")
Back to Journals » OncoTargets and Therapy » Volume 7
The potential roles of hepatocyte growth factor (HGF)-MET pathway inhibitors in cancer treatment
Authors Parikh R, Wang P, Beumer J, Chu E, Appleman L
Received 27 September 2013
Accepted for publication 20 November 2013
Published 11 June 2014 Volume 2014:7 Pages 969—983
DOI https://doi.org/10.2147/OTT.S40241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Rahul A Parikh,1 Peng Wang,2 Jan H Beumer,3 Edward Chu,1 Leonard J Appleman1
1Division of Hematology-Oncology, University of Pittsburgh School of Medicine, Cancer Therapeutics Program, University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA; 2Division of Medical Oncology, University of Kentucky College of Medicine, Markey Cancer Center, Lexington, KY, USA; 3University of Pittsburgh School of Pharmacy, Cancer Therapeutics Program, University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA
Abstract: MET is located on chromosome 7q31 and is a proto-oncogene that encodes for hepatocyte growth factor (HGF) receptor, a member of the receptor tyrosine kinase (RTK) family. HGF, also known as scatter factor (SF), is the only known ligand for MET. MET is a master regulator of cell growth and division (mitogenesis), mobility (motogenesis), and differentiation (morphogenesis); it plays an important role in normal development and tissue regeneration. The HGF-MET axis is frequently dysregulated in cancer by MET gene amplification, translocation, and mutation, or by MET or HGF protein overexpression. MET dysregulation is associated with an increased propensity for metastatic disease and poor overall prognosis across multiple tumor types. Targeting the dysregulated HGF-MET pathway is an area of active research; a number of monoclonal antibodies to HGF and MET, as well as small molecule inhibitors of MET, are under development. This review summarizes the key biological features of the HGF-MET axis, its dysregulation in cancer, and the therapeutic agents targeting the HGF-MET axis, which are in development.
Keywords: MET inhibitor, HGF inhibitor, cancer
Hepatocyte growth factor (HGF)-MET pathway in normal development and tumorigenesis
Nearly 30 years ago, HGF was identified as a modulator of hepatocyte proliferation and regeneration by three independent groups of investigators.1–3 HGF is a heterodimer with a larger α subunit and smaller β subunit with homology to tissue plasminogen.4 HGF is secreted by cells of mesenchymal origin and binds to the MET receptor on epithelial and endothelial cells. Scatter factor (SF), so named due to its ability to scatter mammary epithelial cells in culture, was characterized by complementary deoxyribonucleic acid (cDNA) cloning in 1990; SF was found to be identical to HGF, with indistinguishable ligand activity for the MET receptor.5 HGF is secreted as a precursor polypeptide bound to heparin proteoglycans in the extracellular matrix. The inactive HGF polypeptide is cleaved by a serine protease at sites of tissue injury and by tumor cells/matrix into active HGF. The only known ligand for the MET receptor is HGF, which upon binding to MET activates a downstream cascade, leading to cell survival, division, and motility.6
Activated MET kinase undergoes autophosphorylation on tyrosine residues (Tyr1349 and Tyr1356) in the docking domain, leading to recruitment and transduction of downstream effectors. Activated MET phosphorylates and binds to growth factor receptor-bound protein 2 (GRB2) and GRB2-associated binding protein 1 (GAB1), acting as a scaffold and promoting MET interactions with proteins involved in the phosphoinositol 3-kinase (PI3K)-protein kinase B (Akt) pathway, the signal transducer and activator of transcription factor (STAT) pathway, the mitogen-activated protein kinase (MAPK) pathway, and the nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) pathway.7 All of these pathways form an integral part of the MET-dependent cell growth, survival, and migration-signaling cascade. During embryogenesis, normal expression and function of HGF, MET, and GAB1 are essential for growth and development of hepatocytes,8 placental trophoblasts, and myoblasts.9 After birth, activation of the HGF-MET pathway appears to be involved in epithelial-mesenchymal transition,10 hepatic, renal, and other organ regeneration after injury, and wound healing. Figure 1 illustrates the HGF-MET signaling pathway.
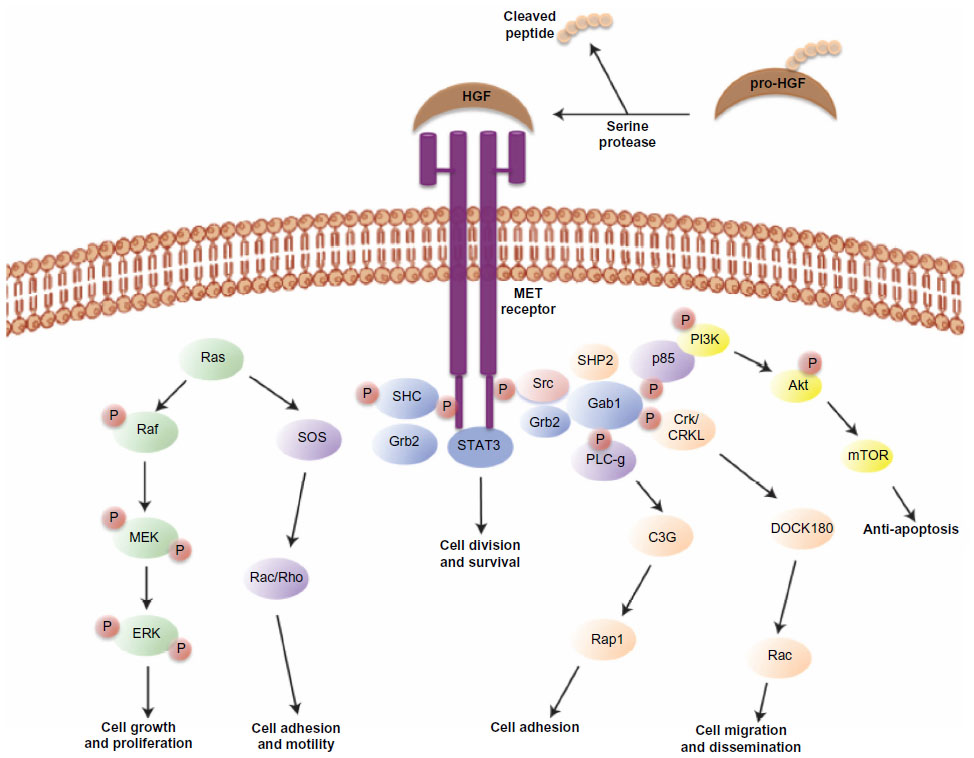 | Figure 1 HGF-MET signaling pathway. |
In a subset of spontaneously transformed NIH-3T3 mouse fibroblasts, amplification and subsequent overexpression of the mouse MET gene was identified. This amplification was not seen in nontransformed NIH-3T3 cells and provided the initial proof of concept that the MET oncogene could act as a transforming factor.11 Subsequently, MET ribonucleic acid (RNA) and protein overexpression was observed in multiple epithelial and mesenchymal tumor cell lines, including breast, thyroid, liver, and kidney cancers.12 Transgenic mice with forced overexpression of HGF were noted to develop multiple epithelial and mesenchymal tumors. This diverse tumorigenesis was associated with MET phosphorylation and autocrine activation. Tumors arising in the tissues of these transgenic mice exhibited morphologic and developmental abnormalities, establishing the role of the HGF-MET pathway in tumorigenesis.13
HGF and MET dysregulation in cancer
In human malignancies, the HGF-MET axis is dysregulated by a number of mechanisms, providing tumor cells with the ability to proliferate and disseminate. The MET gene is activated by point mutations in small-cell lung cancer (SCLC)14 and renal papillary carcinomas.15 MET protein is overexpressed in melanoma and musculoskeletal tumors.16 Fusion of MET with translocated promoter region (TPR) in gastric carcinoma leads to MET overexpression.17–19 Aberrant HGF expression leading to autocrine activation of MET occurs in nearly half of acute myeloid leukemia cell lines, and depletion of HGF or MET leads to inhibition of growth and apoptosis.20 Transgenic mice overexpressing HGF have increased MET expression in tumor cells, providing them with a selective growth advantage; overexpression of HGF in tissues is associated with increased incidence of epithelial and mesenchymal tumors.13 In an elegant study, Lorenzato et al noted that activating somatic MET mutations were infrequent in primary tumors but commonly present at metastatic sites, suggesting that MET mutations are associated with progression rather than initiation of tumorigenesis.21 In colorectal tumors, MET amplification is associated with advanced stages and development of hepatic metastatic disease; gene amplification was observed in 2% (3/177) of localized primary cancers, 9% (6/70) of cancers with distant metastases (P<0.02), and 18% (25/147) of liver metastases (P<0.01).22 Thus, alterations in MET and/or HGF are frequently observed in a wide range of cancers, and their presence appears to confer an increased propensity for a more aggressive clinical behavior manifested by invasion and metastasis.
