is recognized as a potential anticancer drug for its ability to inhibit matrix metalloproteinases. Zhao et al120 showed that CMT-3 reduced the growth and proliferation of HepG2 cells through the inhibition of HMGB1-induced autophagy. Moreover, Wu et al121 also reported that HMGB1-induced autophagy could enhance the resistance of HepG2 cells to irradiation. The autophagy triggered by irradiation could inhibit p53 expression by degrading HMGB1. They suggested that HMGB1-induced autophagy would have great value in the treatment of hepatocellular carcinoma with irradiation. Li et al73 also reported that HMGB1 induces autophagy through interaction with RAGE in HCC. The HMGB1-induced autophagy contributes to the proliferation and sorafenib resistance of HCC. This finding may provide a novel target for the treatment of HCC.
Other tumors
The most common bone tumor in adolescents, osteosarcoma, is associated with a high rate of mortality.122 Studies123,124 have indicated the importance of HMGB1-induced autophagy in the proliferation, migration, and drug resistance of osteosarcoma. HMGB1 binds to Beclin-1 and regulates the formation of the Beclin-1/PI3K-III complex to mediate autophagy in osteosarcoma, consequently contributing to drug resistance.124 Li et al125 revealed the relationship between miR-22 and HMGB1-induced autophagy. miR-22 can directly target the 3′-UTR of HMGB1. Therefore, the overexpression of miR-22 can suppress the HMGB1-induced autophagy against drug resistance. Guo et al123 proved that the interaction between miR-22 and HMGB1 also blocks autophagy in normal osteosarcoma cells and inhibits osteosarcoma cell proliferation, migration, and invasion. In addition, the inhibition of macrophage migration inhibitory factor (MIF), which is an inflammation-related cytokine implicated in the chemoresistance of cancer, can enhance chemosensitivity by decreasing the level of HMGB1-induced autophagy.126 The increase of HMGB1 also facilitates autophagy in human synovial sarcoma cells and consequently decreases the sensitivity to chemotherapy.127 miR-34A is regarded as a tumor suppressor and it was proved to bind with HMGB1 3′-UTR in retinoblastoma cells.128 This leads to the inhibition of autophagy in retinoblastoma cells under stress and promotes the death of tumor cells. In medulloblastoma, miR-let-7f-1 has similar functions in cisplatin resistance.129 Neuroblastoma is another common tumor in childhood. Liu et al130 demonstrated that HMGB1-induced autophagy, which mostly depends on the activation of TLR4, promoted the proliferation of neuroblastoma cells. Wang et al131 further proved that HMGB1 is a critical regulator of autophagy in neuroblastoma and promotes resistance to doxorubicin. In other words, HMGB1-induced autophagy plays a vital role in the development of chemoresistance and tumorigenesis in neuroblastoma.
Discussion
We have reviewed the roles of HMGB1-induced autophagy in human malignancies. HMGB1 is an abundant nonhistone protein that is distributed both inside and outside of cells. It can induce autophagy through various pathways depending on its location. HMGB1-induced autophagy also contributes to tumorigenesis, such as through proliferation and migration. It has been highlighted that HMGB1-induced autophagy plays a vital role in drug resistance in numerous types of cancer, such as lung cancer, hematological malignancies, and breast cancer. This process can also be regulated by several noncoding RNAs which has been summarized in Table 2. Noncoding RNA can be divided into housekeeping RNA and regulatory RNA. MirRNA and lncRNA are typical regulatory RNAs that are strongly involved in carcinogenesis.132,133 LncRNA plays a major role in the autophagy in cancer.134 LncRNA MALAT-1 regulates chemotherapy-induced autophagy in gastric cancer via miR-23b-3p sequestration.135 In our review, lncRNA MALAT-1 was also shown to prevent the degradation of HMGB1 via modification of the ubiquitination of HMGB1.95 Hence, it could upregulate HMGB1-induced autophagy and promote the proliferation of MM. Several studies have revealed the interplay between miRNA and autophagy in cancer.136 MirRNA participates in various steps of autophagy from upstream induction to later stages of autophagosome maturation and lysosome fusion. We summarized that several miRNAs can suppress autophagy through HMGB1 in cancer. Dual-luciferase reporter assay suggested that miRNAs can target 3′-UTR of the HMGB1 gene to inhibit the expression of HMGB1 and reduce the HMGB1-induced autophagy. This effect leads to the promotion of apoptosis and decrease of radioresistance and chemoresistance.
