Back to Journals » OncoTargets and Therapy » Volume 10
The non-small cell lung cancer EGFR extracellular domain mutation, M277E, is oncogenic and drug-sensitive
Authors Yu S, Zhang Y, Pan YJ, Cheng C, Sun YH, Chen HQ
Received 9 January 2017
Accepted for publication 10 June 2017
Published 12 September 2017 Volume 2017:10 Pages 4507—4515
DOI https://doi.org/10.2147/OTT.S131999
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Samir Farghaly
Su Yu,1,2 Yang Zhang,1 Yunjian Pan,1 Chao Cheng,1,3 Yihua Sun,1,3 Haiquan Chen1–4
1Department of Thoracic Surgery, Fudan University Shanghai Cancer Center, Shanghai, China; 2Cancer Research Center, Fudan University Shanghai Cancer Center, Shanghai, China; 3Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China; 4Institutes of Biomedical Sciences, Fudan University, Shanghai, China
Purpose: To identify novel oncogenic mutations in non-small cell lung cancer patient specimens that lack mutations in known targetable genes (“pan-negative” patients).
Methods: Comprehensive mutational analyses were performed on 1,356 lung adenocarcinoma specimens. In this cohort of patients, common lung cancer oncogenic driver mutations were detected in the epidermal growth factor receptor (EGFR) kinase domain, the human epidermal growth factor receptor 2 kinase domain, as well as the KRAS, BRAF, ALK, ROS1 and RET genes. A sub-cohort of pan-negative patient specimens was assayed for mutations in the EGFR extracellular domain (ECD). Additionally, EGFR mutant NIH-3T3 stable cell lines were constructed and assessed for protein content, anchorage-independent growth, and tumor formation in xenograft models to identify oncogenic mutations. BaF3 lymphocytes were also used to test sensitivities of the mutations to tyrosine kinase inhibitors.
Results: In pan-negative lung adenocarcinoma cases, a novel oncogenic EGFR ECD mutation was identified (M277E). EGFR M277E mutations encoded oncoproteins that transformed NIH-3T3 cells to grow in the absence of exogenous epidermal growth factor. Transformation was further evidenced by anchorage-independent growth and tumor formation in immunocompromised xenograft mouse models. Finally, as seen in the canonical EGFR L858R mutation, the M277E mutation conferred sensitivity to both erlotinib and cetuximab in BaF3 cell lines and to erlotinib in xenograft models.
Conclusion: Here, a new EGFR driver mutation, M277E, was identified in the ECD of a lung adenocarcinoma specimen. For patients with M277E-mutant lung adenocarcinoma who experienced disease recurrence, treatment with an EGFR tyrosine kinase inhibitor may predict good prognosis.
Keywords: EGFR extracellular domain mutation, non-small cell lung cancer, oncogene, drug sensitive
Introduction
The human epidermal growth factor receptor (EGFR) gene, a member of the ErbB family of receptor tyrosine kinases, functions in epithelial cell proliferation. Stimulation of the receptor with epidermal growth factor (EGF) or other cognate ligands induces receptor dimerization and auto-phosphorylation.1–4 Somatic mutations in the EGFR kinase domain (KD) are common in lung adenocarcinomas, especially exon 19 deletions and L858R mutations.5–7 Recently, a series of missense mutations in the EGFR extracellular domain (ECD) were reported in colorectal cancer, glioma, and neuroblastoma cases;8–13 there are no reported cases of EGFR ECD mutations in lung cancer. Previously, Vivanco et al compared differential sensitivities of specific glioma and lung cancer mutations to EGFR kinase inhibitors. These data showed that in lung cancer, EGFR was activated by KD mutations, while in glioblastoma (GBM), EGFR was activated by mutations in the ECD.8,10
Here, sequencing results are presented from a comprehensive panel of oncogenic driver mutations performed on a large prospective series of non-small cell lung cancer (NSCLC) specimens collected from patients who received surgical resection. A novel, oncogenic EGFR ECD mutation was identified and examined. Contrary to previous data, the novel EC EGFR lung cancer mutation was sensitive to first-generation EGFR inhibitors. These results were further confirmed using BaF3 drug sensitivity assays. Taken together, these results identify a unique therapeutic opportunity for patients with EC EGFR-mutant lung cancers.
Materials and methods
Patient samples and ethical considerations
All patients who participated in this research provided written informed consent. This study protocol was approved by the Ethical Committee of the Fudan University Shanghai Cancer Center (Reference number: 050432–4-1212B). All procedures were performed in accordance with the ethical standards of the Ethical Committee and followed the Declaration of Helsinki. All of the patients were lung adenocarcinoma cases recruited from April 2007 to May 2013 in the Fudan University Shanghai Cancer Center.
EGFR mutational analyses
Briefly, primers were designed to amplify the EGFR ECD from genomic DNA using standard polymerase chain reaction methodologies. Direct dideoxynucleotide sequencing was used to analyze the amplified products. For newly identified mutations, germline DNA specimens were sequenced from paired non-cancerous lung tissues to demonstrate that mutations were somatically acquired.
Cell culture
Cell lines were purchased from the American Type Culture Collection. NIH-3T3 cells were maintained in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% calf serum (Thermo Fisher Scientific), 100 units/mL penicillin, and 100 mg/mL streptomycin. BaF3 cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 (Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (Gibco) and 10 ng/mL interleukin-3 (IL-3) (PeproTech, Rocky Hill, NJ, USA).