Overexpression and upregulation of the HGF-MET pathway has been shown to be an important escape mechanism for tumor cells to develop resistance to inhibition of the epidermal growth factor receptor (EGFR), RAS-RAF-MEK, and Akt–mTOR (mammalian target of rapamycin) pathways. In a subset (22%) of non-small-cell lung cancers (NSCLC) with activating mutations of the EGFR gene, MET amplification is associated with gefitinib resistance by promoting human epidermal growth factor receptor (HER)-3-mediated activation of PI3K.23 Overexpression of HGF followed by MET phosphorylation in NSCLCs with EGFR-activating mutations reactivates the PI3K-Akt signaling pathway and leads to gefitinib resistance.24 Similarly, resistance to RAS inhibitors is mediated by upregulation and secretion of HGF by the tumor microenvironment.25 MET activation was noted to mediate resistance to HER2 inhibition by lapatinib in HER2-amplified gastric cancer cells.26 In esophageal squamous cell carcinoma, secretion of HGF by fibroblasts is associated with a highly invasive phenotype and early disease progression.27 MET amplification is associated with a highly aggressive phenotype in a subset of gastro-esophageal adenocarcinomas.28 TP53 mutations or p53 deficiency is associated with MET dysregulation and promotes tumor cell mobility and invasion.29 Alterations in the HGF-MET axis can lead to development of resistance to inhibition of a number of different pathways; combining HGF/MET inhibition with targeted EGFR, MEK, or PI3K inhibitors would appear to represent a rational approach to treating these resistant tumors.
Targeting the HGF-MET axis
Currently, a number of strategies targeting the HGF-MET pathway are in development. These approaches include the use of small molecule MET tyrosine kinase inhibitors (TKI), anti-HGF neutralizing antibodies, and anti-MET neutralizing antibodies. Each of these approaches will be reviewed below. The molecular sites of action for agents targeting the HGF-MET pathway are shown in Figure 2. Table 1 summarizes the target receptors, half-life, and characteristics of HGF-MET inhibitors. The development of a number of these agents has advanced to evaluation for efficacy in Phase II and Phase III clinical trials (Table 2). A timeline of significant pathways involving the HGF-MET axis and clinical development of agents targeting the HGF-MET pathway is shown in Figure 3.
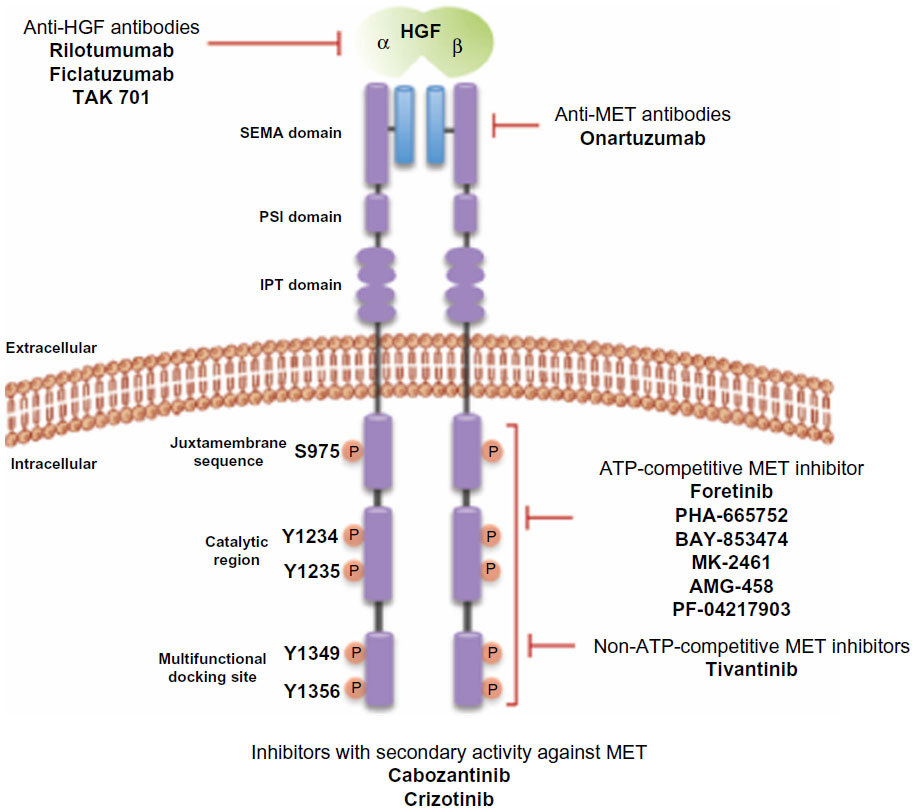 | Figure 2 HGF-MET inhibitors and potential sites of action. |
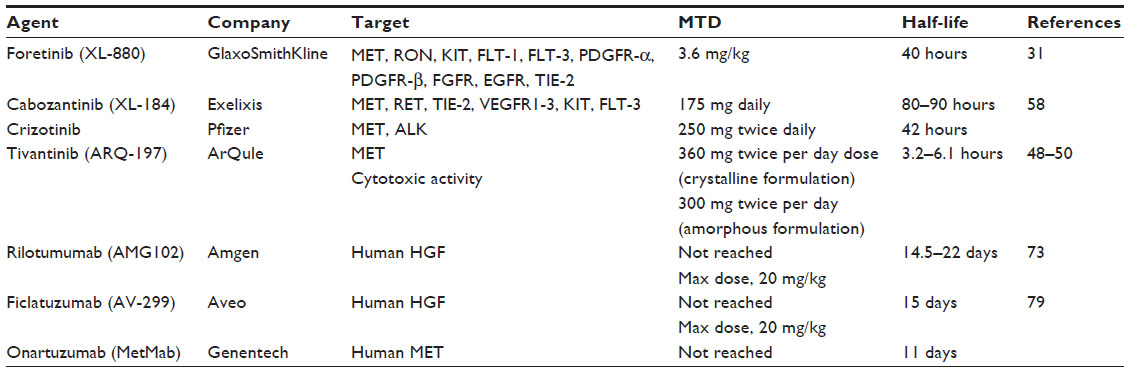 | Table 1 Summary of MET-targeted agents, target receptors, maximal tolerated dose, and half-life for HGF and MET inhibitors |
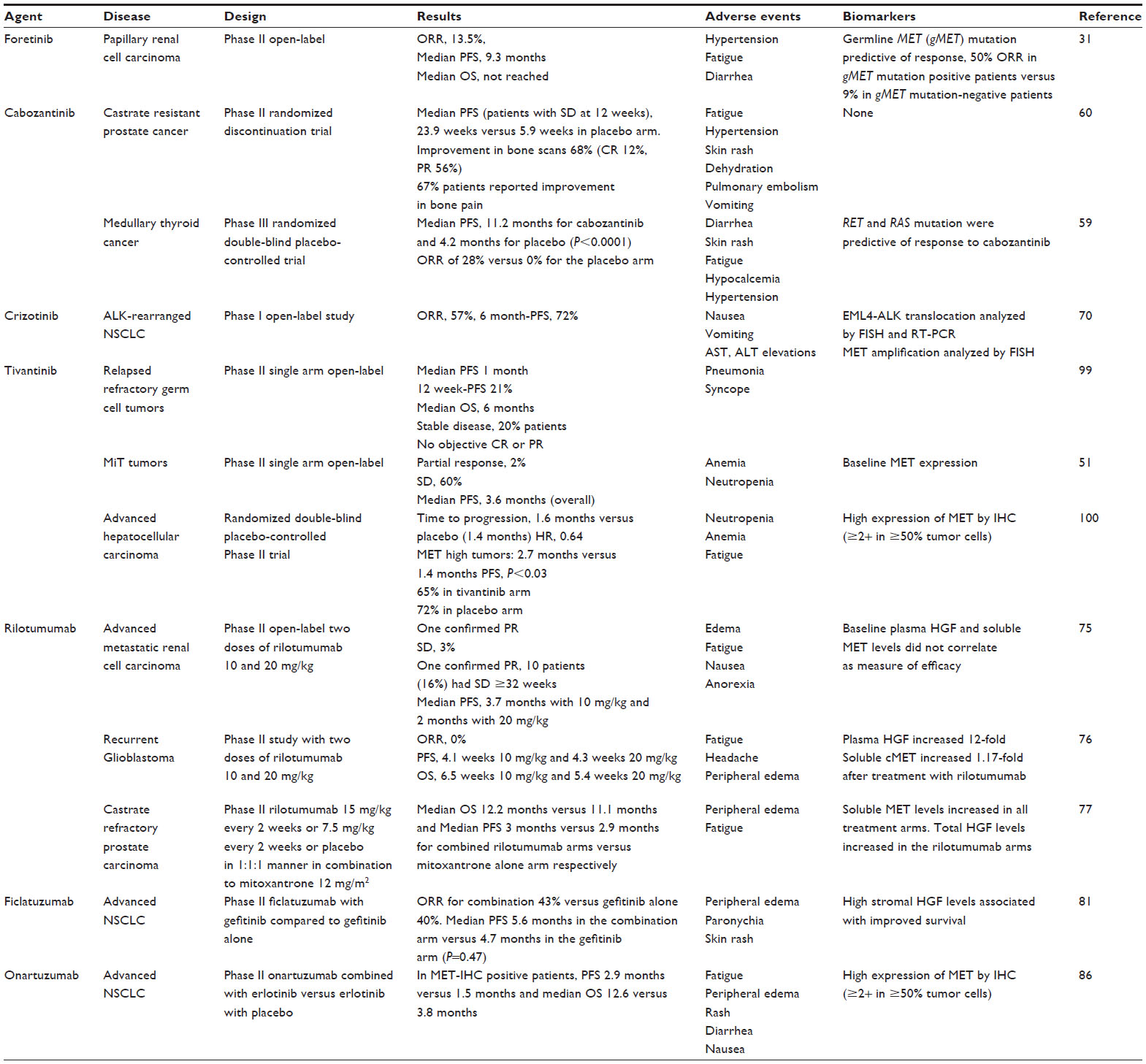 | Table 2 Results of key Phase II/III trials with HGF-MET inhibitors |
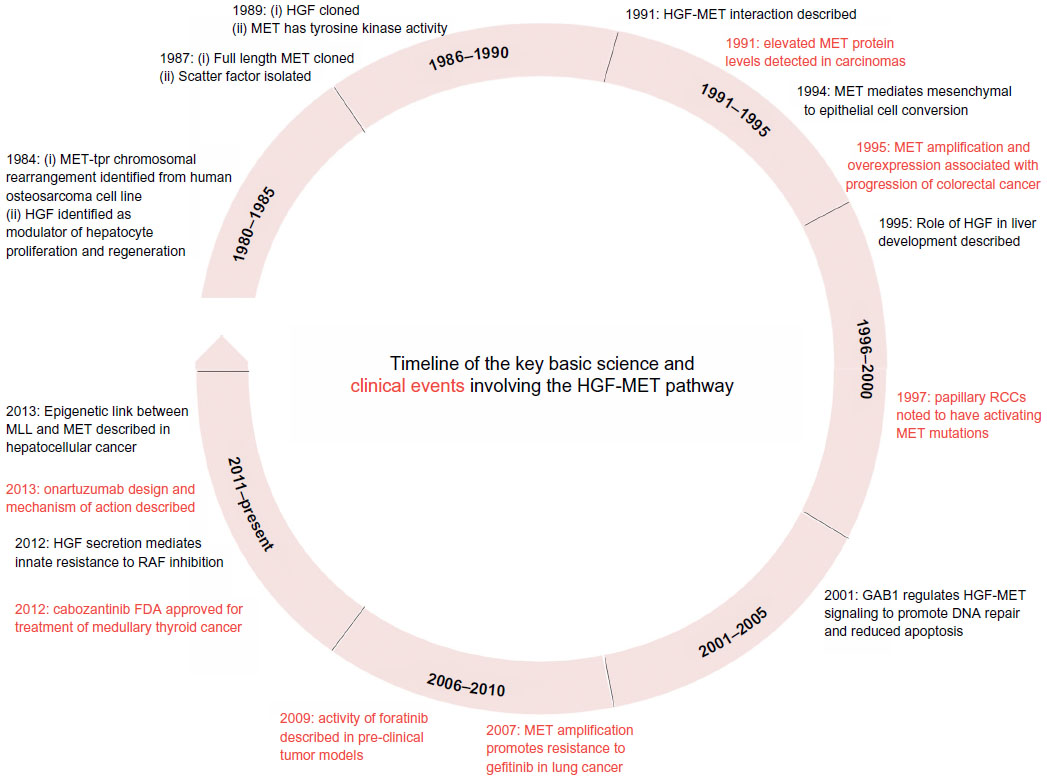 | Figure 3 A timeline of important discoveries related to the HGF-MET pathway. Black represents basic science discoveries and red represents clinical/translational discoveries. |
Adenosine triphosphate (ATP)-competitive MET kinase inhibitors
Foretinib
Foretinib was developed as a small molecular inhibitor of MET and vascular endothelial growth factor (VEGF) receptor 2 (VEGFR-2/kinase insert domain receptor [KDR]) with additional activity against RON, KIT, fms-related tyrosine kinase (FLT)-1, FLT-3, platelet-derived growth factor receptor (PDGFR)-α, PDGFR-β, fibroblast growth factor receptor (FGFR), EGFR, and tyrosine kinase with immunoglobulin-like and EGF-like domains (TIE)-2. Foretinib inhibits MET at a half maximal inhibitory concentration (IC50) of 0.4 nmol/L; subsequently, it also inhibits HGF-mediated MET phosphorylation, migration, and invasion of MET-amplified human gastric cancer cell lines. In a mouse model with lung metastases, oral foretinib produced a significant dose-dependent reduction in both size and number of lung metastases, which led to its further development.30
