")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 15
The Potent Novel CDK4/6 Inhibitor TQB3616 in Hormone Receptor Positive Breast Cancer: Preclinical Characterization with in vitro and Human Tumor Xenograft Models
Authors Hu W , Wang L, Luo J, Zhang J, Li N
Received 11 August 2023
Accepted for publication 26 November 2023
Published 8 December 2023 Volume 2023:15 Pages 899—912
DOI https://doi.org/10.2147/BCTT.S434973
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert Clarke
Wenyu Hu,1,* Lei Wang,1,* JiaLing Luo,1 Jian Zhang,2 Nanlin Li1
1Department of Thyroid, Breast and Vascular Surgery, Xijing Hospital, Air Force Military Medical University, Xi’an, Shaanxi, People’s Republic of China; 2The State Key Laboratory of Cancer Biology, Department of Biochemistry and Molecular Biology, Air Force Military Medical University, Xi’an, Shaanxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Nanlin Li; Jian Zhang, Email [email protected]; [email protected]
Purpose: Inhibition of the cyclin-dependent kinase (CDK) 4/6-retinoblastoma (RB) pathway exerts a considerable inhibitory effect, preventing the spread and metastasis of breast cancer cells and promoting tumor regression. In this study, we examined the antitumor activity of TQB3616, a novel inhibitor of CDK4/6 activity, which showed a greater efficacy improvement in antitumor effects.
Methods: TQB3616 group, abemaciclib group and endocrine or HER-2 targeted combination therapy group were set up respectively. The effects of drugs on cell proliferation activity, cell cycle, apoptosis, downstream protein expression and gene expression of HR positive (T47D, MCF-7) and HER-2 positive (BT474, MDA-MB-361) breast cancer cell lines were studied. The antiproliferative effect of TQB3616 was also measured in vivo.
Results: TQB3616 showed a remarkable inhibitory effect on the proliferation of hormone receptor-positive breast cancer cells in vitro. In addition, TQB3616 combined with endocrine therapy or Human Epidermal Growth Factor Receptor 2 (HER2) targeted therapy showed significant synergistic antitumor activity in estrogen receptor (ER)-positive/HER2-negative or HER2-positive breast cancer. In contrast to abemaciclib, which targets the CDK4/6 pathway with proven efficacy, the oral agent TQB3616 not only induced G1 stalling, leading to a profound reduction in the level of RB protein phosphorylated at Ser807/811, but also showed enhanced tumor killing effects by promoting cell apoptosis. Oral administration of TQB3616 showed more potent antitumor activity than abemaciclib in an in vitro breast cancer xenograft model, causing significant tumor regression associated with sustained target inhibition in tumor tissue and manageable in vivo toxicity.
Conclusion: The results of this study indicate that TQB3616 is a novel CDK4/6 inhibitor, and its highly effective antitumor activity against breast cancer is expected to yield promising therapeutic effects in clinical studies.
Keywords: breast cancer, endocrine therapy, CDK inhibitors, apoptosis, abemaciclib
A Letter to the Editor has been published for this article.
Introduction
Globally, the incidence of female breast cancer is 11.7% according to the latest statistics from the International Agency for Research on Cancer in 2020, and approximately 2.26 million new cases are diagnosed each year. Women worldwide are at risk of developing breast cancer, which is also the most common cause of cancer-related death.1 Breast cancer is a highly heterogeneous disease. The most common subtype of breast cancer comprises estrogen receptor (ER)-positive tumors, which depend on estrogen for survival and proliferation, accounting for approximately 75% of all breast cancer cases.2 Treatments of ER-positive breast cancer include therapies designed to inhibit estrogen production and/or endocrine therapies directly targeting ER activity. The main drugs include ER competitive antagonists, aromatase inhibitors, and cyclin-dependent kinase 4/6 (CDK4/6) inhibitors, which are considered to be the most effective and precisely targeted adjuvant treatments for estrogen-sensitive breast cancer.3–5
The process that controls cell division consists of several distinct phases collectively referred to as the cell cycle.6 Mitogenic stimuli such as estrogen and growth factors initiate the cell cycle and promote its progression by inducing the expression of cyclin and cyclin-dependent kinase (CDK).7 The activities of CDK4 and CDK6 are regulated by the abundance of cyclin D1, and cyclin D1 levels are elevated, affecting cell cycle passage of check points and RB-dependent S-phase entry.8,9 Specifically, CDK4/6 inhibitors block the cyclin D1-CDK4/6-retinoblastoma (RB) protein axis, a well-documented cancer-promoting pathway, by affecting cell cycle progression.10,11 Inhibition of intracellular and mitotic hormone signaling that stimulates cancer cell proliferation to interrupt the proliferation of malignant tumor cells plays a significant role in the therapy of ER-positive breast cancer patients.11,12
An important treatment milestone was reached with the development and approval of CDK4/6 inhibitors for hormone receptor(HR)-positive and human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer. The effectiveness of CDK4/6 inhibitors is enhanced when they are combined with drugs that prevent downstream estrogen-dependent stimulation of cancer cells. The efficacy of combining CDK4/6 inhibitors with endocrine therapy such as fulvestrant or an aromatase inhibitor is impressive.13–15 Oral CDK4/6 inhibitors, including palbociclib, ribociclib, and abemaciclib, have significantly prolonged progression-free survival in patients with breast cancer when administered in combination with endocrine therapy.16 According to clinical trial results, the three aforementioned CDK4/6 inhibitors show differences in efficacy and adverse reactions.17 Several clinical studies have revealed the profound efficacy of abemaciclib combined with fulvestrant in the therapy of early-stage or metastatic HR-positive breast cancer.18,19 Despite compelling evidence showing the efficacy of abemaciclib, it is also associated with adverse effects, notably, diarrhea, during treatment that may lead to dose modification; as a result, among permanently discontinued treatments, abemaciclib is drug with the highest percentage of reports indicating permanent discontinuation.20,21 Moreover, these drugs are expensive or not available in developing countries. Therefore, we plan to optimize the molecular structure of CDK4/6 inhibitors to achieve higher efficacy and reduce the severity of adverse reactions. In this study, we evaluated an alternative to abemaciclib as part of our larger objective to optimize CDK4/6 inhibitor drugs.
