Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
The preparation, characterization, and pharmacokinetic studies of chitosan nanoparticles loaded with paclitaxel/dimethyl-β-cyclodextrin inclusion complexes
Authors Ye Y, Wang Y, Lou K ![]() , Chen Y, Chen R
, Chen Y, Chen R ![]() , Gao F
, Gao F ![]()
Received 25 February 2015
Accepted for publication 20 April 2015
Published 3 July 2015 Volume 2015:10(1) Pages 4309—4319
DOI https://doi.org/10.2147/IJN.S83508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Ya-Jing Ye,1 Yun Wang,1 Kai-Yan Lou,1 Yan-Zuo Chen,1 Rongjun Chen,2 Feng Gao1,3,4
1Department of Pharmaceutics, School of Pharmacy, East China University of Science and Technology, Shanghai, People’s Republic of China; 2Department of Chemical Engineering, Imperial College London, London, United Kingdom; 3Shanghai Key Laboratory of Functional Materials Chemistry, 4Shanghai Key Laboratory of New Drug Design, East China University of Science and Technology, Shanghai, People’s Republic of China
Abstract: A novel biocompatible and biodegradable drug-delivery nanoparticle (NP) has been developed to minimize the severe side effects of the poorly water-soluble anticancer drug paclitaxel (PTX) for clinical use. PTX was loaded into the hydrophobic cavity of a hydrophilic cyclodextrin derivative, heptakis (2,6-di-O-methyl)-β-cyclodextrin (DM-β-CD), using an aqueous solution-stirring method followed by lyophilization. The resulting PTX/DM-β-CD inclusion complex dramatically enhanced the solubility of PTX in water and was directly incorporated into chitosan (CS) to form NPs (with a size of 323.9–407.8 nm in diameter) using an ionic gelation method. The formed NPs had a zeta potential of +15.9–23.3 mV and showed high colloidal stability. With the same weight ratio of PTX to CS of 0.7, the loading efficiency of the PTX/DM-β-CD inclusion complex-loaded CS NPs was 30.3-fold higher than that of the PTX-loaded CS NPs. Moreover, it is notable that PTX was released from the DM-β-CD/CS NPs in a sustained-release manner. The pharmacokinetic studies revealed that, compared with reference formulation (Taxol®), the PTX/DM-β-CD inclusion complex-loaded CS NPs exhibited a significant increase in AUC0→24h (the area under the plasma drug concentration–time curve over the period of 24 hours) and mean residence time by 2.7-fold and 1.4-fold, respectively. Therefore, the novel drug/DM-β-CD inclusion complex-loaded CS NPs have promising applications for the significantly improved delivery and controlled release of the poorly water-soluble drug PTX or its derivatives, thus possibly leading to enhanced therapeutic efficacy and less severe side effects.
Keywords: dimethyl-β-cyclodextrin, inclusion complex, chitosan nanoparticles, drug delivery, sustained release, paclitaxel
Introduction
Paclitaxel (PTX, molecular weight =853.91 g/mol, poorly water-soluble), a hydrophobic (HP) natural plant product, has shown potent antitumor effects against human solid tumors including breast cancer, ovarian cancer, and non-small cell lung cancer.1,2 Usually, PTX is administered to patients via an intravenous route. Due to its low water solubility, some solubilizer or injection oil has to be used, which induces great toxicity such as hypersensitivity and peripheral neuropathy.3 The clinical use of PTX is impaired due to the lack of appropriate delivery vehicles. Therefore, there is a need for the development of a new formulation for the efficient loading, delivery, and sustained release of the poorly water-soluble drug PTX in order to enhance its therapeutic efficacy and to minimize its side effects.
Cyclodextrin (CD) has a hydrophilic outer surface and a HP central cavity. A variety of lipophilic drugs can be loaded into the HP cavity of CD via a HP interaction, thus improving the solubility of lipophilic drugs and the stability of labile drugs.4–6 It has been demonstrated that the use of β-CD can improve drug loading efficiency (LE), stabilize the incorporated drugs, and sustain the drug release of nanoparticles (NPs).7–10 Heptakis (2,6-di-O-methyl)-β-CD (DM-β-CD) is a derivative of β-CD, which not only has a suitable cavity depth and surface activity for drug loading but also shows 25 times higher water solubility than β-CD.11 It was found that DM-β-CD had a dramatic improvement on the solubilization and stabilization of PTX.12,13
NP drug-delivery systems have been widely used to solubilize drugs, protect them from degradation, enhance their absorption and subsequent bioavailability, and modify their tissue distribution profiles.14,15 The biodegradable and biocompatible natural polymer chitosan (CS) has been extensively employed in the preparation of NPs. The mucoadhesive polymer can increase the residual time at the site of absorption, and has favorable controlled drug-release abilities.15 Furthermore, it can be easily chemically crosslinked with multivalent anions such as the phosphate group of tripolyphosphate (TPP) via its ionic amino groups.16 A number of research groups have reported the use of CS NPs for the delivery of poorly water-soluble drugs as well as water-soluble proteins.16–19 A recent tendency is to incorporate CD into CS NPs to further improve the stability and loading of poorly soluble drugs.20–22 These NPs have numerous features required for an ideal novel drug delivery system, such as uniform nanoscale size, biocompatible, biodegradable, stable, as well as high drug-loading and controlled-release capacities.23,24 It has become evident that these NPs could also load hydrophilic molecules with high efficiency and enhance their transport efficiency across mucosal surfaces.7,25 Here, CS, DM-β-CD, and PTX have been combined for the first time to construct a novel NP for the effective delivery of the clinically used, poorly water-soluble anticancer drug PTX, and its potential to minimize the severe side effects of PTX has been investigated. PTX was complexed to DM-β-CD by an aqueous solution-stirring method and the resulting PTX/DM-β-CD inclusion complex was then incorporated into CS to form NPs by an ionic gelation method for the first time. The physicochemical characteristics of the NPs such as particle size, size distribution, entrapment efficiency (EE), LE, and in vitro drug release were determined. The pharmacokinetic parameters of these NPs were also investigated in rats.
Materials and methods
Chemicals and reagents
CS (molecular weight =110 kDa, 90.0% deacetylation degree) was purchased from Yuhuan Ocean Biochemical Co. Ltd (Zhejiang, Peoples’ Republic of China). Heptakis DM-β-CD was purchased from Kaiyang Biotech Co. Ltd (Shanghai, Peoples’ Republic of China). Sodium TPP was purchased from the Shanghai Chemical Reagent Company (Shanghai, Peoples’ Republic of China). PTX (purity >99%) was purchased from Xi’an Qing Yue Biotechnology Co. Ltd (Xi’an, Peoples’ Republic of China). The male Wistar rats used in the experiments were supplied by the Department of Experimental Animals at Fudan University. All other reagents used in this study were high performance liquid chromatography (HPLC) or reagent grade.
