Back to Journals » International Journal of Nanomedicine » Volume 11
The preparation of metal–organic frameworks and their biomedical application
Authors Liu R, Yu T, Shi Z, Wang Z
Received 20 November 2015
Accepted for publication 12 January 2016
Published 23 March 2016 Volume 2016:11 Pages 1187—1200
DOI https://doi.org/10.2147/IJN.S100877
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Rong Liu,1,2 Tian Yu,1 Zheng Shi,1 Zhiyong Wang3
1School of Medicine and Nursing, Chengdu University, Chengdu, 2Antibiotics Research and Re-evaluation Key Laboratory of Sichuan Province, Sichuan Industrial Institute of Antibiotics, Chengdu University, Chengdu, 3Paul C. Lauterbur Research Center for Biomedical Imaging, Shenzhen Key Laboratory for MRI, Institute of Biomedical and Health Engineering, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, People’s Republic of China
Abstract: The development of a safe and targetable drug carrier is a major challenge. An efficient delivery system should protect cargo from degradation and cleanup, and control of drug release in the target site. Metal–organic frameworks (MOFs), consisting of metal ions and a variety of organic ligands, have been applied for drug delivery due to their distinct structure. In this review, we summarized the synthesis strategies of MOFs, especially emphasizing the methods of pore creation in frameworks, which were based on recent literatures. Subsequently, the controlled size, biocompatibility, drug releasing performances, and imaging of MOFs were discussed, which would pave the road for the application in drug-delivery systems.
Keywords: metal-organic frameworks, pore creation, the controlled size, biocompatibility, drug releasing performances, imaging
Introduction
Nanomaterial has been paid a wide attention in drug delivery due to its highly tunable shape, size, and composition.1,2 Owing to the assistance of biotargeted ligands or the enhanced permeability and retention effect, these nanosystems are endowed with the versatile performances to target tumor site in vivo, which could achieve efficient therapy and eliminate the side effects of drug.3,4 Generally, the nanocarriers are usually simplified into two types – organic and inorganic vehicles. The organic nanocarriers5,6 such as micelle,7–9 liposome,10,11 and dendrimer12–14 have been fabricated by tailoring natural and synthetic organic materials. And drugs would be loaded by physical encapsulation and/or chemical conjugation. However, the loading capacities were usually less than 10% most likely due to the limitation of drug adsorptive sites.15,16 In contrast, a variety of state-of-the-art porous inorganic nanoparticles17–19 possess the desirable micro-/mesopores with large specific surface areas, which provide them with higher loading capacity as well as the stimuli-responsive drug controlled release property. Nevertheless, the undesirable degradability and the relative difficulty in post-functionalization are still the main barriers for bioapplication.
Metal–organic frameworks (MOFs) are a kind of organic–inorganic hybrid compound with one-, two-, or three-dimensional (1D, 2D, 3D) structural topologies consisting of inorganic metal ions/clusters and organic ligands.20–23 Because of high surface area, micro-/mesoporosity,24,25 and the potential further chemical functionalization, MOFs have attracted an increasing interest in the biomedical field in recent years.26,27 In this review, the construction strategies of MOFs utilizing carboxylate, phosphonate, and sulfonate ligands, especially, the formation strategies of porous structures in MOFs will be focused. Furthermore, how to control the size, biocompatibility, drug-releasing performances, and imaging of MOFs have been extensively discussed in order to explore their application in the drug delivery field.
The building of MOFs with typical organic ligands
Hitherto, MOFs could be fabricated by tailoring of linear and branched organic ligands to tune the surface area and control the pore size. Typical organic materials with carboxylate, phosphonate, or sulfonate groups have been considered as the three representative coordinating ligands.
Carboxylate ligands
In general, the carboxylic ligands can adopt versatile coordination modes with metal components28 to produce a majority of coordination polymers that exhibit highly thermal stability.29 Thus, many efforts have been paid on metal carboxylates.30 Some MOFs consisting of aryldicarboxylates,31 particularly, are constructed by π–π stacking and hydrogen-bonding interaction in the fascinating crystal structures. For example, benzene carboxylates ligands could bridge metal centers to form a network.32–34 As shown in Figure 1A, tetrahedral Zn2+ ions were linked by 1,4-benzenedicarboxylate (1,4-BDC) molecules to form 1D linear structure.32–34 Accordingly, the 2D-layered structure MOF was formed due to the π–π stacking and hydrogen-bond interactions between polymer chains (Figure 1B).32–35 In succession, benzenecarboxylate ligands could bridge metal centers to form a 3D network structure (Figure 1C). In the 3D network, a single oxygen atom was bonded with four Zn atoms to form a regular [Zn4O]6+ tetrahedron node that was subsequently linked by a –CO2– group of 1,4-BDC to form an octahedral Zn4O(CO2)6 cluster.36 Later, every Zn4O(CO2)6 unit was joined together by benzene linkers. Therefore, an extended 3D framework was eventually produced.