The CDK4/6 inhibitor TQB3616 exhibits high selectivity and effectiveness and was developed by ChiaTai Tianqing Pharmaceutical Company (Jiangsu, China) (Figure 1). Preclinical evaluation of TQB3616 showed antiproliferative activity in most malignant tumor cell lines with high RB protein expression. Furthermore, it has been shown to be effective in vivo against breast cancer and lung cancer xenografts.22 In the current study, TQB3616 was molecularly and biologically characterized and tested in hormone receptor-positive breast tumor cells using abemaciclib as a reference. TQB3616 showed great antitumor activity in cell lines that express hormone receptor in breast cancer. A xenograft study showed that TQB3616 displayed favorable pharmacokinetic and pharmacodynamic properties without causing significant toxicity. Thus, these results inform ongoing clinical trials of TQB3616.
 |
Figure 1 (A). Molecular formula and chemical structure of TQB3616 and abemaciclib; (B). Molecular docking of TQB3616 with CDK4 protein; (C). Molecular docking of TQB3616 with CDK6 protein. |
Materials and Methods
Molecular Modeling Methods
AutoDockTools-1.5.6 (The Scripps Research Institute, La Jolla, CA) and Vina 1.1.2 (same unit as above) and Pymol-2.5 (DeLano Scientific LLC, San Francisco, CA, USA) was used as the molecular modeling software. The X-ray eutectic structure of human CDK4/6 was downloaded from the PDB database (http://www.rcsb.org/; PDB code 7sj3/5L2S). Proteins were pretreated by removal of HOH and removal of metal ions using Pymol. The ligand structures were created using ChemDraw and then conformational optimization was completed by Chem3D. The output data have been saved as pdb files. The complex pdb file was established by pymol and the docking results were checked to determine the binding ability of the analyzed compounds in the active site.
Cell Lines and Cultures
The T47D and MCF-7 human HER2-negative/HR-positive breast cancer cell lines were accessed from the Shanghai Cell Bank (Shanghai, China). The BT474 and MDA-MB-361 human HER2/HR-positive breast cancer cell lines were purchased from the Shanghai Cell Bank (Shanghai, China). DMEM supplemented with 10% FBS was used to maintain T47D cells in culture. The MCF-7 cells were maintained in MEM supplemented with 10% FBS. RPMI 1640 medium supplemented with 10% FBS was used to culture the BT474 and MDA-MB-361 cells.
Chemicals and Antibodies
Abemaciclib (LY2835219) and fulvestrant (ICI-182780) used for in vitro experiments were purchased from Selleck, and abemaciclib (Verzenios) used for in vivo experiments was purchased from Eli Lilly and Company. Both in vitro and in vivo experiments were conducted with TQB3616 provided by ChiaTai Tianqing Pharmaceutical Company (Jiangsu, China). In DMSO, 10 mM of the reagent was dissolved and stored at −80°C. Trastuzumab (Herceptin) required for the experiments was purchased from Roche (USA), and the reagent was dissolved in sterilized water containing 1.1% benzol and stored in the dark at 4°C until further use. Antibodies against Rb, pRb (Ser807/811), FOXM1, Cyclin D1 (Thr286), and β-actin were ordered from Cell Signaling Technology. pRb (Ser 780), FOXM1 (Thr600) antibodies were purchased from Signalway Antibody. The Cyclin D1 antibody was ordered from Proteintech.
Cell Proliferation Measurement
Cell Counting Kit-8 (CCK-8) assays were performed to determine the 50% inhibitory concentrations (IC50 values, Cell 50% inhibitory concentrations) of several cell lines. Cells (from 5000 to 10,000 per well) were seeded into 96-well plates. Twenty-four hours later, abemaciclib and TQB3616, or a combination of both drugs with fulvestrant or trastuzumab, were administered over a period of 72 hours. Abemaciclib and TQB3616 were added to breast cancer cells simultaneously with fulvestrant or trastuzumab. DMSO-treated cells were used as the control group. Then, ten microliters of CCK-8 solution was added in each well and incubated for 2.5 hours. A microplate reader (BioTek, Epoch 2, GA, USA) was used to automatically measure the optical density (OD) of each well at 450 nm. Based on the average OD value (the absorbance) of five wells in each group, the cell inhibition rate(IR) was calculated for each group with the following formula: IR = (1-OD value of experimental group/OD value of negative control group) ×100%.23 The cell inhibition rate was calculated using GraphPad software (GraphPad Prism 8).
Clonal Formation Experiment
Cells were seeded into six-well plate and treated with 0.1% DMSO, different concentrations of abemaciclib and TQB3616 for 72 hours after the cells had attached to the well, 48 hours after seeding. Crystal violet was used to stain the cells cultured from 2 to 3 weeks, and then, distilled water was used to wash the cells.24
Western Blot Analysis
Different concentrations of the drug were used to treat the cells. Inhibitors against proteases and phosphatases were added before adding a cell lysis buffer. The proteins in the lysed cells were separated by 10% SDS‒PAGE, and then transferred to PVDF or nitrocellulose (NC) membranes by electrophoretic transfer. Quantification of proteins was performed using ImageJ software after Western blot imaging (MINICHEMI910, Beijing, China) was completed.