Preparation of PTX/DM-β-CD inclusion complexes
The solid PTX/DM-β-CD inclusion complexes in a stoichiometric molar ratio ranging from 1:1 to 1:70 were prepared by an aqueous solution-stirring method and then dried by lyophilization.26 Typically, the calculated amounts of PTX and DM-β-CD were weighed and dissolved in 50% ethanol, which was mixed thoroughly under magnetic stirring for 24 hours at 20°C or 40°C. After equilibrium was reached, the mixture was evaporated. The PTX/DM-β-CD inclusion complexes were washed with ultrapure water several times and freeze dried. The yield of the inclusion complex was calculated according to Equation 1:
|
|
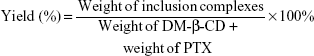
Preparation of CS NPs
CS NPs were prepared by a mild ionic crosslinking method according to the procedure previously developed by Yuan et al.19 Briefly, a CS solution (1 mg/mL) was prepared with glacial acetic acid, 1.75 times the CS weight. Then, the CS solution was adjusted to a pH of 3.8–6.5 with 0.1 N NaOH. The crosslinking agent TPP (1 mg/mL, 1 mL) was added dropwise to the CS solution with magnetic stirring at 800 rpm and continued stirring for a certain period of time (15, 30, 60, or 120 minutes). The NPs were isolated from the solution by centrifugation for 30 minutes at 15,000 rpm and washed with water three times. The purified NPs were freeze dried to obtain dry NPs with 3% mannitol as a lyophilized protection agent. Each sample was prepared in triplicate to obtain the average value of the obtained results and to calculate the error bars.
Preparation of DM-β-CD/CS NPs
DM-β-CD/CS NPs were prepared using the similar mild ionic crosslinking method as described above for preparing CS NPs. CS solution was adjusted to a pH of 4.5 with 0.1 N NaOH, and the final concentration of CS solution was 1 mg/mL. One milliliter of 1 mg/mL TPP containing DM-β-CD (final concentration ranging from 0 to 30 mg/mL) was added dropwise to 5 mL of CS solution with magnetic stirring at 800 rpm and continued stirring for 30 minutes. The NPs were isolated from the solution by centrifugation and washed and freeze dried as described in the “Preparation of CS NPs” section.
Preparation of PTX-loaded CS NPs
PTX-loaded CS NPs were prepared using the similar crosslinking method as described above for preparing the CS NPs.19 CS solution was adjusted to the pH of 4.5 with 0.1 N NaOH, and the final concentration of CS solution was 1 mg/mL. The calculated amount of 2 mg/mL PTX in ethanol was added to 5 mL of CS solution at room temperature. The weight ratios of PTX to CS were 0, 0.7, and 1. PTX-loaded CS NPs were formed by the continuous addition of 1 mg/mL TPP (1 mL) under stirring (800 rpm) at room temperature for 30 minutes. The NPs were washed and freeze dried as described in the “Preparation of DM-β-CD/CS NPs” section.
Preparation of PTX/DM-β-CD inclusion complex-loaded CS NPs
PTX/DM-β-CD inclusion complex-loaded CS NPs were prepared using the similar crosslinking method as described for preparing the CS NPs. A certain amount of PTX/DM-β-CD inclusion complexes were dissolved in 5 mL of a 1 mg/mL CS solution. The pH of the CS solution was then adjusted to 4.5 with 0.1 N NaOH. One milliliter of 1 mg/mL TPP was added dropwise to the CS solution with magnetic stirring at 800 rpm for 30 minutes. The NPs were washed and freeze dried as described in the “Preparation of PTX-loaded CS NPs” section.
The physicochemical and morphological characterization of PTX/DM-β-CD inclusion complexes and NPs
The particle size and the zeta potential of the NPs in the aqueous solution were measured using Delsa Nano dynamic light scattering (DLS) (Beckman Coulter Inc, Brea, CA, USA) equipped with a 4 mW He-Ne laser at a wavelength of 633 nm at 25°C.
The Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet 6700 FTIR spectrometer (Thermo Electron Scientific Instruments Corp, Madison, WI, USA) in the range of 4,000–400 cm−1 using KBr pellets.
The external morphology of PTX, DM-β-CD, and PTX/DM-β-CD inclusion complex was analyzed by scanning electron microscopy. The samples were coated with platinum and observed with a scanning electron microscope (S-3400N, Hitachi, Tokyo, Japan). The morphological evaluation of NPs was performed with a JEM-2010 JEOL transmission electron microscope (TEM) (JASCO, Tokyo, Japan). Samples were stained with 2% (w/v) phosphotungstic acid and then dropped on a copper grid.
Stability study
The CS NPs and DM-β-CD/CS NPs were freshly prepared, isolated and re-suspended in 0.9% (w/v) physiological saline. Samples were incubated at 4°C under agitation (100 rpm) and collected at different time points during a period of 35 days. The sizes of the NPs were measured by DLS. Each experiment was performed in triplicate.
Determination of PTX by HPLC
PTX was determined using a modified reverse-phase HPLC system (Agilent Technologies, Santa Clara, CA, USA). Chromatographic separation was conducted on a Diamonsil® C18 column (4.6×250 mm, 5 μm, Dikma, Beijing, Peoples’ Republic of China). The ternary mobile phase consisted of methanol/acetonitrile/water (40/30/30, v/v/v), which was delivered at an isocratic flow rate of 1 mL/min at 40°C. The detection wavelength was set at 227 nm. The injection volume was 20 μL, and the retention time of PTX was 9.5 minutes.
The entrapment and LEs of PTX/DM-β-CD inclusion complexes and PTX-loaded NPs
For PTX/DM-β-CD inclusion complexes, a certain amount of inclusion complexes were solubilized with methanol and sonicated for 30 minutes. Then, the samples were diluted with methanol and the concentration of PTX was determined by HPLC.
For PTX-loaded NPs, the NPs were centrifuged and the supernatant was collected. The concentration of PTX in the supernatant was determined by HPLC.