 | Figure 1 (A) 1D, (B) 2D, and (C) 3D structures of MOF-5 constructed from 1,4-BDC and Zn2+ ions. |
Although aromatic carboxylic acids, for example, the benzenedicarboxylate, could directly form porosity materials via coordination of metal ions, it is of great significance to enlarge the inner pore for MOFs to achieve the desirable molecule adsorption. Theoretically, the elongation of organic linkers seems to be a convenient approach to expand the inner space of frameworks. However, the elongated organic linkers often resulted in fragile frameworks37 and induced the susceptible self-interpenetration of the large void space, thereby precluding the formation of high porosity.38 Fortunately, there are two simple approaches that have been used to overcome such drawbacks. The first one was to reduce the concentration of reagent since it could affect the formation of the non-interpenetrating and interpenetrating.39 The second approach was to use aromatic tritopic ligands (Figure 2), which would generate a closely related net without a self-dual tiling (referred as qom). Thereby, MOFs-built progress could generate the non-interpenetrating frameworks with porous structure.25
 | Figure 2 The tritopic carboxylate ligands of H3BTB, H3BBC, and H3BTE. |
Different from the aromatic carboxylic ligands, the heterocyclic carboxylic acids have expanded the scope of the preparation of MOFs with various functionalities.40 Herein, the multiple proton donor and acceptor by nitrogen atom and carboxylate oxygen coordinated with metal ions to form exact network structures with specific topologies. For instance, the ligand of 3,5-pyrazoledicarboxylicacid (H3pdc)41 with multiple coordination sites and an asymmetric geometry allowed construction of structures with higher dimensions and acentric crystal structures. It is worth noting that acidity-dependent coordination modes should be formed by protons of H3pdc. The hydrogen of carboxyl group with adjacent nitrogen was easily deprotonated among three hydrogens of H3pdc, so that the adjacent oxygen and nitrogen could form a chelating bond to the metal center. Moreover, two hydrogens attached to carboxylic oxygen atoms were easier to be deprotonated than that linked to the nitrogen. Deprotonation from H3pdc could affect the formation of coordination networks in different acid condition. Generally, higher pH value would lead to high connectivity (μ) of ligands and dimensional structures. It was reported that three calcium compounds formed three different coordination modes for Hpdc2− (Figure 3A–C). At a pH value of 2.5, the connectivity of the 1D structures of [Ca(Hpdc)(H2O)4]·2H2O (Figure 3A) adopted μ3. The Hpdc2− tended to form five-member chelating rings with metal centers. If the basic solvent was further added, the connectivity of the 1D structures of [Ca(Hpdc)(H2O)4]·H2O (Figure 3B) would be changed to μ4, although the final pH value of reaction solution was still the same. The Hpdc2− tended to form four-member chelating rings with metal centers. When the final pH value was increased to 6, the connectivity for the 3D structure of Ca(Hpdc)(H2O)4 (Figure 3C) was up to μ6. Whereas, the dicarboxylate ligands have four coordination modes in two strontium compounds, and there was μ1 and two μ2 connectivity in [Sr(H3pdc)(H2pdc)2(H2O)3]·2(H3pdc)·4H2O (Figure 3D–F) with 1D structure at relatively low pH, and a μ7 in Sr(Hpdc)(H2O) (Figure 3G) at high pH. In the coordination mode of Hpdc2− of Sr(Hpdc)(H2O), the strontium metals in the neighboring planes were respectively bridged by two carboxylate groups of Hpdc2− in tridentate (η1, η2) and tetradentate (η2, η2) fashion, which would result in the formation of a 3D structure. In the case of the two barium compounds, a 1D structure of [Ba(H2pdc)2(H2O)4]·2H2O (Figure 3H) was produced at low pH, and the ligand Hpdc2− had one coordination mode with a connectivity number of μ3. When a basic solvent was added to the reaction, a 3D structure of Ba(Hpdc)(H2O) with μ7 (Figure 3I) and μ8 (Figure 3J) connectivity was well fabricated. MOFs, based on alkaline-earth metal elements and H3pdc, formed structures with higher dimensionality, because alkaline-earth metal elements did not coordinate with water molecules at high pH value, thereby preventing the formation of structure with low dimensionality.41
 | Figure 3 The connectivities (μn, n represents connecting number) between the metal cations and ligands of H3pdc regulated by pH values. |
Phosphonate ligands
Monophosphonic acids or linear bisphosphonic acids42 were also used as building ligands, and the layered structure or a pillared-layered structure without void space would be formed due to the efficient packing43 between phosphonic groups. Furthermore, several nonlinear phosphonic ligands have been applied to form MOFs. Because free phosphonic acid molecule occupied the space between phosphonate layers, only a limited porosity formed. For example, 1,1′-binaphthyl-derived ligands could be functionalized at specific positions of the 1,1′-binaphthyl moiety and has served as the chiral backbone.44 In Figure 4A and B, the phosphonic acid was introduced onto 6,6′ positions of 1,1′-binaphthyl moiety containing crown ether groups45 or diethoxy.46 And the phosphonic ligands, namely, 2,2′-pentaethyleneglycol-1,1′-binaphthyl-6,6′-bis(phosphonic acid) and 2,2′-diethoxy-1,1′-binaphthyl-6,6′-bis(phosphonic acid) (L1 and L2), were respectively synthesized. Then, the linkers of L1 or L2 were coordinated with lanthanide metals to construct homochiral lanthanide bisphosphonate MOFs with a formula of [Ln2(L1-H)2(MeOH)8]·(L1-H4)·(HCl)3·(H2O)6 and [Ln(L2-H2)(L2-H3)(H2O)4]·×H2O. In [Ln2(L1-H)2(MeOH)8]·(L1-H4)·(HCl)3·(H2O)6 structure, the organic group from chiral crown ether groups existed in the layer of lanthanide phosphonate, which selectively interacted with organic cations, such as the protonated amino acids. Therefore, the rest of the space only accommodated three hydrogen chlorides and six water molecules per formula unit, and the limited porosity has precluded its application in chiral separations. Additionally, a similar result was acquired in [Ln(L2-H2)(L2-H3)(H2O)4]·×H2O.
 | Figure 4 Phosphonic ligands for the construction of metal-phosphonate MOFs. |
Accordingly, some efforts were applied to generate porosity in such metal–phosphonate system. Phosphate ester with the same effective length as the phosphonate was added into the system of layered metal phosphonates in order to replace some original phosphonates. When the phosphate ester pillar was selectively hydrolyzed, the interlayer space between metal phosphonates would form.47 Dines et al47 took advantage of this method to build mixed-component-layered metal phosphonates/phosphates, Zr(O3PO(CH2)6OPO3)0.5(O3P(CH2)8PO3)0.5. When the phosphate ester of Zr(O3PO(CH2)6OPO3)0.5(O3P(CH2)8PO3)0.5 was selectively hydrolyzed, Zr(O3POH)(O3P(CH2)PO3)0.5 with available void space was prepared. After and before the hydrolysis, the BET surface area ranged from 49 to 209 m2·g−1. However, in mixed-component-layered metal phosphonates/phosphates, irregular pores would form due to irregular substitute of phosphate ester pillars for phosphonates.48,49
In order to construct regular pores and avoid the formation of simple layers structure, polyphosphonate ligands43,50 were usually used in the metal–phosphonate system. An important study showcased that 1,3,5-benzenetriphosphonic acid (BTP) multidentate ligand (L3) (Figure 4C) was chosen to react with Cu(ClO4)2 in the presence of 4,4′-bipyridine and 4,4′-trimethylenedipyridine. {Cu2C6H3[PO2(OH)]2[PO3](C13H14 N2)}·3H2O·0.5HCON(CH3)2 with a large channel-sized 3D framework was formed by tetra copper units connecting with BTP ligands and 4,4′-trimethylenedipyridine linkers in different direction. And the size of large channels was up to 1.4×0.91 nm, which allowed the lattice water and acetone to enter.51 Likewise, 1,3,5-tris(4-phosphonophenyl)benzene, L4, (Figure 4D) with a rigid trigonal core was used to synthesize Sr2(H2L4)(CH3OH)(H2O)4 with permanent porosity. However, its BET surface area was low due to a twofold interpenetrated microporous structure. If the phenyl spacer in L4 was prolonged, higher degrees of interpenetration could be prevented, thereby further increasing the porosity in materials.52 Taking 1,3,5,7-tetrakis(4-phosphonato phenyl) adamantine (L5) (Figure 4E) as the case in point, it was used to build [Cu3(H3L5)(OH)(H2O)3]·H2O·MeOH with an open channel structure. Although twofold interpenetration was observed in the desolvated material with permanent porosity, a higher degree of interpenetration was prevented for steric requirements of the cluster nodes.43
Sulfonate ligands
Sulfonate ligands could coordinate with metal cations to form MOF materials with diverse structures because of the coordinative flexibility of sulfonate groups.53 Comparing with phosphonate ligands, the coordinative tendency of sulfonate groups with metal ions is weaker, so it is more difficult to form a porous solid with sulfonate groups.54
Some efforts have been carried out to produce porosity in metal–sulfonate MOF materials. A “bent” pillar strategy55 was suggested by Cote and Shimizu. As shown in Figure 5, a “bent” pillar with orientation of sulfonate groups at 120° was incorporated into the interlayer of sulfonate-bridged metal centers. Since the “bent” pillar packed inefficiently in the interlayer, the porosity was generated afterward. In a report, 4,5-dihydroxybenzene-1,3-disulfonate dihydrate was used as a pillar to bridge adjacent layers in metal sulfonate network.55 Consequently, a 16-membered ring open space was constructed, and the 16-membered rings aligned along the a-axis to form channels with approximately 0.4 nm in diameter.