Cell Cycle Analysis
Seeded into six-well plates, cells were starved in serum-free medium for 24 hours allowed adherence to the wells. The cells were treated with 0.1% DMSO, abemaciclib and TQB3616 at various concentrations and incubated for one day before being fixed with a 75% ethanol solution for 12–24 hours. A flow cytometer (BD FACS Calibur, BD Biosciences, San Jose, CA, USA) was utilized to determine the phase of the cell cycle for the cells.25
Apoptosis Analysis
The cells were seeded in six-well plates at a concentration of 1.2×104 cells per well. After 24 h for adherence, the cells were treated with 0.1% DMSO and different concentrations of abemaciclib and TQB3616 and then cultured for 72 h. The apoptosis of centrifuged cells was determined by flow cytometry using annexin V/PI assay after they had been resuspended in PBS.26,27
Transmission Electron Microscopy
After cells adhered to the 6-well plates, they were treated with 0.1% DMSO, abemaciclib and TQB3616, cultured for 24 h and then cells were fixed with methanol for more than 24 h. Ultrastructures in the cells were observed with transmission electron microscopes.28
Gene Sequencing and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis of Breast Cancer Cells
Gene sequencing was performed with T47D cells treated with abemaciclib (LY2835219), TQB3616 or DMSO. R software (version 4.1.1), including the “bioconductor” add-on package, was used to compare differentially expressed genes (DEGs) between the treatment and control groups. A linear model was created with microarray data using the “limma” R package to identify differences between the treatment and control groups of T47D cells. Significance analysis of microarray (SAM) was performed with a false discovery rate (FDR) < 0.01 and an |log2- FC| ≥ 2 chosen as the threshold values. Furthermore, we explored the key pathway by which abemaciclib (LY2835219) and TQB3616 exert effects on HER2-negative and HR-positive breast cancer. According to the key pathways enriched with differentially expressed genes, the Bioconductor-based “ggplot2” package and “pathview” package were used to visualize the results of a KEGG pathway enrichment analysis.
Real-Time Quantitative PCR
TRIzol reagent was used for RNA extraction from homogenized frozen xenograft tumor tissue. A PrimeScriptTM II 1st Strand cDNA Synthesis Kit (Vazyme Biotech Co., Ltd.) was used to synthesize cDNA with mRNA-specific primers. A Bio–Rad C1000 real-time PCR machine was used to perform quantitative real-time PCR. The 2−ΔΔCT method was used to determine the relative expression of the target gene. The mean expression of at least three replicates of tumor samples for each transcript. Supplementary Table S1 includes primer sequences.
Xenograft Tumors
After 6–8 weeks of quarantine, NOD-SCID mice aged 6–8 weeks were confined to specific-pathogen-free (SPF) environments for 5–7 days. The surgical area was cleaned, and the hair was removed. Estrogen in rods designed for sustained hormone release were implanted into the back of the mice. Two washes of PBS were performed on MCF-7 cells after two weeks in logarithmic growth phase. After detachment and centrifugation, the cells were mixed with PBS, and then, the same amount of matrix glue was added to the mix thoroughly. The concentration of the matrix glue was adjusted to be 1× 107/200 µL, and the cell and matrix mixture was placed on ice. The cells were injected at a concentration of 1×107 into the site of the mammary fat pad of the NOD-SCID mice. After the tumor volume reached approximately 300 mm3, the mice were randomly assigned to three groups and given once-daily doses of abemaciclib (50 mg/kg), TQB3616 (50 mg/kg), or distilled water (control). After 33 days of continuous administration, tumor growth inhibition was observed.
Results
Molecular Docking Results of TQB3616
We compared the chemical formula of TQB3616 (C24H27FN8C4H4O4) with that of abemaciclib (C27H32F2N8) (Figure 1A). Compared with abemaciclib, TQB3616 is indazolyl at the right end and replaced by F at the seventh position, and piperazine at the left end is directly connected to the main body, and C4H4O4 chemical group is added. In order to confirm the interaction between TQB3616and CDK4/6, the molecular docking of TQB3616 and CDK4/6 was performed using AutoDockTools 1.5.6 program. The fast gradient optimized conformation search algorithm was used for molecular docking study, and the optimal conformation was selected according to the binding energy scoring ranking. PyMOL procedures were carried out to study the optimal conformation and the interaction between CDK4/6. The results showed that TQB3616 could form a strong interaction with CDK4 and CDK6 with stable binding. TQB3616 was designed and constructed to bind selectively and efficiently to CDK4 and CDK6 proteins (Figure 1B and C).