The EE and the LE were calculated as follows:
|
|

|
|
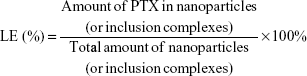
The in vitro drug-release profiles of the NPs
The drug release from the PTX/DM-β-CD inclusion complex-loaded CS NPs was investigated. The NPs (25 mg) and 2 mL of release medium were added to a dialysis tube, which was then placed in 50 mL of phosphate-buffered saline (PBS, pH 7.4) with continuous stirring at 100 rpm (37.0°C±0.5°C). At a specific time interval (5, 15, 30 minutes, 1, 2, 4, 8, 12, 24, 36, and 48 hours), a 2 mL sample was collected from the bulk PBS solution and replaced by an equal volume of fresh PBS. The sample was filtered through a 0.22 μm filter unit and assayed using a UV spectrophotometer (Thermo Fisher, Waltham, MA, USA) at 227 nm. The accumulative release percentage was calculated at each time point. The analysis was performed in triplicate for each sample.
Animal testing
Six male Wistar rats (250±15 g, fasting overnight) were randomly divided into two groups; Group A was treated with reference formulation (Taxol®) and Group B was treated with the PTX/DM-β-CD inclusion complex-loaded CS NPs. The reference formulation (Taxol®) and the PTX/DM-β-CD inclusion complex-loaded CS NPs were diluted in physiological saline up to 0.5 mL and immediately injected into the tail vein of rats at a dose of 5 mg PTX/kg. Blood samples (0.2 mL) were drawn from the rat’s tail vein at specific time intervals and centrifuged to obtain plasma (100 μL). Methyl tert-butyl ether (1 mL) was added to the plasma and mixed for 3 minutes. The mixture was centrifuged at 12,000 rpm for 5 minutes, and the upper part (0.9 mL) was transferred and dried by nitrogen gas. The residue was dissolved in 200 μL of the mobile phase, and the concentration of PTX in the plasma was assayed by HPLC. All of the animal experiments were carried out in accordance with the guidelines evaluated and approved by the ethics committee of East China University of Science and Technology.
Statistical analysis
Multiple group comparisons were conducted using one-way analysis of variance and then by least significant difference using statistical software (SPSS, Chicago, IL, USA). All data are presented as a mean value with its standard deviation (mean ± SD). P-values less than 0.05 were considered as statistically significant.
Results and discussion
PTX/DM-β-CD inclusion complexes
The PTX/DM-β-CD inclusion complex was prepared by an aqueous solution-stirring method. It was found that the molar ratio of PTX to DM-β-CD had a significant effect on the EE and the LE (Table 1). When the molar ratio was controlled as high as 1, the encapsulation of drugs into the HP cavity of DM-β-CD was not successful. Both the EE and the LE were significantly increased by decreasing the molar ratio down to 0.02 and then leveled off when the molar ratio was further decreased. This could be attributed to the relatively high molecular weight of PTX (853.906 g/mol) and its relatively complex structure, leading to one PTX molecule not being able to be totally incorporated into one CD cavity. Two functional moieties of PTX have been reported to contribute to its interaction with CD: one is the C-3′N benzoyl moiety (B-ring), and the other is a HP cluster site among the C-3′ phenyl (C-ring), C-2 benzoate (A-ring), and C-4 acetoxy moieties (Figure 1).27 These moieties may randomly enter the HP cavity of different CD molecules. In addition, the drug encapsulation can be further enhanced by increasing the temperature from 20°C to 40°C. This might be due to the accelerated molecular motion of PTX at higher temperatures, thus reducing the repulsive force between the hydrophilic groups on the surface of DM-β-CD and the HP groups of PTX. This, as a result, could facilitate the loading of the poorly water-soluble PTX into the HP cavity of DM-β-CD.28 However, PTX might be decomposed after extended complexation with DM-β-CD at sufficiently high temperatures. The optimal PTX/DM-β-CD inclusion complexes, which were made at a temperature of 40°C and a complexation time of 24 hours, showed an EE of 82.1%±1.4% and an LE of 9.6%±1.5% and were used to prepare drug-delivery NPs.
 | Table 1 Physicochemical properties of the PTX/DM-β-CD inclusion complexes |
 | Figure 1 Chemical structure of paclitaxel with a numbering scheme. |
Evidence for complexation formation of a guest molecule with CDs can be obtained by IR spectroscopy. A change in the characteristic absorption peaks of the guest molecule will be observed upon complexation. The IR spectra of PTX, DM-β-CD, PTX/DM-β-CD inclusion complex, and physical mixture of PTX and DM-β-CD are shown in Figure 2. IR spectrum of PTX (Figure 2A) showed absorption band at 1,475 cm−1 that was assigned to benzene ring. IR spectrum of DM-β-CD (Figure 2B) showed characteristic absorption peaks of C-O-C and methyl group at 1,040 and 2,940 cm−1. IR spectrum of the physical mixture (Figure 2C) corresponded to the superposition of the spectra of the two individual components, and it retained the benzene ring absorption band of PTX at 1,475 cm−1. However, the benzene ring absorption band of PTX at 1,475 cm−1 was masked in the PTX/DM-β-CD inclusion complex (Figure 2D). The −OH band at 3,400 cm−1 of PTX/DM-β-CD inclusion complex became broadened, suggesting the formation of hydrogen bonding between the drug and DM-β-CD. Moreover, the fingerprint spectra of physical mixture and PTX/DM-β-CD inclusion complex at 600–1,200 cm−1 were quite different. These observations led to the conclusion that the PTX/DM-β-CD inclusion complexes were successfully obtained.