Western blotting
NIH-3T3 cells were washed with cold PBS prior to lysis (category number 9803; Cell Signaling Technology, Inc., Danvers, MA, USA). A phenylmethylsulfonyl fluoride protease and phosphatase inhibitor cocktail (Merck 524625) was added immediately prior to use. Proteins were separated with 4%–12% Tris–glycine sodium dodecyl sulfate polyacrylamide gel electrophoresis gels (Invitrogen, Carlsbad, CA, USA) and transferred to polyvinylidene difluoride membranes. Antibody binding was detected using the ImageQuant LAS 4000 imaging system. Antibodies against phospho-EGFR (Tyr-1068, 2234), EGFR (4267), phospho-Akt (Ser 473, 4060), Akt (4691), phospho-Erk1/2 (Thr202/Tyr204, 9101), and Erk1/2 (p44/42 MAPK, 4695) were obtained from Cell Signaling Technology, Inc.
Mutagenesis, virus production, and stable-expressing cell line generation
Retroviral EGFR expression constructs containing puromycin (pBabe-puro-EGFR) were used for site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). All constructs were fully sequenced and ecotropic viruses were produced by cotransfection of the pBabe-puro-EGFR constructs with pCL-Eco into 293T cells. NIH-3T3 cells were infected with EGFR mutation-carrying viruses and 8 μg/mL polybrene (hexadimethrine bromide; Sigma, St Louis, MO, USA). Infected cells were selected with 2 μg/mL puromycin (InvivoGen, San Diego, CA, USA) 2 days after infection. The same viral stocks and 8 μg/mL polybrene were used to infect BaF3 cells while spinning at 800× g for 90 min at 25°C. Cells were re-infected 24 h later. Infected BaF3 cells were selected using 2 μg/mL puromycin 24 h after the second infection in the presence of IL-3. IL-3 was serially reduced over 2 weeks to select for IL-3-independent cells.
Soft agar anchorage-independent growth assays
Solutions of 0.6% agar in mediums were plated into 6-well plates and solidified. Aliquots of 1×105 NIH-3T3 cells expressing EGFR mutations were suspended with or without EGF (10 ng/mL) in 0.3% agar and 10% calf serum solutions and plated into the agar-coated 6-well plates. Plates were incubated for 3 weeks, and then photographed at 4× using the QuickCapture imaging system (Logitech, Lausanne, Switzerland). Numbers of colonies were counted in triplicate wells from 5 fields photographed with 4× magnification.
Cell growth inhibition assays
BaF3 is an IL-3-dependent pro-B cell line that has been widely used to study oncogenic activity of genes and development of drugs that target oncogenic drivers.14–17 BaF3 can grow independent of IL-3 cytokines when oncogenic drivers are expressed in cellular DNA. BaF3 cells have been adapted for compound profiling high-throughput assays and for assays predicting clinical resistance.
To detect sensitivities of EGFR mutations to inhibitors, EGFR mutant constructs were expressed in BaF3 cells, and IL-3 independent cell lines were derived.15,18 BaF3 cells expressing each EGFR mutant construct were selected using puromycin and a gradual reduction of IL-3 concentrations. For growth inhibition assays, BaF3 cells (5,000/well) were plated into 96-well plates (Corning). After 24 h incubation in standard conditions, cells were treated with either erlotinib (Tarceva®, Roche, San Francisco, CA, USA) or cetuximab (Erbitux®, Lilly, Indianapolis, IN, USA) at the indicated concentrations and then incubated for another 72 h. Viable cell numbers were measured using Cell Counting Kits (Dojindo, Rockville, MD, USA). Data were expressed as percentages of growth relative to untreated control cells. Data were calculated by the multi-scan spectrum function and plotted in Prism Software (GraphPad Software, Inc., La Jolla, CA, USA).
Xenograft experiments
The Institutional Animal Care and Use Committee of SLAC (IACUC) approved all animal protocols used in this study (Reference number: SLAC-2016092801). All animal experiments in this research were carried out according to the institutional guidelines for animal safety. Animals were treated humanely for each experimental procedure. All animals were lawfully acquired and their retention and use were in compliance with federal, state, and local laws and regulations, and in accordance with the guidelines of the IACUC guide for care and use of laboratory animals.
Immunocompromised mice were injected with NIH-3T3 cells stably expressing exogenous pBabe-puro vector constructs, or pBabe-puro vectors containing: EGFR wild-type (WT), M277E, or L858R mutant isoforms. Expression levels of EGFR in each NIH-3T3 cell line were confirmed using western blot methodologies. Mice were injected subcutaneously with 1×106 cells per mouse. Experimental mice were divided into 4 groups of 5 mice receiving either: pBabe-puro vector, pBabe-puro-EGFR WT, pBabe-puro-EGFR M277E, or pBabe-puro-EGFR L858R. After 2 weeks of incubation, tumors were evaluated every 3 days for tumor volumes, calculated using the formula: (mm3) =0.5× length × width × width. Standard deviations were calculated using Prism Software. Animals in which tumor volumes grew to sizes of 200–300 mm3 were treated with erlotinib (150 mg/kg) or vehicle daily for 2 weeks. In each group, 3 mice were treated with erlotinib and 2 were treated with vehicle. Tumor sizes were measured during the treatment period every 3 days.