 | Figure 5 A scheme depicting a layered network with porosity by “bent” pillars (G represents guest molecules). |
Another approach, so called “rigid adamantine-based unit as a spacer” strategy, was employed to generate an open channel–layered structure.56 For instance, 1,3,5,7-tetrakis (4-sulfophenyl) adamantine (L)56 was chosen to fabricate the [Ba2 (L) (H2O)4 (dioxane)]·(H2O)3(dioxane)3 with a 2D open grid structure. Ba4 clusters bridged six sulfonate groups stemming from six different ligands, resulting in the formation of the layers. Each ligand employed three of four sulfonate groups to coordinate with different Ba2+ centers, while the fourth sulfonate group was not coordinated with any of the Ba2+ centers but away from the layer. The open channel–framework structure was formed by the only three sulfonate groups of each ligand.
“Using metalloligands” was the third strategy to generate pores in metal-sulfonate coordination frameworks.57–59 Anionic metalloligands as cluster nodes were constructed by the reaction of a few of equivalents of ligands with lanthanide metals. And then, the metalloligands interacted with other metal cations such as alkali/alkaline earth metals57 to form metal-sulfonate solids. As illustrated in Figure 6A, Chandler et al58 built a nonsymmetrical, unsaturated metalloligand of [ML3(H2O)2]3− by the reaction of three equivalents of 4,4′-disulfo-2,2′-bipyridine-N,N′-dioxide (L) and one equivalent of Ln3+(M). Subsequently, Ba2 (H2O)4[EuL3(H2O)2](H2O)5.44Cl with microporous metal-sulfonate frameworks was constructed by cross-linking metalloligands with barium cations. Similarly, an anionic cubic metalloligands (Figure 6B) of [Ln(L)4]5− was reported, which was constructed by 4,4′-disulfo-2,2′-bipyridine-N, N′-dioxide (L) and lanthanide ions (Ln).59 The cluster of Na4Cl3+ consisted of three sodium ions and one sodium chloride was employed as the cross-linker. A coordination framework with 3D structure of Na6(H2O)6[Ln(L)4](H2O)nCl was formed by cross-linking of the sulfonate groups of metalloligands and Na4Cl3+. When the cubic metalloligands were spaced by Na4Cl3+ aggregates, a robust and open framework generated, and the average pore width was up to 1.2 nm.
 | Figure 6 The schemes of metalloligands of (A) [LnL3(H2O)2]3− and (B) [LnL4]5− formed via the coordination of organic ligand (L, 4,4′-disulfo-2,2′-bipyridine-N, N′-dioxide) and lanthanide metal (Ln). |
The fourth strategy was to make use of the “charge-assisted hydrogen bonds”.54 In this strategy, polyvalent metal cation and organosulfonate anions were used to construct a permanently porous network. In the network, the hexagonal H-bonding sheet formed, which was sustained by charge-assisted H-bonds between polyvalent metal cation and organosulfonate anion. The H-bonded sheets were propped apart by the rigid organosulfonate anions to form porosity. For example, Dalrymple and Shimizu54 chose Ni(tame)22+ as polyvalent metal cation and BSEB2− as organosulfonate anion to build a robust, regular, and permanently porous solid, {[Ni(tame)2]1(BSEB)1}∞ (tame =1,1,1-tris(aminomethyl)ethane, BSEB =4,4′-bis(sulfoethynyl)bi-phenyl). A porous framework was formed because a layer of Ni(tame)2 units were pillared by BSEB anions (Figure 7).
 | Figure 7 Layers of Ni (tame) units (C: black) pillared by BSEB anions (C: gray) to form the porous structure of {[Ni(tame)2]1(BSEB)1}∞. (Ni, cyan; N, blue; H, white; S, yellow; O, red). |
Various MOFs with permanent and regular pores would be designed by different constructing-pore strategies. It was favorable for MOFs to possess pores, because these pores made them adsorb and release molecules, which laid the foundation for the application of MOFs in the field of drug delivery.
MOFs-based drug-delivery systems
An efficient drug delivery system would minimize side effects and enhance the therapeutic outcome. However, some fundamental factors affect the transportation of nanovehicle in biological systems such as nanoscale, biocompatibility, and the capability of controlling drug release. These factors may be the key points to evade reticuloendothelial system cleanup, protect the carrier from degradation, and improve the effect of drug treatment in targeted delivery process.3,5,60,61 Hence, it is essential to develop a biosafe and biofunctional nanoplatform.
Size control of MOFs
The size of nanoparticles influences both the clearance and biodistribution in vivo. Small particles with 20–30 nm are usually eliminated by renal excretion. Comparably, larger particles with 30–300 nm can be rapidly taken up by the mononuclear phagocytic system cells and are found mainly in the liver, the spleen and the bone marrow.62 Therefore, how to control particles at the nanoscale is of great importance for drug-delivery carriers.
For MOFs, there were two regulations to reduce their size down to the nanoregime.63 The first regulation was that crystal growth inhibitors were added in reaction system to retard the nucleation growth process. Iron (III) acetate, as a crystal growth inhibitor, has been introduced by Horcajada et al64 during the preparation process of porous iron muconate MOFs. In this reaction system, there was a competition between a monofunctional carboxylate of iron acetate and bifunctional carboxylate of iron muconate, which reduced the crystal growth process. Similarly, Cho et al65 utilized pyridine to modulate the growth rates of different crystal facets (Figure 8). In the absence of pyridine, hexagonal rods occurred with an average width of ~1.75 μm and length of ~16.3 μm. In the presence of a small amount of pyridine (2 equivalents), the shape of particles changed from hexagonal rods to hexagonal lumps. When the amount of pyridine increased to 25 equiv, average width and length were reduced to ~0.97 μm and ~0.43 μm, respectively. Wherein, compared to their widths, the lengths of particles were dramatically reduced. Excess pyridine effectively controlled crystal growth in the direction of the hexagonal facet, which acted as a crystal growth inhibitor.