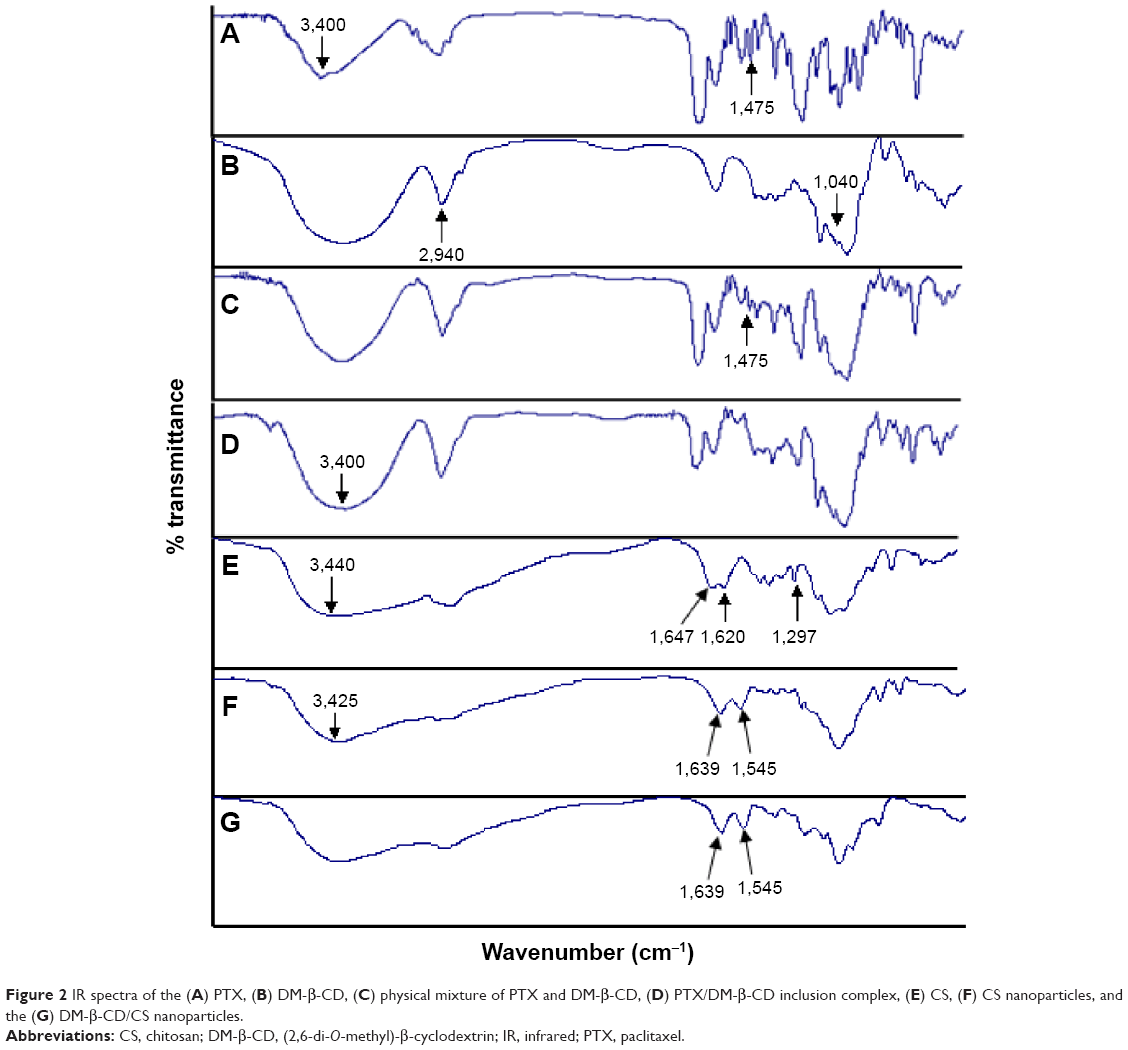 | Figure 2 IR spectra of the (A) PTX, (B) DM-β-CD, (C) physical mixture of PTX and DM-β-CD, (D) PTX/DM-β-CD inclusion complex, (E) CS, (F) CS nanoparticles, and the (G) DM-β-CD/CS nanoparticles. |
The scanning electron microphotographs of PTX, DM-β-CD, and PTX/DM-β-CD inclusion complex are shown in Figure 3. PTX crystals appeared as aggregates of thin needles (Figure 3A),29 while DM-β-CD exhibited somewhat spherical shape with holes on the surface (Figure 3B).30 When PTX was incorporated into DM-β-CD, the resulting PTX/DM-β-CD inclusion complex showed a drastic change in the shape and morphological features of the original PTX and DM-β-CD, which tended to be amorphous (Figure 3C). The change in the surface morphology of the inclusion complex was indicative of the presence of a new solid phase, which might be due to the molecular encapsulation of PTX in the DM-β-CD.
 | Figure 3 SEM photographs of the (A) PTX, (B) DM-β-CD, and (C) PTX/DM-β-CD inclusion complex. |
CS and DM-β-CD/CS NPs
CS NPs were prepared via the ionic crosslinking of CS with TPP, and the effects of the pH, the weight ratio of CS to TPP, and the crosslinking time on the particle size and zeta potential were investigated. As shown in Table 2, the solution pH played a critical role in the formation of CS NPs as CS is a weakly basic polysaccharide, with an average of 0.837 amino groups per disaccharide unit, and is insoluble at neutral or alkaline pH values. Precipitation of aggregates was observed at a pH of 6.5, which could be due to the reduced electrostatic repulsion between the formed NPs resulting from a low level of CS protonation. Upon further acidification to the pH range of 4.0–6.0, more amine groups of CS were positively charged. The resulting CS NPs showed the gradually increased zeta potential from 29.8 to 42.9 mV with a decreasing pH from 6.0 to 4.0. The NPs had a typical hydrodynamic size ranging from 238.5 to 391.2 nm in diameter and displayed good colloidal stability. However, when the CS was further acidified to a pH of 3.8, no NPs were formed successfully. This is consistent with the argument that the formation of CS NPs and their surface charge densities are strongly dependent on the solution pH.31 Moreover, it was found that the formation of CS NPs was only possible for a certain weight ratio of CS to TPP within the range of 3–7. The smallest particle size was obtained at a weight ratio of 5 (ie, when the mole ratio of the CS unit to TPP was 9.9). Two CS units could be linked by one TPP molecule, which means that almost all of the amino groups of CS were involved in crosslinking and would result in more stable CS NPs. In addition, a marginal increase in particle size was observed when the crosslinking time was increased from 15 to 120 minutes.
