Back to Journals » Vascular Health and Risk Management » Volume 19
The Potential of Single Nucleotide Polymorphisms (SNPs) as Biomarkers and Their Association with the Increased Risk of Coronary Heart Disease: A Systematic Review
Authors Sitinjak BDP ![]() , Murdaya N
, Murdaya N ![]() , Rachman TA, Zakiyah N
, Rachman TA, Zakiyah N ![]() , Barliana MI
, Barliana MI ![]()
Received 16 January 2023
Accepted for publication 30 April 2023
Published 5 May 2023 Volume 2023:19 Pages 289—301
DOI https://doi.org/10.2147/VHRM.S405039
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Pietro Scicchitano
Bernap Dwi Putra Sitinjak,1,* Niky Murdaya,1,* Tiara Anisya Rachman,1,* Neily Zakiyah,1,2 Melisa Intan Barliana2,3
1Department of Pharmacology and Clinical Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Bandung, West Java, Indonesia; 2Center of Excellence for Pharmaceutical Care Innovation, Universitas Padjadjaran, Bandung, West Java, Indonesia; 3Department of Biological Pharmacy, Biotechnology Pharmacy Laboratory, Faculty of Pharmacy, Universitas Padjadjaran, Bandung, West Java, Indonesia
*These authors contributed equally to this work
Correspondence: Neily Zakiyah, Faculty of Pharmacy, Universitas Padjadjaran, Jl. Raya Bandung Sumedang KM 21, Jatinangor, Sumedang, 45363, Indonesia, Tel/Fax +62-22-7796200, Email [email protected]
Abstract: Human genetic analyses and epidemiological studies showed a potential association between several types of gene polymorphism and the development of coronary heart disease (CHD). Many studies on this pertinent topic need to be investigated further to reach an evidence-based conclusion. Therefore, in this current review, we describe several types of gene polymorphisms that are potentially linked to CHD. A systematic review using the databases EBSCO, PubMed, and ScienceDirect databases was searched until October of 2022 to find relevant studies on the topic of gene polymorphisms on risk factors for CHD, especially for the factors associated with single nucleotide polymorphisms (SNPs). The risk of bias and quality assessment was evaluated by Joanna Briggs Institute (JBI) guidelines. From keyword search results, a total of 6243 articles were identified, which were subsequently narrowed to 14 articles using prespecified inclusion criteria. The results suggested that there were 33 single nucleotide polymorphisms (SNPs) that can potentially increase the risk factors and clinical symptoms of CHD. This study also indicated that gene polymorphisms had a potential role in increasing CHD risk factors that were causally associated with atherosclerosis, increased homocysteine, immune/inflammatory response, Low-Density Lipoprotein (LDL), arterial lesions, and reduction of therapeutic effectiveness. In conclusion, the findings of this study indicate that SNPs may increase risk factors for CHD and SNPs show different effects between individuals. This demonstrates that knowledge of SNPs on CHD risk factors can be used to develop biomarkers for diagnostics and therapeutic response prediction to decide successful therapy and become the basis for defining personalized medicine in future.
Keywords: coronary heart disease, single nucleotide polymorphisms, risk factor, personalized medicine
Introduction
Coronary heart disease (CHD) is a constriction of the coronary arteries caused by the accumulation of atherosclerotic plaques in the epicardial blood vessels, which blocks blood flow and reduces oxygen supply to the heart.1,2 CHD cases continue to be the leading cause of death from cardiovascular disease worldwide. According to the World Health Organization (WHO), 17.5 million people died because of cardiovascular disease in 2012, contributing for 31% of all deaths. CHD is projected to be responsible for 7.4 million mortalities, while stroke is responsible for 6.7 million mortalities.3 According to data published by Roth et al in 2019, cases of CHD ranked first in the globe as the leading cause of cardiovascular death (49.2%), followed by ischemic stroke (17.7%). Global Burden Disease (GBD) has recorded 197 million cases of CHD-related deaths in 2019.4
Heart disease is caused by a variety of factors, one of them is an unhealthy lifestyle. High-fat diet, low physical activity, smoking, and drinking alcohol can all contribute to an increased risk in cardiovascular disease incidents.4 Furthermore, cardiovascular disease is also influenced by genetic factors.5,6 According to Kessler et al, genetics accounts for the majority of CHD risk factors (50–60%).7 Genetic factors combined with an unhealthy lifestyle increase the risk of heart disease. Genetic variables, as previously indicated, enhance the risk of having CHD by up to 91% when compared to healthy people eating the same diet.8 This demonstrates that genetic factors, as well as lifestyle, might contribute to increasing the risk of CHD.
Several investigations have been conducted in the last ten years to identify the genetic variations implicated in the development of cardiovascular disease. Numerous genetic polymorphisms have been shown to correlate with an increased risk of CHD by previous genetic association studies. Genetic screening could be an effective way of predicting increased risk of CHD. Single nucleotide polymorphisms (SNPs) are a type of genetic variation that has recently become increasingly investigated. SNPs are DNA variants caused by changes in a single nucleotide base. SNPs can induce changes such as noncoding variations, which can change amino acids and increase the expression of CHD-triggering genes.9 In 2007, The Wellcome Trust Case Control Consortium as part of the Genome-Wide Association Study (GWAS) published information on SNPs linked with an increased incidence of developing cardiovascular disease and conditions that contribute to increasing the risk of cardiovascular diseases, such as blood lipid levels, obesity, and hypertension. Nikpay et al found SNPs at 56 gene loci that significantly enhanced the risk of CHD in meta-analysis research using 1000 samples from GWAS.10 In addition, Morieri’s 2018 research, in which more than 160 gene loci connected to CHD genetic variables were obtained, supports this conclusion.11 According to these previous studies, it was suggested that in addition to an unhealthy lifestyle, genetic heterogeneity in atherosclerosis has the potential to contribute to a large number of CHD cases.
The influence of gene polymorphisms on body physiology has offered insights into biomarkers that contribute to CHD risk factors. Several studies have linked genetic and lifestyle aspects with changes in a biomarker of CHD. This causes DNA to be modified, resulting in greater obesity responses, unbalanced lipid metabolism, increased inflammatory activity, and atherosclerotic plaques.12 In general, genetic biomarkers are thought to be superior to non-genetic indicators for risk assessment. This is due to the fact that the diagnosis is more specific, and the approach is more personalized. Even if the condition is asymptomatic, genetic biomarkers can be used to determine risk factors. This allows for the implementation of lifestyle changes and therapies to begin earlier.13–15 Therefore, it provides diagnostic and therapeutic opportunities in personalized medicine.
