Back to Journals » Journal of Inflammation Research » Volume 19
The NLRP3–α-Synuclein Circuit: A Core Driver and Therapeutic Target in Parkinson’s Disease
Authors Liu K, He Z, Zhang L, Wu YM, Xiao L
Received 3 February 2026
Accepted for publication 19 May 2026
Published 28 May 2026 Volume 2026:19 597280
DOI https://doi.org/10.2147/JIR.S597280
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dharmappa Krishnappa
Keting Liu,1,2 Zemin He,3 Lihao Zhang,2 Yong-ming Wu,2 Li Xiao4
1Department of Neurology, Chengdu Seventh People’s Hospital, Affiliated Cancer Hospital of Chengdu Medical College, Chengdu, Sichuan, 610213, People’s Republic of China; 2Department of Neurology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong Province, 510515, People’s Republic of China; 3Department of Thoracic Surgery, The First People’s Hospital of Shuangliu District, Chendu, Sichuan Province, People’s Republic of China; 4Department of Human Anatomy, Aging Mechanisms and Interventions Key Laboratory of Sichuan Province, Chengdu Medical College, Chengdu, Sichuan Province, 610500,People’s Republic of China
Correspondence: Li Xiao, Department of Human Anatomy, Aging Mechanisms and Interventions Key Laboratory of Sichuan Province, Chengdu Medical College, No. 783, Xindu Avenue, Xindu District, Chengdu, Sichuan Province, 610500, People’s Republic of China, Email [email protected]
Abstract: The neuroinflammatory mechanisms underlying Parkinson’s disease (PD) have evolved from a view of localized central events to a systemic network perspective involving “brain-periphery” interactions. The NLRP3 inflammasome, as a core component of the innate immune system, plays a pivotal role within this network. This review aims to systematically elucidate NLRP3’s multifaceted roles in PD: First, it delves into the “self-amplifying” vicious cycle between α-synuclein pathology and NLRP3 activation, supports the hypothesis that this cycle may serve as a core engine. Second, it transcends central nervous system limitations to systematically examine how the central-peripheral immune interaction network—comprising gut microbiota and peripheral immune cells—regulates this cycle. Third, it objectively evaluates the potential and current limitations of NLRP3 pathway-associated molecules as biomarkers for disease diagnosis and progression monitoring. Fourth, it comprehensively reviews therapeutic strategies targeting NLRP3 and critically examines key challenges in their clinical translation. This paper provides an integrated perspective for understanding PD as a systemic inflammatory disease and offers a theoretical framework for future biomarker development and precision treatment strategies based on this pathway.
Keywords: Parkinson’s disease, NLRP3 inflammasome, neuroinflammation, pyroptosis, α-synuclein, therapeutic targets
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by progressive loss of dopaminergic neurons in the substantia nigra of the midbrain. Beyond the classic pathology of protein misfolding and aggregation, neuroinflammation has been demonstrated to be a core factor driving the onset and progression of PD.1,2 Within the complex neuroinflammatory network of PD, the NLRP3 inflammasome—a key component of the innate immune system—directly participates in and amplifies neuroinflammatory cascades by mediating the mature release of interleukin-1β (IL-1β) and IL-18, as well as inducing pyroptosis.1,3 Notably, pathological α-synuclein aggregates—the core pathological protein in Parkinson’s disease—can serve as endogenous danger signals (DAMPs) to directly activate NLRP3 inflammasomes in microglia.4–6 The active caspase-1 generated by NLRP3 activation not only promotes the maturation of inflammatory factors such as IL-1β but also directly cleaves α-synuclein to accelerate its abnormal aggregation.3 Crucially, NLRP3-mediated pyroptosis induces the release of intracellular α-synuclein, allowing its dissemination to neighboring cells.3–6 These processes collectively form a self-amplifying vicious cycle: α-synuclein aggregate → NLRP3 activation → increased α-synuclein aggregation and seeding.3,7 This positive feedback loop has been validated in multiple PD experimental models, such as MPTP mice and α-synuclein transgenic models.3,6–8 It is considered one of the “core engines” driving the continuous advancement of PD.3,7,9 Furthermore, dopamine depletion resulting from dopaminergic neuronal degeneration releases its endogenous inhibition of NLRP3 activation,5,9 thereby further amplifying neuroinflammatory responses. This may represent a key mechanism underlying the specific vulnerability exhibited by this neuronal population.