Results
Novel EGFR somatic alterations detected in NSCLC cases
A total of 1,356 lung adenocarcinoma cases collected from April 2007 to May 2013 were sequenced for EGFR KD mutations, KRAS mutations, HER2 KD mutations, BRAF mutations, ALK fusions, ROS1 fusions, RET fusions, and AKT1 mutations.19 All of the oncogenic driver mutations were mutually exclusive. From this population, 143 were considered “pan-negative” (not possessing known oncogenic driver mutations). These specimens were selected to test for EGFR ECD mutations. Nine EGFR ECD mutations were detected, including: V159E (n=1, 0.70%), Q181H (n=1, 0.70%), M277E (n=1, 0.70%), R324L (n=1, 0.70%), T363N (n=1, 0.70%), A508V (n=1, 0.70%), N528D (n=1, 0.70%), and V592I (n=2, 1.40%; Figure 1). The sample from which the EGFR M277E mutation was identified came from a 70-year-old female patient without a history of smoking. The DNA sequence of this mutation was altered from ATG to GAG, resulting in the EGFR M277E mutation (Figure S1). A case report of this patient is provided in the Supplementary materials.
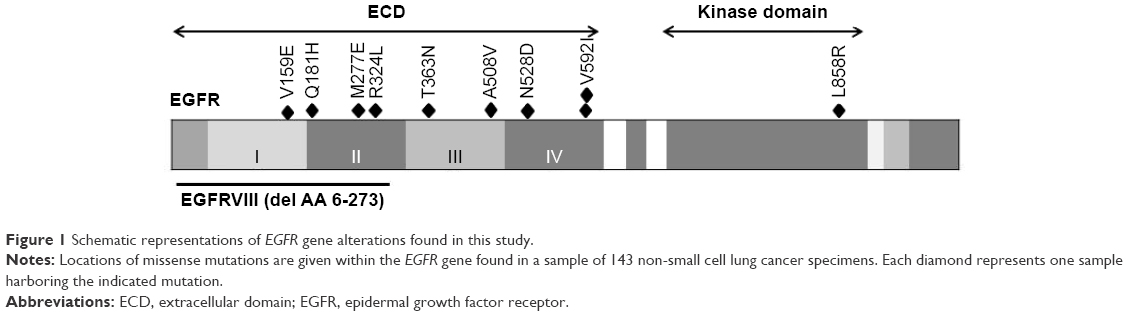 | Figure 1 Schematic representations of EGFR gene alterations found in this study. |
The EGFR M227E mutation induced EGFR auto-phosphorylation and promoted transformation in vitro
Overexpression of WT EGFR can transform NIH-3T3 cells when supplemented with EGF; oncogenic mutations transform NIH-3T3 cells in the absence of exogenous EGF stimulation.1,3–7 The substitution mutation EGFR M277E was tested to determine whether NIH-3T3 cells were transformed in the absence of EGF using soft agar assays to monitor anchorage-independent growth. The EGFR L858R mutation is an established mutation that occurs in cases of lung adenocarcinoma; it was used as a positive control in these experiments (Figure 2A and B). EGFR M277E mutant proteins were hyperphosphorylated in NIH-3T3 cells, similar to EGFR L858R mutant proteins in the same system (Figure 2C).15,16 This mechanism of constitutive activity due to constitutive hyperphosphorylation is consistent with mechanisms driving the ERBB2 S310F in lung adenocarcinoma.16
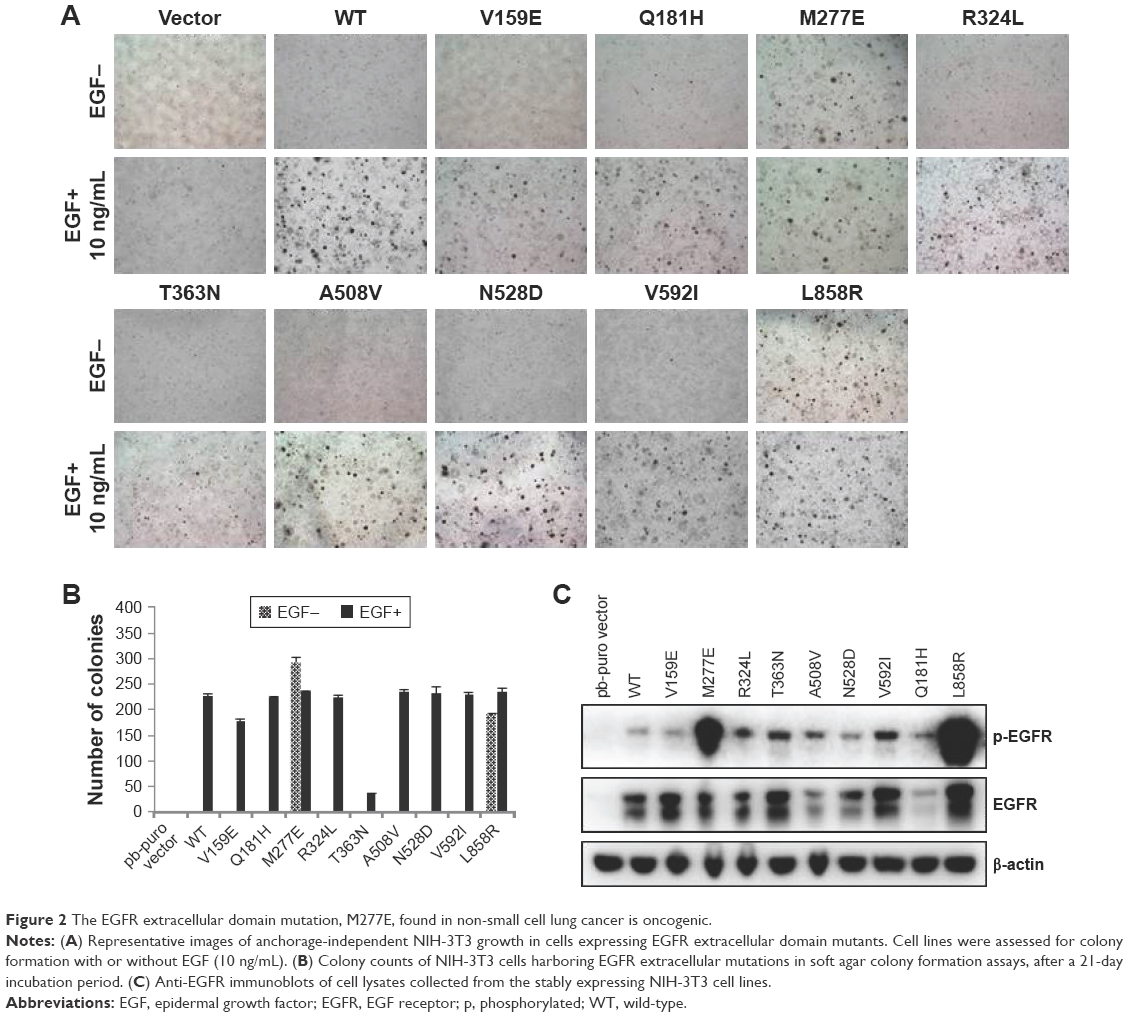 | Figure 2 The EGFR extracellular domain mutation, M277E, found in non-small cell lung cancer is oncogenic. |
EGFR M277E and L858R mutant-expressing NIH 3T3 cells had enhanced formation of spindle morphologies and were more refractive under light microscopes. These phenotypes were similar to those observed in oncogenic KRAS (G12V) transformed NIH 3T3 cells (data was not shown). NIH 3T3 cells transformed with vector and EGFR WT constructs grew in monolayers and had no changes in refraction or spindle formation (Figure S2A and B).21 These results suggest that EGFR M227E mutations transformed NIH-3T3 cells by constitutively activating EGFR, independent of exogenous EGF stimulation.