Previous systematic reviews have been conducted and mainly focused on one variant such as rs10757274 of Cyclin-dependent Kinase inhibitor 2B Antisense RNA (CDKN2BAS) gene,16 nitric oxide,17 Interleukin-6 (IL-6),18 or one risk factor, ie, inflammatory cytokine.19 Therefore, the aim of current review was to comprehensively investigate the genetic heterogeneity of CHD and the genetic risks that are potentially associated with CHD and provide genetic polymorphism understanding in a broader aspect.
Methods
This systematic review followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) checklist.20 The details of the method were conducted as follows:
Literature Search Strategy
Literature searches were performed using several databases, ie, the EBSCO, PubMed, and ScienceDirect. The search was conducted to find relevant studies on the topic of gene polymorphisms on risk factors for CHD, especially for the factors associated with SNPs. Search strategies included the use of the following terms: “Coronary Artery Disease” OR “Coronary Heart Disease” OR “Ischemic Heart Disease” AND “Gene Polymorphism” AND “Biomarker Disorder”. The selection of keywords refers to the PECO (Population, Exposure, Comparator, and Outcome) method. The selected population consisted of individuals with CHD. The observed exposure included the gene polymorphism and the outcome focused on the biomarker disorder or physiological changes related to polymorphism.
Literature Selection
The screening process was carried out in two stages. The initial screening is based on title and abstract, followed by full-text screening. Only articles published from 2017 to 2022 were selected. Those years were chosen because there were no researchers who have reviewed articles between 2017 and October of 2022 and research on SNPs has increased from previous years. The screening continued based on the title and abstract. Lastly, the third screening was based on the full article. Inclusion criteria were used for the literatures screening process are human studies with CHD; published between 2017 and October of 2022, as this topic has been extensively assessed over the last five years; both experimental studies (eg, randomized controlled trials) and observational studies (cross-sectional, case-control, retrospective/prospective cohort, etc.); and focused on the details of genotypes that undergo polymorphism and their physiological effects. Irrelevant studies, reviews, animal studies or in vitro studies were excluded from the literature selection.
Data Extraction
From each included study, data were extracted on study characteristics, including author, year of publication, study design, SNPs variant, gene expression, clinical manifestations and conclusions, as well as the number and characteristics of the population. To streamline data extraction, a spreadsheet was used to collect the results from the articles obtained. Then, to facilitate data synthesis, identification of clinical manifestations was carried out first. These manifestations were based on the physiological changes discussed in each of the articles included. Then, the physiological changes caused by each variant of the SNPs were discussed in detail narratively.
Risk of Bias and Quality Assessment
Risk of bias and quality assessment evaluated by Joanna Briggs Institute (JBI) consists of case control,21 cross-sectional,22 Mendelian randomized controlled,23 cohort study,24 and RCT study design.21 The JBI checklist for case control, cohort, and RCT study design are divided into three risks of bias classes (score ≤49 = low risk; 50–69% = moderate risk; and 70% = low risk); for cross-sectional studies (score 0–3 = high risk; score 4–6 = some concerns; and score 7–8 = low risk of bias).22 For the Mendelian study, there is no checklist guideline to assess the risk of bias. Based on Davies et al, 2018 the Mendelian study must include 3 requirements that are genetic variation associated with CHD, no unmeasured confounder factors of the association between genetic variation and outcome, and genetic variants only affecting CHD risk factors.23
Results
Systematic Search
Based on the search results on the EBSCO, PubMed, and ScienceDirect databases using predetermined keywords, 6243 articles were obtained, with details of 65 articles on EBSCO, 1169 articles on PubMed, and 5009 articles on Science Direct. All articles were published from 2017 to 2022. A total of 2395 articles were collected to enter the title and abstract screening stage. At this stage, the exclusion criteria of nonhuman trial and review articles were applied, resulting in 181 articles. Then, these articles proceed to undergo the full text screening stage. Articles that do not use CHD population, not related to SNPs, and not relevant to the research topic because they do not discuss the increase in risk factors were eliminated. Furthermore, as many as 167 articles were excluded and the final 14 articles were obtained. After data extraction and analysis of the results, 33 variants of SNPs were obtained (Figure 1).
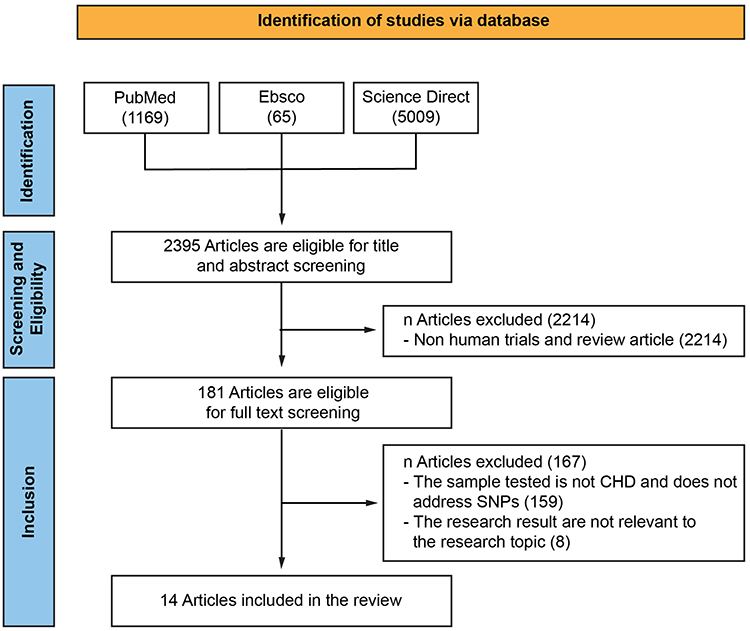 |
Figure 1 Schematic of Selection of Articles from Database with PRISMA Flowchart Method. Abbreviations: CHD, Coronary Heart Disease; RCT, Randomized Controlled Trial. Notes: PRISMA figure adapted from Page MJ, McKenzie JE, Bossuyt PM et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. Creative Commons.20 |
Main Findings
The use of the word biomarker disorder aims to narrow literature search so that the articles obtained are considered comprehensive in discussing physiological effects on CHD. There were several clinical manifestations obtained in data extraction, like atherosclerosis, therapeutic effectiveness, increased homocysteine, immune/inflammatory response, Low-Density Lipoprotein (LDL), and arterial lesions. The results of data extraction can be seen in Table 1. According to all literature, SNP variants show the impact of increasing the risk of CHD through changing gene expression pathways. Changes in gene expression trigger some protein and are usable for parameters in CHD biomarkers. Other results show alteration caused by SNPs impacts therapy effectiveness, which triggers worsening of CHD. This result provides information as a marker for personalized therapy options.