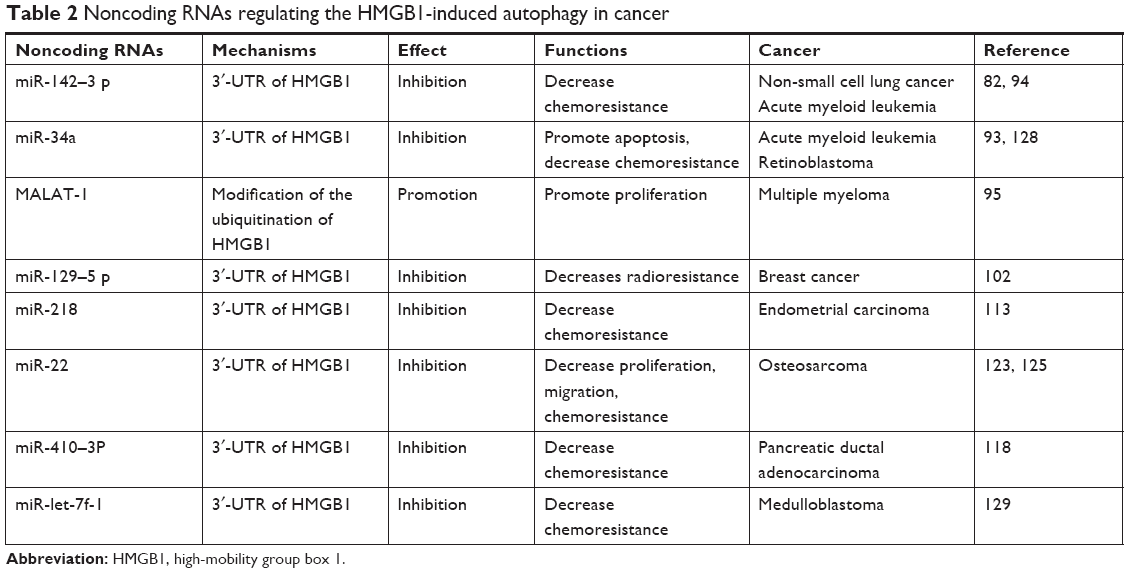 | Table 2 Noncoding RNAs regulating the HMGB1-induced autophagy in cancer |
As mentioned above, HMGB1-induced autophagy plays an important role in tumorigenesis, especially in resistance to chemoradiotherapy. Autophagy inhibitors such as hydroxychloroquine have now been proved to improve clinical outcomes of cancer in various clinical trials.12 We also have some strategies to regulate HMGB1 directly or indirectly for cancer treatment. Liver metastasis of colorectal cancer can be prevented by the administration of anti-HMGB1 antibody.137 Soluble RAGE can also act as a decoy to block the HMGB1/RAGE axis and suppress tumor growth and metastasis.138 We believe that cites such as 3′-UTR and ubiquitination of HMGB1 can act as targets for clinical treatment. HMGB1-induced autophagy can be regulated by noncoding RNAs, which may help us with resistance to cancer treatment such as chemoradiotherapy.
However, we still know little about the complex regulatory network underlying HMGB1-induced autophagy. As mentioned above, there is a close relationship between HMGB1 and autophagy in cancer. HMGB1 is an undisputed regulator of autophagy.139 An increasing number of studies have proved that autophagy also regulates HMGB1 release to exert corresponding functions.140 Thorburn et al141 revealed that autophagy in dying tumor cells could induce the selective release of HMGB1 without classical necrosis or damage to the cell membrane. Then, HMGB1 could function as an immune modulator. Moreover, Zhan et al142 reported that the release of HMGB1 occurred as a consequence of autophagy in gastric cancer cells and it is essential for the autophagy-mediated inhibition of apoptosis induced by vincristine. Zhang et al70 reported that the autophagy-related release of HMGB1 could induce the proliferation of gastric cancer cells.