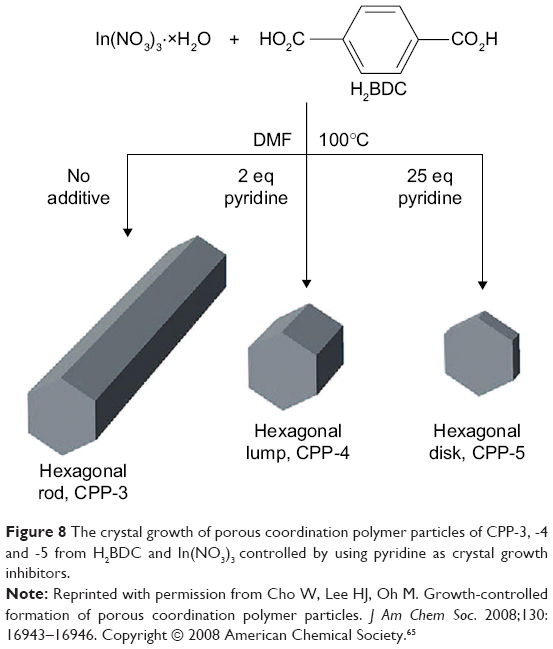 | Figure 8 The crystal growth of porous coordination polymer particles of CPP-3, -4 and -5 from H2BDC and In(NO3)3 controlled by using pyridine as crystal growth inhibitors. |
Another efficient approach introduced in the research was to construct “confined space” by using nanoscale templates. In this process, surfactant, solvent, and/or hydrotrope were chosen to build the limited reaction space of metal ions and organic ligands, resulting in the formation of particles with nanoscale. This template-confining space synthesis method for MOFs has been reported by several groups. Specifically, Gd-based MOF66 nanoparticles were prepared by “confined space” constructed with cationic cetyltrimethylammonium bromide, isooctane, 1-hexanol, and water (Figure 9). Wherein, the size of Gd-based MOF nanoparticles could be controlled by adjusting the molar ratio of water/surfactant. For instance, with an increase in molar ratio, the size increased. Moreover, it could be seen clearly in Figure 9 that after hydrotrope (eg, sodium salicylate) was further added into the reaction system, the morphology and size of nanoscale templates were dramatically altered, along with the size change of Gd-based MOF nanoparticles.67,68
 | Figure 9 The control of a confined space by adding hydrotrope in oil and water phases containing the surfactant of CTAB. |
Biocompatibility of MOFs
To minimize nonspecific protein adhesion and inflammatory events is another critical consideration for MOFs. For retained physicochemical properties of MOFs, a better control over their toxicity and biocompatibility is required.69
To avoid MOFs’ toxic side effects, the choice of low-toxic metal ions and ligands has been identified as a possible solution to this issue.26,70 Previous papers have reported that the toxicity of raw materials was a key factor to effect on the cytotoxicity of the resulting MOFs. Imaz et al71 used Zn2+ metal ions and 1,4-bis(imidazol-l-ylmethyl)benzene organic ligands with low toxicity to structure Zn-based MOFs. It was concluded that these nanoparticles did not show any cytotoxicity in vitro, after they were evaluated on human promyelocytic leukemia cells (HL60) for 24 and 48 hours. Similarly, Horcajada et al70 have assessed the cytotoxicity of several Fe-based MOFs such as MIL-88A and MIL-100, which were separately synthesized by low toxicity of fumarate and trimesate as ligands. The highest dose was 220 mg·kg−1 for MIL-88A and MIL-100 on Wistar female rats via intravenous injection. Compared to the control group, the spleen and liver weights of the test rats had a slight increase. Fortunately, after 1–3 months, the weights of all the body organs returned to normal without significant cytotoxic effects. In continuation of previous research, coating a biocompatible material on MOFs was found to be another possible solution.72 An excellent report demonstrated that Gd-based MOFs were decorated with biocompatible random copolymers of PNIPAM-co-PNAOS-co-PFMA(poly(N-isopropylacrylamide)-poly(N-acryl-oxysuccinimide)-poly(fluorescein O-met-hacrylate)) to reduce the toxicity of Gd nanoparticles.73,74 In this process, the thiocarbonylthio end group of triblock copolymers was reduced to thiolate under basic conditions and then attached to the surface of Gd-MOF nanoparticles through coordination to form a coating layer of about 9 nm.75 By the modification of the homopolymers, Gd MOFs nanoparticles possessed good biocompatibility.
Drug release from MOFs
Currently, many anticancer drugs can be loaded into MOFs by the high porosity of framework and π–π interaction, along with a high drug loading content. For example, the loading content of the azidothymidine triphosphate and cidofovir in MIL-101_NH2 (amino terephthalic acid as ligand, Fe3+ as the metal cation) was about 42.0% and 41.9%, respectively.70 However, it is not enough for an excellent carrier to just have a high drug loading but it is also required that it possesses a good capability of controlling drug release.
Recent research has concentrated on developing the methods of better control over drug release from MOFs. Three main strategies have been found including the “drug-matrix-interaction-controlled drug release”, the “coating-controlled drug release”, and the “cation-triggered drug release”. In the first case, the release of drugs from MOFs with high and regular porosity was affected by the interaction between MOFs and drugs. For example, in the ibuprofen-loaded flexible MIL-53(Fe)74 carrier, because geometrical and energetical characteristics of ibuprofen were accorded with that of the flexible tailor-made container of MIL-53(Fe), the interaction between the carrier and drug was strengthened, which prolonged the drug release time. The result showed that the release time of ibuprofen from MIL-53(Fe) was prolonged to approximately 20 days, when the accumulated release rate was close to 100%. Additionally, Horcajada et al70 have investigated the controlled release of three drugs, doxorubicin, cidofovir, and azidothymidine triphosphate from the MIL-100(Fe) nanoparticles. It was demonstrated that all the three drugs released slowly without “burst effect” over several days because hydrogen-bonding and π–π interaction between the drugs and the frameworks of MIL-100(Fe) prolonged the time of drug release.