EGFR M227E is susceptible to EGFR blockade in vitro
Multiple agents that target constitutively activated EGFR proteins are approved for lung cancer treatments. Erlotinib, a tyrosine kinase inhibitor (TKI), is widely used to treat lung adenocarcinomas with EGFR L858R and exon 19 deletions. Cetuximab is an anti-EGFR monoclonal antibody that binds to the EGFR ECD, blocks dimerization, and results in apoptosis of tumor cells.22 To determine whether EGFR M277E mutations were sensitive to erlotinib and cetuximab, NIH-3T3 EGFR-mutant-expressing stable cell lines were tested for phosphorylated EGFR protein contents. Cell lines expressing EGFR M277E and L858R mutant proteins had decreased levels of phosphorylated EGFR after treatment with either erlotinib or cetuximab (Figure 3A and B). After erlotinib treatment, EGFR mutant NIH-3T3 cells appeared to have reduced AKT phosphorylation levels. Treatment with either erlotinib or cetuximab did not inhibit pERK activation in EGFR M227E mutant cells; pERK was inhibited in EGFR L858R mutant cells (Figure 4B and C). Thus, EGFR M227E mutant NIH 3T3 cells were less sensitive to EGFR inhibitors than EGFR L858R positive controls. The EGFR M277E was less sensitive to EGFR inhibitors and pERK was not inhibited by the inhibitors, which may be a correlation, but the molecular mechanism is not clear. In cetuximab-treated EGFR M277E mutant cells, neither phosphorylated AKT nor phosphorylated ERK levels were inhibited. While the mechanistic underpinnings of these agents in the context of the EGFR M277E mutation are unknown, the results showed that erlotinib and cetuximab inhibited EGFR hyperactivity induced by the M277E mutation.
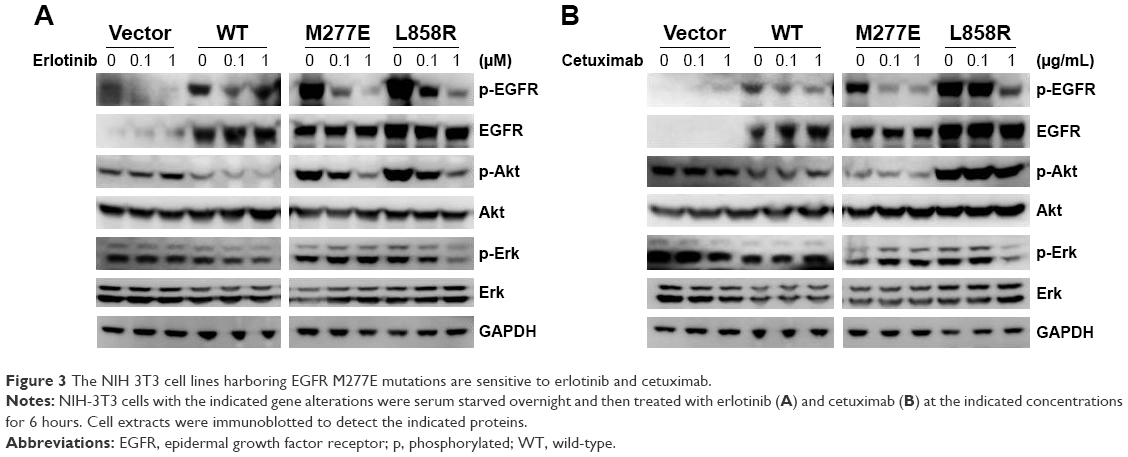 | Figure 3 The NIH 3T3 cell lines harboring EGFR M277E mutations are sensitive to erlotinib and cetuximab. |
Next, BaF3 cells were transformed with the EGFR M277E and control constructs. Overexpression of EGFR EC and KD mutations conferred IL-3 independence to the BaF3 cells, which were not found in BaF3 cells transformed with vector control and WT EGFR genes (data not shown). Proliferations of BaF3 cell lines expressing EGFR mutant constructs were detected upon IL-3 withdrawal. Conversely, BaF3 cells expressing EGFR WT constructs did not grow in the absence of IL-3 (Figure 4A). Next, EGFR M277E- and L858R-transformed BaF3 cells were treated with erlotinib and cetuximab. Treatments resulted in abrogation of cell survival rates in the EGFR M277E and L858R expressing lines (Figure 4B and C). Together, these results suggest that EGFR M277E mutations may be treated by erlotinib or cetuximab in lung cancer patients.