Cancer-associated fibroblasts (CAFs) as the major components of the niche of breast cancer initiating cells play a vital role in the progression of breast cancer. CAFs with high autophagy activity can release HMGB1, which activates the TLR4 receptor on luminal breast cancer cells and enhances the stemness and tumorigenicity of these cells.143 In addition, Luo et al68 reported an interesting phenomenon regarding energy metabolism induced by the autophagy-released HMGB1 in the context of cancer. HMGB1 was increased in serum and colonic mucosa in a mouse model of colon carcinogenesis, along with increases in autophagy and glutamine, and a decrease in PKM1 levels. The autophagy-released HMGB1 from cancer cells induced the transfer of 13C-glutamine of muscle into cancer cells and the consolidation of 13C-glutamine into acetyl-CoA. This effect was proved to be associated with RAGE and TLR4 receptors. HMGB1 could decrease the level of active mTOR via RAGE and active Beclin-1 through TLR4. The autophagy in tumor-hosting tissue was induced by the autophagy-released HMGB1 and consequently glutamine was recruited to cancer cells as an energy source. Su et al69 also proposed the existence of positive feedback of the RAGE-HMGB1-Erk1/Erk2 pathway in HMGB1-induced autophagy in Lewis cells. Hence, HMGB1 can also be involved downstream of autophagy in cancer. In addition, the autophagy-released HMGB1 has the ability to induce further autophagy in other cells. There may be positive feedback between autophagy and HMGB1 in tumors, which warrants further investigation. An understanding of this is necessary for further clinical applications of HMGB1-induced autophagy.
Conclusion
In this review, we have summarized regulatory networks known to be behind the HMGB1-induced autophagy. HMGB1-induced autophagy plays an important role in cancer biology, such as proliferation, migration and chemoresistance. Further investigation of novel molecular mechanisms involved in this is needed because there is clearly great potential to exploit HMGB1-induced autophagy in efforts to combat cancer from different perspectives, especially in terms of its drug resistance. A better understanding of HMGB1-induced autophagy should provide additional insight into ways of reversing drug resistance in cancer therapy.
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (No. 81672307, 81871871), the Medical Innovation Team Foundation of the Jiangsu Provincial Enhancement Health Project (No. CXTDA2017021), and the “333 high class Talented Man Project” (No. BRA2016509).
Disclosure
The authors report no conflicts of interest in this work.
References
Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. | ||
Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–1930. | ||
White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21(22):5037–5046. | ||
Goldsmith J, Levine B, Debnath J. Autophagy and cancer metabolism. Methods Enzymol. 2014;542:25–57. | ||
Mowers EE, Sharifi MN, Macleod KF. Autophagy in cancer metastasis. Oncogene. 2017;36(12):1619–1630. | ||
Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14(1):48. | ||
Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci. 2017;18(6):E1279. | ||
Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85(6):830–838. | ||
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015;34(7):856–880. | ||
Fulda S. Autophagy in cancer therapy. Front Oncol. 2017;7:128. | ||
Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27(1):107–132. | ||
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–542. | ||
Sun X, Tang D. HMGB1-dependent and -independent autophagy. Autophagy. 2014;10(10):1873–1876. | ||
Martinotti S, Patrone M, Ranzato E. Emerging roles for HMGB1 protein in immunity, inflammation, and cancer. Immunotargets Ther. 2015;4:101–109. | ||