As research deepens, the academic community’s understanding of the inflammatory mechanisms underlying Parkinson’s disease (PD) is undergoing a paradigm shift from a localized to a systemic perspective. Traditional views have predominantly focused on localized inflammatory responses mediated by microglia within the central nervous system. However, recent evidence indicates that activation of the peripheral immune system and its dynamic interactions with the central nervous system profoundly influence the pathological progression of PD.10,11 Specifically, systemic factors such as dysbiosis of the gut microbiota, peripheral immune cell infiltration, and blood-brain barrier dysfunction have been demonstrated to significantly influence the activation status of NLRP3 inflammasomes within the central nervous system. These factors collectively constitute the complex “gut-brain axis” and systemic immune-inflammatory network.10,11 The introduction of this systemic perspective offers significant insights for understanding the clinical heterogeneity of PD and exploring novel therapeutic targets.
However, existing reviews predominantly focus on summarizing individual mechanisms or therapeutic strategies, with insufficient systematic integration of the following interrelated key questions: (1) What are the detailed molecular mechanisms underlying the bidirectional vicious cycle between α-synuclein and NLRP3 inflammasome, and do they constitute the core engine driving disease progression? (2) Through which specific interfaces do the central and peripheral immune systems achieve functional coupling and jointly regulate NLRP3 activation? (3) What practical bottlenecks—regarding target specificity, long-term safety, and patient selection—do NLRP3 pathway-targeting therapies face in clinical translation? Against this backdrop of unresolved questions, this review aims to systematically address and critically examine these issues with NLRP3 inflammasome as the pivotal hub. This paper will first focus on the bidirectional vicious cycle between α-synuclein and NLRP3, elucidating its mechanism as the “core engine” driving disease progression. Subsequently, it will systematically describe how the central-peripheral immune interaction network regulates this cycle, providing evidence for understanding PD as a systemic inflammatory disease. Following this, the biomarker value and limitations of NLRP3-associated molecules will be objectively evaluated. Finally, we comprehensively review therapeutic strategies targeting NLRP3, delving into their mechanism heterogeneity, controversies, and future directions in clinical translation. This paper aims to provide an integrated perspective for deepening the understanding of PD’s inflammatory mechanisms and to establish a theoretical framework for developing future precision intervention strategies.
Structural Features, Activation Mechanisms, and Core Role of the NLRP3 Inflammasome in Neuroinflammation
Structural Features and Activation Mechanisms of the NLRP3 Inflammasome
The NLRP3 inflammasome is a cytoplasmic multiprotein complex belonging to the nucleotide-binding and oligomerization domain (NOD)-like receptor family, capable of recognizing microbial ligands and endogenous danger signals.12 The NLRP3 inflammasome core structure comprises the sensor protein NLRP3, the adaptor protein ASC (apoptosis-related speck-like protein), and the effector protein caspase-1.13 Structurally, NLRP3 comprises three characteristic domains:13 the N-terminal pyrophosphate-containing domain (PYD) responsible for binding to ASC; the central nucleotide-binding and oligomerization domain (NACHT) possessing ATPase activities that drive protein oligomerization; and the C-terminal leucine-rich repeat (LRR) domain responsible for recognizing danger signals and maintaining a self-inhibited conformation in the resting state. ASC acts as a “molecular bridge”, binding to oligomerized NLRP3 through its PYD domain while recruiting pro-caspase-1 via its CARD domain. During activation, it forms large “ASC plaques” to amplify inflammatory signals. Caspase-1 exists as an inactive zymogen; upon recruitment near ASC, it activates through self-cleavage.