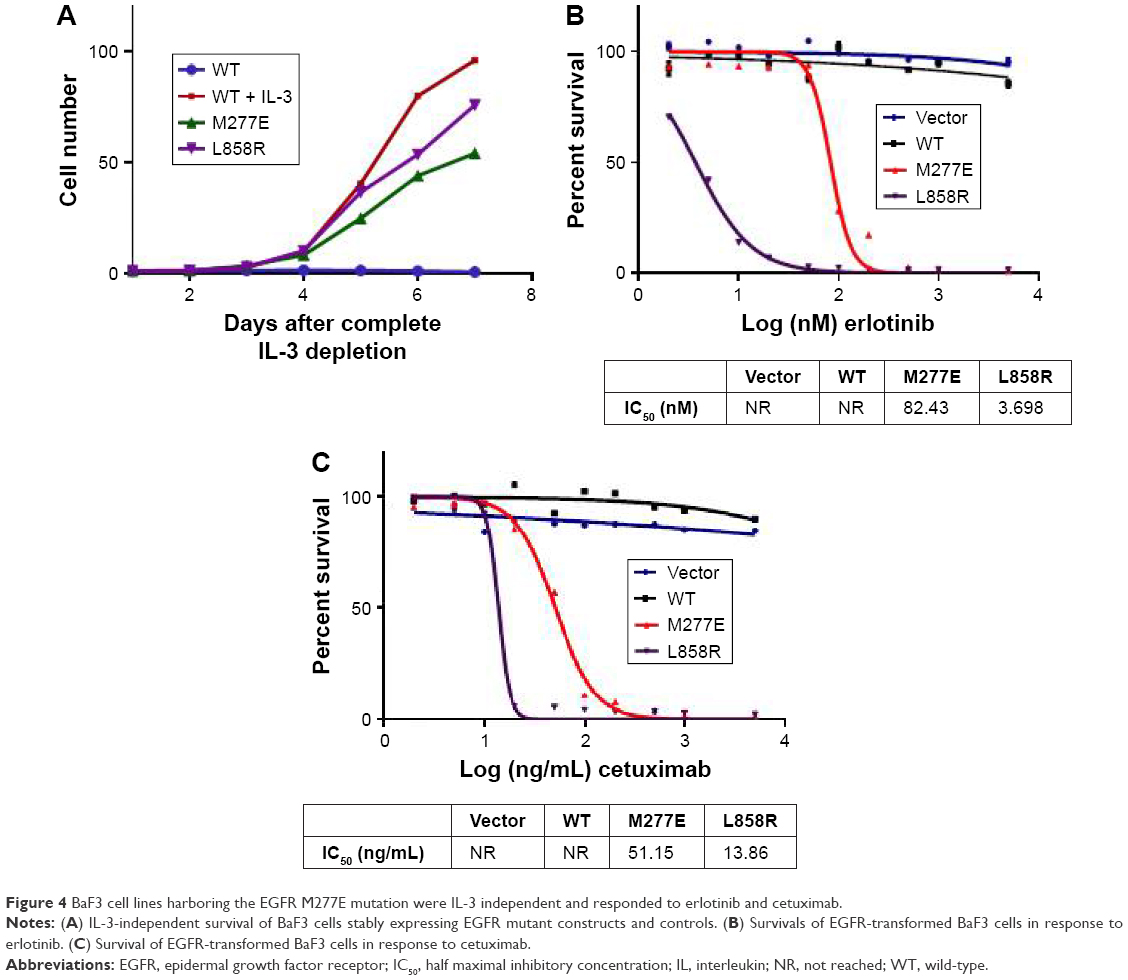 | Figure 4 BaF3 cell lines harboring the EGFR M277E mutation were IL-3 independent and responded to erlotinib and cetuximab. |
EGFR M227E mutations conferred tumor growth capacity and sensitivity to inhibitors in vivo
NIH 3T3 cells expressing EGFR M277E, EGFR L858R, EGFR WT or vector constructs were subcutaneously injected into nude mice and tumor growth was monitored every 3 days. Both EGFR M277E and L858R mutations drove tumor formation (Figure 5A). Interestingly, tumors driven by overexpression of the EGFR M277E mutation were larger than EGFR L858R tumors (Figure 5A). These results demonstrate that NIH 3T3 cells harboring EGFR M277E mutations had oncogenic potential in vivo. When the tumors reached ~200–300 mm3 in the oncogenic xenograft models, the subjects were treated with either erlotinib or vehicle. At the end of the study time-course, tumor burdens in the erlotinib and vehicle treated groups were noticeably distinct. In fact, erlotinib treatment resulted in dramatic antitumor responses in subjects harboring EGFR M277E mutations, and complete response in subjects with EGFR L858R mutant tumors (Figure 5B and C). These data indicate that EGFR M227E mutations in NIH-3T3 cells had transformational capacities in vivo and were sensitive to EGFR tyrosine kinase inhibition.