In the initial Phase I trial of foretinib in patients with metastatic or unresectable solid tumors, 40 patients were enrolled in eight dose cohorts. All patients received foretinib for 5 days orally every 14 days. The maximum tolerated dose (MTD) was 3.6 mg/kg and dose limiting toxicities included aspartate aminotransferase and lipase elevations. Responses were seen in two patients with papillary renal cell carcinoma and one patient with medullary thyroid carcinoma. The recommended Phase II dose (RP2D) of foretinib was determined to be 240 mg daily for 5 days every 14 days.31 The half-life for foretinib was determined to be approximately 40 hours, and no pharmacokinetic differences were observed based on age, sex, or body weight.31
In a Phase II study of foretinib in patients with papillary renal cell carcinoma, 74 patients were enrolled into two cohorts comparing daily to intermittent foretinib. A total of 37 patients in cohort A received 240 mg foretinib on days 1–5 every 14 days, while 37 patients in cohort B received 80 mg daily foretinib. The overall response rate was 13.5% and median progression-free survival was 9.3 months. Five of ten patients with a germline MET mutation responded to foretinib, while five of 57 patients without germline MET mutation responded to foretinib. The most common side effects of this agent were fatigue, hypertension, and gastrointestinal (GI) toxicity.32,33 This study is unique as the authors clearly stratified patients based on germline or acquired MET mutations, and they noted that germline MET mutations were a stronger predictive marker of response to foretinib compared to acquired MET mutations.31,32
Seventy-four patients were enrolled in a Phase II study of foretinib in patients with metastatic gastric adenocarcinoma. Stable disease was seen in ten of 49 patients on intermittent dosing (240 mg/day, for 5 days, every 2 weeks) and in five of 25 patients on continuous dosing daily (80 mg/day). Median duration of stable disease was 3.2 months, and the study failed to meet its primary endpoint of an objective response rate (ORR) >25%. Of 67 patients with tumor samples, three had MET amplification, one of whom had disease stabilization.34
PHA-665752
PHA-665752 is an ATP-competitive small-molecule inhibitor of the MET kinase with additional activity against RON and VEGF2. In gastric carcinoma mouse-xenograft models, PHA-665752 inhibited MET phosphorylation and tumor growth.35 Treatment with PHA-665752 induced apoptosis in five of five gastric cancer cell lines with MET amplification but none of twelve cell lines without MET amplification.36 The addition of PHA-665752 to gefitinib, an EGFR TKI, reversed acquired resistance to gefitinib in lung cancer cell lines with MET amplification.23
BAY-853474
BAY-853474 is a selective, potent, oral small-molecule inhibitor of the MET kinase. BAY-853474 inhibits MET autophosphorylation in vitro and inhibits proliferation and growth of MET-dependent tumor xenograft models in vivo.37 BAY-853474 use was associated with reduced tumor burden in glioblastoma, NSCLC, and gastric cancer xenograft models. The inhibition of MET phosphorylation was dose-dependent, and soluble plasma biomarkers HGF, VEGF, interleukin-8, and MET-ectodomain predicted response to treatment.38
MK-2461
MK-2461 is an inhibitor of MET with additional activity against RON, FLT-1, FGFR, PDGFR, and other receptor tyrosine kinases. In MET-amplified human GTL16 gastric cancer cell lines and a murine xenograft model of MET-dependent gastric cancer, oral MK-2461 inhibited MET signaling and cell growth.39 Genomic amplification of MET and constitutive activation of MET, FGFR2, or PDGFR2 were noted to be predictive of response to MK-2461.39
AMG-458
AMG-458 is a potent inhibitor of the MET, VEGFR-2, and insulin-like growth factor 1 receptor signaling pathways.40 It significantly inhibited tumor growth in the NIH-3T3 cells transfected with TPR-MET, leading to constitutive activation of MET and U-87 MG human glioblastoma xenograft models.41 There is currently no clinical trial information available for AMG-458.