The classical activation of NLRP3 inflammasome follows a “dual signaling model”.13,14 Signal 1 (initiator signal) activates the NF-κB pathway via Toll-like receptors (TLRs) or cytokine receptors, inducing the transcriptional expression of NLRP3 and pro-IL-1β.13–15 Signal 2 (activation signal) is triggered by multiple stimuli (such as ATP, reactive oxygen species, and lysosomal rupture). Through mechanisms including potassium ion efflux,13,14 mitochondrial dysfunction (mtROS production),5 and lysosomal damage.7 It promotes NLRP3 to undergo conformational changes and oligomerization. In this process, NEK7 acts as a key regulatory protein by interacting with the LRR domain of NLRP3, thereby promoting its oligomerization and assembly with ASC.13,14 Activated caspase-1 cleaves pro-IL-1β and pro-IL-18 to generate mature inflammatory factors, while simultaneously cleaving gasdermin D (GSDMD) to induce pyroptosis.12,16 This series of processes is precisely regulated by protein interactions, post-translational modifications (such as ubiquitination and phosphorylation), and subcellular localization.13,14
Specific Regulatory Patterns in Neuroinflammation
Within the central nervous system, the regulation of NLRP3 inflammasomes exhibits a more complex pattern of tissue specificity and cell type dependency. It is primarily expressed in microglia,17 while astrocytes may also participate in activation. For instance, in ischemic stroke models, astrocyte-derived lipoprotein-2 (LCN2) activates NLRP3 through the receptor 24p3R.18 This cell type-specific regulation, together with the unique signaling interaction network within the neural microenvironment, collectively constitutes the complex landscape of NLRP3 activation in the central nervous system.
At the regulatory mechanism level, NLRP3 activation in neuroinflammation undergoes multi-tiered precision control. Regarding ion channel regulation, the THIK-1 potassium channel participates in NLRP3 activation in microglia by modulating K⁺ efflux; its inhibition or genetic knockout significantly reduces IL-1β release.19 At the level of immune receptor interactions, TREM2 negatively regulates pyroptosis by activating β-catenin signaling to suppress NLRP3 expression and complex assembly.20 Mitochondrial stress serves as a central hub, activated by environmental toxins such as cadmium through the Sirt3-mtROS axis to trigger NLRP3,21 while compounds like cryptotanshinone inhibit its activation by blocking calcium signaling and mtROS production.22
Furthermore, endogenous danger signals in the neuropathological environment (such as Aβ and α-synuclein) can directly serve as damage-associated molecular patterns (DAMPs) to trigger NLRP3 assembly.12,23 These signals interact with the aforementioned regulatory network to jointly determine the activation threshold and intensity of NLRP3 within the central nervous system. Notably, post-translational modifications (such as ubiquitination and phosphorylation) and subcellular localization (such as the endoplasmic reticulum-mitochondrial interface) further constitute the fine-tuned regulatory hierarchy of NLRP3 activation,13,14 ensuring its equilibrium between pathogen clearance and excessive inflammation. Dysregulation of this balance may drive neurodegenerative diseases.
Pyroptosis: A Core Effect Mechanism Linking Neuronal Inflammation and Neurodegeneration
Pyroptosis, the terminal event of NLRP3 inflammasome activation, is not only a product of neuroinflammation but also a key effector driving neurodegeneration, forming a direct causal pathway from inflammatory signaling to neuronal death.
From the molecular execution mechanism perspective, activated caspase-1 cleaves gasdermin D (GSDMD), releasing its N-terminal domain and forming pores in the cell membrane.12 This subsequently leads to cell permeabilization and lysis, massive release of inflammatory factors, and leakage of intracellular contents (including pathological proteins).12,16 This process not only directly causes cell death but, more importantly, releases pathological proteins and danger signals into the extracellular space. These activate nearby immune cells, triggering a cascading amplification of secondary inflammatory responses and forming a positive feedback loop of “pyroptosis-inflammation-more pyroptosis”.