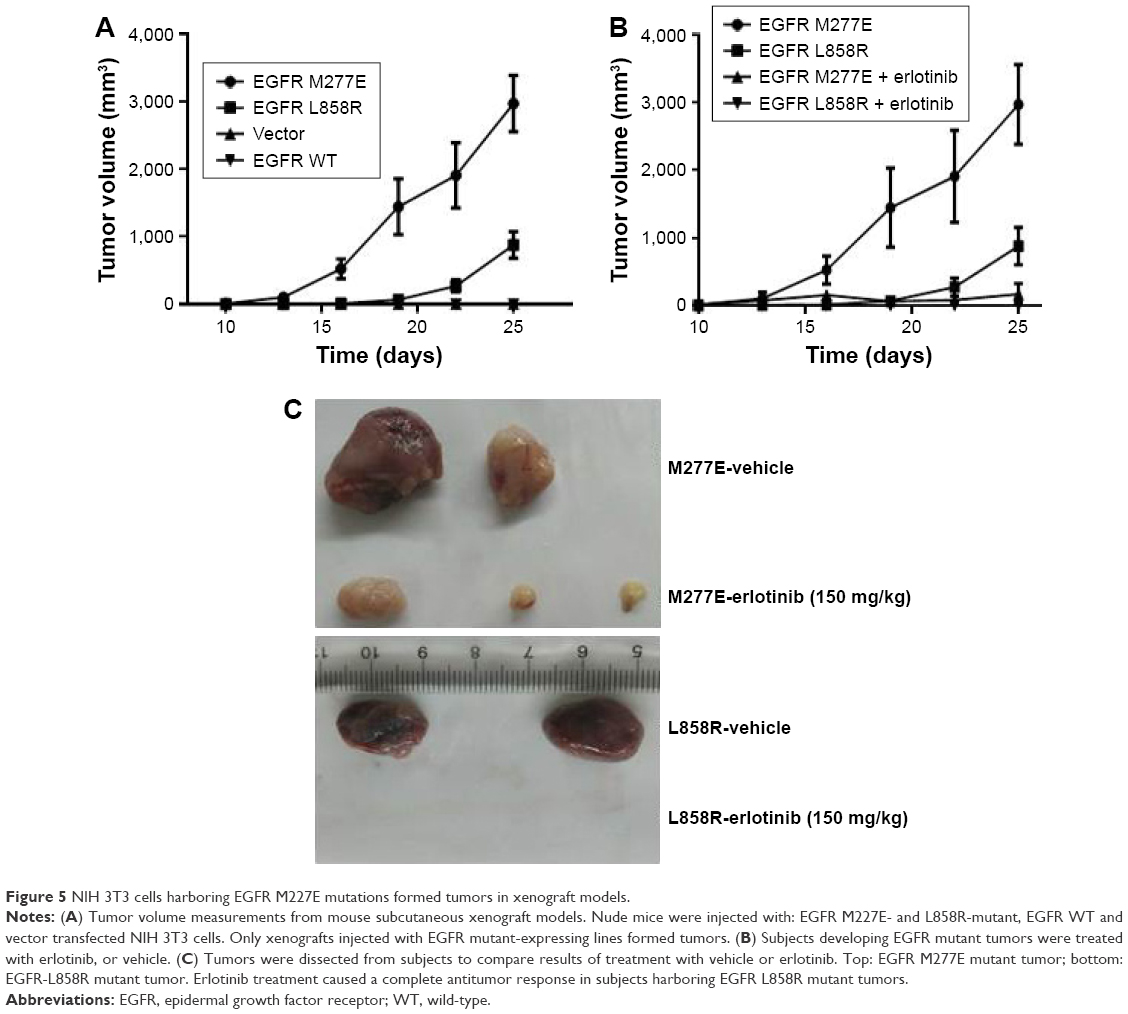 | Figure 5 NIH 3T3 cells harboring EGFR M227E mutations formed tumors in xenograft models. |
Discussion
EGFR plays critical roles in many biological processes and is a driver of tumorigenesis.23 Mutation test in the EGFR KD is commonly recommended in the management of NSCLC.5,24 Identifying such mutations can predict which patients will benefit from EGFR TKI therapies. EGFR mutations that are sensitive to TKI agents include: exon 19 deletions (~45%), L858R (~40%), G719X (~3%), L861Q (~2%), exon 18 indels, exon 19 insertions, EGFR kinase domain duplication (duplications of exons 18–25), and others.18,25–29 Mutations that are resistant to TKI agents include: exon 20 insertions (~5%~10%) and T790M (<1%).26,30,31 Previous studies have identified 2 EGFR ECD mutations (A289D and R324L), but no independently oncogenic EGFR ECD mutations have been reported in NSCLC.19
Previously, Liao et al discovered that oncogenic mutations in the ECD of the fibroblast growth factor receptor (FGFR) occurred in squamous cell lung carcinomas.20 Another group found that FGFR2 ECD insertions were both oncogenic and sensitive to FGFR kinase inhibitors in lung cancer cases.32 Additionally, oncogenic ECD mutations of ERBB2 have been reported in lung cancers.16–15 It was also reported that EGFR ECD missense mutations occurred in 10% to 15% of GBM cases. Vivanco et al found that EGFR ECD mutations are more common in GBMs and KD mutation are more common in lung cancers. Nonetheless, TKI-targeted therapies were identified to treat ECD mutations in GBM.8,9,33,34 A novel EGFR ECD mutant (EGFRdelta768) was discovered in neuroblastoma cases.12 Additionally, a novel heterozygous mutation in the FGF1 ECD, associated with Hartsfield syndrome,35 and ERBB3 EC mutations, were identified in human cancers.15 Thus, ECD mutations are relatively common oncogenic human kinase mutations. The present research was the first to identify an independently oncogenic EGFR ECD mutation in NSCLC.