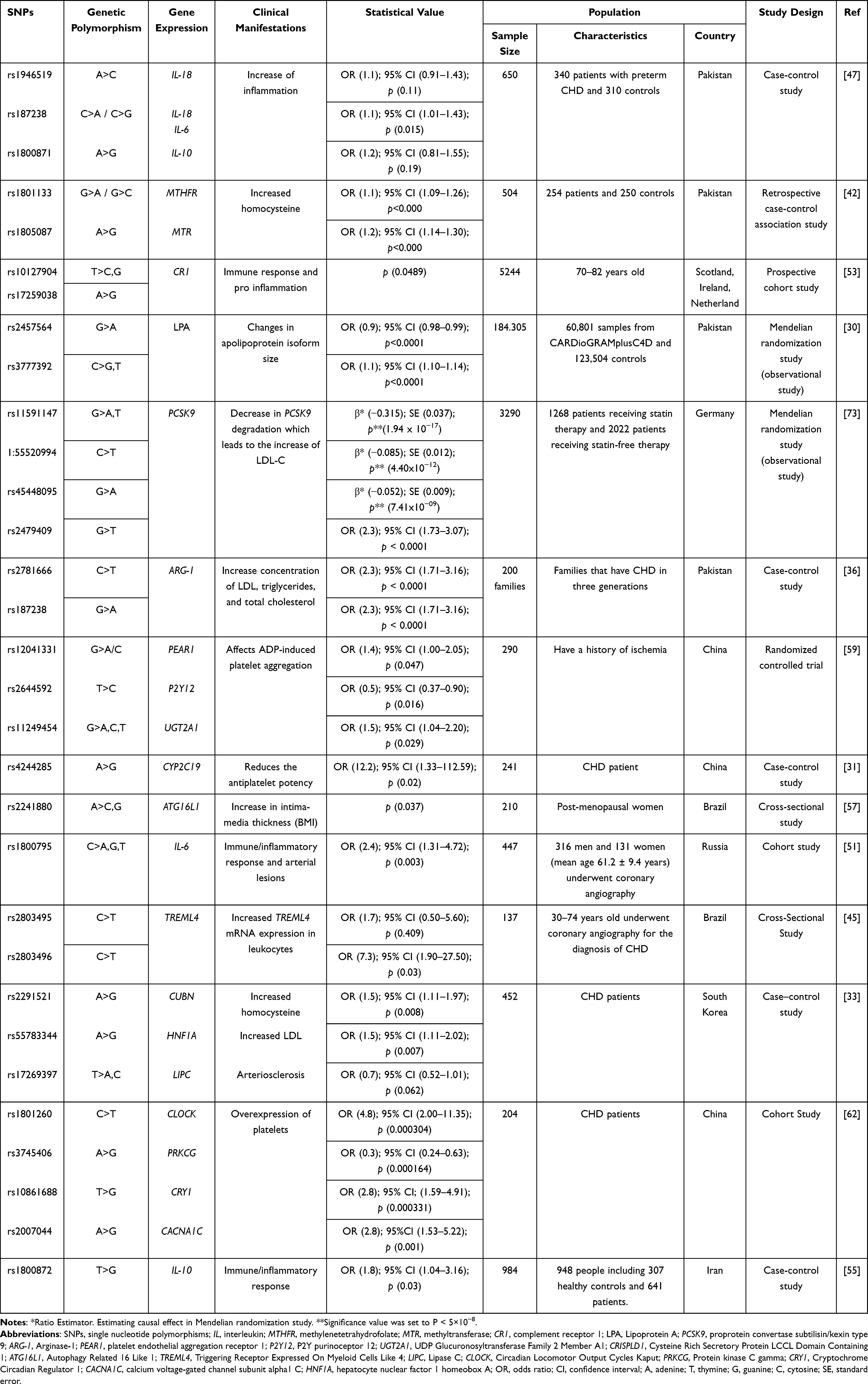 |
Table 1 Data Extraction Results |
Risk of Bias Assessment
From the risk of bias assessment using JBI critical appraisal tools, it was shown that 5 out of 12 articles (42%) had low risk of bias, 4 out of 12 (33%) articles had medium risk of bias, and 1 out of 12 (8%) articles had high risk of bias. Meanwhile, for Mendelian study, all articles included in review are valid due to genetic variation affecting the outcome of CHD risk factors and there are no measurement confounders of the association between genetic variation and outcome.
Discussion
This systematic review describes the effect of polymorphisms on CHD risk factors and their potential as biomarkers. This review defines the potential side effects triggered by genetic changes between individuals that affect increasing risk factors of CHD and the effectiveness of therapy (Table 1). Overall, the results suggest that several types of genetic polymorphisms have the potential to increase the risk factors and incidence of CHD even though the variation in severity varies between individuals with the same polymorphism. These studies related to the observed polymorphisms have opportunities to develop biomarkers and personalized medicine in the future.25
Among all the SNPs discussed in this review (Table 1), 9/33 types of SNPs trigger the inflammatory response and SNPs that trigger plaque found 7/33 SNPs. This result correlates with the epidemiological literature by Roth et al 2019 which stated that these two factors were essential in increasing CHD cases.4 The risk of CHD will be even more significant when internal genetic factors are associated with an unhealthy lifestyle. In this review, the result shows that genetics contribute to immune-inflammatory and chronic inflammatory processes.19 Some SNPs related with the atherosclerotic cardiovascular change showed the prevalence of hypercholesterolemia was significantly higher than among the general population.26
Atherosclerosis is plaque deposits that restrict or thicken blood arteries, causing blood flow to be interrupted. Some genetic alterations at the Chr9p21 gene locus can affect coronary heart risk factors.27 In this case, LDL contributed to the development and progression of atherosclerosis.28 Several causes can cause rising LDL, including elevation of lipoprotein concentration, which acts as a lipid transporter, and changes in the isoform of apolipoproteins, which are protein components in lipoproteins. Furthermore, Lp-PLA2 (lipoprotein) plays a role in the hydrolysis of phospholipids, specifically LDL, which might result in the production of pro-inflammatory components in atherosclerosis.29 Effect of SNPs rs2457564 in the apolipoprotein gene can reduce the size of apolipoprotein. On the other hand, the rs3777392 variant of SLC22A1 (Solute Carrier Family 22 Member 1) can increase lipoprotein concentrations, similar mechanism with rs2457564. According to this study, the concentration of lipoprotein and apolipoprotein influenced by SNPs may affect the increased risk of CHD and myocardial infarction.30
Park et al 2020 used Whole-Exome Sequencing database and discovered 15 SNPs outside the Chr9p21 region that were linked to CHD and then separated them into three groups: Cubilin (CUBN), Hepatocyte Nuclear Factor 1 Homeobox A (HNF1A), and Lipase C (LIPC). These proteins are involved in homocysteine, folate, vitamin B12, High-Density Lipoprotein (HDL), and LDL expression. A variant CUBN (rs2291521) caused a change in the G→A allele, leading to a higher rise in LDL shown with adjusted odds ratio (AOR) and confidence interval (CI) (AOR = 3.109; 95% CI 1.201–8.045). This is related to a rise in cubilin synthesis, which facilitated endocytosis of apolipoproteins and HDL into cells. Changes in the C→T allele at HNF1A (rs55783344) led to an increase in LDL level (AOR = 3.924; 95% CI 1.416–10,875). Mutation in the A→G allele in LIPC (rs17269397) led to a drop in HDL level (AOR = 2.551; 95% CI 1.389–4.684).28,29,31,32 Previous research using GWAS catalog samples to assess coronary heart population has shown similar results.32 Because SNPs have a contribution to these rules, these three proteins have the potential to be constructed in CHD diagnostic systems.33
Alteration in nitric oxide (NO) also impacts atherosclerosis through the altered action of Arginase-1 (ARG-1) other than increasing lipid concentration. Hepatocyte cells usually produce ARG-1 and are also expressed in several types of vascular and endothelial cells.34 In various populations, some variants of the ARG-1-expressing gene have been corresponding to cardiovascular disease.35 Shah et al 2019 reported a 2.7–4.7 U/L increase in Arginase-1 activity in CHD patients, compared to healthy controls with 1.12–1.4 U/L.36 NO level seems to be significantly lower in coronary heart patients than in healthy people. NO production would decrease when an increase in ARG-1 levels occurred.37 ARG-1 is predicted to be a new therapeutic target in cardiac patients because it has a role in the development of atherosclerosis through endothelial cell dysfunction.38
Alteration of ARG-1 allele G→T (rs2781666) resulted in an increased risk factor higher than healthy controls (OR = 2.3; 95% CI = 1.73–3.07; p 0.0001), similarly occurred changes C→T in the variation rs2781667. Furthermore, these SNPs alter the expression of CRP protein, which is one of the important indicators of inflammation in CHD. In a GWAS assessment performed by Vinayagamoorthy et al in 2014, a favorable connection between high CRP and genetic variation in ARG1 was identified. According to these results, the variant in ARG1 has the potential to provide information for the early identification and treatment of CHD in the hereditary family since these two SNPs may be inherited to offspring.39