In the second case, the release of drugs from MOFs was controlled by the coating thickness of the biocompatible materials on the carrier. In their study, Rieter et al72 prepared silica-decorated Pt-based nanoscale coordination polymer. Concretely speaking, Pt-based nanoscale coordination polymer (Pt-based NCP) was built by the reaction of Tb3+ ions and c,c,t-(diamminedichlorodisuccinato) Pt (IV) ligands, and then it was functionalized by polyvinyl pyrollidone, along with the envelopment of amorphous silica layer with a thickness of 2 or 7 nm to ultimately form NCP-1′-a or NCP-1′-b. It can been seen clearly in Figure 10 that with the increase in the thickness of silica shell, the release rate of cisplatin from the carrier gradually slowed down. Moreover, when accumulated release rate was up to 50%, the drug release time from NCP-1, NCP-1′-a, and NCP-1′-b was 1, 5.5, and 9 hours, respectively. This work suggested that there was better control over the drug release rate from carrier by the coating thickness. Lin et al demonstrated further insights into the effect of coating thickness on drug release by the amino-functionalized MIL-101(Fe)@ SiO2.76 Firstly, the amino-functionalized MIL-101(Fe),1a, (Figure 11A) was synthesized by adding 17.4 mol% 2-aminoterephthalic acids (NH2-BDC) into the reaction system containing terephthalic acids (BDC) and Fe3+ ions. Then, ethoxysuccinatocisplatin (ESCP) prodrug was chemically linked to 1a by amino end group, thereby forming 1b (Figure 11A). Finally, a thin silica was coated to 1b by Na2SiO3 as the silica source to build 1b@SiO2 (Figure 11A). Interestingly, for the existence of uniform silica shell, the release time of BDC-NH-ESCP was lengthened to a t1/2 of ~14 hours, compared to 1b with a t1/2 of ~1.2 hours (Figure 11B).
 | Figure 10 Release profiles of cisplatin from Pt-based NCP with the thickness of silica layer of 0 nm (NCP-1), 2 nm (NCP-1′-a), and 7 nm (NCP-1′-b). |
 | Figure 11 (A) The intracellular release of BDC-NH-ESCP from a silica-coated MOF particle (1b@SiO2, where 1b represents ESCP-loaded amino-functionalized MIL-101(Fe)). (B) Release profiles of BDC-NH-ESCP from the MOF particle of 1b (red) and 1b@SiO2 (blue) in PBS buffer at 37°C. |
In the third case, cations were used to trigger the release of cationic drugs from anionic MOFs. An important study on it was made by An et al. Anionic MOFs,77 Zn8(ad)4(BPDC)6O·2Me2NH2·8DMF·11H2O, was constructed by using biphenyl-dicarboxylic acid, adenine, and zinc acetate dehydrate. And then procainamide, a cationic drug, was introduced into MOFs through cation exchange between the drug and dimethylammonium cations in the pores of MOFs. The drug loaded amount was up to 0.22 g·g−1 material after 15 days of incubation. The drug release experiment (Figure 12) revealed that in 0.1 M PBS buffer (pH=7.4), the electrostatic repulsion between the metal ions (eg, K+ and Na+) and cationic drugs led to a steady release of procainamide (Figure 12A). The amount of drug released was almost up to 100% after 72 hours, while in the deionized water, the release of procainamid was very slow due to the absence of ionic interaction and only 20% of the drug was released from MOFs over 72 hours (Figure 12B).
 | Figure 12 (A) A scheme of cation-triggered release of procainamide from bio-MOF-1. (B) The release profiles of procainamide from the bio-MOF-1 in PBS buffer and deionized water (blue, PBS buffer; red, deionized water). |
Imaging tracking of MOFs drug-delivery system
One of the most acute challenges in drug delivery is how to monitor the vehicle and evaluate the therapeutic effect. Molecular imaging, an emerging medical technique, has been proposed to noninvasively assess the pharmacokinetics and the curative process with the assistance of contrast agents. MOFs nanoparticle with tunable size and surface properties provides a functional platform to perform the simultaneous image-guided therapy. By conjugating or loading multiple components such as imaging agents, drugs, and targeted ligands, not only can these multifunctional nanoparticles carry out treatment but they can also become visible.
Magnetic resonance (MR) imaging, a safe molecular imaging method, provides excellent special resolution and deep tissue penetration without high-energy radiation. This imaging modality is according to the calculation of the hydrogen atoms’ relaxation time from the excited state returning to an equilibrium state under altering resonance frequency. Ideally, sensitive agents should improve the proton’s longitudinal (1/T1, R1) and/or transverse (1/T2, R2) relaxation rates at the targeted site to obtain a high signal-to-noise ratio. It is worth to note that the quantity and the mobility of the metal-coordinated water affect the relaxation time. Now, paramagnetic and superparamagnetic materials have been used as imaging components, for example, the iron oxide nanoparticle and gadolinium (Gd) chelates. Recently, nontoxic porous iron(III) carboxylates framework with magnetic property has promised an approach for MR imaging.70 The iron MOF nanocarriers MIL-88A (fumarate as ligand, Fe3+ as the metal cation) and MIL-100 (trimesic acid as ligand, Fe3+ as the metal cation) were synthesized to associate therapeutics and visibility. The T2 relaxivities of these iron based MOFs are 56 and 73 s−1·mM−1 for MIL-88A and MIL-100 respectively, under a 9.4 T magnetic field. After modified with poly(ethylene glycol) (PEG), the numbers of water molecules coordinated to the Lewis acid metal sites were increased from one to two. Therefore, these nanoparticles, PEG-MIL-88A and PEG-MIL-100, showed slightly higher imaging performance, which is represented in the increased relaxivities (95 and 92 s−1·mM−1 respectively). In addition to the nanostructures’ high drug-loading capacity, they also exhibited anticancer therapy effect. However, the higher blood-to-background noise level is the drawback of T2 contrast agents. Meanwhile, Gd-based T1 imaging probe produces a high longitudinal relaxivity (r1) without the misevaluation effect. In a recent report, Gd MOFs73 were applied as visible nanodevices. A functional copolymer, poly(N-isopropylacrylamide)-co-poly(N-acryloxysuccinimide)-co-poly(fluorescein O-methacrylate) tailoring with targeting peptide (glycine–arginine–glycine–aspartic