Evidence regarding pathological protein dissemination further reveals the amplification effect of pyroptosis. In brain tissue from Alzheimer’s disease (AD) patients, cleaved GSDMD is widely expressed in microglia, astrocytes, and neurons. Moreover, the number of GSDMD-positive neurons shows a significant positive correlation with the degree of neuronal loss in the hippocampal CA1 region.24 More critically, NLRP3-positive microglia are frequently located adjacent to Aβ plaques, suggesting that pyroptotic cell lysis directly promotes the release of pathological proteins from intracellular compartments into the extracellular space, thereby accelerating their dissemination throughout the brain parenchyma.24
The Pervasive Role of NLRP3 Inflammasomes in Neurodegenerative Diseases
Interventional studies across multiple neurodegenerative models provide functional validation for this causal pathway: In MPTP-induced Parkinson’s disease (PD) models, inhibition of the NLRP3/caspase-1 pathway significantly mitigates dopaminergic neuronal damage.25 Confirming that the activation of this pathway is an upstream driver of neuronal death. Cadmium exposure induces pyroptosis in hippocampal neurons and cognitive impairment by activating the NLRP3-GSDMD axis,21 indicating that environmental toxins can directly cause neurological damage through this pathway. In vascular dementia (VaD) models, NLRP3 inhibitor AMS-17 mitigates neuronal degeneration by suppressing caspase-1 activation,25 further supporting the causal role of this pathway.
The above evidence collectively suggests that NLRP3 inflammasome-mediated pyroptosis not only directly causes neuronal death and neural network disruption,15 but also drives its dissemination within the brain by releasing pathological proteins. This transforms localized initial injury into a progressively worsening global pathological process. This causal chain renders key molecules in the pyroptosis pathway—such as caspase-1 and GSDMD—highly promising therapeutic intervention targets.
The Bidirectional Vicious Cycle Between α-Synuclein and NLRP3
The abnormal aggregation of α-synuclein and the activation of the NLRP3 inflammasome are not isolated pathological events. Existing research indicates that the two can mutually drive each other through multiple mechanisms (as shown in Figure 1), forming a self-amplifying vicious cycle. This cycle is considered one of the core engines driving the progressive progression of Parkinson’s disease.
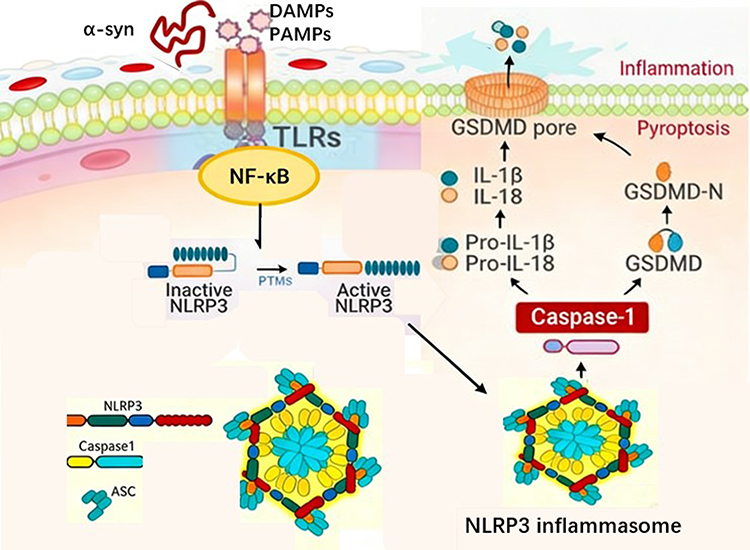 |
Figure 1 Schematic Illustration of the NLRP3 Inflammasome Signaling Pathway Activated by α-Synuclein in Parkinson’s Disease. The NLRP3 inflammasome comprises the sensor protein NLRP3, the adaptor protein ASC, and the effector protein caspase-1. Its classical activation follows a two-step model: an initiator signal upregulates NLRP3 and cytokine precursor expression via the TLR/NF-κB pathway; an activator signal triggers NLRP3 oligomerization and completes inflammasome assembly. Activated caspase-1 performs a dual function: it cleaves pro-IL-1β/18 to generate mature inflammatory factors while simultaneously cleaving the GSDMD protein. The GSDMD N-terminal domain forms pores in the cell membrane, directly inducing pyroptosis accompanied by massive release of inflammatory mediators. |
Molecular Mechanism of Pathological α-Synuclein Activation of NLRP3