TQB3616 Inhibits the Proliferation of Breast Cancer Cells in vitro
To identify a breast cancer treatment alternative to abemaciclib, we first compared the in vitro inhibitory effects of TQB3616 or abemaciclib on the T47D and MCF-7 HER2-negative and HR-positive breast cancer cell proliferation. We compared the in vitro inhibitory effects of TQB3616 or abemaciclib on the T47D and MCF-7 HER2-negative and HR-positive breast cancer cell proliferation. The results showed that both TQB3616 and abemaciclib inhibited tumor cell proliferation (in the T47D cells the IC50 for TQB3616 was 82.4 nM, and the IC50 for abemaciclib was 56.0 nM; in MCF-7 cells, the IC50 for TQB3616 was 115.5 nM, and the IC50 for abemaciclib was 124.2 nM) (Figure 2A and B). Next, we compared the synergistic inhibition of TQB3616 or abemaciclib with the commonly used endocrine treatment agent fulvestrant in the HER2−/HR-positive breast cancer cells, and the results were similar for both drugs (Figure 2E and F). A comparison of the inhibitory effects of TQB3616 and abemaciclib was also performed with HER2-positive /HR-positive BT474 and MDA-MB-361 breast cancer cells. TQB3616 showed better inhibitory activity in the HER2-positive /HR-positive breast cancer cells than abemaciclib (in the BT474 cells, the IC50 for TQB3616 was 136.4 nM, and the IC50 for abemaciclib was 1190.0 nM; in the MDA-MB-361 cells, the IC50 for TQB3616 was 870.4 nM, and the IC50 for abemaciclib was 1005.0 nM) (Figure 2C and D). We combined TQB3616 or abemaciclib with commonly used HER2-negative-targeted therapies and compared the potential synergistic inhibitory effect on HER2-negative and HR-positive breast cancer cells. The inhibitory effect on these cells in both treatment groups was similar (Figure 2G and H). It is worth noting that TQB3616 can maximally inhibit the proliferation of breast cancer cells compared with all other drugs.
 |
Figure 2 Effects of TQB3616, abemaciclib, fulvestrant, trastuzumab and their combination on the proliferation of HR-positive breast cancer cell lines. (A–D) The proliferation rates of T47D, MCF-7, BT474, and MDA-MB-361 cell lines were measured by CCK-8 assay after treatment for 72 hours with various concentrations of TQB3616, abemaciclib, and fulvestrant. (E and F) T47D and MCF-7 cells were treated with different concentrations of TQB3616 or abemaciclib in combination with fulvestrant for 72 hours. (G and H) TQB3616 or abemaciclib was combined with trastuzumab to inhibit BT474 and MDA-MB-361 cells for 72 hours. |
TQB3616 Reduces the Colony Formation Rate of HER2-Negative/HR-Positive Breast Cancer Cells in vitro
After 72 h of treatment with TQB3616 or abemaciclib, HR-positive breast cancer cells were significantly less likely to form colonies than the control group cells, and a greater reduction in the colony formation rate was observed in the group treated with TQB3616. A similar pattern was observed with the T47D and BT474 cell lines. When the treatment concentration of TQB3616 was 1.5 µM, the colony formation of T47D and BT474 cells was completely abrogated (Figure 3A and B).
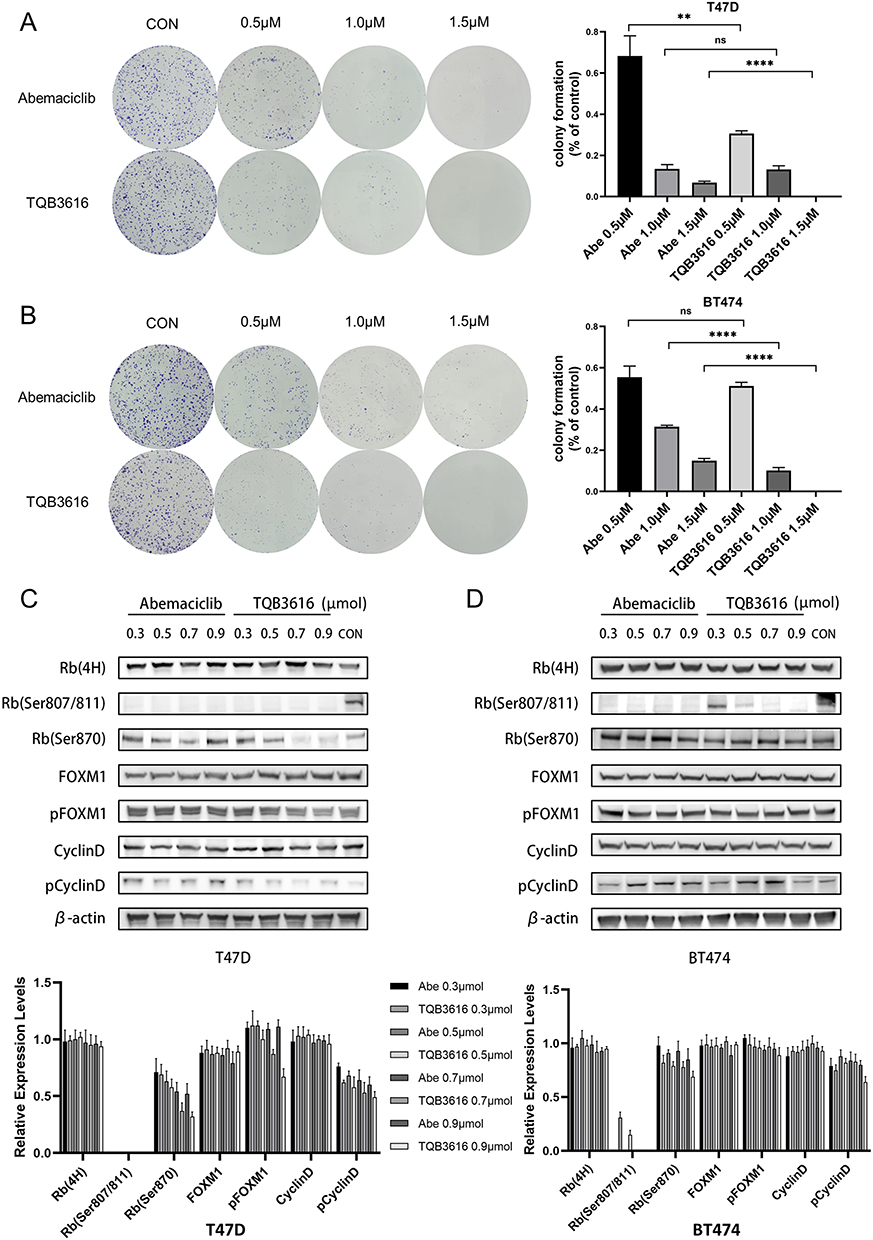 |
Figure 3 Effect of abemaciclib or TQB3616 on colony-formation- and cell cycle-associated proteins in breast cancer cell lines. (A and B) T47D cells and BT474 cells were treated with 0.1% DMSO, 0.5 µM abemaciclib, 1.0 µM abemaciclib, 1.5 µM abemaciclib, 0.5 µM TQB3616, 1.0 µM TQB3616, or 1.5 µM abemaciclib, and colony formation was assessed 2 to 3 weeks later as described in the Materials and methods section. Crystal violet staining of colonies is shown in the images. The means and standard deviations were determined on the basis of three independent experiments. (C and D) The T47D and BT474 cell lines were treated with abemaciclib or TQB3616 to inhibit cell cycle-related protein phosphorylation. ns, no statistical difference. **p<0.01, ****P<0.0001. |
Inhibitory Effect of TQB3616 on the Levels of Cell Cycle-Related Proteins
A Western blot was carried out to assess the protein levels in cells that were treated with TQB3616 or abemaciclib (Figure 3C and D). We found that both TQB3616 and abemaciclib reduced the levels of phosphorylated Rb. In T47D cells, the inhibitory effect of TQB3616 on the phosphorylation of the Rb protein was similar to that of abemaciclib. However, in BT474 cells, abemaciclib inhibited Rb protein phosphorylation to a greater extent than TQB3616. The abnormal activation of phosphorylated Cyclin D protein was inhibited by TQB3616 at high concentrations.