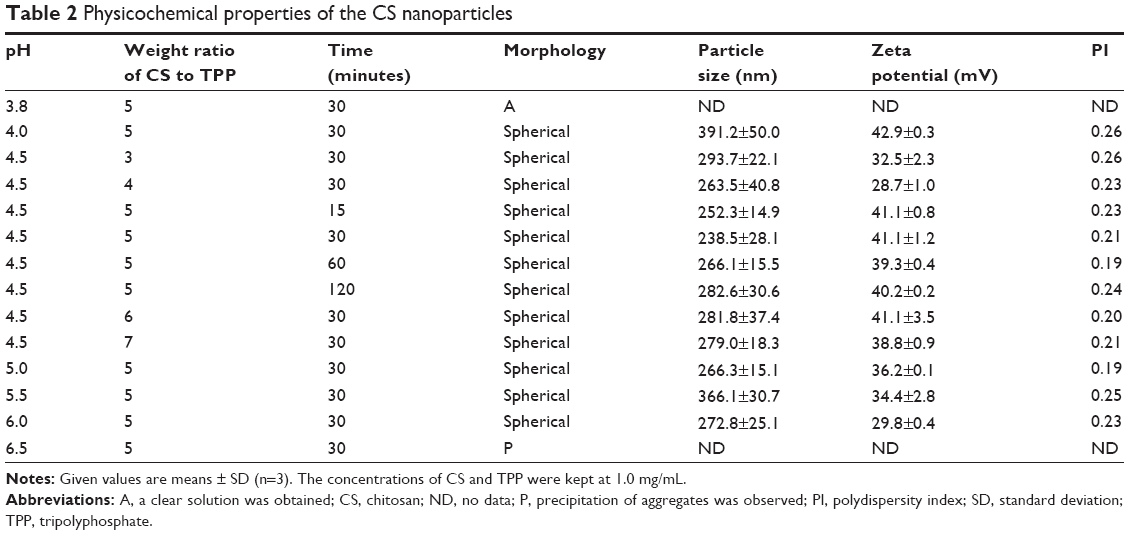 | Table 2 Physicochemical properties of the CS nanoparticles |
DM-β-CD/CS NPs were prepared using the similar ionic crosslinking method and showed an increased hydrodynamic size and zeta potential compared with the control CS NPs (Table 3). The addition of smaller amounts of DM-β-CD (weight ratios of DM-β-CD to CS in the range of 0.5–1.5) resulted in the formation of larger particles, with diameters ranging from 440.2 to 521.4 nm. By increasing the weight ratio of DM-β-CD to CS further to the range of 2.0–6.0, the diameters of the NPs decreased to within the range of 275.7–356.6 nm. The reason might be that low concentration of DM-β-CD could facilitate the incorporation process into CS and form larger particles, whereas high concentration of DM-β-CD might lead to a more compact and smaller nanostructure due to a series of weak intermolecular forces such as hydrogen bond/HP interactions between DM-β-CD and CS. The TEM image (Figure 4) showed the spherical morphology of the CS and DM-β-CD/CS NPs. Because of the presence of DM-β-CD, the size of the DM-β-CD/CS NPs had a minor increase compared with that of the CS NPs. However, no noticeable change in the appearance of the NPs was observed. The particle size determined by TEM in a dry state was smaller than the hydrodynamic size determined by DLS.
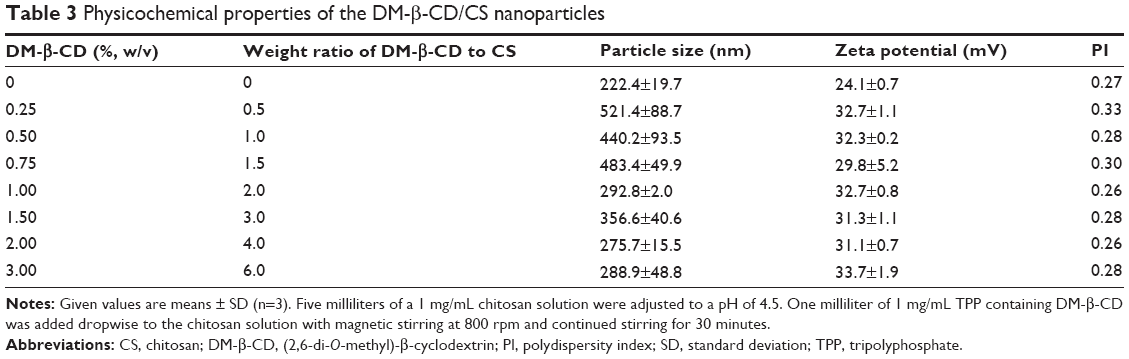 | Table 3 Physicochemical properties of the DM-β-CD/CS nanoparticles |
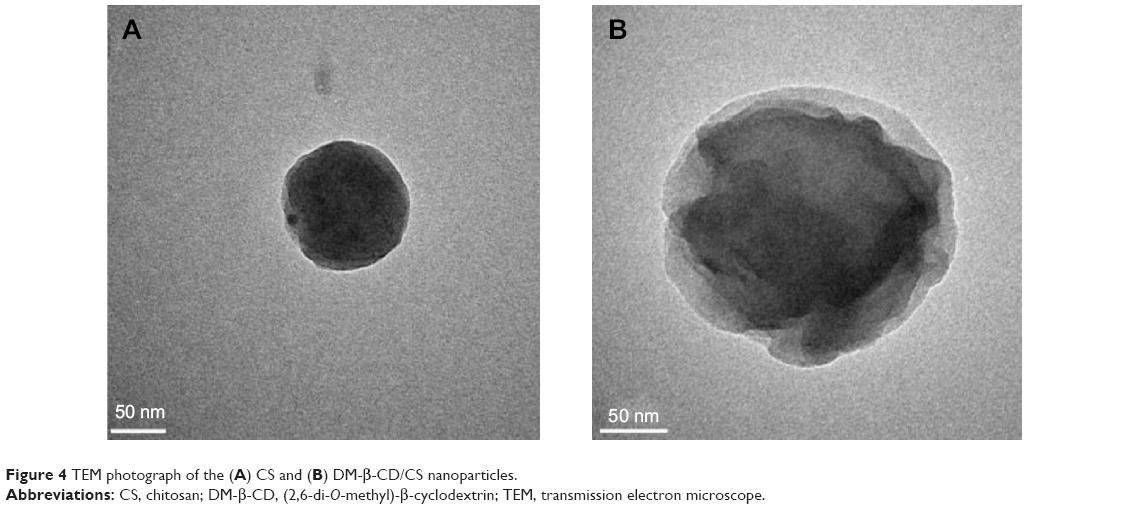 | Figure 4 TEM photograph of the (A) CS and (B) DM-β-CD/CS nanoparticles. |
The structure, composition, and stability of the CS and DM-β-CD/CS NPs were investigated. Figure 2B, E, and F shows the FT-IR spectra of the DM-β-CD, CS, and CS NPs, respectively. The −OH stretching vibration shifted from 3,440 to 3,425 cm−1. The −NH2 bending vibration shifted from 1,620 to 1,545 cm−1, and a new peak appeared at 1,639 cm−1, indicative of the successful crosslinking interaction between TPP and the amino groups of CS within the CS NPs (Figure 2F). Figure 2G shows the FT-IR spectrum of the DM-β-CD/CS NPs, which was similar to that of the CS NPs (Figure 2F) but distinct from that of the DM-β-CD (Figure 2B). The peak of 3,425 cm−1 became wider, indicating that hydrogen bonding was enhanced between CS and DM-β-CD.20 Figure 5 shows the time-dependent stability of the CS and DM-β-CD/CS NPs. No significant change in particle size was observed up to 35 days, indicating that both NPs were stable in 0.9% (w/v) physiological saline.