Misfolded and aggregated α-synuclein, acting as an endogenous damage-associated molecular pattern (DAMP), effectively activates the NLRP3 inflammasome.26,27 This process primarily follows the classical “dual signaling” model. α-synuclein oligomers or fibrils activate the NF-κB signaling pathway through pattern recognition receptors such as Toll-like receptors (eg, TLR2, TLR4),26–28 thereby upregulating the transcriptional expression of NLRP3 and pro-IL-1β. Signal 2 (activation signal) is triggered when α-synuclein aggregates are phagocytosed by microglia.6,26,27 This process involves lysosomal membrane rupture and cathepsin B release,7 or by inducing mitochondrial dysfunction that generates substantial reactive oxygen species (mtROS).5,7,28 These cellular stress events collectively constitute the activation signal, triggering NLRP3 oligomerization.10,27 Research suggests that α-synuclein may activate NLRP3 through the CD36-Fyn kinase pathway, with NEK7 playing a crucial role as a key regulatory protein in NLRP3 oligomerization and inflammasome assembly.10
It is noteworthy that some studies suggest α-synuclein-driven NLRP3 activation may occur independently of classical activators such as ATP, involving distinct upstream pathways.29 In the substantia nigra region of Parkinson’s disease patients, α-synuclein has been observed to interact with Toll-like receptors, potentially contributing to NLRP3 activation. This process triggers NF-κB nuclear translocation and the release of proinflammatory factors,4 providing clinical-pathological evidence for this mechanism.
NLRP3 Activation Promotes Aggregation and Dissemination of α-Synuclein
Activation of the NLRP3 inflammasome may not only be a consequence of the inflammatory response but also serve as the starting point of a positive feedback loop. In addition to cleaving the IL-1β precursor, activated caspase-1 may directly or indirectly influence the aggregation state of α-synuclein, as suggested by some studies.27,30 Experiments demonstrate that caspase-1 deficiency suppresses α-synuclein-induced microglial activation and dopaminergic neuronal damage.15 In α-synuclein A53T transgenic mice, activation of the NLRP3/caspase-1 pathway correlates with enhanced α-synuclein aggregation, while the NLRP3 inhibitor MCC950 attenuates this process,7 suggesting this pathway may participate in regulating α-synuclein pathology.
More critically, NLRP3-activated pyroptosis may serve as a key mechanism for pathological protein dissemination. Activated caspase-1 cleaves GSDMD protein, inducing inflammatory programmed cell death in microglia and leading to the release of cellular contents—including mature IL-1β, IL-18, and pathological α-synuclein.8,28 These released pathological proteins may be taken up by neighboring cells, thereby promoting their dissemination among neurons.8,30 Concurrently, IL-1β released during pyroptosis can further activate surrounding glial cells, amplifying the inflammatory response and potentially exacerbating the intercellular transfer of α-synuclein.25 In an MPTP-induced Parkinson’s disease model, caspase-1-dependent pyroptosis was found to contribute to dopaminergic neuron degeneration and motor dysfunction,25 providing preliminary functional validation for this mechanism.
Experimental Evidence and Theoretical Models of Vicious Cycles
The bidirectional interaction between α-synuclein and the NLRP3 inflammasome has been validated to varying degrees across multiple experimental models, gradually forming a coherent theoretical framework.
Experimental evidence indicates that overexpression of α-synuclein enhances NLRP3 activation, while dopamine receptor DRD1 agonists inhibit this process, suggesting dopaminergic signaling may participate in regulating this circuit.9 In Parkinson’s disease patients, NLRP3-associated molecules in peripheral blood mononuclear cells positively correlate with plasma α-synuclein levels, and both are significantly associated with disease severity (UPDRS III score).10 Dopamine depletion within microglia leads to NLRP3 inhibition reversal, sustaining enhanced inflammation and potentially accelerating α-synuclein aggregation.25 These observations provide multifaceted support for the existence of a vicious cycle.