Xia Q, Xu J, Chen H, et al. Association between an elevated level of HMGB1 and non-small-cell lung cancer: a meta-analysis and literature review. Onco Targets Ther. 2016;9:3917–3923. | ||
Seidu RA, Wu M, Su Z, Xu H. Paradoxical role of high mobility group box 1 in glioma: a suppressor or a promoter? Oncol Rev. 2017;11(1):325. | ||
Cebrián MJ, Bauden M, Andersson R, Holdenrieder S, Ansari D. Paradoxical role of HMGB1 in pancreatic cancer: tumor suppressor or tumor promoter? Anticancer Res. 2016;36(9):4381–4390. | ||
Ju LL, Zhao CY, Ye KF, Yang H, Zhang J. Expression and clinical implication of Beclin1, HMGB1, p62, survivin, BRCA1 and ERCC1 in epithelial ovarian tumor tissues. Eur Rev Med Pharmacol Sci. 2016;20(10):1993–2003. | ||
Bianchi ME, Falciola L, Ferrari S, Lilley DM. The DNA binding site of HMG1 protein is composed of two similar segments (HMG boxes), both of which have counterparts in other eukaryotic regulatory proteins. Embo J. 1992;11(3):1055–1063. | ||
Sánchez-Giraldo R, Acosta-Reyes FJ, Malarkey CS, Saperas N, Churchill MEA, Campos JL. Two high-mobility group box domains act together to underwind and kink DNA. Acta Crystallogr D Biol Crystallogr. 2015;71(7):1423–1432. | ||
Hamid KM, Nejati A, Shoja Z, et al. Quantitative evaluation of BAFF, HMGB1, TLR 4 AND TLR 7 expression in patients with relapsing remitting multiple sclerosis. Iran J Allergy Asthma Immunol. 2016;15(1):75–81. | ||
Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93(6):865–873. | ||
Ding J, Cui X, Liu Q. Emerging role of HMGB1 in lung diseases: friend or foe. J Cell Mol Med. 2017;21(6):1046–1057. | ||
Prasad S, Thakur MK. Distribution of high mobility group proteins in different tissues of rats during aging. Biochem Int. 1990;20(4):687–695. | ||
Müller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255(3):332–343. | ||
Begum N, Pash JM, Bhorjee JS. Expression and synthesis of high mobility group chromosomal proteins in different rat skeletal cell lines during myogenesis. J Biol Chem. 1990;265(20):11936–11941. | ||
Čabart P, Kalousek I, Jandová D, Hrkal Z. Differential expression of nuclear HMG1, HMG2 proteins and H1 (zero) histone in various blood cells. Cell Biochem Funct. 1995;13(2):125–133. | ||
Nestl A, Von Stein OD, Zatloukal K, et al. Gene expression patterns associated with the metastatic phenotype in rodent and human tumors. Cancer Res. 2001;61(4):1569–1577. | ||
Ueda T, Yoshida M. HMGB proteins and transcriptional regulation. Biochim Biophys Acta. 1799;2010(1–2):114–118. | ||
Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res. 2016;111:534–544. | ||
Bonaldi T, Talamo F, Scaffidi P. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J. 2003;22(20):5551–5560. | ||
Lu B, Antoine DJ, Kwan K, et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A. 2014;111(8):3068–3073. | ||
Yang Z, Li L, Chen L, et al. PARP-1 mediates LPS-induced HMGB1 release by macrophages through regulation of HMGB1 acetylation. J Immunol. 2014;193(12):6114–6123. | ||
Zhang X, Wheeler D, Tang Y, et al. Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J Immunol. 2008;181(7):5015–5023. | ||
Kang R, Zhang Q, Zeh HJ, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19(15):4046–4057. | ||
He SJ, Cheng J, Feng X, Yu Y, Tian L, Huang Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget. 2017;8(38):64534–64550. | ||
Ugrinova I, Pasheva E. HMGB1 protein: a therapeutic target inside and outside the cell. Adv Protein Chem Struct Biol. 2017;107:37–76. | ||
Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol. 2015;6:93. | ||
Tang D, Kang R, Cheh CW, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29(38):5299–5310. | ||
Messer JS. The cellular autophagy/apoptosis checkpoint during inflammation. Cell Mol Life Sci. 2017;74(7):1281–1296. | ||
Hori O, Brett J, Slattery T, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270(43):25752–25761. | ||
Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. | ||
Orlova VV, Choi EY, Xie C, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. Embo J. 2007;26(4):1129–1139. | ||
Mou K, Liu W, Han D, Li P. HMGB1/RAGE axis promotes autophagy and protects keratinocytes from ultraviolet radiation-induced cell death. J Dermatol Sci. 2017;85(3):162–169. | ||
Liu X, Cao H, Li J, et al. Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ. 2017;24(4):683–693. | ||
Liu L, Gao F, Ye Y, et al. Influence of HMGB1/MAPK/m-TOR signaling pathway on cell autophagy and chemotherapy resistance in K562 cells. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2016;41(10):1016–1023. | ||
Dajon M, Iribarren K, Cremer I. Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology. 2017;222(1):89–100. | ||
Chiba S, Baghdadi M, Akiba H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832–842. | ||
Kuil J, Buckle T, van Leeuwen FW. Imaging agents for the chemokine receptor 4 (CXCR4). Chem Soc Rev. 2012;41(15):5239–5261. | ||
Schiraldi M, Raucci A, Muñoz LM, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. 2012;209(3):551–563. | ||
Kew RR, Penzo M, Habiel DM, Marcu KB. The IKKα-dependent NF-κB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J Immunol. 2012;188(5):2380–2386. | ||
Thankam FG, Dilisio MF, Dietz NE, Agrawal DK. TREM-1, HMGB1 and RAGE in the shoulder tendon: dual mechanisms for inflammation based on the coincidence of glenohumeral arthritis. PLoS One. 2016;11(10):e0165492. | ||
El Mezayen R, El Gazzar M, Seeds MC, Mccall CE, Dreskin SC, Nicolls MR. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett. 2007;111(1):36–44. | ||
Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. | ||
Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 2015;116(8):1477–1490. | ||
Kulikov AV, Luchkina EA, Gogvadze V, Zhivotovsky B. Mitophagy: link to cancer development and therapy. Biochem Biophys Res Commun. 2017;482(3):432–439. | ||
Kuramitsu Y, Wang Y, Kitagawa T, et al. High-mobility group box 1 and mitogen-activated protein kinase activated protein kinase-2 are up-regulated in gemcitabine-resistant pancreatic cancer cells. Anticancer Res. 2015;35(7):3861–3865. | ||
Tang D, Kang R, Livesey KM, et al. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13(6):701–711. | ||
Kang R, Livesey KM, Zeh HJ, Lotze MT, Tang D. Metabolic regulation by HMGB1-mediated autophagy and mitophagy. Autophagy. 2011;7(10):1256–1258. | ||
Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37(6):315–324. | ||
Liang XH, Kleeman LK, Jiang HH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72(11):8586–8596. | ||
Toton E, Lisiak N, Sawicka P, Rybczynska M. Beclin-1 and its role as a target for anticancer therapy. J Physiol Pharmacol. 2014;65(4):459–467. | ||
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. | ||
Høyer-Hansen M, Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14(9):1576–1582. | ||
Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. | ||
Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892. | ||
Kang R, Livesey KM, Zeh HJ, Loze MT, Tang D. HMGB1: a novel Beclin 1-binding protein active in autophagy. Autophagy. 2010;6(8):1209–1211. | ||
Luo Y, Yoneda J, Ohmori H, et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res. 2014;74(1):330–340. | ||
Su Z, Wang T, Zhu H, et al. HMGB1 modulates Lewis cell autophagy and promotes cell survival via RAGE-HMGB1-Erk1/2 positive feedback during nutrient depletion. Immunobiology. 2015;220(5):539–544. | ||
Zhang Q-Y, Wu LQ, Zhang T, Han YF, Lin X. Autophagy-mediated HMGB1 release promotes gastric cancer cell survival via RAGE activation of extracellular signal-regulated kinases 1/2. Oncol Rep. 2015;33(4):1630–1638. | ||
Wang Z, Wang N, Liu P, Xie X. AMPK and Cancer. EXS. 2016;107:203–226. | ||
Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121–129. | ||
Li J, Wu PW, Zhou Y, et al. Rage induces hepatocellular carcinoma proliferation and sorafenib resistance by modulating autophagy. Cell Death Dis. 2018;9(2):225. | ||
White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. | ||
Sooro MA, Zhang N, Zhang P. Targeting EGFR-mediated autophagy as a potential strategy for cancer therapy. Int J Cancer. 2018;143(9):2116–2125. | ||
Das CK, Mandal M, Kögel D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018;37(5). | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Booth L, Roberts JL, Poklepovic A, Dent P. PDE5 inhibitors enhance the lethality of [pemetrexed + sorafenib]. Oncotarget. 2017;8(8):13464–13475. | ||
Booth L, Roberts JL, Poklepovic A, Gordon S, Dent P. PDE5 inhibitors enhance the lethality of pemetrexed through inhibition of multiple chaperone proteins and via the actions of cyclic GMP and nitric oxide. Oncotarget. 2017;8(1):1449–1468. | ||
Booth L, Roberts JL, Poklepovic A, Dent P. [pemetrexed + sildenafil], via autophagy-dependent HDAC downregulation, enhances the immunotherapy response of NSCLC cells. Cancer Biol Ther. 2017;18(9):705–714. | ||
Zhang R, Li Y, Wang Z, Chen L, Dong X, Nie X. Interference with HMGB1 increases the sensitivity to chemotherapy drugs by inhibiting HMGB1-mediated cell autophagy and inducing cell apoptosis. Tumour Biol. 2015;36(11):8585–8592. | ||
Chen Y, Zhou X, Qiao J, Bao A. MiR-142-3p overexpression increases chemo-sensitivity of NSCLC by inhibiting HMGB1-mediated autophagy. Cell Physiol Biochem. 2017;41(4):1370–1382. | ||
Pan B, Chen D, Huang J, et al. HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol Cancer. 2014;13(1):165. | ||
Dong Z, Liang S, Hu J, Jin W, Zhan Q, Zhao K. Autophagy as a target for hematological malignancy therapy. Blood Rev. 2016;30(5):369–380. | ||
Yu Y, Xie M, Kang R, Livesey KM, Cao L, Tang D. HMGB1 is a therapeutic target for leukemia. Am J Blood Res. 2012;2(1):36–43. | ||
Sehgal AR, Konig H, Johnson DE, et al. You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia. 2015;29(3):517–525. | ||
Kong Q, Xu LH, Xu W, Fang JP, Xu HG. HMGB1 translocation is involved in the transformation of autophagy complexes and promotes chemoresistance in leukaemia. Int J Oncol. 2015;47(1):161–170. | ||
Liu L, Yang M, Kang R, et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 2011;25(1):23–31. | ||
Yang L, Yu Y, Kang R, et al. Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk Lymphoma. 2012;53(2):315–322. | ||
Zhao M, Yang M, Yang L, et al. HMGB1 regulates autophagy through increasing transcriptional activities of JNK and ERK in human myeloid leukemia cells. BMB Rep. 2011;44(9):601–606. | ||
Yang L, Chai W, Wang Y, et al. Reactive oxygen species regulate the differentiation of acute promyelocytic leukemia cells through HMGB1-mediated autophagy. Am J Cancer Res. 2015;5(2):714–725. | ||
Trino S, Lamorte D, Caivano A, et al. MicroRNAs as new biomarkers for diagnosis and prognosis, and as potential therapeutic targets in acute myeloid leukemia. Int J Mol Sci. 2018;19(2):460. | ||
Liu L, Ren W, Chen K. MiR-34a Promotes apoptosis and inhibits autophagy by targeting HMGB1 in acute myeloid leukemia cells. Cell Physiol Biochem. 2017;41(5):1981–1992. | ||
Zhang Y, Liu Y, Xu X. Upregulation of miR-142-3p Improves drug sensitivity of acute myelogenous leukemia through reducing P-glycoprotein and repressing autophagy by targeting HMGB1. Transl Oncol. 2017;10(3):410–418. | ||
Gao D, Lv AE, Li HP, Han DH, Zhang YP. LncRNA MALAT-1 elevates HMGB1 to promote autophagy resulting in inhibition of tumor cell apoptosis in multiple myeloma. J Cell Biochem. 2017;118(10):3341–3348. | ||
Yang YH, Chen K, Li B, et al. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway. Apoptosis. 2013;18(11):1363–1375. | ||