The Malignancy of HR-Positive Breast Cancer Cell Lines is Further Decreased by the TQB3616 Effect in Promoting G1/S Phase Stalling and Apoptosis
TQB3616 and abemaciclib inhibit cell proliferation, promote apoptosis, or exert a combined inhibitory effect on cell growth. The effects of TQB3616 and abemaciclib treatment on cell cycle progression and apoptosis were evaluated based on data obtained by flow cytometry.
The results of a cell cycle assay showed that in T47D and BT474 breast cancer cell lines, both TQB3616 and abemaciclib induced G1/S phase arrest to a large extent and their efficacy increased with increasing dose. No significant differences were found between their effects (Figure 4A, B and E). Apoptosis assays confirmed the proapoptotic effect of TQB3616, as shown by RNA sequencing (RNA-seq). TQB3616 was more effective than abemaciclib in promoting apoptosis in T47D and BT474 breast cancer cells. A high concentration (10 µM) of TQB3616 induced the most HR-positive breast cancer cells to undergo early or late apoptosis (Figure 4C, D and F).
 |
Figure 4 Abemaciclib and TQB3616 effects on the apoptosis rate and cell cycle progression. (A and B) After 24 hours of incubation with PBS (CON), abemaciclib (0.5 μM, 1.0 μM, or 1.5 μM), or TQB3616 (0.5 μM, 1.0 μM, or 1.5 μM) treatment, PI staining and flow cytometry were used to determine the proportion of T47D and BT474 cells in each phase of the cell cycle. The cell cycle analysis was performed with ModFit. (C and D) By double staining cells with annexin V-FITC/PI and analyzing the flow cytometry results, apoptotic T47D and BT474 cells were identified. TQB3616 (0.5 μM, 1.0 μM, or 1.5 μM) was incubated with cells for 24 hours. (E) Histograms show representative results. TQB3616 and abemaciclib induced clear G1/S phase arrest, in contrast to that observed for the control group. (F) Histograms show representative results. Apoptosis was and cellular stress was induced in HR-positive breast cancer cell lines by TQB3616 or abemaciclib, in contrast to the control cells. The means and standard deviations were calculated on the basis of three independent experiments. |
Effect of TQB3616 and Abemaciclib Treatment on Cellular Ultrastructures
Cells treated with TQB3616 or abemaciclib at high concentrations were fixed and analyzed 24 hours after treatment (Figure 5). The results showed that after administration of 5 μM abemaciclib, vacuoles of varying sizes appeared within the cell matrix, and these vacuoles contained undigested organelle remnants and other cellular debris, as observed by transmission electron microscopy. The formation of these vacuoles may be related to lysosomes. The necrosis of individual cells was seen in the field of view. With an increasing concentration of abemaciclib, the number of vacuoles within the cell matrix was increased, and they were larger and more pronounced.29 Notably, administration of 5 μM TQB3616 increased the number, size and density of lysosomes in the cell matrix. A few endoplasmic reticula and Golgi vesicles were enlarged. Some of these cells underwent necroptosis, as indicated by heterochromatin accumulation on one side of a cell with chromosomes near the plasma membrane, and apoptotic bodies were observed in the visual field. With an increasing concentration of TQB3616, necroptosis was observed in a large number of cells within the field of view, and cells appeared in early, middle and late stages of necroptosis. The necroptotic cells in the early stage were more obvious, and the number increased. Numerous ER and Golgi vesicles were enlarged.
 |
Figure 5 Morphological characteristics of abemaciclib-treated and TQB3616-treated T47D cells as observed by electron microscopy. |
The Mechanisms Underlying the Effect of TQB3616 on the Apoptosis Rate of HR-Positive Breast Cancer Cells
To compare the molecular mechanisms of TQB3616 and abemaciclib in HR-positive breast cancer cells, T47D cells were treated with TQB3616 (1 µM), abemaciclib (1 µM), or 0.1% DMSO for 72 h, with 3 replicates established for each group. Then, RNA sequencing was performed on the T47D cells from three different treatment groups.