PF-04217903
PF-04217903 showed excellent in vitro activity against the MET-amplified human GTL16 gastric carcinoma and H1993 NSCLC cell lines. PF-04217903 partially inhibited proliferation in U-87 MG human glioblastoma cells and two colon cancer cell lines (SW620, HT29). PF-04217903 inhibited MET phosphorylation and tumor proliferation in both a GTL16 xenograft athymic mouse model with MET amplification and in a U-87 MG human glioblastoma xenograft model with an activated HGF-MET autocrine loop.42,43 A Phase I study of PF-04217903 in advanced cancer was terminated by Pfizer in early 2012.
Non-ATP-competitive MET kinase inhibitors
Tivantinib
Tivantinib (ARQ 197) is currently undergoing Phase II clinical development based upon preclinical cytotoxic activity and ATP non-competitive inhibition of MET with a minimal IC50 of 0.1 μM44 by in vitro assay of recombinant kinase protein. Tivantinib binds to a hydrophobic cleft in the kinase domain, stabilizing the inactive conformation and inhibiting MET autophosphorylation.45 Tivantinib is orally bioavailable, and it is metabolized by the liver cytochrome P450 (CYP) CYP2C19, and to a lesser extent by CYP3A4. Polymorphisms in CYP2C19 (CYP2C19*2 and CYP2C19*3) have been identified that are associated with decreased metabolism of tivantinib, resulting in significantly increased drug exposure.46 These polymorphisms are most commonly seen in Asians (35% allele frequency), but also occur in African-Americans (17%) and Caucasians (15%).47
In vitro, tivantinib induced caspase-dependent apoptosis in MET-expressing human cancer cell lines, including the HT29 (colon adenocarcinoma), MKN-45 (gastric cancer), and MDA-MB-231 (breast cancer) cell lines; this small molecule demonstrated antitumor activity in a wide range of human cancer xenograft models.48
The clinical pharmacology and safety profile of tivantinib have been investigated in several Phase I studies.48–50 The main dose-limiting toxicities have been leukopenia, neutropenia, thrombocytopenia, vomiting, and dehydration. At a dose of 400 mg daily, febrile neutropenia was observed in two subjects, one of whom also experienced grade 3 mucositis, palmar-plantar erythrodysesthesia, and hypokalemia.50 All toxicities were reversible upon discontinuation of tivantinib. Drug treatment was associated with a decrease in phosphorylated MET, significant decreases in total MET and phosphorylated focal adhesion kinase, and a significant increase in the level of apoptosis, as measured by a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.50 The RP2D for tivantinib was established as 300 mg twice daily of the amorphous formulation, which is equivalent to 360 mg twice daily of the crystalline formulation.
In a Phase II study, 47 patients with advanced microphthalmia transcription factor (MITF)-associated tumors (MiT) were treated with 360 mg twice daily tivantinib.51 The study concluded that tivantinib was safe and tolerable in patients with MiT tumors, but antitumor activity was relatively modest, with a partial response in one clear cell sarcoma patient (2%) and stable disease in 28 patients (60%).51
In a randomized, controlled Phase II study, the combination of tivantinib with erlotinib, an EGFR TKI, showed a trend towards improved progression-free survival (PFS) and overall survival (OS) compared with placebo with erlotinib; these results were found in a subset of patients with non-squamous histology, as well as in patients with wild-type EGFR. Interestingly, there was a significant benefit in PFS and OS in all 15 patients with mutated KRAS. Although the study did not meet its primary endpoint, evidence of activity was demonstrated, particularly among patients with KRAS mutations.52 MARQUEE was a randomized, double-blind, controlled study evaluating tivantinib versus placebo, in combination with erlotinib. Patients in the study had locally advanced or metastatic, non-squamous NSCLC, and had received prior treatment. However, the study was stopped early; at the planned interim analysis, there was no improvement in overall survival, the primary study endpoint. The combination of erlotinib with tivantinib demonstrated a statistically significant improvement in PFS, with no safety concerns in the intent-to-treat population.53
Two recent preclinical studies have called into question the mechanism of tivantinib action. A series of experiments demonstrated that the cytotoxic effect of tivantinib on cell lines was independent of MET genetic status (wild-type, amplified, and/or kinase-deleted).54,55 Both studies observed that tivantinib caused G2 arrest, in sharp contrast to other MET inhibitors, which cause arrest at the G1 checkpoint. Both studies observed alterations in microtubule dynamics; in one study, tivantinib was noted to stabilize microtubules, while the other report observed that tivantinib destabilized microtubules. The potential implications for further clinical development of tivantinib as a MET inhibitor are discussed in a recent review.56
Multitargeted inhibitors of MET and other kinases
Cabozantinib
Cabozantinib is an oral small molecule inhibitor of RET, MET, and VEGF-2, with additional activity against VEGF-1, VEGF-3, KIT, and FLT-3.57 Eighty-five patients with advanced solid malignancies, including an expansion cohort of 37 patients with medullary thyroid cancer (MTC), were enrolled in a Phase I dose-escalation study of oral cabozantinib. The MTD for cabozantinib was 175 mg, and dose-limiting toxicities included palmar-plantar erythrodysesthesia, mucositis, aspartate aminotransferase, alanine transaminase, and lipase elevations. Ten of 35 evaluable patients with MTC had a partial response; an additional 15 patients with MTC had stable disease for at least 6 months. Activating RET mutations were seen in 81% of patients with MTC and the presence of these mutations appeared to correlate with response to cabozantinib.58 In a Phase III EXAM study, 330 patients with locally advanced or metastatic MTC were assigned in a 2:1 ratio to receive cabozantinib 160 mg daily or placebo, respectively. Median progression-free survival, the primary endpoint, was 11.2 months for cabozantinib and 4.2 months for placebo (P<0.0001), with an ORR of 28% versus 0% for the placebo arm.59 Common grade 3 or greater adverse events associated with cabozantinib were diarrhea (15.9% versus 1.8%), palmar-plantar erythrodysesthesia (12.6% versus 0%), fatigue (9.3% versus 2.8%), hypocalcemia (9.3% versus 0%), and hypertension (7.9% versus 0%).59
Based on these results, in late 2012 cabozantinib was approved by the US Food and Drug Administration (FDA) for the treatment of MTC. In the EXAM study, patients with RET mutations had a significantly longer median PFS (60 weeks) compared to RET mutation-negative patients (25 weeks). In the RET mutation-negative subgroup, patients with RAS mutations had a median PFS of 47 weeks, accounting for most of the benefit observed in this subgroup of patients.59 Thus, RET and RAS mutations in MTC are predictive of response to cabozantinib.