Metabolic disorders have contributed to the risk of coronary artery disease such as the rise in homocysteine. The increase of homocysteine causes oxidative stress and endothelial damage, which leads to atherothrombotic disease.40 C→T mutation at rs1801133 of the methylenetetrahydrofolate reductase (MTHFR) gene may alter the amino acid sequence at 222 from alanine to valine. As a result of this change, the production of MTHFR enzymes was decreasing, and homocysteine levels were increasing. The risk of CHD with every 5mol/L increase in homocysteine (OR 1.42; 95% CI 1.11–1.84).41 It conforms with the results that show MTHFR variation might cause an increase in coronary heart risk factors (OR = 1.1749). Furthermore, the variant of rs1805087 from the MTHFR gene causes a higher risk of CAD and progresses to incidence of vascular disease (OR = 1.9020).42
Atherosclerosis has been characterized as inflammation caused by the ruptured plaque inside the arterial wall that promotes elevated cytokine levels, monocyte recruitment, and macrophage infiltration in arterial.43 Inflammation of the artery can stimulate more procoagulants and proinflammatory factors, causing severe CHD symptoms.44 Several receptors are expressing neutrophil and monocyte-mediated inflammatory reactions, including Triggering Receptor Expressed on Myeloid Cells Like 4 (TREML4). Because TREML4 expression corresponds to the atherosclerotic process, it can be recognized as a cardiovascular risk biomarker.45 Variants rs2803496 and rs2803495 (change in the C allele) can affect transcription factors, resulting in a 1.4-fold increase in TREML4 expression. Sen et al found similar results, specifically the influence of rs2803496 on Coronary Artery Calcification (CAC), another positive indicator for coronary heart disease. There was a 6.5-fold increased risk of CAC at the in vitro research and the variant of rs2803495 did not demonstrate a significant increase.46
In CHD, some pro-inflammatory and anti-inflammatory cytokines have been discovered with gene variations influencing some of them. Ansari et al 2017 looked for polymorphisms in IL-18, TNF-α, IL-6, and IL-10 expression genes to evaluate the distribution of SNPs in premature CHD cases in Pakistan and the impact of these gene polymorphisms on serum cytokine imbalance in the sample population.47 Increasing IL-18 correlated with inflammation in DMT2, atherosclerotic lesions, hypertension, metabolic syndrome, and CHD. The IL-18 gene (rs187238) variant at CHD population could increase IL-18 levels.48 This is due to a mutation in the sequenced genotype allele C→G, which caused an increase in the transcription factor IL-18 and elevated risk of coronary heart disease (OR 1.13; 95% CI 1.01 to 1.43; p = 0.015). Another variant IL-18, such as rs1946519 with a change 656 sequence A→C, showed an increase of IL-18 expression but it was not significant (OR 1.13; 95%Cl 0.91–1.43 p = 0.11).47 In addition, an in vitro investigation in IL-18 promoter transfected-HeLa-229 cells found that activation of the base change C→G at rs187238 boosted transcriptional activity.49
Other inflammatory mediators, such as IL-6, have a role in promoting inflammation. The IL-6 expression gene polymorphism occurred on chromosome 7p21-24 in rs1800795, rs1800796, and rs1800797. These alterations usually boost gene transcription by stimulating the IL-6 gene promoter, which increases circulating IL-6 levels. The IL-6 rs1800795 enhanced the risk of CHD (OR 1.32; 95%Cl 1.12–1.55, p = 0.021) significantly. However, rs1800796 and rs1800797 of IL-6 did not have an impactful increase in risk factors. Variant rs1800795 exhibits a genetic–environmental interaction beside increasing risk. Alcohol users and smokers with rs1800795 showed a considerably greater risk of CHD. The rs1800795 variant interacted substantially with the CC or CG genotype rs1800795 in smokers (OR = 3.22; 95%Cl = 2.45–3.94) and alcohol users (OR = 3; 95%Cl = 2.20–4.24) according to generalized multifactor dimensionality reduction (GMDR) analysis (a tool for analyzing gene-environment interactions).50 Changes in the G allele at rs1800795 have varied effects depending on gender. The expression of rs1800795 in women induces a rise in High-sensitivity C-reactive protein (hsCRP), a cardiovascular disease marker. However, rs1800795 has more expression at coronary artery disease in men. Men expressed more G allele (OR 2.45; 95% CI 1.31–4.72; P = 0.003), indicating that arterial lesions were larger in males than females.51
Elevating Complement Reactive Protein (CRP) in the bloodstream was known as a sign of coronary heart disease. The amino acid changes of Pro to Arg (C→G at 5507 positions) by variant of rs3811381 are associated with CRP levels. The same condition occurred at changes of His to Arg (A→G in the positions of 3650) at rs2274567.52 CRP is the protein with the function to reduce immune complexes and lipoproteins that contribute to atherosclerosis. Using the PROSPER technique, changes in alleles were detected with various variants on 5244 samples and suggest that rs10127904 and rs17259038 correspond to increased CRP levels, which implies atherosclerosis and causing more inflammation.53 As the previous interleukins discussed before, alteration in the promoter of IL-10 rs1800872 and rs1800871 give impact to inflammation in CHD. IL-10 is in the human genome at 1q31 and human genome at 1q32 regions.54 IL-10 substantially elevated (OR: 1.8, 95% CI: 1.04–3.16, p = 0.03) in these variants. These SNPs caused CA allele alteration at the 592 of position. In addition, gender and age have an influential impact on the expression of rs1800872 on coronary heart risk. Men have a greater risk than women (p 0.001; OR = 5.821; 95% CI = 2.831–11.970) and show higher risk after 40 years (P 0.001; OR = 5.049; 95% CI = 2414–10,561). On the other hand, changes in IL-10 at rs1800871 affected increasing the risk of inflammation in the coronary heart population, but were less significant (OR = 1.16; 95% CI = 0.81 to 1.55).55