Cluster maps showing the sequenced samples according to gene expression levels revealed some similarity in gene expression between the breast cancer cells treated with TQB3616 and abemaciclib (Figure 6A). After removing batch differences and normalizing the data, 3062 DEGs were obtained; 1425 of these DEGs showed upregulated expression with 1637 of these DEGs showing downregulated expression according to the critical thresholds (a p value < 0.01 and an |logFC| ≥ 2) (Figure 6B). Gene expression in the cells treated with TQB3616 and abemaciclib compared to that in the control cells showed that TQB3616 and Abemaciclib together downregulated the expression of 6413 genes (Figure 6C). KEGG pathway and GO enrichment analysis were performed to identify DEG functions. DEGs in the TQB3616 and abemaciclib treatment groups inhibited the occurrence and progression of breast cancer, as indicated by the pathways and terms that they enriched (Figure 6D and E). Moreover, visualization of the RNA-seq results shows the inhibited expression of key molecules between different treatment groups (Figure 6F). Specifically, the KEGG analysis revealed that, compared with abemaciclib, TQB3616 downregulated the expression of genes involved apoptosis signaling pathways. The KEGG map shows that TQB3616 downregulated the expression of genes in FOXO, mTOR, and P53 signaling pathways and cell cycle signaling pathway (Figure 6G).
 |
Figure 6 Inhibition of breast cancer cell growth by TQB3616 may have been enhanced by increased apoptosis. (A) The colors in the cluster map showing the sequenced samples indicate the gene expression levels as indicated by the correlation coefficients. (B) A volcano plot showing gene expression with red lines representing upregulated genes and blue lines representing downregulated genes. (C) Intersection of common downregulated genes in the abemaciclib or TQB3616 groups compared with the blank control group. (D) WikiPathways enrichment analysis of top 20 Down in abemaciclib group compared with blank control group (Pathway entries correspond to more than 2 differentially expressed genes). E WikiPathways enrichment analysis of top 20 Down in TQB3616 group compared with blank control group (Pathway entries correspond to more than 2 differentially expressed genes). (D–E) WikiPathways enrichment analysis top 20 (Pathway entries corresponding to more than 2 differentially expressed genes). (F) Subset of genes commonly altered by CDK4/6-targeted therapy in MCF-7 breast cancer cell line xenograft studies. Values represent the mean percentage of gene expression inhibition in the abemaciclib and TQB3616 groups compared to the vehicle control group. (G) TQB3616-induced DEG enrichment in KEGG pathways in T47D cells compared to control cells. |
HER2-Negative/HR-Positive Breast Cancer Xenograft Tumor Growth is Inhibited by TQB3616
A xenograft mouse model was used to compare the inhibitory effects of TQB3616 and abemaciclib on HER2-negative and HR-positive breast cancer in vivo. The MCF-7 cell line was used to establish the xenograft tumor model in mice, and the model mice were divided into three groups at random: a vehicle group, a TQB3616 group (50 mg/kg/day) and an abemaciclib group (50 mg/kg/day). We found that TQB3616 inhibited MCF-7 tumor growth better than abemaciclib (Figure 7A–C). However, in the evaluation of the growth and weight of the mice, the body weight of the mice in the TQB3616 group was generally lower than that of the mice in the abemaciclib group, indicating that although the tumor killing effect of TQB3616 was greater, the negative effect of TQB3616 on the body may also have been greater (Figure 7D). The lower mouse weights may indicate a negative effect on the tumor-killing efficacy of TQB3616 to a certain extent. Compared to abemaciclib, TQB3616 exhibited more potent anticancer activity against HER2-negative and HR-positive breast cancer xenograft tumors. In xenografts, TQB3616 was more effective than abemaciclib in inhibiting some genes that regulate cell cycle or apoptosis, such as CCNB1, E2F1, E2F2, and CDKN1A etc. (Figure 7E). The inhibitory effect of TQB3616 on RB (Ser807/811) protein in the cyclin D1-CDK4/6-retinoblastoma (RB) protein pathway was consistent with the results in vitro (Figure 7F).