One hundred and seventy one men with castrate refractory prostate adenocarcinoma were enrolled in a Phase II randomized discontinuation trial with cabozantinib. The patients received 100 mg of cabozantinib for a 12-week run-in period, followed by randomization of patients with stable disease to cabozantinib or placebo. Due to the efficacy of cabozantinib, the randomization was halted early. The ORR at 12 weeks, a co-primary endpoint, was 5%, while 75% of patients had stable disease. In 31 patients with stable disease at 12 weeks who were randomly assigned to cabozantinib or placebo, the median progression free survival was 23.9 months with cabozantinib and 5.9 months with placebo, resulting in a hazard ratio of 0.12. The most common dose limiting toxicities were fatigue, hypertension, and hand-foot syndrome.60
Cabozantinib has a black-box warning for gastrointestinal perforations (3%), fistula formation (1%), and severe hemorrhage, which occurred in 3% of patients in the EXAM trial. Additional side effects include venous and arterial thrombosis, hypertension, and palmo-plantar erythrodysesthesia. Cabozantinib is highly protein-bound (99.7%) and has a long half-life of approximately 55 hours. Grapefruit juice and other grapefruit products can inhibit CYP enzymes and thus should be avoided with cabozantinib. A high-fat meal increased cabozantinib Cmax by 41% and area under curve by 57%; patients are thus recommended to not eat from 2 hours before until 1 hour after cabozantinib use. Concomitant ketoconazole use (a CYP inhibitor) in healthy subjects increased single-dose cabozantinib exposure by 38%, while rifampin use (a CYP inducer) reduced single-dose cabozantinib exposure by 77%. Thus coadministration of strong CYP3A4 inhibitors (ketoconazole, itraconazole, voriconazole, clarithromycin, atazanavir, ritonavir) or CYP3A4 inducers (phenytoin, dexamethasone, carbamazepine, rifampin, phenobarbital, St John’s Wort) should be avoided. Presently, there is limited data for cabozantinib use in hepatic impairment, and it should be avoided in patients with moderate to severe impairment. No dose adjustment is required for cabozantinib in patients with mild or moderate renal insufficiency.61 Cabozantinib is under investigation in multiple other tumor types and has shown preliminary activity in metastatic breast cancer,62 melanoma,63 hepatocellular carcinoma,64 renal cell carcinoma,65 NSCLC,66 ovarian cancer,67 and urothelial carcinoma.68
Crizotinib
Crizotinib is a small molecule inhibitor of the anaplastic lymphoma kinase (ALK) with additional activity against the MET, ROS, and RON receptors at clinically relevant doses. After evaluation of a panel of more than 120 kinases in biochemical and phosphorylation assays, crizotinib was identified to be nearly 20-fold more selective for both ALK and MET than other kinases.69 Based on the results of an open-label, two-part Phase I study in 82 patients, crizotinib was approved for use in ALK-rearranged advanced NSCLC. The overall response rate was 57% (46 partial and one complete response in 82 patients) with crizotinib use in this select group of patients with ALK rearrangements.70 Crizotinib was also noted to be active in patients with NSCLC with MET amplification but no ALK rearrangements and in patients with MET-amplified esophagogastric adenocarcinoma.28,71
Anti-HGF monoclonal antibodies
Rilotumumab
Rilotumumab (AMG 102) is a fully human anti-HGF neutralizing immunoglobulin G subclass 2 (IgG2) monoclonal antibody. In preclinical tests, a combination of rilotumumab and temozolomide, an oral cytotoxic alkylating agent, displayed growth inhibitory effects in a human glioblastoma cell line U-87 MG. In xenograft models, combining rilotumumab with temozolomide or docetaxel (a microtubule inhibitor, with cytotoxic activity) induced significant tumor growth inhibition compared to either agent alone.72 In Phase I studies, rilotumumab demonstrated linear kinetics with a mean half-life of 18 days. It was well-tolerated at the maximum planned dose of 20 mg/kg.73
In the Phase I clinical trial with rilotumumab, 40 patients with refractory advanced solid malignancies were enrolled in six sequential dose-escalation cohorts and one dose expansion cohort. In the dose-escalation cohort, patients were administered doses of 0.5, 1, 3, 5, 10, or 20 mg/kg every 2 weeks until side-effects or disease progression were observed. Nine additional patients received rilotumumab at 20 mg/kg in the dose expansion cohort. Rilotumumab was well-tolerated with fatigue (13%), anorexia (8%), constipation (8%), and nausea (8%) being the most commonly observed toxicities. Sixteen of 23 evaluable patients (70%) had stable disease with PFS ranging from 7.9 to 40 weeks, with no actual responses.73 A Phase Ib study in patients with advanced solid malignancies combined rilotumumab with bevacizumab, a humanized VEGF-A monoclonal antibody (12 patients), or motesanib, a VEGF, PDGFR, and stem cell factor receptor inhibitor (2 patients). The patients were sequentially enrolled into four cohorts receiving 3, 10, or 20 mg/kg rilotumumab with 10 mg/kg of bevacizumab, or 3 mg/kg of rilotumumab intravenous every 2 weeks with 75 mg of oral daily motesanib. Enrollment into the motesanib arm was suspended early due to concern over cholecystitis caused by motesanib. Fatigue (72%), nausea (58%), constipation (42%), and peripheral edema (42%) were the most common treatment-related side effects. Eight of ten patients had reductions in the size of their tumors, with four patients demonstrating stable disease at week 24 after administration, and PFS ranged from 8–122 weeks.74
A single arm, open-label, two-stage Phase II study enrolled 61 patients with advanced or metastatic renal cell carcinoma, with 40 patients receiving rilotumumab 10 mg/kg every 2 weeks, and 21 patients receiving the 20 mg/kg dose. The median PFS was 3.7 months at 10 mg/kg of rilotumumab and 2 months at the 20 mg/kg dose; one patient with a confirmed partial response remained on maintenance therapy for 2.5 years. Peripheral edema (46%), fatigue (38%), and nausea (28%) were the most common side effects associated with rilotumumab.75 In patients with relapsed or recurrent glioblastoma, 40 patients received rilotumumab 10 mg/kg every 2 weeks while 20 patients received rilotumumab at a dose of 20 mg/kg every 2 weeks as a part of a Phase II clinical trial. There were no objective responses based on central assessment, and median OS and PFS were 6.5 months and 4.1 weeks, respectively, in the 10 mg/kg dose cohort, and 5.4 months and 4.3 weeks in the 20 mg/kg dose cohort, respectively. Prior bevacizumab treatment had no effect on the study endpoints. Fatigue (38%), headache (33%), and peripheral edema (23%) were the most common side effects.76
A double-blind Phase II randomized study was performed in patients with metastatic castrate-resistant prostate adenocarcinoma and who had progressed on prior taxane therapy. A total of 144 patients were assigned to receive rilotumumab 15 mg/kg every 2 weeks, 7.5 mg/kg every 2 weeks, or placebo in 1:1:1 manner in combination with mitoxantrone 12 mg/m2 with prednisone. Median OS was 12.2 months in the combined rilotumumab arms versus 11.1 months in the placebo arm, and median PFS was 3 months compared to 2.9 months in the placebo arm. The combined rilotumumab arms demonstrated a higher number of patients with peripheral edema (24%) compared to the placebo arm (8%).77 A Phase III trial (RILOMET-1, NCT01697072)78 of epirubicin, cisplatin, and capecitabine (ECX) combined with rilotumumab or placebo for untreated, advanced, MET-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma is currently ongoing.