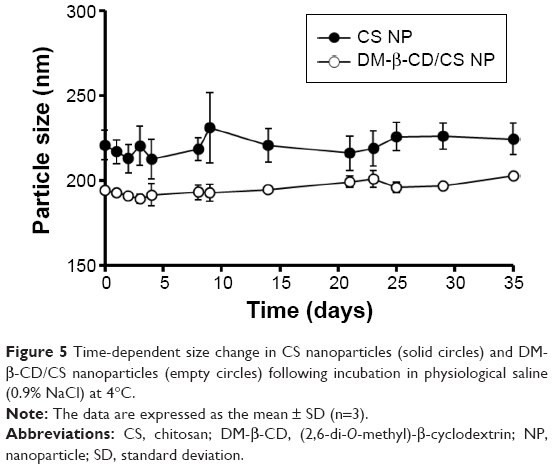 | Figure 5 Time-dependent size change in CS nanoparticles (solid circles) and DM-β-CD/CS nanoparticles (empty circles) following incubation in physiological saline (0.9% NaCl) at 4°C. |
PTX-loaded CS NPs and PTX/DM-β-CD inclusion complex-loaded CS NPs
The PTX-loaded CS NPs and the PTX/DM-β-CD inclusion complex-loaded CS NPs were prepared via the ionic crosslinking method, and the influence of the weight ratio of PTX to CS on their properties was investigated (Table 4). The hydrodynamic size of the NPs increased with the addition of PTX to CS. As shown in Table 4, the PTX-loaded CS NPs had low EEs (≤9.13%) and low LEs (≤0.15%) when the weight ratio of PTX to CS was in the range of 0–1. PTX could not be efficiently incorporated into CS NPs due to the precipitation of the poorly water-soluble drugs in the CS solution. In the same range of the weight ratio of PTX to CS, PTX/DM-β-CD inclusion complex-loaded CS NPs exhibited significantly enhanced EEs and LEs up to 82.74% and 3.97%, respectively. With the same weight ratio of PTX to CS at 0.7, the LE of PTX/DM-β-CD inclusion complex-loaded CS NPs was 30.3-fold higher than that of the PTX-loaded CS NPs. This is because HP PTX was efficiently loaded into the HP cavity of DM-β-CD, which has a hydrophilic outer surface and could be incorporated into the CS NPs easily, therefore leading to the significantly increased encapsulation of PTX into DM-β-CD/CS NPs. In the later study, the weight ratio of PTX to CS was set at 0.7 to prepare the PTX/DM-β-CD inclusion complex-loaded CS NPs.
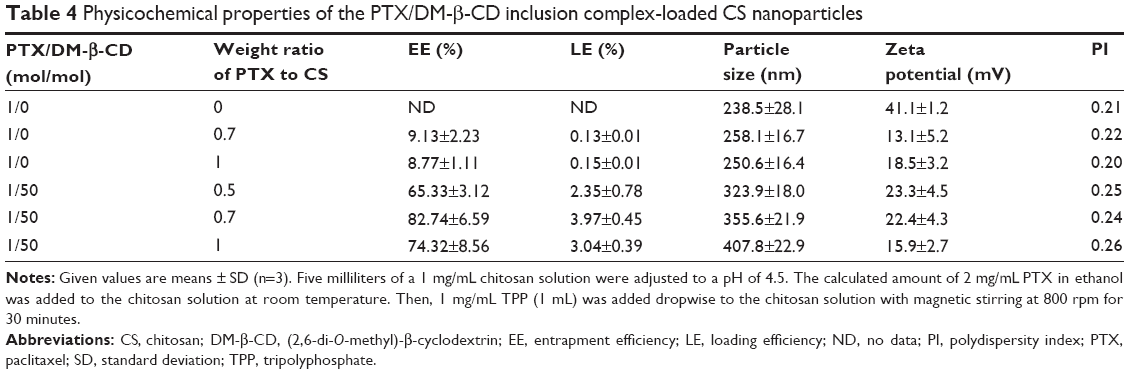 | Table 4 Physicochemical properties of the PTX/DM-β-CD inclusion complex-loaded CS nanoparticles |
The in vitro drug-release profile of the PTX/DM-β-CD inclusion complex-loaded CS NPs was investigated in PBS (pH 7.4) at 37°C. As shown in Figure 6, free PTX in PBS solution was released rapidly from the dialysis tube and the cumulative release reached 99.5% at 10 minutes. The PTX/DM-β-CD inclusion complex-loaded CS NPs displayed a sustained drug-release profile. It was a biphasic release process with an initial burst release phase followed by a delayed release phase. Compared with free PTX in PBS solution, the PTX/DM-β-CD inclusion complex-loaded CS NPs had a much smaller burst release with the cumulative release of PTX of 37.8% within the first 0.5 hour, which could be due to the desorption of the surface-bound or adsorbed drugs. In the delayed release phase, more drug was released from the incorporated PTX/DM-β-CD inclusion complexes or the inside of the NPs.
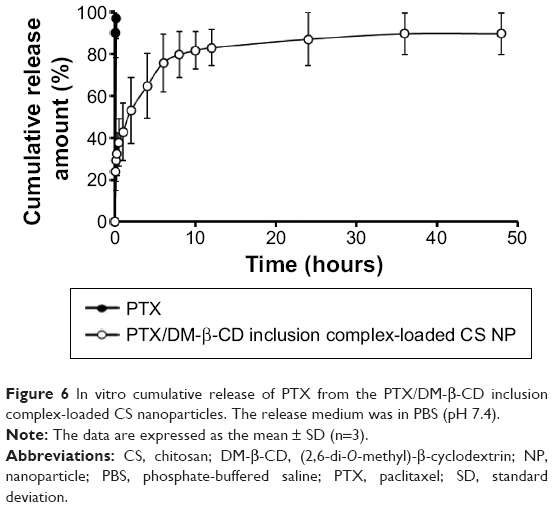 | Figure 6 In vitro cumulative release of PTX from the PTX/DM-β-CD inclusion complex-loaded CS nanoparticles. The release medium was in PBS (pH 7.4). |
Pharmacokinetics in rats
The blood plasma concentration of PTX in rats was determined at various time points after the intravenous injection of a single dose of reference formulation (Taxol®) or the PTX/DM-β-CD inclusion complex-loaded CS NPs (equivalent 5 mg/kg PTX). As shown in Figure 7, the maximum plasma concentration of PTX after the intravenous administration of the PTX/DM-β-CD inclusion complex-loaded CS NPs was reached at 1 minute post-injection, and then a biphasic profile with a rapid initial drug elimination phase was observed within the first 30 minutes, followed by a slower elimination phase. At 15 minutes post-injection, the blood plasma concentration of PTX in the rats treated with the PTX/DM-β-CD inclusion complex-loaded CS NPs was 18.1 μg/mL, while that in the rats treated with reference formulation (Taxol®) was only 7.5 μg/mL. In the extended post-injection period up to 24 hours, the PTX plasma concentration in the rats treated with the PTX/DM-β-CD inclusion complex-loaded CS NPs was 3.6-fold higher than that in the rats treated with reference formulation (Taxol®).