In this study, a new EGFR ECD somatic mutation, M277E, was identified in lung adenocarcinoma. This mutation promoted oncogenesis in the absence of the EGFR ligand, EGF. In contrast with other EGFR ECD mutations found in this research, the M277E (ECD) and L858R (KD) mutants transformed NIH-3T3 cell lines, leading to EGFR auto-phosphorylation that was independent of stimulation. The EGFR M277E mutation was sensitive to erlotinib treatment in BaF3 cell lines and in xenograft models. These results paralleled those seen with the well-described EGFR oncogenic driver mutation, L858R, suggesting similar mechanistic underpinnings for the 2 mutations. This finding is unusual because ECD kinase mutations may strongly alter oncogenic mechanisms depending on the location of the mutation. For example, M277E mutations may change the structure of EGFR, potentially influencing the dimerization pattern of the protein. Elucidating the mechanism of oncogenesis is important in future studies.
In this paper, a unique therapeutic opportunity for patients with EGFR M277E mutant lung cancers was discovered. Additional EGFR ECD mutations may exist in lung adenocarcinomas. This study design may be a useful reference for future work.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81330056, 81401886, 81572264, 81422029, 81472173, 81502480, and 81372525), the Key Project of Science and Technology Commission of the Shanghai Municipality (JGGG1302), the Shen-Kang Center Project (SKMB1201), and a grant from the Science and Technology Commission of the Shanghai Municipality (14495810800).
Disclosure
The authors report no conflicts of interest in this work.
References
Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19(13):3159–3167. | ||
Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl 4):S3–S8. | ||
Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and −resistant EGFR mutants. PLoS Med. 2005;2(11):e313. | ||
Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 2006;16(12):649–656. | ||
Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | ||
Rosell R, Taron M, Reguart N, Isla D, Moran T. Epidermal growth factor receptor activation: how exon 19 and 21 mutations changed our understanding of the pathway. Clin Cancer Res. 2006;12(24):7222–7231. | ||
Valley CC, Arndt-Jovin DJ, Karedla N, et al. Enhanced dimerization drives ligand-independent activity of mutant epidermal growth factor receptor in lung cancer. Mol Biol Cell. 2015;26(22):4087–4099. | ||
Ymer SI, Greenall SA, Cvrljevic A, et al. Glioma specific extracellular missense mutations in the first cysteine rich region of epidermal growth factor receptor (EGFR) initiate ligand independent activation. Cancers (Basel). 2011;3(2):2032–2049. | ||
Lee JC, Vivanco I, Beroukhim R, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006;3(12):e485. | ||
Vivanco I, Robins HI, Rohle D, et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2(5):458–471. | ||
Sanchez-Martin FJ, Bellosillo B, Gelabert-Baldrich M, et al. The first-in-class anti-EGFR antibody mixture Sym004 overcomes cetuximab resistance mediated by EGFR extracellular domain mutations in colorectal cancer. Clin Cancer Res. 2016;22(13):3260–3267. | ||
Keller J, Nimnual AS, Varghese MS, VanHeyst KA, Hayman MJ, Chan EL. A novel EGFR extracellular domain mutant, EGFRdelta768, possesses distinct biological and biochemical properties in neuroblastoma. Mol Cancer Res. 2016;14(8):740–752. | ||
Arena S, Bellosillo B, Siravegna G, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. 2015;21(9):2157–2166. | ||
Jia Y, Yun CH, Park E, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–132. | ||
Jaiswal BS, Kljavin NM, Stawiski EW, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23(5):603–617. | ||
Greulich H, Kaplan B, Mertins P, et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci U S A. 2012;109(36):14476–14481. | ||