Ficlatuzumab
Ficlatuzumab (AV-299) is a humanized monoclonal anti-HGF IgG1 antibody that binds to HGF, thereby inhibiting the HGF-MET interaction. In a Phase I study, 24 patients with solid malignancies were treated with single agent ficlatuzumab at 2, 5, 10, and 20 mg/kg, respectively, every 2 weeks, while 13 patients received the combination of ficlatuzumab 20 mg/kg every 2 weeks and erlotinib 150 mg daily. Grade 3 mucositis was seen in one patient on single-agent ficlatuzumab. Rash and diarrhea were common adverse events seen with combination therapy.79 The half-life of single-agent ficlatuzumab was observed to be approximately 15 days, and no MTD was determined. Ficlatuzumab therapy was associated with elevated serum HGF levels but no changes in serum soluble-MET levels.79
A Phase Ib study combined gefitinib 250 mg daily with two dose levels of ficlatuzumab (10 and 20 mg/kg every 2 weeks) in Asian patients with unresectable NSCLC. Of the 15 patients enrolled in the study, none had dose-limiting toxicities, while common adverse events included cough, rash, poor appetite, and diarrhea. At the 20 mg/kg dose, five patients had a partial response and four had stable disease at 12 weeks of treatment.80 A multicenter, randomized open-label Phase II study evaluated a combination of ficlatuzumab with gefitinib versus gefitinib alone in 188 patients of Asian ethnicity with stage IIIB or stage IV NSCLC. The overall response rate was comparable for the combination (43%) and the gefitinib alone arms (40%). Median PFS was 5.6 months in the ficlatuzumab plus gefitinib arm compared to 4.7 months in the gefitinib only arm (P=0.47). The combination of ficlatuzumab with gefitinib showed a trend for improved ORR and PFS in a subgroup of patients with EGFR-sensitizing mutations and low MET biomarker levels. Preliminary results suggested improved survival with the combination in patients with high stromal HGF (P=0.03), but no difference in the overall population. Paronychia (47%), acne (27%), peripheral edema (38%), and eczema (17%) were the most common side effects associated with the combination therapy.81
TAK701
TAK701 is a humanized anti-HGF monoclonal antibody that was found to overcome gefitinib resistance in EGFR-mutated human NSCLC cell lines both in vitro and in xenograft mouse models.82 A Phase I study with TAK701 in advanced solid malignancies revealed no specific dose-limiting toxicities, with cough, fatigue, and constipation as the common adverse events.83
Anti-MET monoclonal antibodies
Onartuzumab
Onartuzumab (MetMAb) is a humanized monovalent monoclonal antibody which binds to the extracellular SEMA domain of MET. As a result, onartuzumab blocks the binding of the HGF α-chain to the MET receptor.84 In an orthotopic mouse xenograft model with KP4 pancreatic cancer cells, onartuzumab inhibited tumor growth, reduced MET phosphorylation with a concomitant decrease in Ki-67 index, and improved survival of the mice.85 A Phase I dose-escalation study evaluated single agent onartuzumab and a combination of onartuzumab plus bevacizumab in advanced solid malignancies in 43 patients. The half-life for onartuzumab was 11 days, and no adverse pharmacokinetic interactions with bevacizumab were observed. The most common treatment-related adverse events included fatigue, peripheral edema, and hypoalbuminemia. In a Phase II study of erlotinib in combination with onartuzumab in patients with advanced NSCLC after initial therapy, 69 patients were randomized to receive onartuzumab 15 mg/kg intravenously every 3 weeks in combination with erlotinib 150 mg daily, and 68 patients received placebo intravenously every 3 weeks with erlotinib 150 mg daily. In patients with positive MET immunohistochemical staining (defined as ≥50% tumor cells with moderate or strong staining intensity by immunohistochemistry [IHC]), the addition of onartuzumab to erlotinib resulted in a significant improvement in PFS from 1.5 to 2.9 months and median overall survival from 3.8 to 12.6 months.86 A Phase III clinical trial evaluating the combination of erlotinib with onartuzumab in NSCLC (MetLUNG study) is currently ongoing.87 A Phase II randomized double-blind, placebo-controlled clinical trial comparing cytotoxic chemotherapy with oxaliplatin, 5-fluorouracil, and leucovorin (mFOLFOX-6) combined with bevacizumab and placebo to mFOLFOX-6 with bevacizumab and onartuzumab is also currently ongoing.88
Biomarkers for MET inhibitors
In the era of targeted agents, it is critically important to identify biomarkers which predict response to a specific class of agents. Although a number of predictive biomarkers to HGF-MET inhibitors are currently being evaluated, to date none have yet been validated or FDA-approved. Potential biomarkers explored in clinical trials to predict response to HGF-MET inhibitors include MET amplification (MET/Centromere enumeration probe 7 [CEP7] ratio >2 in 200 interphase nuclei), MET translocation, MET mutations, quantitative IHC for total MET or phosphorylated MET, plasma levels of HGF, and soluble MET receptor. In preclinical studies with PHA-665752, a MET inhibitor, the investigators demonstrated that MET amplification was an excellent predictor of PHA-665752 sensitivity in 41 human NSCLC cell lines.89 BAY-853474, another specific MET inhibitor, was able to reduce the tumor burden in glioblastoma, NSCLC, and gastric cancer xenograft models. The inhibition of MET phosphorylation was dose-dependent, and the soluble plasma biomarkers HGF, VEGF, and interleukin-8, as well as the MET-ectodomain, could potentially be used to monitor the response to treatment.38 However, clinical samples showed only moderately elevated levels of these biomarkers even with MET amplification.38 This study supported the use and development of IHC-based analysis of MET phosphorylation in addition to plasma biomarkers for monitoring response.
Treatments with ficlatuzumab and rilotumumab have been shown to result in an increase in plasma total HGF and soluble MET concentrations from baseline. Elevation of plasma total HGF and soluble MET concentrations were reported as pharmacodynamic biomarkers.76,80 In a Phase Ib/II clinical trial combining rilotumumab with panitumumab (a human monoclonal antibody to EGFR) for KRAS wild-type metastatic colorectal carcinomas, response rate to the combination correlated with high cytoplasmic staining for MET in tumor cells.90 Exploratory biomarker analyses performed on patients with advanced NSCLC (n=128) receiving onartuzumab in combination with erlotinib demonstrated an OS benefit in MET-positive patients (n=65), defined as ≥50% of tumor cells staining ≥2+ intensity for MET by IHC; HR =0.37, P=0.002). A subgroup analysis suggested IHC analysis of MET expression was a more sensitive predictor than MET amplification as assessed by FISH.91 In a multicenter Phase II randomized controlled trial, MET-expression-positive patients (defined as MET ≥2+ in >50% of tumor at IHC) with unresectable hepatocellular carcinoma showed a significant benefit with tivantinib in the second-line setting.92 Interestingly, another Phase II trial with tivantinib demonstrated no obvious relationship of treatment outcome with biomarkers, including MET gene amplification, phospho-MET and HGF expression in tumor, and serum HGF.93 Recently, the use of gene expression profiling has allowed for the identification of specific gene signatures which can be used to classify and predict tumor responses to targeted agents or chemotherapy. Such signatures are currently being developed and tested for colorectal carcinomas.94 Thus, future clinical trials need to better identify and refine biomarkers, which will allow for better prediction of response to MET inhibitors.
Mechanisms of resistance to HGF-MET inhibitors
Since solid malignancies are comprised of highly heterogeneous groups of cells, the use of targeted inhibitors may select a malignant clone of cells, which are inherently resistant to blockage of the HGF-MET pathway. Acquired resistance in cancer cells can develop by point mutations, which inhibit the interaction and/or binding of a molecule to its target receptor. Another mechanism of acquired resistance is activation of compensatory signaling pathways or mechanisms, which can bypass the effects of targeted agents. In a preclinical model, exposure to MET inhibitors PHA-665752 and PF-2341066 led to MET resistance through an acquired mutation in the MET activation loop (Y1230H/C).95 In addition, overexpression of TGF-α, a ligand for EGFR, resulted in activation of the EGFR pathway and subsequent resistance to MET inhibitors.95 In a MET-amplified gastric cancer cell line GTL16, prolonged exposure to PF-04217903 resulted in 7q32-34 amplification and overexpression of a novel Staphylococcal Nuclease And Tudor Domain Containing 1-v-raf murine sarcoma viral oncogene homolog B (SND1-BRAF) fusion protein. The constitutively active SND1-BRAF fusion protein resulted in upregulation and activation of the mitogen-activated protein kinase pathway, which conferred resistance to MET inhibition.96 After prolonged exposure to small molecule MET inhibitors, resistant cells developed MET and KRAS amplification. KRAS amplification was associated with loss of MET dependence and resistance to MET inhibitors.96 A patient with MET-mutated papillary renal cell carcinoma treated with PF-04217903 was noted to have an acquired tandem duplication of the mutated MET gene and developed resistance to PF-04217903-mediated MET inhibition.97 Strategies to overcome HGF-MET resistance would involve targeting of multiple compensatory pathways simultaneously either by using multitargeted agents such as cabozantinib (MET, VEGF), crizotinib (MET, ALK), or by combining targeted agents like onartuzumab or ficlatuzumab with erlotinib/gefitinib (MET/HGF and EGFR inhibitors respectively). The use of inhibitors of heat shock protein 90 (HSP90), a molecular chaperone to MET, and a wide range of other key cellular proteins may offer another approach to overcome resistance to MET inhibition.89,98
Summary
The HGF-MET axis is frequently dysregulated in cancer, especially in advanced or metastatic disease. Upregulation of the HGF-MET axis can promote resistance to small molecule inhibition of several important cellular signaling pathways, including the EGFR, RAS-RAF-MEK, and Akt-mTOR pathways. A number of potential predictive biomarkers including MET amplification (MET/CEP7 ratio >2 in 200 interphase nuclei), MET translocation, MET mutation analysis, quantitative IHC for total MET and phosphorylated MET, plasma levels of HGF, and soluble MET receptor, are currently being evaluated in clinical studies. HGF-MET inhibitors hold promise as novel molecules that can be used either as monotherapy or as part of combination therapy with EGFR, RAS-RAF-MEK, and Akt-mTOR inhibitors in the treatment of various human cancers. To conclude, in the era of personalized medicine, there is an increased need to validate biomarkers in specific tumor types to identify patients who will benefit from HGF-MET inhibitors.