 |
Figure 7 HR-positive breast cancer xenograft models were treated with abemaciclib and TQB3616, and their respective anticancer effects in vivo were analyzed. Over 33 days, immunodeficient mice were administered PBS (vehicle), abemaciclib (50 mg/kg/day), or TQB3616 (50 mg/kg/day). (A) Photographs showing resected tumors. (B and C) Tumor growth curve and tumor weights. (D) A change in weight was observed in mice within three days of the first treatment. (E) The percentage of inhibited mRNA expression of 12 cell cycle-related or apoptosis-regulating genes in tumors collected from each treatment group and compared to that in the empty vector group. (F) Protein expression levels in xenograft tumors from each treatment group. **p<0.01, ***p<0.001, ****P<0.0001. |
Discussion
Cancerous tumors have been closely associated with the dysregulation of the cyclin-CDK-Rb pathway in the cell cycle mechanism. The Rb pathway is dysregulated in more than four of five human cancers and plays a key role in the dysregulation of cell proliferation.30,31 The active cyclin D/CDK4 complex targets retinoblastoma proteins to phosphorylate them, thereby releasing the transcription factor E2F, which activates G1/ S phase gene expression.32 On the other hand, the Ras/Raf/MEK/MAPK pathway can phosphorylate cyclin D1 at Thr286, enhancing its ubiquitination and proteasomal degradation to promote tumor formation.33 In the field of breast cancer, select CDK4/6 inhibitors targeting the cyclin-CDK-Rb pathway have demonstrated great promise in preclinical studies and in clinical trials. Molecular targeting of this pathway has become an effective therapy for malignant tumors and has been widely recommended for the treatment of hormone receptor-positive breast cancers.16,34,35 However, although clinically effective, there is still a certain chance of severe neutropenia, tumor recurrence and drug resistance during drug use. Current CDK4/6 inhibitors can also improve cell-killing efficacy and toxicity via adjustments in their molecular structure.36,37 Therefore, the development of new CDK4/6 inhibitors with improved pharmacological characteristics, enhanced efficacy, and ability to delay drug resistance is of great clinical significance for patients. Inhibitors of CDK4/6 can be predicted by the degree of RB phosphorylation inhibition.38,39 Molecular docking results showed the excellent characteristics of TQB3616. Here, we evaluated the antitumor activity of TQB3616, a novel highly potent and selective small-molecule CDK4/6 inhibitor, in hormone receptor-positive models of different subtypes of breast cancer. When administered from low concentrations to high concentrations, TQB3616 significantly reduced the expression of the phosphorylated RB protein in hormone receptor-positive cell lines, and these effects were associated with reduced cell cycle progression and subsequent effects on cell senescence and cell proliferation inhibition. Notably, at the highest concentration, TQB3616 completely inhibited breast cancer cell proliferation in all experimental groups, reflecting its excellent killing effect on breast cancer cells. Subsequently, we combined TQB3616 or abemaciclib with fulvestrant (an ER antagonist) in ER-positive/HER2-negative cell lines and with trastuzumab (an HER2-negative-targeting agent) in ER-positive/HER2-positive cell lines, and the results showed that TQB3616 was superior to abemaciclib in the combination therapy. In a colony formation assay, our data showed that both of the analyzed CDK4/6 inhibitors reduced the colony formation rate of HR-positive breast cancer cells in vitro, and in these experiments, TQB3616 was significantly more effective than abemaciclib.
A KEGG enrichment analysis of the two drug groups showed that TQB3616 performed well in multiple CDK4/6 common inhibitory signaling pathways, such as cell cycle, apoptosis, p53 signaling pathway and cell senescence, and showed a good inhibitory effect on CDK4/6 downstream signaling molecules, such as E2F and FOXM1. Breast cancer cells in which FOXM1 expression is loss are more sensitive to endocrine therapy, as FOXM1 is not expressed in most normal tissues.40,41 Apoptosis and senescence pathways were positively influenced by TQB3616 in the analyzed cells. As shown with electron microscopy, TQB3616 induced a wide range of apoptosis features in cancer cells, and its effect was greater than that of abemaciclib. Specifically, many apoptotic bodies appeared in the cancer cells, indicating that they were gradually fragmenting and disintegrating. A proapoptotic effect of TQB3616 was also observed in an apoptosis assay performed by flow cytometry. This compound inhibited the progression of cell cycle in the G1/S phase, as with other CDK4/6 inhibitors, along with increasing apoptosis rates in tumor cells.
Our in vitro results showed that TQB3616 exhibited superior anticancer activity compared to that of abemaciclib, a well-recognized CDK4/6 inhibitor. In addition, treatment of an identical number of nude mice with TQB3616 and abemaciclib showed that TQB3616 was superior to Abemaciclib in reducing tumors. Although the body weight of the mice in the TQB3616 group was generally lower than that of the mice in the abemaciclib group, the difference was not significant. In summary, our findings indicate that TQB3616 is a highly potent and selective small-molecule CDK4/6 inhibitor, showed more potent antitumor activity than abemaciclib with manageable toxicity. The results of this study suggests the possibility that TQB3616 can be translated into the clinic and provide a solid scientific basis for clinical trials of TQB3616. Clinical trials testing the efficacy and adverse effects of this potent CDK4/6 inhibitor in humans are ongoing (NCT03850873, NCT05365178, NCT05375461).
Conclusion
As a result of this study, TQB3616 has been identified as a novel CDK4/6 inhibitor, and its highly effective antitumor activity against breast cancer is expected to yield promising therapeutic effects in clinical studies.