acid–serine) and antineoplastic drug methotrexate, was prepared to modify Gd-MOF nanoparticles. The MR imaging result demonstrated these nanoparticles behaved as positive contrast agents to reduce the T1 relaxation time regardless of whether modified with polymer or not. Nevertheless, the hydrophilic polymer coating on MOF nanoparticles increased water retention time that caused imaging sensitivity to raise approximately triple times (polymer-modified Gd-MOF nanoparticles: r1= 33.4 mM−1·s−1; unmodified Gd-MOF nanoparticles: r1= 9.86 mM−1·s−1). Furthermore, these nanoparticles combined with dual-modal MR/fluorescence imaging and showed active targeting and treatment capacity to the avb3-integrin overexpressed FITZ-HAS tumor cells. More importantly, the synthesis of these functional MOFs is very straightforward. A method, lately, provided a simple route to synthesize Gd-MOFs by incorporating the hydrotropes – 5-methyl salicylic acid, sodium salicylate (NaSal), and salicylic acid. It is easier to control the nanoparticle size, which was necessary for the biological applications.67 Moreover, the structures and compositions of MOF nanoparticles can be readily tuned via different organic linkers and metal ions. Thus, manganese (II), which possessed five unpaired electrons and less toxicity than Gd3+, was applied for enhanced MRI application. Taylor et al used terephthalic acid and trimesic acid bridging ligands to synthesize a kind of Mn-based MOF nanoparticles. And the nanoparticles, which functionalized with silica shell, cyclic arginine-glycine-aspartate (RGD) peptide and a fluorophore, exhibited cancer cells’ target-specific MR imaging effect.78
Optical imaging, another powerful modality, has been widely employed for molecular imaging. Lately, a kind of zeolitic imidazolate framework was constructed with Zn2+ ions and 2-methylimidazole.79 These nanoparticles could encapsulate fluorescent carbon nanodots (C-dots) and 5-fluorouracil (5-FU) to serve as a novel class of nanocarrier for simultaneous fluorescence imaging and pH-responsive drug delivery in HeLa cancer cells. Furthermore, the size and imaging sensitivity of the nanoparticles can be tuned by varying the amount and the concentration of components in the synthesis progress. However, each kind of imaging technology possesses unique strengths and weaknesses; thus, bimodal or multimodal imaging could provide complementary information. Fortunately, the chem-physical property of nanoMOF is able to be turned. For example, some rare metal iron-based MOF nanostructure, Gd0.95(BDC)1.5(H2O)2:Eu0.05 and Gd0.95(BDC)1.5(H2O)2:Tb0.05, which were highly luminescent with characteristic red and green light from Eu3+ and Tb3+, respectively, could be used as potential MRI contrast agents for multimodal imaging.66
These MOFs nanosystems, incorporating imaging agents, therapeutic drug, and targeting ligands, have the potential to improve the therapeutic outcome and develop as the next generation of personalized patient medicines.
Conclusion and future directions
Porosity is a very important feature of MOFs, especially for the loading and release of drugs. However, MOFs possess pores, when the network-sustaining interactions for MOFs are of sufficient strength to pay the energetic penalty for existence of the pores themselves.80 A series of approaches, such as “reducing the concentration of reaction system” strategy, “choosing aromatic tritopic carboxylate ligands” strategy, “choosing polyphosphonate ligands” strategy, “metalloligands” strategy, and “charge-assisted hydrogen bonds” strategy, have been adopted to build porous MOFs with the use of carboxylates, phosphonates, and sulfonates as ligands. Besides porosity, MOFs should be fabricated with nanoscale size, good biocompatibility, and better control over drug release. Methods of adding crystal growth inhibitors or constructing “confined space” have been used to reduce the size of MOFs to nanoscale. Low-toxic metal ions and ligands as well as the coating of biocompatible materials have been applied to improve the biocompatibility of MOFs. To control the release of drugs from MOFs, the drug-matrix-interaction-controlled route, the coating-controlled route, and the cation-triggered route have been adopted. Certainly, it is also important to monitor the vehicle and evaluate the therapeutic effect. By conjugating or loading multiple components such as imaging agents, drugs, and targeted ligands, not only can these multifunctional nanoparticles carry out treatment but also they become visible. Although considerable progress has been made, the development of nanoscale porous MOFs as drug-delivery carriers is yet in the take-off stage compared to other nanocarriers. The corresponding investigation for porous and biocompatible nanosized MOFs on in vivo, biodistribution, metabolism, or degradation mechanisms should be made further for their better application in future drug-delivery systems.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (51203157, 51203177), the National High Technology Research and Development Program of China (SS2014AA020708), and Antibiotics Research and Re-evaluation Key Laboratory Foundation of Sichuan Province (ARRLKF15-04).
Disclosure
The authors report no conflict of interest in this work.
References
Peer D, Karp JM, Hong S, Faro K, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. | ||
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. | ||
Dufort S, Sancey L, Coll JL. Physico-chemical parameters that govern nanoparticles fate also dictate rules for their molecular evolution. Adv Drug Deliver Rev. 2012;64:179–189. | ||
Son YJ, Jang JS, Cho YW, et al. Biodistribution and anti-tumor efficacy of doxorubicin loaded glycol-chitosan nanoaggregates by EPR effect. J Control Release. 2003;91:135–145. | ||
Liu R, Li D, He B, et al. Anti-tumor drug delivery of pH-sensitive poly(ethylene glycol)-poly(L-histidine-)-poly(L-lactide) nanoparticles. J Control Release. 2011;152:49–56. | ||
Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Nanomedicine-challenge and perspectives. Angew Chem Int Edit. 2009;48:872–897. | ||
Zhang J, Wang LQ, Wang HJ, Tu KH. Micellization phenomena of amphiphilic block copolymers based on methoxy poly(ethylene glycol) and either crystalline or amorphous poly(caprolactone-b-lactide). Biomacromolecules. 2006;7:2492–2500. | ||