Activation of Autophagy-related 16-like 1 (ATG16L1) has caused inflammatory events in the autophagy pathway of endothelial cells, which leads to immunological stimulation for apoptosis.56 The SNPs rs2241880 of ATG16L1 caused a change in the amino acid threonine to alanine at the 300 positions. This change caused a decrease in the activation of ATG16L1 and accumulation of cholesterol in cells. An increase in ATG16L1 rs2241880 affected HDL levels and had an association to CHD risk (OR = 2.18 95% CI = 1.00–4.78 p = 0.050). The ATG16L1 polymorphism is also associated with age and menopause.57
Genetic variations predicted have contributed to the considerable interindividual variability in platelet therapy. Instance platelet activation and aggregation play a role in cardiovascular events.58 Therefore, it is necessary to identify genetic markers that can predict the response to therapy effectiveness. In 2018, Zhang et al investigated the association between Platelet Endothelial Aggregation Receptor 1 (PEAR1), P2Y purinoceptor 12 (P2Y12), and UDP Glucuronosyltransferase Family 2 Member A1 (UGT2A1) gene polymorphisms to determine platelet reactivity as measured by thromboelastographic (TEG) in 290 patients with ischemic events. This research uses a case-control association study method between the High Platelet Reactivity (HPR) and Normal Platelet Reactivity (NPR) groups that aims to find potential predictors to guide the use of clopidogrel and aspirin in clinical practice. The result showed those who receive antiplatelet therapy exhibit high HPR and affect the effectiveness of therapy compared to patients without SNPs. The mean Inhibitor Platelet Activity (IPA) was significantly lower in patients suffering from recurrent ischemic events than patients without recurrence (p = 0.048).59
Platelet reactivity was linked to PEAR1 rs12041331, rs2644592, and rs57731889, P2Y12 rs16863356 and rs7634096, and UGT2A1 rs11249454 in the research were founded in the intron region of gene, the nonfunctional domain. The rs11264580 is a synonymous mutation found in the exon region of PEAR1. The major G allele at rs12041331 is found in the PEAR1 intron and correlated to enhanced platelet aggregation via PEAR1 protein expression upregulation.60 Herrera et al found that the CC genotype of rs2768759 in the promoter region of the PEAR1 gene is associated with greater platelet reactivity following low-dose aspirin administration in a 2008 study.61 Intron or promoter variants can function as binding sites and influence gene expression, suggesting that variations in this gene at various places may have distinct consequences on platelet reactivity. PEAR1, P2Y12, and UGT2A1 genetic variation were substantially associated with adenosine diphosphate (ADP)-induced platelet aggregation.59
According to Wang et al 2018 the CYP2C19*2 gene polymorphism increased the incidence of high on-treatment platelet reactivity (HTPR), which indicates clopidogrel resistance therapeutic in CHD.36 CYP2C19*2 rs4244285 and rs4986893 variation causing lower levels of clopidogrel metabolite compounds, resulting in clopidogrel resistance.31 CYP2C19*2 has been linked to an increased risk of HTPR when using clopidogrel. SNPs in Circadian Locomotor Output Cycles Kaput (CLOCK), Cryptochrome Circadian Regulator 1 (CRY1), Calcium Voltage-Gated Channel Subunit Alpha1 C (CACNA1C), and Protein Kinase C Gamma (PRKCG) affect platelet activity in clopidogrel-treated patients via the circadian rhythm system, according to Su et al 2021.62 The circadian rhythm system keeps track of the body cycle time and serves as the foundation for physiological and biochemical tasks. Circadian rhythm genes have a role in several heart-related disorders.63 Several investigations have discovered that the CLOCK gene is involved in age-related cardiovascular disorders (such as ischemic heart disease and cardiomyopathy) and that variations in expression impact myocardial cell regeneration.64 CLOCK mRNA expression was high (p = 0.034) in individuals with an AG genotype of rs1801260. It affects blood glucose homeostasis, which changes the reaction to clopidogrel.62
The TC genotype rs3745406 in PRKCG can reduce clopidogrel resistance-causing mRNA expression. PRKCG is a gene that plays a significant role in behavioral and emotional disorder susceptibility.65 Major depressive illness, unpleasant life experiences, anxiety, and depression, all of which are predictors of endothelium and platelet reactivity, can be altered by PRKCG rs3745406.66 As a result, changing PRKCG expression may affect clopidogrel response by affecting mood. Polymorphisms of CRY1 and CACNA1C can also alter the response to clopidogrel, although not via mRNA expression.67 In one investigation, the CRY1 variant rs10861688 correlates with depression. CACNA1C rs2007044 impacts left Inferior Frontal Gyrus (IFG) activation, leading to behavioral and emotional vulnerability, specifically schizophrenia, according to other studies.68 As a result, emotional changes often impact the efficacy of clopidogrel/antiplatelet treatment. As a result, these SNPs have the potential as markers to guide individual treatment by assessing cardiovascular risk and considering antiplatelet therapy in the future.