The Ethics Statement
All animal procedures were approved by Welfare and Ethics Committee of the Experimental. Animal center of the Fourth Military Medical University. The procedures conformed to the guidelines of NC3Rs ARRIVE. This study followed the Guidelines for Ethical Review of Laboratory Animal Welfare in China.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81472598). The authors acknowledged the assistance of ChiaTai Tianqing Pharmaceutical Company (Jiangsu, China) for offering the chemical compounds.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. DeSantis CE, Ma J, Gaudet MM, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438–451. doi:10.3322/caac.21583
3. Osborne CK, Wood AJJ. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339(22):1609–1618. doi:10.1056/NEJM199811263392207
4. Boér K. Fulvestrant in advanced breast cancer: evidence to date and place in therapy. Ther Adv Med Oncol. 2017;9(7):465–479. doi:10.1177/1758834017711097
5. Piezzo M, Cocco S, Caputo R, et al. Targeting Cell Cycle in Breast Cancer: CDK4/6 Inhibitors. Int J Mol Sci. 2020;22(1):21. doi:10.3390/ijms22010021
6. Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1(3):222–231. doi:10.1038/35106065
7. de Dueñas EM, Gavila-Gregori J, Olmos-Antón S, et al. Preclinical and clinical development of palbociclib and future perspectives. Clin Transl Oncol. 2018;20(9):1136–1144. doi:10.1007/s12094-018-1850-3
8. Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15(5):2672–2681. doi:10.1128/MCB.15.5.2672
9. Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672–1677. doi:10.1126/science.274.5293.1672
10. Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13(1):261–291. doi:10.1146/annurev.cellbio.13.1.261
11. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18(2):753–761. doi:10.1128/MCB.18.2.753
12. Scott SC, Lee SS, Abraham J. Mechanisms of therapeutic CDK4/6 inhibition in breast cancer. Semin Oncol. 2017;44(6):385–394. doi:10.1053/j.seminoncol.2018.01.006
13. Piezzo M, Chiodini P, Riemma M, et al. Progression-free survival and overall survival of CDK 4/6 inhibitors plus endocrine therapy in metastatic breast cancer: a systematic review and meta-analysis. Int J Mol Sci. 2020;21(22):1.
14. Finn RS, Martin M, Rugo HS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925–1936. doi:10.1056/NEJMoa1607303
15. Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209–219. doi:10.1056/NEJMoa1505270
16. Spring LM, Wander SA, Andre F, Moy B, Turner NC, Bardia A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet. 2020;395(10226):817–827. doi:10.1016/S0140-6736(20)30165-3
17. Braal CL, Jongbloed EM, Wilting SM, Mathijssen RHJ, Koolen SLW, Jager A. Inhibiting CDK4/6 in breast cancer with palbociclib, ribociclib, and abemaciclib: similarities and differences. Drugs. 2021;81(3):317–331. doi:10.1007/s40265-020-01461-2
18. Sledge GW, Toi M, Neven P, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875–2884. doi:10.1200/JCO.2017.73.7585
19. Johnston SRD, Harbeck N, Hegg R, et al. Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2-, node-positive, high-risk, early breast cancer (monarchE). J Clin Oncol. 2020;38(34):3987–3998. doi:10.1200/JCO.20.02514
20. Goetz MP, Toi M, Campone M, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35(32):3638–3646. doi:10.1200/JCO.2017.75.6155
21. Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–1748. doi:10.1056/NEJMoa1609709
22. Xu ZB, Hu LH, Liu YC, et al. Preclinical evaluation of TQB3616, a highly potent and selective small-molecule CDK4/6 inhibitor. Cancer Res. 2018;78:2.
23. van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol. 2011;731:237–245.
24. Aapro MS, Eliason JF, Krauer F, Alberto P. Colony formation in vitro as a prognostic indicator for primary breast cancer. J Clin Oncol. 1987;5(6):890–896. doi:10.1200/JCO.1987.5.6.890
25. Nunez R. DNA measurement and cell cycle analysis by flow cytometry. Curr Issues Mol Biol. 2001;3(3):67–70.
26. Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1(3):1458–1461. doi:10.1038/nprot.2006.238
27. Crowley LC, Scott AP, Marfell BJ, Boughaba JA, Chojnowski G, Waterhouse NJ. Measuring cell death by propidium iodide uptake and flow cytometry. Cold Spring Harb Protoc. 2016;2016:1.
28. Novikoff AB. Electron microscopy: cytology of cell fractions. Science. 1956;124(3229):969–972. doi:10.1126/science.124.3229.969
29. Hino H, Iriyama N, Kokuba H, et al. Abemaciclib induces atypical cell death in cancer cells characterized by formation of cytoplasmic vacuoles derived from lysosomes. Cancer Sci. 2020;111(6):2132–2145. doi:10.1111/cas.14419
30. Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin Cancer Res. 2014;20(13):3379–3383. doi:10.1158/1078-0432.CCR-13-1551
31. Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602(1):73–87. doi:10.1016/s0304-419x(02)00037-9
32. Lukas J, Bartkova J, Bartek J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G 1 checkpoint. Mol Cell Biol. 1996;16(12):6917–6925. doi:10.1128/MCB.16.12.6917
33. Shao J, Sheng H, DuBois RN, Beauchamp RD. Oncogenic Ras-mediated cell growth arrest and apoptosis are associated with increased ubiquitin-dependent cyclin D1 degradation. J Biol Chem. 2000;275(30):22916–22924. doi:10.1074/jbc.M002235200
34. Gelbert LM, Cai S, Lin X, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014;32(5):825–837. doi:10.1007/s10637-014-0120-7
35. Long F, He Y, Fu H, et al. Preclinical characterization of SHR6390, a novel CDK 4/6 inhibitor, in vitro and in human tumor xenograft models. Cancer Sci. 2019;110(4):1420–1430. doi:10.1111/cas.13957
36. Schwartz GK, LoRusso PM, Dickson MA, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011;104(12):1862–1868. doi:10.1038/bjc.2011.177
37. Flaherty KT, Lorusso PM, Demichele A, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;8(2):568–576. doi:10.1158/1078-0432.CCR-11-0509
38. Patnaik A, Rosen LS, Tolaney SM, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6(7):740–753. doi:10.1158/2159-8290.CD-16-0095
39. Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77.
40. Wonsey DR, Follettie MT. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005;65(12):5181–5189. doi:10.1158/0008-5472.CAN-04-4059
41. Bergamaschi A, Madak-Erdogan Z, Kim YJ, Choi YL, Lu H, Katzenellenbogen BS. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. 2014;16(5):436. doi:10.1186/s13058-014-0436-4
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.