Kang N, Perron ME, Prud’homme RE, Zhang YB, Gaucher G, Leroux JC. Stereocomplex block copolymer micelles: core-shell nanostructures with enhanced stability. Nano Lett. 2005;5:315–319. | ||
Kim TY, Kim DW, Chung JY, et al. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10:3708–3716. | ||
Linderoth L, Peters GH, Madsen R, Andresen TL. Drug delivery by an enzyme-mediated cyclization of a lipid prodrug with unique bilayer-formation properties. Angew Chem Int Edit. 2009;48:1823–1826. | ||
Volodkin DV, Skirtach AG, Mohwald H. Near-IR remote release from assemblies of liposomes and nanoparticles. Angew Chem Int Edit. 2009;48:1807–1809. | ||
de Groot FMH, Albrecht C, Koekkoek R, Beusker PH, Scheeren HW. “Cascade-release dendrimers” liberate all end groups upon a single triggering event in the dendritic core. Angew Chem Int Edit. 2003;42:4490–4494. | ||
Gillies ER, Jonsson TB, Frechet JMJ. Stimuli-responsive supramolecular assemblies of linear-dendritic copolymers. J Am Chem Soc. 2004;126:11936–11943. | ||
Xu XH, Yuan H, Chang J, He B, Gu ZW. Stimuli-responsive supramolecular assemblies of linear-dendritic copolymers. Angew Chem Int Edit. 2012;51:3130–3133. | ||
Tong R, Cheng JJ. Paclitaxel-initiated, controlled polymerization of lactide for the formulation of polymeric nanoparticulate delivery vehicles. Angew Chem Int Edit. 2008;47:4830–4834. | ||
Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug delivery by PEGylated drug conjugates. Adv Drug Deliver Rev. 2003;55:217–250. | ||
Yang YN, Niu YT, Zhang J, et al. Biphasic synthesis of large-pore and well-dispersed benzene bridged mesoporous organosilica nanoparticles for intracellular protein delivery. Small. 2015;11:2743–2749. | ||
Yang PP, Gai SL, Lin J. Functionalized mesoporous silica materials for controlled drug delivery. Chem Soc Rev. 2012;41:3679–3698. | ||
Tang SH, Huang XQ, Chen XL, Zheng NF. Hollow mesoporous zirconia nanocapsules for drug delivery. Adv Funct Mater. 2010;20:2442–2447. | ||
Serre C, Millange F, Surble S, Ferey G. A route to the synthesis of trivalent transition-metal porous carboxylates with trimeric secondary building units. Angew Chem Int Edit. 2004:43:6286–6289. | ||
Rowsell JL, Millward AR, Park KS, Yaghi OM. Hydrogen sorption in functionalized metal-organic frameworks. J Am Chem Soc. 2004;126:5666–5667. | ||
Ferey G, Latroche M, Serre C, Millange F, Loiseau T, Percheron-Guegan A. Hydrogen adsorption in the nanoporous metal-benzenedicarboxylate M(OH)(O2C–C6H4–CO2)(M = Al3+, Cr3+), MIL-53. Chem Commun. 2003;2976–2977. | ||
Seo JS, Whang D, Lee H, et al. A homochiral metal–organic porous material for enantioselective separation and catalysis. Nature. 2000;404:982–986. | ||
Britt D, Tranchemontagne D, Yaghi OM. Metal-organic frameworks with high capacity and selectivity for harmful gases. Proc Natl Acad Sci U S A. 2008;105:11623–11627. | ||
Furukawa H, Ko N, Go YB, et al. Ultrahigh porosity in metal-organic frameworks. Science. 2010;329:424–428. | ||
Horcajada P, Gref R, Baati T, et al. Metal–organic frameworks in biomedicine. Chem Rev. 2012;112:1232–1268. | ||
Keskin S, Kizilel S. Biomedical applications of metal organic frameworks. Ind Eng Chem Res. 2011;50:1799–1812. | ||
Wang PF, Wu GZ, Wang RX, Wang X. Two d(10) coordination polymers based on the substitutional aromatic multicarboxylate and n-donor co-ligands: syntheses, structures and luminescent properties. J Inorg Organomet Polymer Mater. 2012;22:1028–1033. | ||
Lin X, Blake AJ, Wilson C, et al. A porous framework polymer based on a zinc(II) 4,4′-bipyridine-2,6,2′,6′-tetracarboxylate: synthesis, structure, and “zeolite-like” behaviors. J Am Chem Soc. 2006;128:10745–10753. | ||
Latroche M, Surble S, Serre C, et al. Hydrogen storage in the giant-pore metal–organic frameworks MIL-100 and MIL-101. Angew Chem Int Edit. 2006;45:8227–8231. | ||
Rosi NL, Eckert J, Eddaoudi M, et al. Hydrogen storage in microporous metal-organic frameworks. Science. 2003;300:1127–1129. | ||
Rodrigues MO, de Paula MV, Wanderley KA, Vasconcelos IB, Alves S, Soares TA. Metal organic frameworks for drug delivery and environmental remediation: a molecular docking approach. Int J Quantum Chem. 2012;112:3346–3355. | ||
Edgar M, Mitchell R, Slawin AMZ, Lightfoot P, Wright PA. Solid-state transformations of zinc 1,4-benzenedicarboxylates mediated by hydrogen-bond-forming molecules. Chem-Eur J. 2001;7:5168–5175. | ||
Guilera G, Steed JW. Topological control in coordination polymers by non-covalent forces. Chemical communications. Chem Commun. 1999;1563–1564. | ||
Li H, Eddaoudi M, O’Keeffe M, Yaghi OM. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature. 1999;402:276–279. | ||
Yaghi OM, O’Keeffe M, Ockwig NW, Chae HK, Eddaoudi M, Kim J.Reticular synthesis and the design of new materials. Nature. 2003; 423:705–714. | ||
Schnobrich JK, Koh K, Sura KN, Matzger AJ. A framework for predicting surface areas in microporous coordination polymers. Langmuir. 2010;26:5808–5814. | ||
Chen B, Eddaoudi M, Hyde ST, O’Keeffe M, Yaghi OM. Interwoven metal-organic framework on a periodic minimal surface with extra-large pores. Science. 2001;291:1021–1023. | ||
Eddaoudi M, Kim J, Rosi N, et al. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science. 2002;295:469–472. | ||
Mahata P, Natarajan S. Pyridine- and imidazoledicarboxylates of zinc: hydrothermal synthesis, structure, and properties. Eur J Inorg Chem. 2005;11:2156–2163. | ||
Pan L, Frydel T, Sander MB, Huang XY, Li J. The effect of pH on the dimensionality of coordination polymers. Inorg Chem. 2001;40:1271–1283. | ||
Tripuramallu BK, Mukherjee S, Das SK. Mechanistic aspects for the formation of copper dimer bridged by phosphonic acid and extending its dimensionality by organic and inorganic linkers: synthesis, structural characterization, magnetic properties, and theoretical studies. Cryst Growth Des. 2012;12:5579–5597. | ||
Taylor JM, Mahmoudkhani AH, Shimizu GKH. A tetrahedral organophosphonate as a linker for a microporous copper framework. Angew Chem Int Edit. 2007;46:795–798. | ||
Ma LQ, Lin WB. Designing metal-organic frameworks for catalytic applications. Top Curr Chem. 2010;293:175–205. | ||
Ngo HL, Lin WB. Chiral crown ether pillared lamellar lanthanide phosphonates. J Am Chem Soc. 2002;124:14298–14299. | ||