Finally, SNPs as biomarkers could assess illness susceptibility, disease screening, diagnostics, prognosis, patient stratification (based on patient response or side effects/adverse medication responses), drug dose, monitoring drug response, and predicting drug resistance.69 Genetic influences may affect medication metabolism, transport, effects, and side effects, resulting in differences in treatment response across individuals.70 Combining and exploiting multiple types of individual genetic information can be used to personalize therapy.71 On a bigger scale, the research found that over 3,000,000 SNPs are associated with population differences depending on location and ethnicity, known as population, differentiated SNPs (pdSNPs). Then, it found that 1443 drugs were affected by at least one pdSNP. It is a matter of consideration whether a drug is qualified to be effective in a particular population. Can it be administered to another without additional testing? Should the drug be retested in another population before being administered?72 Based on this case, SNPs can be tools for diagnostics, determining, or predicting the effectiveness of therapy.
Furthermore, our strength lies in providing comprehensive information on the potential of SNPs as biomarkers and their effects on risk factors for coronary heart disease, systematically. However, this review has some limitations. First, we included a large variety of study designs and methods due to the limited number of articles that were published on this specific topic. However, we overcome the heterogeneity of the included studies, by conducting the review in a narrative way and outlining the current evidence. Second, both reporting and publication bias may exist, because we only included published studies. Inevitably, positive findings have a higher chance of being published, rather than negative results. We also did not consider grey literature to ensure comparability among included studies.
Conclusion
The findings of this study indicate that gene polymorphisms or SNPs may increase risk factors for coronary heart disease. SNPs show different effects between individuals. The existence of SNPs causes alterations in the physiological processes of the body, which leads to increased LDL, blood vessel inflammation, atherosclerosis, homocysteine, and decrease therapeutic efficiency. This demonstrates that knowledge of SNP on CHD risk factors can be used to develop biomarkers for diagnostics and therapeutic response prediction to decide successful therapy and become the basis for defining personalized medicine in future.
Acknowledgments
Bernap Dwi Putra Sitinjak, Niky Murdaya and Tiara Anisya Rachman share joint first authorship.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114(12):1852–1866. doi:10.1161/CIRCRESAHA.114.302721
2. Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111(25):3481–3488. doi:10.1161/CIRCULATIONAHA.105.537878
3. Baihaqi WM, Setiawan NA, Ardiyanto I. Rule extraction for fuzzy expert system to diagnose Coronary artery disease.
4. Roth GA, Mensah GA, Johnson CO, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. 2020;76(25):2982–3021. doi:10.1016/j.jacc.2020.11.010
5. Sayols-Baixeras S, Lluís-Ganella C, Lucas G, Elosua R. Pathogenesis of coronary artery disease: focus on genetic risk factors and identification of genetic variants. Appl Clin Genet. 2014;7(7):15. doi:10.2147/TACG.S35301
6. Orho-Melander M. Genetics of coronary heart disease: towards causal mechanisms, novel drug targets and more personalized prevention. J Intern Med. 2015;278(5):433–446. doi:10.1111/joim.12407
7. Kessler T, Erdmann J, Schunkert H. Genetics of coronary artery disease and myocardial infarction - 2013. Curr Cardiol Rep. 2013;15(6):1–8. doi:10.1007/s11886-013-0368-0
8. Khera AV, Emdin CA, Drake I, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375(24):2349–2358. doi:10.1056/NEJMoa1605086
9. Zeng L, Talukdar HA, Koplev S, et al. Contribution of regulatory-gene networks to heritability of coronary artery disease. J Am Coll Cardiol. 2019;73(23):2946. doi:10.1016/j.jacc.2019.03.520
10. Nikpay M, Goel A, Won HH, et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121.
11. Morieri ML, Gao H, Pigeyre M, et al. Genetic tools for coronary risk assessment in type 2 diabetes: a cohort study from the ACCORD clinical trial. Diabetes Care. 2018;41(11):2404–2413. doi:10.2337/dc18-0709
12. Sumi MP, Mahajan B, Sattar RSA, et al. Elucidation of epigenetic landscape in coronary artery disease: a review on basic concept to personalized medicine. Epigenet Insights. 2021:14. doi:10.1177/2516865720988567
13. Dent THS. Predicting the risk of coronary heart disease. II: the role of novel molecular biomarkers and genetics in estimating risk, and the future of risk prediction. Atherosclerosis. 2010;213(2):352–362. doi:10.1016/j.atherosclerosis.2010.06.021
14. Qamar T, Mukherjee S. Genetic approaches for the diagnosis and treatment of rheumatoid arthritis through personalized medicine. Gene Rep. 2021;23(1):1–9.
15. Hindi NN, Alenbawi J, Nemer G. Pharmacogenomics variability of lipid-lowering therapies in familial hypercholesterolemia. J Pers Med. 2021;11(9):877. doi:10.3390/jpm11090877
16. Xu LB, Zhang YQ, Zhang NN, et al. Rs10757274 gene polymorphisms in coronary artery disease. Medicine. 2020;99(3). doi:10.1097/MD.0000000000018841
17. Li X, Lin Y, Zhang R. Associations between endothelial nitric oxide synthase gene polymorphisms and the risk of coronary artery disease: a systematic review and meta-analysis of 132 case-control studies. Eur J Prev Cardiol. 2019;26(2):160–170. doi:10.1177/2047487318780748
18. Salari N, Mansouri K, Hosseinian-Far A, et al. The effect of polymorphisms (174G> C and 572C> G) on the Interleukin-6 gene in coronary artery disease: a systematic review and meta-analysis. Genes Environ. 2021;43(1). doi:10.1186/s41021-021-00172-8
19. Tabaei S, Motallebnezhad M, Tabaee SS. Systematic review and meta-analysis of association of polymorphisms in inflammatory cytokine genes with coronary artery disease. Inflamm Res. 2020;69(10):1001–1013. doi:10.1007/s00011-020-01385-3
20. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;2021:372.
21. Aromataris E, Munn Z. JBI Manual for Evidence Synthesis; 2020; Available from: https://wiki.jbi.global/display/MANUAL/11.1+Introduction+to+Scoping+reviews.