Based on the above evidence, researchers proposed two complementary theoretical models: The bidirectional feedback loop model depicts a cyclical amplification process where “abnormal α-synuclein activates NLRP3 → caspase-1 activation promotes α-synuclein aggregation → pyroptosis releases pathological proteins → dissemination to neighboring cells triggers a new round of inflammation”. 8,30 Metabolic regulation models reveal that Parkin deficiency may impair NLRP3 ubiquitination and degradation, with accumulated mitochondrial reactive oxygen species (mitoROS) further amplifying inflammatory signaling and promoting α-synuclein pathology within neurons.31,32 These two models elucidate the potential molecular basis for the bidirectional vicious cycle between α-synuclein and NLRP3 from distinct perspectives.
Central-Peripheral Immune Interaction Network
Neuroinflammation in PD extends beyond the central nervous system (CNS), involving complex central-peripheral immune interactions. The NLRP3 inflammasome, as a core regulatory component of the innate immune system, plays a pivotal role in this interactive network. It synergistically drives the pathological progression of PD through the gut-brain axis, peripheral immune cell infiltration, and systemic inflammatory effects.
The Bridging Role of NLRP3 in the Gut-Brain Axis
The gut-brain axis serves as a bidirectional communication pathway linking the gut and brain, with NLRP3 inflammasomes playing a pivotal bridging role. Preclinical studies indicate that dysbiosis of the gut microbiota can simultaneously activate NLRP3 inflammasomes in both the colon and brain, compromising the integrity of the intestinal barrier and blood-brain barrier (BBB). This leads to the dissemination of peripheral inflammatory factors to the central nervous system, ultimately triggering cognitive dysfunction.33 In the context of PD, existing evidence supports the existence of a “microbiota-gut-inflammasome-brain axis” pathological mechanism, wherein gut microbiota dysbiosis mediates peripheral-central immune interactions by activating NLRP3 inflammasomes, thereby contributing to neuroinflammation and neurodegeneration.34 Environmental stressors (such as heat stress) can also indirectly affect CNS function by altering the composition of the gut microbiota and its metabolites, thereby disrupting the homeostasis of the microbiota-gut-brain axis.35 However, there is currently a lack of direct clinical evidence confirming the causal relationship of NLRP3 in the gut-brain axis of PD.34
Effects of Gut Microbiota Dysbiosis on NLRP3 Activation
Dysbiosis of the gut microbiota is a key trigger for activating NLRP3 inflammasomes. Animal models confirm that stress states such as chronic sleep deprivation can lead to gut dysbiosis, significantly upregulating NLRP3 expression in the colon and brain. This is accompanied by increased permeability of the intestinal barrier and blood-brain barrier (BBB), ultimately inducing neuroinflammation and neuronal damage.33 Downregulating NLRP3 expression in the intestine effectively protects intestinal barrier function, reduces peripheral inflammatory factor levels, and decreases NLRP3 activation in the brain, thereby improving neurological deficits.33 In PD, gut microbiota dysbiosis may continuously activate the NLRP3 pathway in intestinal and central immune cells by releasing pathogen-associated molecular patterns (PAMPs) or metabolites, forming a vicious cycle of “gut-derived inflammation-central neurodegeneration”.34
Peripheral Immune Cell Infiltration and Blood-Brain Barrier Disruption
Peripheral immune cell infiltration and BBB dysfunction constitute the core components of central-peripheral immune interactions in PD. Research findings indicate: 1. Peripheral immune activation promotes central inflammation. PD patients exhibit significantly elevated gene expression of NLRP3, ASC, and caspase-1 in peripheral blood mononuclear cells (PBMCs). Furthermore, plasma IL-1β levels correlate positively with motor symptom severity, suggesting peripheral immune activation may influence central pathology via the circulatory system.10 2. NLRP3-Driven BBB Impairment: In cerebral ischemia models, NLRP3 inflammasome activation promotes cerebral edema and blood-brain barrier (BBB) disruption. Its inhibitor