Evans OR, Ngo HL, Lin WB. Chiral porous solids based on lamellar lanthanide phosphonates. J Am Chem Soc. 2001;123:10395–10396. | ||
Dines MB, Cooksey RE, Griffith PC, Lane RH. Mixed-component layered tetravalent metal phosphonates phosphates as precursors for microporous materials. Inorg Chem. 1983;22:1003–1004. | ||
Alberti G, Costantino U, Marmottini F, Vivani R, Zappelli P. Zirconium phosphite (3,3′,5,5′-tetramethylbiphenyl) diphosphonate, a microporous, layered, inorganic-organic polymer. Angew ChemInt Edit. 1993;32:1357–1359. | ||
Alberti G, Costantino U, Vivani R, Zappelli P. Preparation of zirconium diphosphonate-phosphites with a narrow distribution of mesopores. MRS Proc. 1991;233:101–106. | ||
Stock N, Guillou N, Senker J, Ferey G, Bein T. Synthesis and characterization of the tetraphosphonic acid ester (Et2O3PCH2)(4)C6H2 and the open-framework cadmium tetra-phosphonate, Cd-2[(HO3PCH2)(4)C6H2]. Z Anorg Allg Chem. 2005;631:575–581. | ||
Kong D, Zon J, McBee J, Clearfield A. Rational design and synthesis of porous organic-inorganic hybrid frameworks constructed by 1,3,5-benzenetriphosphonic acid and pyridine synthons. Inorg Chem. 2006;45:977–986. | ||
Vaidhyanathan R, Mahmoudkhani AH, Shimizu GKH. A microporous alkaline-earth phosphonate sustained by one-dimensional inorganic and organic units. Can J Chem. 2009;87:247–253. | ||
Yu JO, Cote AP, Enright GD, Shimizu GKH. The first nonlayered metal sulfonate structure: a 1-D Ba2+ network incorporating hydrophobic channels. Inorg Chem. 2001;40:582–583. | ||
Dalrymple SA, Shimizu GKH. Crystal engineering of a permanently porous network sustained exclusively by charge-assisted hydrogen bonds. J Am Chem Soc. 2007;129:12114–12116. | ||
Cote AP, Shimizu GKH. The first example of a functional pillared metal sulfonate network. Chem Commun. 2001;251–252. | ||
Hoffart DJ, Cote AP, Shimizu GKH. An adamantane-based coordination framework with the first observation of discrete metal sulfonate clusters. Inorg Chem. 2003;42:8603–8605. | ||
Sun HY, Huang CH, Jin XL, Xu GX. The synthesis, crystal-structure and synergistic fluorescence effect of a heteronuclear lanthanide complex (HLC) {Na3TbLa2(C7H3SO6)4·26H2O}n. Polyhedron. 1995;14:1201–1206. | ||
Chandler BD, Cramb DT, Shimizu GK. Microporous metal-organic frameworks formed in a stepwise manner from luminescent building blocks. J Am Chem Soc. 2006;128:10403–10412. | ||
Chandler BD, Yu JO, Cramb DT, Shimizu GKH. Series of lanthanide-alkali metal-organic frameworks exhibiting luminescence and permanent microporosity. Chem Mater. 2007;19:4467–4473. | ||
Kwon GS, Kataoka K. Block-copolymer micelles as long-circulating drug vehicles. Adv Drug Deliver Rev. 1995;16:295–309. | ||
Kwon GS, Okano T. Polymeric micelles as new drug carriers. Adv Drug Deliver Rev. 1996;21:107–116. | ||
Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69:1–9. | ||
Yang WT, Feng J, Zhang HJ. Facile and rapid fabrication of nanostructured lanthanide coordination polymers as selective luminescent probes in aqueous solution. J Mater Chem. 2012;22:6819–6823. | ||
Horcajada P, Serre C, Grosso D, Boissiere C, Perruchas S, Sanchez C, Férey G. Colloidal route for preparing optical thin films of nanoporous metal-organic frameworks. Adv Mater. 2009;21:1931–1935. | ||
Cho W, Lee HJ, Oh M. Growth-controlled formation of porous coordination polymer particles. J Am Chem Soc. 2008;130:16943–16946. | ||
Rieter WJ, Taylor KML, An HY, Lin WL, Lin WB. Nanoscale metal-organic frameworks as potential multimodal contrast enhancing agents. J Am Chem Soc. 2006;128:9024–9025. | ||
Hatakeyama W, Sanchez TJ, Rowe MD, Serkova NJ, Liberatore MW, Boyes SG. Synthesis of gadolinium nanoscale metal-organic framework with hydrotropes: manipulation of particle size and magnetic resonance imaging capability. ACS Appl Mater Inter. 2011;3:1502–1510. | ||
Bhat M, Gaikar VG. Characterization of interaction between butylbenzene sulfonates and cetyl pyridinium chloride in a mixed aggregate system. Langmuir. 2000;16:1580–1592. | ||
Prabhakar PK, Raj S, Anuradha PR, Sawant SN, Doble M. Biocompatibility studies on polyaniline and polyaniline-silver nanoparticle coated polyurethane composite. Colloid Surface B. 2011;86:146–153. | ||
Horcajada P, Chalati T, Serre C, et al. Porous metal-organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat Mater. 2010;9:172–178. | ||
Imaz I, Rubio-Martinez M, Garcia-Fernandez L, Garcia F, Ruiz-Molina D, Hernando J, Puntes V, Maspoch D. Coordination polymer particles as potential drug delivery systems. Chem Commun. 2010;46: 4737–4739. | ||
Rieter WJ, Pott KM, Taylor KML, Lin WB. Nanoscale coordination polymers for platinum-based anticancer drug delivery. J Am Chem Soc. 2008;130:11584–11585. | ||
Rowe MD, Thamm DH, Kraft SL, Boyes SG. Polymer-modified gadolinium metal-organic framework nanoparticles used as multifunctional nanomedicines for the targeted imaging and treatment of cancer. Biomacromolecules. 2009;10:983–993. | ||
Horcajada P, Serre C, Maurin G, et al. Flexible porous metal-organic frameworks for a controlled drug delivery. J Am Chem Soc. 2008;130:6774–6780. | ||
Rowe MD, Chang CC, Thamm DH, et al. Tuning the magnetic resonance imaging properties of positive contrast agent nanoparticles by surface modification with RAFT polymers. Langmuir. 2009;25:9487–9499. | ||
Taylor-Pashow KM, Rocca JD, Xie Z, Tran S, Lin WB. Postsynthetic modifications of iron-carboxylate nanoscale metal-organic frameworks for imaging and drug delivery. J Am Chem Soc. 2009;131:14261–14263. | ||
An JY, Geib SJ, Rosi NL. Cation-triggered drug release from a porous zinc-adeninate metal-organic framework. J Am Chem Soc. 2009;131:8376–8377. | ||
Taylor KML, Rieter WJ, Lin WB. Manganese-based nanoscale metal-organic frameworks for magnetic resonance imaging. J Am Chem Soc. 2008;130:14358–14359. | ||
He L, Wang TQ, An JP, et al. Carbon nanodots@zeolitic imidazolate framework-8 nanoparticles for simultaneous pH-responsive drug delivery and fluorescence imaging. Cryst Eng Comm. 2014;16:3259–3263. | ||
Thomas-Gipson J, Beobide G, Castillo O, et al. Porous supramolecular compound based on paddle-wheel shaped copper(II)-adenine dinuclear entities. Crystengcomm. 2011;13:3301–3305. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.