Wang R, He W, Li Z, Chang W, Xin Y, Huang T. Caveolin-1 functions as a key regulator of 17β-estradiol-mediated autophagy and apoptosis in BT474 breast cancer cells. Int J Mol Med. 2014;34(3):822–827. | ||
Ladoire S, Enot D, Andre F, Zitvogel L, Kroemer G. Immunogenic cell death-related biomarkers: impact on the survival of breast cancer patients after adjuvant chemotherapy. Oncoimmunology. 2016;5(2):e1082706. | ||
Ladoire S, Penault-Llorca F, Senovilla L, et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy. 2015;11(10):1878–1890. | ||
Ladoire S, Enot D, Senovilla L, et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy. 2016;12(5):864–875. | ||
Amornsupak K, Insawang T, Thuwajit P, O-Charoenrat P, Eccles SA, Thuwajit C. Cancer-associated fibroblasts induce high mobility group box 1 and contribute to resistance to doxorubicin in breast cancer cells. BMC Cancer. 2014;14(1):955. | ||
Luo J, Chen J, He L. mir-129-5p Attenuates irradiation-induced autophagy and decreases radioresistance of breast cancer cells by targeting HMGB1. Med Sci Monit. 2015;21:4122–4129. | ||
Malats N, Real FX. Epidemiology of bladder cancer. Hematol Oncol Clin North Am. 2015;29(2):177–189. | ||
Feuerstein MA, Goenka A. Quality of life outcomes for bladder cancer patients undergoing bladder preservation with radiotherapy. Curr Urol Rep. 2015;16(11):75. | ||
Liao H, Xiao Y, Hu Y, Xiao Y, Yin Z, Liu L. Suppression of cellular proliferation and invasion by hmgb1 knockdown in bladder urothelial carcinoma cells. Oncol Res. 2015;22(5):235–245. | ||
Shrivastava S, Mansure JJ, Almajed W, et al. The role of HMGB1 in radioresistance of bladder cancer. Mol Cancer Ther. 2016;15(3):471–479. | ||
Leow JJ, Martin-Doyle W, Rajagopal PS, et al. Adjuvant chemotherapy for invasive bladder cancer: a 2013 updated systematic review and meta-analysis of randomized trials. Eur Urol. 2014;66(1):42–54. | ||
Yin H, Yang X, Gu W, et al. HMGB1-mediated autophagy attenuates gemcitabine-induced apoptosis in bladder cancer cells involving JNK and ERK activation. Oncotarget. 2017;8(42):71642–71656. | ||
Lin HJ, Liu HH, Lin CD, et al. Cytolethal distending toxin enhances radiosensitivity in prostate cancer cells by regulating autophagy. Front Cell Infect Microbiol. 2017;7:223. | ||
Zhang YX, Yuan YQ, Zhang XQ, Huang DL, Wei YY, Yang JG. HMGB1-mediated autophagy confers resistance to gemcitabine in hormone-independent prostate cancer cells. Oncol Lett. 2017;14(5):6285–6290. | ||
Xu Y, Chen Z, Zhang G, et al. HMGB1 overexpression correlates with poor prognosis in early-stage squamous cervical cancer. Tumour Biol. 2015;36(11):9039–9047. | ||
Xia J, Yu X, Song X, Li G, Mao X, Zhang Y. Inhibiting the cytoplasmic location of HMGB1 reverses cisplatin resistance in human cervical cancer cells. Mol Med Rep. 2017;15(1):488–494. | ||
Ran X, Yang J, Liu C, Zhou P, Xiao L, Zhang K. MiR-218 inhibits HMGB1-mediated autophagy in endometrial carcinoma cells during chemotherapy. Int J Clin Exp Pathol. 2015;8(6):6617–6626. | ||
Hsieh MJ, Hsieh YH, Lin CW, Chen MK, Yang SF, Chiou HL. Transcriptional regulation of Mcl-1 plays an important role of cellular protective effector of vincristine-triggered autophagy in oral cancer cells. Expert Opin Ther Targets. 2015;19(4):455–470. | ||
Liu W, Zhang Z, Zhang Y, et al. HMGB1-mediated autophagy modulates sensitivity of colorectal cancer cells to oxaliplatin via MEK/ERK signaling pathway. Cancer Biol Ther. 2015;16(4):511–517. | ||
Boone BA, Zeh HJ, Bahary N. Autophagy inhibition in pancreatic adenocarcinoma. Clin Colorectal Cancer. 2018;17(1):25–31. | ||
Søreide K, Sund M. Epidemiological-molecular evidence of metabolic reprogramming on proliferation, autophagy and cell signaling in pancreas cancer. Cancer Lett. 2015;356(2 Pt A):281–288. | ||