22. Conway R, Grimshaw AA, Konig MF, et al. SARS–CoV-2 infection and COVID-19 outcomes in rheumatic diseases: a systematic literature review and meta-analysis. Arthritis Rheumatol. 2022;74(5):766–775. doi:10.1002/art.42030
23. Davies NM, Holmes MV, Smith Davey G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:1.
24. Goplen CM, Verbeek W, Kang SH, et al. Preoperative opioid use is associated with worse patient outcomes after Total joint arthroplasty: a systematic review and meta-analysis. BMC Musculoskelet Disord. 2019;20(1). doi:10.1186/s12891-019-2619-8
25. Tada H, Fujino N, Nomura A, et al. Personalized medicine for cardiovascular diseases. J Hum Genet. 2021;66(1):67–74. doi:10.1038/s10038-020-0818-7
26. Hu P, Dharmayat KI, Stevens CAT, et al. Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: a systematic review and meta-analysis. Circulation. 2020;141(22):1742–1759. doi:10.1161/CIRCULATIONAHA.119.044795
27. Dandona S, Stewart AFR, Chen L, et al. Gene dosage of the common variant 9p21 predicts severity of coronary artery disease. J Am Coll Cardiol. 2010;56(6):479–486. doi:10.1016/j.jacc.2009.10.092
28. Nordlie MA, Wold LE, Kloner RA. Genetic contributors toward increased risk for ischemic heart disease. J Mol Cell Cardiol. 2005;39:4. doi:10.1016/j.yjmcc.2005.06.006
29. Tomkin H. GLDL as a cause of atherosclerosis. Open Atheroscler Thromb J. 2012;5(1):13–21.
30. Saleheen D, Haycock PC, Zhao W, et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: a Mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017;5(7):524–533. doi:10.1016/S2213-8587(17)30088-8
31. Wang JY, Zhang YJ, Li H, et al. CRISPLD1 rs12115090 polymorphisms alters antiplatelet potency of clopidogrel in coronary artery disease patients in Chinese Han. Gene. 2018;678:226–232. doi:10.1016/j.gene.2018.08.027
32. Alliey-Rodriguez N, Grey TA, Shafee R, et al. NRXN1 is associated with enlargement of the temporal horns of the lateral ventricles in psychosis. Transl Psychiatry. 2019;9(1):1–7. doi:10.1038/s41398-019-0564-9
33. Park HS, Kim IJ, Kim EG, et al. A study of associations between CUBN, HNF1A, and LIPC gene polymorphisms and coronary artery disease. Sci Rep. 2020;10(1):1–10. doi:10.1038/s41598-019-56847-4
34. Teupser D, Burkhardt R, Wilfert W, Haffner I, Nebendahl K, Thiery J. Identification of macrophage arginase I as a new candidate gene of atherosclerosis resistance. Arterioscler Thromb Vasc Biol. 2006;26:2. doi:10.1161/01.ATV.0000195791.83380.4c
35. Meroufel D, Dumont J, Médiène-Benchekor S, et al. Characterization of arginase 1 gene polymorphisms in the Algerian population and association with blood pressure. Clin Biochem. 2009;42(10–11):1178–1182. doi:10.1016/j.clinbiochem.2009.03.004
36. Shah SFA, Khan MJ, Iqbal T, et al. Arginase-1 variants and the risk of familial coronary artery disease in subjects originating from Pakistan. Genet Test Mol Biomarkers. 2019;23(1):32–38. doi:10.1089/gtmb.2018.0227
37. Yang Z, Ming XF, Conway R, Grimshaw AA, Konig MF. Functions of arginase isoforms in macrophage inflammatory responses: impact on cardiovascular diseases and metabolic disorders. Front Immunol. 2014;5. doi:10.3389/fimmu.2014.00533
38. Pernow J, Jung C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res. 2013;98(3):334–343. doi:10.1093/cvr/cvt036
39. Vinayagamoorthy N, Hu HJ, Yim SH, et al. New variants including ARG1 polymorphisms associated with C-reactive protein levels identified by genome-wide association and pathway analysis. PLoS One. 2014;9(4):e95866. doi:10.1371/journal.pone.0095866
40. Jakubowski H. Pathophysiological consequences of homocysteine excess. J Nutr. 2006;136(6):1741S–1749S. doi:10.1093/jn/136.6.1741S
41. Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ. 2002;325(7374):1202. doi:10.1136/bmj.325.7374.1202
42. Masud R, Baqai HZ. The communal relation of MTHFR, MTR, ACE gene polymorphisms and hyperhomocysteinemia as conceivable risk of coronary artery disease. Appl Physiol Nutr Metab. 2017;42(10):1009–1014. doi:10.1139/apnm-2017-0030
43. Alie N, Eldib M, Fayad ZA, Mani V. Inflammation, atherosclerosis, and coronary artery disease: PET/CT for the evaluation of atherosclerosis and inflammation. Clin Med Insights Cardiol. 2015;8(Suppl 3):13–21. doi:10.4137/CMC.S17063
44. Auer J, Weber T, Berent R, Lassnig E, Lamm G, Eber B. Genetic polymorphisms in cytokine and adhesion molecule genes in coronary artery disease. Am J Pharmacogenomics. 2003;3(5):317–328. doi:10.2165/00129785-200303050-00003
45. Duarte VHR, Miranda CT, De OF, et al. TREML4 mRNA expression and polymorphisms in blood leukocytes are associated with atherosclerotic lesion extension in coronary artery disease. Sci Rep. 2019;9(1). doi:10.1038/s41598-019-43745-y
46. Sen SK, Boelte KC, Barb JJ, et al. Integrative DNA, RNA, and protein evidence connects TREML4 to coronary artery calcification. Am J Hum Genet. 2014;95(1):66–76. doi:10.1016/j.ajhg.2014.06.003
47. Ansari WM, Humphries SE, Naveed AK, Khan OJ, Khan DA. Influence of cytokine gene polymorphisms on proinflammatory/anti-inflammatory cytokine imbalance in premature coronary artery disease. Postgrad Med J. 2017;93(1098):209–214. doi:10.1136/postgradmedj-2016-134167
48. Mitrokhin V, Nikitin A, Brovkina O, et al. Association between IL-18/18R gene polymorphisms and coronary artery disease: influence of IL-18/18R genetic variants on cytokine expression. J Inflamm Res. 2018;11:1–9. doi:10.2147/JIR.S153370
49. Giedraitis V, He B, Huang WX, Hillert J. Cloning and mutation analysis of the human IL-18 promoter: a possible role of polymorphisms in expression regulation. J Neuroimmunol. 2001;112(1–2):146–152. doi:10.1016/S0165-5728(00)00407-0
50. Chen H, Ding S, Liu X, Wu Y, Wu X. Association of interleukin-6 genetic polymorphisms and environment factors interactions with coronary artery disease in a Chinese han population. Clin Exp Hypertens. 2018;40(6):514–517. doi:10.1080/1064196320171403618
51. Klimushina MV, Gumanova NG, Kutsenko VA, et al. Association of common polymorphisms in IL-6 and IL6ST genes with levels of inflammatory markers and coronary stenosis. Meta Gene. 2019;21:100593. doi:10.1016/j.mgene.2019.100593
52. Zorzetto M, Ferrarotti I, Trisolini R, et al. Complement receptor 1 gene polymorphisms are associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168(3):330–334. doi:10.1164/rccm.200302-221OC
53. de Vries MA, Trompet S, Mooijaart SP, et al. Complement receptor 1 gene polymorphisms are associated with cardiovascular risk. Atherosclerosis. 2017;257:16. doi:10.1016/j.atherosclerosis.2016.12.017
54. Koch W, Kastrati A, Böttiger C, Mehilli J, Von Beckerath N, Schömig A. Interleukin-10 and tumor necrosis factor gene polymorphisms and risk of coronary artery disease and myocardial infarction. Atherosclerosis. 2001;159(1):137–144. doi:10.1016/S0021-9150(01)00467-1
55. Ghalandari M, Jamialahmadi K, Nik MM, et al. Association of Interleukin-10-592 C > A gene polymorphism with coronary artery disease: a case-control study and meta-analysis. Cytokine. 2021;139:5.