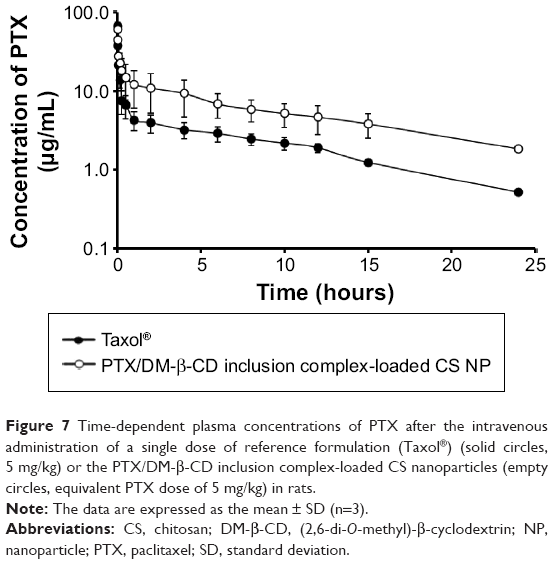 | Figure 7 Time-dependent plasma concentrations of PTX after the intravenous administration of a single dose of reference formulation (Taxol®) (solid circles, 5 mg/kg) or the PTX/DM-β-CD inclusion complex-loaded CS nanoparticles (empty circles, equivalent PTX dose of 5 mg/kg) in rats. |
The pharmacokinetic parameters of reference formulation (Taxol®) and the PTX/DM-β-CD inclusion complex-loaded CS NPs in rats after intravenous administration are shown in Table 5. The PTX/DM-β-CD inclusion complex-loaded CS NPs showed an extended half-life of the elimination phase t1/2β (6.10 hours) compared with reference formulation (Taxol®) (4.56 hours), while the elimination rate Kel constant of the NPs (0.54 h−1) was less than one-third that of reference formulation (Taxol®). This suggested that the clearance rate of PTX in the blood plasma decreased significantly when PTX was incorporated into the DM-β-CD/CS NPs. Moreover, the PTX/DM-β-CD inclusion complex-loaded CS NPs in rats showed a 2.7-fold increase in AUC0→24h (the area under the plasma drug concentration–time curve over the period of 24 hours) and a 1.4-fold increase in the mean residence time compared with the reference formulation (Taxol®). The pharmacokinetic results showed that the PTX/DM-β-CD inclusion complex-loaded CS NPs could enable the drug payload to be released in a more stable and sustained manner.
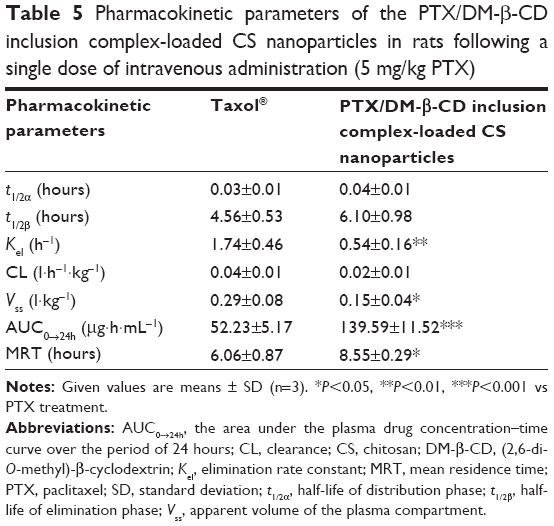 | Table 5 Pharmacokinetic parameters of the PTX/DM-β-CD inclusion complex-loaded CS nanoparticles in rats following a single dose of intravenous administration (5 mg/kg PTX) |
Conclusion
The novel PTX/DM-β-CD inclusion complex-loaded CS NPs have been successfully prepared using biocompatible and biodegradable natural materials by an ionic gelation method. This work represented the first-known successful example of using DM-β-CD/CS as a carrier for the delivery of poorly water-soluble PTX. It was demonstrated in vitro that the drug-delivery system with a uniform particle size possessed a much higher drug encapsulation efficiency than PTX-loaded CS NPs and a lower burst release rate than reference formulation (Taxol®). The high AUC0→24h value indicated that this nano-system had a sustained drug-release profile. Therefore, the DM-β-CD/CS NPs hold great potential as a biocompatible and biodegradable drug carrier for the significantly enhanced delivery and sustained release of poorly water-soluble drugs including PTX. This, as a result, would possibly enhance therapeutic efficacy and reduce the severe side effects of drugs.
Acknowledgments
The authors acknowledge financial support from the Fundamental Research Funds for the Central Universities (WY1213013 ECUST) and the Science and Technology Commission of Shanghai Municipality (STCSM, contract Nos 11DZ2260600 and 10DZ2220500). R Chen acknowledges financial support from the Imperial College of London.
Disclosure
The authors report no conflicts of interest in this work.