Xiong J, Wang D, Wei A, et al. MicroRNA-410-3p attenuates gemcitabine resistance in pancreatic ductal adenocarcinoma by inhibiting HMGB1-mediated autophagy. Oncotarget. 2017;8(64):107500–107512. | ||
Yang M, Liu L, Xie M, et al. Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy. 2015;11(2):214–224. | ||
Zhao L, Xu J, Jiao Y, Wang H, Fan S. Novel mechanisms involving chemically modified tetracycline 3 cytotoxicity. Anticancer Drugs. 2014;25(10):1165–1174. | ||
Jh W, Guo JP, Shi J, et al. CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J Gastroenterol. 2017;23(13):2308–2317. | ||
Friebele JC, Peck J, Pan X, Abdel-Rasoul M, Mayerson JL. Osteosarcoma: a meta-analysis and review of the literature. Am J Orthop. 2015;44(12):547–553. | ||
Guo S, Bai R, Liu W, et al. miR-22 inhibits osteosarcoma cell proliferation and migration by targeting HMGB1 and inhibiting HMGB1-mediated autophagy. Tumour Biol. 2014;35(7):7025–7034. | ||
Huang J, Ni J, Liu K, et al. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012;72(1):230–238. | ||
Li X, Wang S, Chen Y, Liu G, Yang X. miR-22 targets the 3′ UTR of HMGB1 and inhibits the HMGB1-associated autophagy in osteosarcoma cells during chemotherapy. Tumour Biol. 2014;35(6):6021–6028. | ||
Liu Y, Zhao L, Ju Y, et al. A novel androstenedione derivative induces ROS-mediated autophagy and attenuates drug resistance in osteosarcoma by inhibiting macrophage migration inhibitory factor (MIF). Cell Death Dis. 2014;5(8):e1361. | ||
Cai Y, Xu P, Yang L, et al. HMGB1-mediated autophagy decreases sensitivity to oxymatrine in SW982 human synovial sarcoma cells. Sci Rep. 2016;6(1):37845. | ||
Liu K, Huang J, Xie M, et al. MIR34A regulates autophagy and apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy. 2014;10(3):442–452. | ||
Pannuru P, Dontula R, Khan AA, et al. miR-let-7f-1 regulates SPARC mediated cisplatin resistance in medulloblastoma cells. Cell Signal. 2014;26(10):2193–2201. | ||
Liu Y, Song L. HMGB1-induced autophagy in Schwann cells promotes neuroblastoma proliferation. Int J Clin Exp Pathol. 2015;8(1):504–510. | ||
Wang L, Zhang H, Sun M, Yin Z, Qian J. High mobility group box 1-mediated autophagy promotes neuroblastoma cell chemoresistance. Oncol Rep. 2015;34(6):2969–2976. | ||
Romano G, Veneziano D, Acunzo M, Croce CM. Small non-coding RNA and cancer. Carcinogenesis. 2017;38(5):485–491. | ||
Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77(15):3965–3981. | ||
Yang L, Wang H, Shen Q, Feng L, Jin H. Long non-coding RNAs involved in autophagy regulation. Cell Death Dis. 2017;8(10):e3073. | ||
Yiren H, Yingcong Y, Sunwu Y, et al. Long noncoding RNA MALAT1 regulates autophagy associated chemoresistance via miR-23b-3p sequestration in gastric cancer. Mol Cancer. 2017;16(1):174. | ||
Gozuacik D, Akkoc Y, Ozturk DG, Kocak M. Autophagy-regulating microRNAs and cancer. Front Oncol. 2017;7(12):65. | ||
Luo Y, Ohmori H, Fujii K, et al. HMGB1 attenuates anti-metastatic defence of the liver in colorectal cancer. Eur J Cancer. 2010;46(4):791–799. | ||
Ohmori H, Luo Y, Kuniyasu H. Non-histone nuclear factor HMGB1 as a therapeutic target in colorectal cancer. Expert Opin Ther Targets. 2011;15(2):183–193. | ||
Skinner M. Autophagy: in the hands of HMGB1. Nat Rev Mol Cell Biol. 2010;11(11):756–757. | ||
Thorburn J, Frankel AE, Thorburn A. Regulation of HMGB1 release by autophagy. Autophagy. 2009;5(2):247–249. | ||
Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009;16(1):175–183. | ||
Zhan Z, Li Q, Wu P, et al. Autophagy-mediated HMGB1 release antagonizes apoptosis of gastric cancer cells induced by vincristine via transcriptional regulation of Mcl-1. Autophagy. 2012;8(1):109–121. | ||
Zhao XL, Lin Y, Jiang J, et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol. 2017;243(3):376–389. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.