Warmuth M, Kim S, Gu XJ, Xia G, Adrian F. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol. 2007;19(1):55–60. | ||
Banno E, Togashi Y, Nakamura Y, et al. Sensitivities to various EGFR-TKIs of uncommon EGFR mutations L861Q and S768I: what is the optimal EGFR-TKI? Cancer Sci. 2016;107(8):1134–1140. | ||
Wang R, Zhang Y, Pan Y, et al. Comprehensive investigation of oncogenic driver mutations in Chinese non-small cell lung cancer patients. Oncotarget. 2015;6(33):34300–34308. | ||
Liao RG, Jung J, Tchaicha J, et al. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 2013;73(16):5195–5205. | ||
Starczynowski DT, Lockwood WW, Delehouzee S, et al. TRAF6 is an amplified oncogene bridging the RAS and NF-kappaB pathways in human lung cancer. J Clin Invest. 2011;121(10):4095–4105. | ||
Martinelli E, De Palma R, Orditura M, De Vita F, Ciardiello F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin Exp Immunol. 2009;158(1):1–9. | ||
Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65(19):8968–8974. | ||
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. | ||
Gaughan EM, Costa DB. Genotype-driven therapies for non-small cell lung cancer: focus on EGFR, KRAS and ALK gene abnormalities. Ther Adv Med Oncol. 2011;3(3):113–125. | ||
Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015;16(7):830–838. | ||
Ackerman A, Goldstein MA, Kobayashi S, Costa DB. EGFR delE709_T710insD: a rare but potentially EGFR inhibitor responsive mutation in non-small-cell lung cancer. J Thorac Oncol. 2012;7(10):e19–e20. | ||
Gallant JN, Sheehan JH, Shaver TM, et al. EGFR kinase domain duplication (EGFR-KDD) is a novel oncogenic driver in lung cancer that is clinically responsive to afatinib. Cancer Discov. 2015;5(11):1155–1163. | ||
He M, Capelletti M, Nafa K, et al. EGFR exon 19 insertions: a new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res. 2012;18(6):1790–1797. | ||
Wu JY, Yu CJ, Chang YC, Yang CH, Shih JY, Yang PC. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res. 2011;17(11):3812–3821. | ||
Gazdar A, Robinson L, Oliver D, et al. Hereditary lung cancer syndrome targets never smokers with germline EGFR gene T790M mutations. J Thorac Oncol. 2014;9(4):456–463. | ||
Tanizaki J, Ercan D, Capelletti M, et al. Identification of oncogenic and drug-sensitizing mutations in the extracellular domain of FGFR2. Cancer Res. 2015;75(15):3139–3146. | ||
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. | ||
Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. | ||
Takagi M, Miyoshi T, Nagashima Y, et al. Novel heterozygous mutation in the extracellular domain of FGFR1 associated with Hartsfield syndrome. Hum Genome Var. 2016;3:16034. |
Supplementary materials
Case report of lung adenocarcinoma patient harboring an EGFR M277E mutation in her tumor
Figure S1 gives the sequence tracing of an EGFR ECD mutation in a NSCLC adenocarcinoma specimen. This mutation was identified in specimens collected from a 70-year-old female patient who was a non-smoker. The patient had a family history of lung cancer; her mother also had the disease. It was unknown whether her mother harbored the same mutation. The patient underwent thoracoscopic lobectomy surgery at the Fudan University Shanghai Cancer Center in 2011. Pathological analyses showed that it was a Stage 1A tumor of the solid adenocarcinoma subtype. This patient had a good post-surgical prognosis. She has not experienced any tumor recurrences.
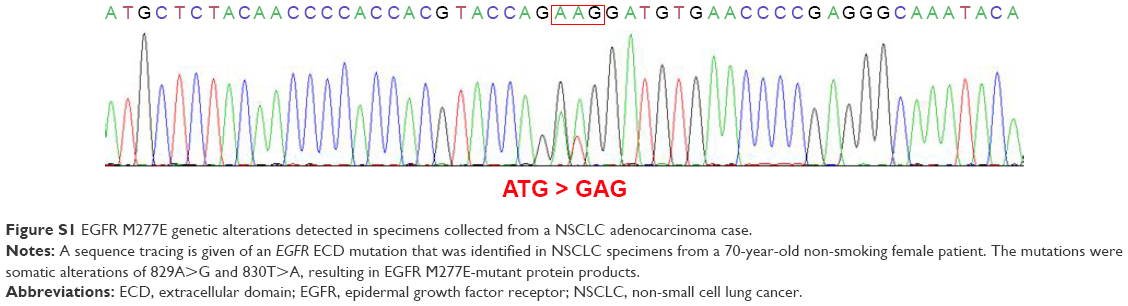 | Figure S1 EGFR M277E genetic alterations detected in specimens collected from a NSCLC adenocarcinoma case. |
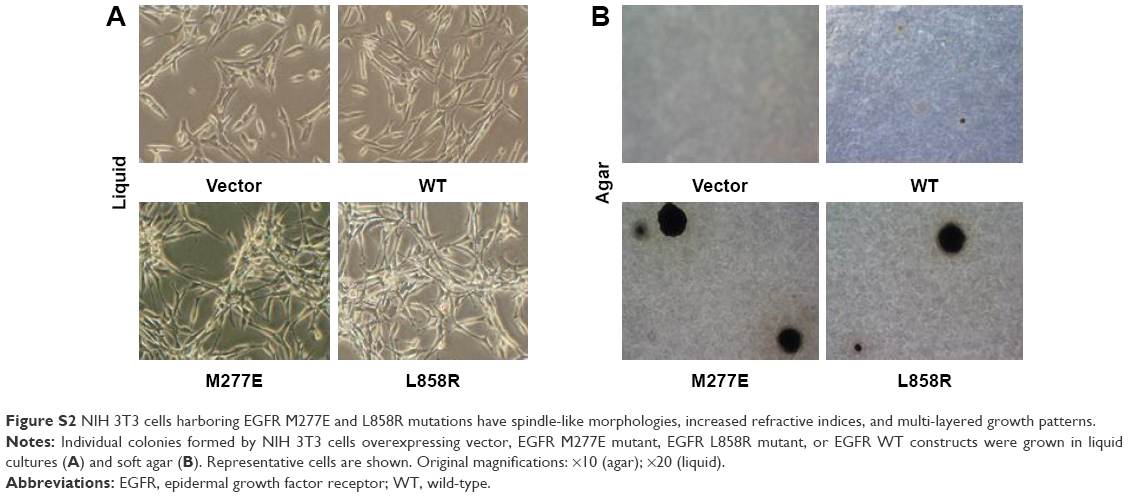 | Figure S2 NIH 3T3 cells harboring EGFR M277E and L858R mutations have spindle-like morphologies, increased refractive indices, and multi-layered growth patterns. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.