Acknowledgment
This review was supported, in part, by the Phase I U01-CA099168 grant from the National Institutes of Health/National Cancer Institute.
Disclosure
L Appleman received research funding from Amgen, Bayer, and Exelixis. The authors report no other conflicts of interest in this work.
References
Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122(3):1450–1459. | |
Michalopoulos G, Houck KA, Dolan ML, Leutteke NC. Control of hepatocyte replication by two serum factors. Cancer Res. 1984;44(10):4414–4419. | |
Russell WE, McGowan JA, Bucher NL. Partial characterization of a hepatocyte growth factor from rat platelets. J Cell Physiol. 1984;119(2):183–192. | |
Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342(6248):440–443. | |
Naldini L, Weidner KM, Vigna E, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991;10(10):2867–2878. | |
Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802–804. | |
Boccaccio C, Andò M, Tamagnone L, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391(6664):285–288. | |
Schmidt C, Bladt F, Goedecke S, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373(6516):699–702. | |
Uehara Y, Minowa O, Mori C, et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373(6516):702–705. | |
Tsarfaty I, Rong S, Resau JH, Rulong S, da Silva PP, Vande Woude GF. The Met proto-oncogene mesenchymal to epithelial cell conversion. Science. 1994;263(5143):98–101. | |
Cooper CS, Tempest PR, Beckman MP, Heldin CH, Brookes P. Amplification and overexpression of the met gene in spontaneously transformed NIH3T3 mouse fibroblasts. EMBO J. 1986;5(10):2623–2628. | |
Di Renzo MF, Narsimhan RP, Olivero M, et al. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene. 1991;6(11):1997–2003. | |
Takayama H, LaRochelle WJ, Sharp R, et al. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc Natl Acad Sci U S A. 1997;94(2):701–706. | |
Ma PC, Kijima T, Maulik G, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63(19):6272–6281. | |
Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16(1):68–73. | |
Wallenius V, Hisaoka M, Helou K, et al. Overexpression of the hepatocyte growth factor (HGF) receptor (Met) and presence of a truncated and activated intracellular HGF receptor fragment in locally aggressive/malignant human musculoskeletal tumors. Am J Pathol. 2000;156(3):821–829. | |
Kijima Y, Hokita S, Yoshinaka H, et al. Amplification and overexpression of c-met gene in Epstein-Barr virus-associated gastric carcinomas. Oncology. 2002;62(1):60–65. | |
Soman NR, Wogan GN, Rhim JS. TPR-MET oncogenic rearrangement: detection by polymerase chain reaction amplification of the transcript and expression in human tumor cell lines. Proc Natl Acad Sci U S A. 1990;87(2):738–742. | |
Soman NR, Correa P, Ruiz BA, Wogan GN. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc Natl Acad Sci U S A. 1991;88(11):4892–4896. | |
Kentsis A, Reed C, Rice KL, et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med. 2012;18(7):1118–1122. | |
Lorenzato A, Olivero M, Patanè S, et al. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res. 2002;62(23):7025–7030. | |
Zeng ZS, Weiser MR, Kuntz E, et al. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 2008;265(2):258–269. | |
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | |
Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68(22):9479–9487. | |
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | |
Chen CT, Kim H, Liska D, Gao S, Christensen JG, Weiser MR. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol Cancer Ther. 2012;11(3):660–669. | |
Grugan KD, Miller CG, Yao Y, et al. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc Natl Acad Sci U S A. 2010;107(24):11026–11031. | |
Lennerz JK, Kwak EL, Ackerman A, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011; 29(36):4803–4810. | |
Hwang CI, Matoso A, Corney DC, et al. Wild-type p53 controls cell motility and invasion by dual regulation of MET expression. Proc Natl Acad Sci U S A. 2011;108(34):14240–14245. | |
Qian F, Engst S, Yamaguchi K, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69(20):8009–8016. | |
Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15(7):2207–2214. | |
Choueiri TK, Vaishampayan U, Rosenberg JE, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31(2):181–186. | |
Posadas EM, Figlin RA. Understanding the role of MET kinase in cancer therapy. J Clin Oncol. 2013;31(2):169–170. | |
Shah MA, Wainberg ZA, Catenacci DV, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PloS One. 2013;8(3):e54014. | |
Christensen JG, Schreck R, Burrows J, et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003;63(21):7345–7355. | |
Smolen GA, Sordella R, Muir B, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A. 2006;103(7):2316–2321. | |
Zopf DK, Kissel M, Mamounas M, et al. The novel highly selective and efficacious MET inhibitor BAY853474: mode of action, basic in vitro characteristics and preclinical pharmacology. EJC Supplements. 2010;8(7):53–54. | |
Klotz M, Schmid E, Steiner-Hahn K, et al. Preclinical evaluation of biomarkers for response monitoring to the MET inhibitor BAY-853474. Biomarkers. 2012;17(4):325–335. | |
Pan BS, Chan GK, Chenard M, et al. MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor. Cancer Res. 2010;70(4):1524–1533. | |
Liu L, Norman MH, Lee M, et al. Structure-based design of novel class II c-Met inhibitors: 2. SAR and kinase selectivity profiles of the pyrazolone series. J Med Chem. 2012;55(5):1868–1897. | |
Liu L, Siegmund A, Xi N, et al. Discovery of a potent, selective, and orally bioavailable c-Met inhibitor: 1-(2-hydroxy-2-methylpropyl)-N-(5-(7-methoxyquinolin-4-yloxy)pyridin-2-yl)-5-meth yl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (AMG 458). J Med Chem. 2008;51(13):3688–3691. | |
Cui JJ, McTigue M, Nambu M, et al. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1 -yl)ethanol (PF-04217903) for the treatment of cancer. J Med Chem. 2012;55(18):8091–8109. | |
Zou HY, Li Q, Lee JH, et al. Sensitivity of selected human tumor models to PF-04217903, a novel selective c-Met kinase inhibitor. Mol Cancer Ther. 2012;11(4):1036–1047. | |
Munshi N, Jeay S, Li Y, et al. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9(6):1544–1553. | |
Eathiraj S, Palma R, Volckova E, et al. Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J Biol Chem. 2011;286(23):20666–20676. | |
Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006;25(11):1679–1691. | |
Desta Z, Zhao X, Shin JG, Flockhart DA. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet. 2002;41(12):913–958. | |
Rosen LS, Senzer N, Mekhail T, et al. A phase I dose-escalation study of Tivantinib (ARQ 197) in adult patients with metastatic solid tumors. Clin Cancer Res. 2011;17(24):7754–7764. | |
Goldman JW, Laux I, Chai F, et al. Phase 1 dose-escalation trial evaluating the combination of the selective MET (mesenchymal-epithelial transition factor) inhibitor tivantinib (ARQ 197) plus erlotinib. Cancer. 2012;118(23):5903–5911. | |
Yap TA, Olmos D, Brunetto AT, et al. Phase I trial of a selective c-MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol. 2011;29(10):1271–1279. | |
Wagner AJ, Goldberg JM, Dubois SG, et al. Tivantinib (ARQ 197), a selective inhibitor of MET, in patients with microphthalmia transcription factor-associated tumors: results of a multicenter phase 2 trial. Cancer. 2012;118(23):5894–5902. | |
Sequist LV, von Pawel J, Garmey EG, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29(24):3307–3315. | |
Scagliotti GV, Novello S, Schiller JH, et al. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer. 2012;13(5):391–395. | |
Basilico C, Pennacchietti S, Vigna E, et al. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res. 2013;19(9):2381–2392. | |
Katayama R, Aoyama A, Yamori T, et al. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013;73(10):3087–3096. | |
Michieli P, Di Nicolantonio F. Targeted therapies: Tivantinib – a cytotoxic drug in MET inhibitor’s clothes? Nat Rev Clin Oncol. 2013; 10(7):372–374. | |
Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–2308. | |
Kurzrock R, Sherman SI, Ball DW, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29(19):2660–2666. | |