56. Smolková B, Bonassi S, Buociková V, et al. Genetic determinants of quantitative traits associated with cardiovascular disease risk. Mutat Res. 2015;778:18–25. doi:10.1016/j.mrfmmm.2015.05.005
57. Orsatti CL, Sobreira ML, Sandrim VC, Nahas-Neto J, Orsatti FL, Nahas EAP. Autophagy-related 16-like 1gene polymorphism, risk factors for cardiovascular disease and associated carotid intima-media thickness in postmenopausal women. Clin Biochem. 2018;61:12–17. doi:10.1016/j.clinbiochem.2018.09.006
58. Lange RA, Hillis LD. Antiplatelet therapy for ischemic heart disease. N Engl J Med. 2004;350(3):277–280. doi:10.1056/NEJMe038191
59. Zhang S, Zhu J, Li H, et al. Study of the association of PEAR1, P2Y12, and UGT2A1 polymorphisms with platelet reactivity in response to dual antiplatelet therapy in Chinese patients. Cardiology. 2018;140(1):21–29. doi:10.1159/000488101
60. Faraday N, Yanek LR, Yang XP, et al. Identification of a specific intronic PEAR1 gene variant associated with greater platelet aggregability and protein expression. Blood. 2011;118(12):3367–3375. doi:10.1182/blood-2010-11-320788
61. Enrique Herrera-Galeano J, Becker DM, Wilson AF, et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol. 2008;28(8):1484–1490. doi:10.1161/ATVBAHA.108.168971
62. Su J, Yu Q, Yang J, et al. The association of polymorphisms in related circadian rhythm genes and clopidogrel resistance susceptibility. Basic Clin Pharmacol Toxicol. 2021;129(3):196–209. doi:10.1111/bcpt.13622
63. Schroder EA, Lefta M, Zhang X, et al. The cardiomyocyte molecular clock, regulation of Scn5a, and arrhythmia susceptibility. Am J Physiol Cell Physiol. 2013;304:10. doi:10.1152/ajpcell.00383.2012
64. Alibhai FJ, LaMarre J, Reitz CJ, et al. Disrupting the key circadian regulator CLOCK leads to age-dependent cardiovascular disease. J Mol Cell Cardiol. 2017;105:24–37. doi:10.1016/j.yjmcc.2017.01.008
65. Schlaepfer IR, Clegg HV, Corley RP, et al. The human protein kinase C gamma gene (PRKCG) as a susceptibility locus for behavioral disinhibition. Addict Biol. 2007;12(2):200–209. doi:10.1111/j.1369-1600.2007.00063.x
66. Zafar MU, Paz-Yepes M, Shimbo D, et al. Anxiety is a better predictor of platelet reactivity in coronary artery disease patients than depression. Eur Heart J. 2010;31(13):1573–1582. doi:10.1093/eurheartj/ehp602
67. Drago A, Monti B, de Ronchi D, Serretti A. CRY1 variations impacts on the depressive relapse rate in a sample of bipolar patients. Psychiatry Investig. 2015;12(1):118. doi:10.4306/pi.2015.12.1.118
68. Zhang Z, Wang Y, Zhang Q, et al. The effects of CACNA1C gene polymorphism on prefrontal cortex in both schizophrenia patients and healthy controls. Schizophr Res. 2019;204:193–200. doi:10.1016/j.schres.2018.09.007
69. Laing E, Hess P, Shen Y, Wang J, Hu S. The role and impact of SNPs in pharmacogenomics and personalized medicine. Curr Drug Metab. 2011;12(5):460–486. doi:10.2174/138920011795495268
70. Vrablik M, Dlouha D, Todorovova V, Stefler D, Hubacek JA. Genetics of cardiovascular disease: how far are we from personalized CVD risk prediction and management? Int J Mol Sci. 2021;22:4182. doi:10.3390/ijms22084182
71. Ho DSW, Schierding W, Wake M, Saffery R, O’Sullivan J. Machine learning SNP based prediction for precision medicine. Front Genet. 2019;10:267. doi:10.3389/fgene.2019.00267
72. Bachtiar M, Ooi BNS, Wang J, et al. Towards precision medicine: interrogating the human genome to identify drug pathways associated with potentially functional, population-differentiated polymorphisms. Pharmacogenomics J. 2019;19(6):516–527. doi:10.1038/s41397-019-0096-y
73. Pott J, Schlegel V, Teren A, et al. Genetic regulation of PCSK9 (proprotein convertase subtilisin/kexin type 9) plasma levels and its impact on atherosclerotic vascular disease phenotypes. Circ Genom Precis Med. 2018;11(5). doi:10.1161/CIRCGEN.117.001992
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Development and External Validation of Nomogram to Identify Risk Factors for CHD in T2DM in the Population of Northwestern China
Meng Q, Yang J, Wang F, Li C, Sang G, Liu H, Shen D, Zhang J, Jiang S, Yusufu A, Du G
Diabetes, Metabolic Syndrome and Obesity 2023, 16:1271-1282
Published Date: 4 May 2023
Association Between AIM2 and Pycard Genes Polymorphisms and Susceptibility to Periodontitis with Coronary Heart Disease
Ali Daily Z, Al-Ghurabi BH, Al-Qarakhli AMA, Hussein HM
Clinical, Cosmetic and Investigational Dentistry 2023, 15:307-320
Published Date: 22 November 2023