References
Yao HJ, Ju RJ, Wang XX, et al. The antitumor efficacy of functional paclitaxel nanomicelles in treating resistant breast cancers by oral delivery. Biomaterials. 2011;32(12):3285–3302. | ||
Zhao D, Zhao X, Zu Y, et al. Preparation, characterization, and in vitro targeted delivery of folate-decorated paclitaxel-loaded bovine serum albumin nanoparticles. Int J Nanomedicine. 2010;5:669–677. | ||
Wang Y, Li X, Wang L, Xu Y, Cheng X, Wei P. Formulation and pharmacokinetic evaluation of a paclitaxel nanosuspension for intravenous delivery. Int J Nanomedicine. 2011;6:1497–1507. | ||
Liu L, Zhu S. A study on the supramolecular structure of inclusion complex of β-cyclodextrin with prazosin hydrochloride. Carbohydr Lett. 2007;68(3):472–476. | ||
Marques HMC, Hadgraft J, Kellaway IW. Studies of cyclodextrin inclusion complexes. I. The salbutamol-cyclodextrin complex as studied by phase solubility and DSC. Int J Pharm. 1990;63(3):259–266. | ||
Ye Y, Sun Y, Zhao H, et al. A novel lactoferrin-modified β-cyclodextrin nanocarrier for brain-targeting drug delivery. Int J Pharm. 2013;458(1):110–117. | ||
Krauland AH, Alonso MJ. Chitosan/cyclodextrin nanoparticles as macro-molecular drug delivery system. Int J Pharm. 2007;340(1–2):134–142. | ||
Boudad H, Legrand P, Lebas G, Cheron M, Duchêne D, Ponchel G. Combined hydroxypropyl-β-cyclodextrin and poly(alkylcyanoacrylate) nanoparticles intended for oral administration of saquinavir. Int J Pharm. 2001;218(1–2):113–124. | ||
Maestrelli F, Garcia-Fuentes M, Mura P, Alonso MJ. A new drug nanocarrier consisting of chitosan and hydoxypropylcyclodextrin. Eur J Pharm Biopharm. 2006;63(2):79–86. | ||
Zhang J, Ma PX. Cyclodextrin-based supramolecular systems for drug delivery: recent progress and future perspective. Adv Drug Deliv Rev. 2013;65(9):1215–1233. | ||
Gamal AE, Terada K, Yamamoto K. Molecular behavior, dissolution characteristics and chemical stability of aspirin in the ground mixture and in the inclusion complex with di-O-methyl-β-cyclodextrin. Int J Pharm. 1986;31(1–2):25–31. | ||
Bouquet W, Ceelen W, Fritzinger B, et al. Paclitaxel/beta-cyclodextrin complexes for hyperthermic peritoneal perfusion-formulation and stability. Eur J Pharm Biopharm. 2007;66(3):391–397. | ||
Hamada H, Ishihara K, Masuoka N, Mikuni K, Nakajima N. Enhancement of water-solubility and bioactivity of paclitaxel using modified cyclodextrins. J Biosci Bioeng. 2006;102(4):369–371. | ||
Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168–182. | ||
Agnihotri SA, Mallikarjuna NN, Aminabhavi TM. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J Control Release. 2004;100(1):5–28. | ||
Jameela SR, Latha PG, Subramoniam A, Jayakrishnan A. Antitumor activity of mitoxantrone-loaded chitosan microspheres against Ehrlich ascites carcinoma. J Pharm Pharmacol. 1996;48(7):685–688. | ||
Mitra S, Gaur U, Ghosh PC, Maitra AN. Tumor targeted delivery of encapsulated dextran-doxorubicin conjugate using chitosan nanoparticles as carrier. J Control Release. 2001;74(1–3):317–323. | ||
Yousefpour P, Atyabi F, Vasheghani-Farahani E, Movahedi AA, Dinarvand R. Targeted delivery of doxorubicin-utilizing chitosan nanoparticles surface-functionalized with anti-her2 trastuzumab. Int J Nanomedicine. 2011;6:1977–1990. | ||
Yuan Z, Ye Y, Gao F, et al. Chitosan-graft-β-cyclodextrin nanoparticles as a carrier for controlled drug release. Int J Pharm. 2013;446(1–2):191–198. | ||
Ji JG, Hao SL, Liu WQ. Preparation and evaluation of O-carboxymethyl chitosan/cyclodextrin nanoparticles as hydrophobic drug delivery carriers. Polym Bull. 2011;67(7):1201–1213. | ||
Amber V, Shailendra S, Swarnlata S. Encapsulation of cyclodextrin complexed simvastatin in chitosan nanocarriers: a novel technique for oral delivery. J Incl Phenom Macrocycl Chem. 2010;66(3–4):251–259. | ||
Mahmoud AA, El-Feky GS, Kamel R, Awad GE. Chitosan/sulfobutylether-β-cyclodextrin nanoparticles as a potential approach for ocular drug delivery. Int J Pharm. 2011;413(1–2):229–236. | ||
Trapani A, Garcia-Fuentes M, Alonso MJ. Novel drug nanocarriers combining hydrophilic cyclodextrins and chitosan. Nanotechnology. 2008;19(18):185101. | ||
Jingou J, Shilei H, Weiqi L, Danjun W, Tengfei W, Yi X. Preparation, characterization of hydrophilic and hydrophobic drug in combine loaded chitosan/cyclodextrin nanoparticles and in vitro release study. Colloids Surf B Biointerfaces. 2011;83(1):103–107. | ||
Trapani A, Lopedota A, Franco M, et al. A comparative study of chitosan and chitosan/cyclodextrin nanoparticles as potential carriers for the oral delivery of small peptides. Eur J Pharm Biopharm. 2010;75(1):26–32. | ||
Lee S, Seo D, Kim HW, Jung S. Investigation of inclusion complexation of paclitaxel by cyclohenicosakis-(1→2)-(beta-D-glucopyranosyl), by cyclic-(1→2)-beta-D-glucans (cyclosophoraoses), and by cyclomaltoheptaoses (beta-cyclodextrins). Carbohydr Res. 2001;334(2):119–126. | ||
Kim H, Choi J, Kim HW, Jung S. Monte Carlo docking simulations of cyclomaltoheptaose and dimethyl cyclomaltoheptaose with paclitaxel. Carbohydr Res. 2002;337(6):549–555. | ||
He L, Wang GL, Zhang Q. An alternative paclitaxel micro-emulsion formulation: hypersensitivity evaluation and pharmacokinetic profile. Int J Pharm. 2003;250(1):45–50. | ||
Agüeros M, Ruiz-Gatón L, Vauthier C, et al. Combined hydroxypropyl-β-cyclodextrin and poly(anhydride) nanoparticles improve the oral permeability of paclitaxel. Eur J Pharm Sci. 2009;38(4):405–413. | ||
Wang DW, Ouyang CB, Liu Q, Yuan HL, Liu XH. Inclusion of quinestrol and 2,6-di-O-methyl-β-cyclodextrin: preparation, characterization, and inclusion mode. Carbohydr Polym. 2013;93(2):753–760. | ||
Gan Q, Wang T, Cochrane C, McCarron P. Modulation of surface charge, particle size and morphological properties of chitosan-TPP nanoparticles intended for gene delivery. Colloid Surface B. 2005;44(2–3):65–73. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.