Schoffski P, Elisei R, Müller S, et al; EXAM Study Group. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients (pts) with documented RECIST progression at baseline. J Clin Oncol. 2012;30(Suppl; abstr 5508). | |
Smith DC, Smith MR, Sweeney C, et al. Cabozantinib in Patients with Advanced Prostate Cancer: Results of a Phase II Randomized Discontinuation Trial. J Clin Oncol. 2013;31(4):412–419. | |
Cometriq (Cabozantinib) [FDA package insert]. South San Francisco, CA: Exelixis; 2013. | |
Winer EP, Tolaney S, Nechushtan H, et al. Activity of cabozantinib (XL184) in metastatic breast cancer (MBC): Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol (Meeting Abstracts). 2012;30(Suppl 535). | |
Gordon MS, Kluger HM, Shapiro G, et al. Activity of cabozantinib (XL184) in metastatic melanoma: Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol (Meeting Abstracts). 2012;30(Suppl 8531) | |
Verslype C, Cohn AL, Kelley RK, et al. Activity of cabozantinib (XL184) in hepatocellular carcinoma: Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol (Meeting Abstracts). 2012;30(Suppl 4007). | |
Choueiri TK, Pal SK, McDermott DF, et al. Efficacy of cabozantinib (XL184) in patients (pts) with metastatic, refractory renal cell carcinoma (RCC). J Clin Oncol (Meeting Abstracts). 2012;30(Suppl 4504). | |
Hellerstedt BA, Edelman G, Vogelzang NJ, et al. Activity of cabozantinib (XL184) in metastatic NSCLC: Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol (Meeting Abstracts). 2012;30(Suppl 7514). | |
Buckanovich RJ, Berger R, Sella A et al. Activity of cabozantinib (XL184) in advanced ovarian cancer patients (pts): Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol (Meeting Abstracts). 2011;29(Suppl 5008). | |
Apolo AB, Parnes HL, Madan RA, et al. A phase II study of cabozantinib in patients (pts) with relapsed or refractory metastatic urothelial carcinoma (mUC). J Clin Oncol (Meeting Abstracts). 2014;32(Suppl 4:307) | |
Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67(9):4408–4417. | |
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | |
Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6(5):942–946. | |
Jun HT, Sun J, Rex K, et al. AMG 102, a fully human anti-hepatocyte growth factor/scatter factor neutralizing antibody, enhances the efficacy of temozolomide or docetaxel in U-87 MG cells and xenografts. Clin Cancer Res. 2007;13(22 Pt 1):6735–6742. | |
Gordon MS, Sweeney CS, Mendelson DS, et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res. 2010;16(2):699–710. | |
Rosen PJ, Sweeney CJ, Park DJ, et al. A phase Ib study of AMG 102 in combination with bevacizumab or motesanib in patients with advanced solid tumors. Clin Cancer Res. 2010;16(9):2677–2687. | |
Schöffski P, Garcia JA, Stadler WM, et al. A phase II study of the efficacy and safety of AMG 102 in patients with metastatic renal cell carcinoma. BJU Int. 2011;108(5):679–686. | |
Wen PY, Schiff D, Cloughesy TF, et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 2011;13(4):437–446. | |
Ryan CJ, Rosenthal M, Ng S, et al. Targeted MET inhibition in castration-resistant prostate cancer: a randomized phase II study and biomarker analysis with rilotumumab plus mitoxantrone and prednisone. Cancer Res. 2013;19(1):215–224. | |
Amgen. A Phase 3, Multicenter, Randomized, Double-Blind, Placebo Controlled Study of Rilotumumab (AMG102) With Epirubicin, Cisplatin, and Capecitabine (ECX) as First-line Therapy in Advanced MET-Positive Gastric or Gastroesophageal Junction Adenocarcinoma. Available from: http://clinicaltrials.gov/show/NCT01697072. NLM Identifier: NCT01697072. Accessed September 27, 2013. | |
Patnaik A, Weiss GJ, Papadopoulos K, et al. Phase I study of SCH 900105 (SC), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb), as a single agent and in combination with erlotinib (E) in patients (pts) with advanced solid tumors. J Clin Oncol. 2010; 28(Suppl; abstr 2525). | |
Tan EP, Park K, Lim WT, et al. Phase Ib study of ficlatuzumab (formerly AV-299), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb) in combination with gefitinib (G) in Asian patients (pts) with NSCLC. J Clin Oncol (Meeting Abstracts). 2011;29(15 Suppl 7571). | |
Mok TSK, Park K, Geater SL, et al. A randomized phase 2 study with exploratory biomarker analysis of ficlatuzumab a humanized hepatocyte growth factor inhibitory MAB in combination with gefitinib versus gefitinib in Asian patients with lung adenocarcinoma. Ann Oncol. 2012;23(abstr 1198P). | |
Okamoto W, Okamoto I, Tanaka K, et al. TAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol Cancer Ther. 2010;9(10):2785–2792. | |
Jones SF, Cohen RB, Bendell JC, et al. Safety, tolerability, and pharmacokinetics of TAK-701, a humanized anti-hepatocyte growth factor (HGF) monoclonal antibody, in patients with advanced nonhematologic malignancies: First-in-human phase I dose-escalation study. J Clin Oncol. 2010;28(15 Suppl abstr 3081). | |
Merchant M, Ma X, Maun HR, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A. 2013;110(32):E2987–E2996. | |
Jin H, Yang R, Zheng Z, et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. C Cancer Res. 2008;68(11):4360–4368. | |
Spigel DR, Ervin TJ, Ramlau R, et al. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J Clin Oncol. 2011;29(Suppl; abstr 7505). | |
Spigel DR, Edelman MJ, Mok T, et al. The MetLUNG study: A randomized, double-blind, phase III study of onartuzumab (MetMAb) plus erlotinib versus placebo plus erlotinib in patients with advanced, MET-positive non-small cell lung cancer (NSCLC). J Clin Oncol. 2012;30(Suppl; abstr TPS7616). | |
Bendell JC, Ervin TJ, Gallinson D, et al. Treatment Rationale and Study Design for a Randomized, Double-Blind, Placebo-Controlled Phase II Study Evaluating Onartuzumab (MetMAb) in Combination With Bevacizumab Plus mFOLFOX-6 in Patients With Previously Untreated Metastatic Colorectal Cancer. Clin Colorectal Cancer. 2013;12(3):218–222. | |
Matsubara D, Ishikawa S, Oguni S, Aburatani H, Fukayama M, Niki T. Molecular predictors of sensitivity to the MET inhibitor PHA665752 in lung carcinoma cells. J Thorac Oncol. 2010;5(9):1317–1324. | |
Eng C, Van Cutsem E, Nowara A, et al. A randomized, phase Ib/II trial of rilotumumab (AMG 102; ril) or ganitumab (AMG 479; gan) with panitumumab (pmab) versus pmab alone in patients (pts) with wild-type (WT) KRAS metastatic colorectal cancer (mCRC): Primary and biomarker analyses. J Clin Oncol. 2011;29(Suppl; abstr 3500). | |
Yu W, Pandita A, Penuel E, et al. Exploratory biomarker analyses from OAM4558g: A placebo-controlled phase II study of erlotinib with or without MetMAb in patients with advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2011;29(Suppl; abstr 7529). | |
Rimassa LPC, Borbath I, Daniele B, et al. Tivantinib (ARQ 197) versus placebo in patients (Pts) with hepatocellular carcinoma (HCC) who failed one systemic therapy: Results of a randomized controlled phase II trial (RCT). J Clin Oncol. 2012;(Suppl; abstr 4006). | |
Muro KRM, Yasui H, Nishina T, et al. A phase II study of tivantinib monotherapy in patients with previously treated advanced or recurrent gastric cancer. J Clin Oncol. 2012;30(Suppl; abstr 4082). | |
Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19(5):619–625. | |
Qi J, McTigue MA, Rogers A, et al. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 2011;71(3):1081–1091. | |
Lee NV, Lira ME, Pavlicek A, et al. A novel SND1-BRAF fusion confers resistance to c-Met inhibitor PF-04217903 in GTL16 cells though MAPK activation. PloS One. 2012;7(6):e39653. | |
Diamond JR, Salgia R, Varella-Garcia M, et al. Initial clinical sensitivity and acquired resistance to MET inhibition in MET-mutated papillary renal cell carcinoma. J Clin Oncol. 2013;31(16):e254–e258. | |
Bachleitner-Hofmann T, Sun MY, Chen CT, et al. Antitumor activity of SNX-2112, a synthetic heat shock protein-90 inhibitor, in MET-amplified tumor cells with or without resistance to selective MET Inhibition. Clin Cancer Res. 2011;17(1):122–133. | |
Feldman DR, Einhorn LH, Quinn DI, et al. A phase 2 multicenter study of tivantinib (ARQ 197) monotherapy in patients with relapsed or refractory germ cell tumors. Invest New Drugs. 2013;31(4):1016–1022. | |
Santoro A, Rimassa L, Borbath I, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14(1):55–63. | |
Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.