MCC950 mitigates BBB damage and downregulates aquaporin-4 (AQP4) expression.36 Notably, in Parkinson’s disease models, the NLRP3 inhibitor MCC7840 has also been shown to improve BBB permeability,8 suggesting that NLRP3-mediated BBB damage mechanisms may share cross-disease commonalities.8 3. Immune cell barrier-crossing migration: In α-synuclein transgenic mice, NLRP3 activation significantly correlates with CD4+ and CD8+ T cell infiltration in the substantia nigra region; NLRP3 inhibition reduces T cell infiltration and microglial activation, thereby protecting dopaminergic neurons.7 Furthermore, bone marrow-derived macrophages undergo NLRP3 inflammasome activation upon MPP⁺ stimulation, and these activated immune cells may enter the CNS through a compromised blood-brain barrier.37
Synergistic Effects of Systemic Inflammation and Central Nervous System Inflammation
Systemic inflammation and central nervous system inflammation form a synergistic amplification effect through the NLRP3 pathway: 1. Liver-brain axis interaction: In MPTP-induced PD mouse models, inhibiting hepatic NLRP3 inflammasomes reduces peripheral proinflammatory cytokine release, thereby mitigating midbrain microglial activation and dopaminergic neuron loss. This confirms that hepatogenic inflammation exacerbates central neurodegeneration via systemic pathways.38 2. Circulating Inflammatory Mediators and Vesicular Transport: Extracellular vesicles enriched with NLRP3 were detected in the plasma of PD patients, with their levels significantly correlated with disease status. This suggests that circulating vesicles may serve as carriers mediating the transmission of peripheral inflammatory signals to the central nervous system.39 3. Inflammatory Cascade Amplification: Local inflammation triggered by NLRP3 activation in the gut can compromise the intestinal barrier, allowing endotoxins (such as LPS) to enter the bloodstream and activate systemic immune responses. Concurrently, disruption of the blood-brain barrier facilitates the entry of peripheral inflammatory cells and factors into the CNS, forming a positive feedback loop with NLRP3 activation in microglia. This ultimately amplifies neuroinflammation and neuronal damage.33,38,39
Clinical Research Advances on NLRP3-Related Molecules as Biomarkers for PD
The pivotal role of NLRP3 inflammasomes and their associated molecules in the pathological progression of Parkinson’s disease (PD) provides a theoretical basis for their use as biomarkers for clinical diagnosis and disease monitoring. Current research primarily focuses on the diagnostic value of NLRP3 pathway-related molecules in bodily fluids and peripheral blood mononuclear cells (PBMCs).
Clinical Value of Body Fluid Biomarkers
Cerebrospinal fluid (CSF) testing directly reflects the inflammatory state of the central nervous system. Studies indicate that levels of NLRP3 downstream effector molecules IL-1β, IL-6, and TNF-α are significantly elevated in the CSF of PD patients, while neurofilament light chain (NfL), a marker of axonal damage, also shows an upward trend.40 However, cerebrospinal fluid acquisition is invasive and unsuitable for routine screening and dynamic monitoring, limiting its clinical application.
Blood testing is more aligned with clinical practice due to its convenience and reproducibility. Serological studies have revealed significant differences in serum levels of TNF and neutrophil gelatinase-associated lipocalin (NGAL) between PD patients and healthy controls.40 Serum IL-8 levels showed a positive correlation with PD clinical scores, suggesting that inflammatory cytokines may serve as supplementary indicators for assessing disease severity.41 The monocyte-specific marker sCD163 is elevated in the cerebrospinal fluid of patients with late-stage Parkinson’s disease and correlates with cognitive decline, potentially serving as an early warning indicator for cognitive deterioration.42
Diagnostic Value of Peripheral Blood Mononuclear Cells
Peripheral blood mononuclear cells (PBMCs) provide a convenient window for exploring peripheral immune activation, as their collection is non-invasive and reproducible, making them suitable for clinical application. Studies have found that gene expression and protein levels of NLRP3, ASC, and caspase-1 in PBMCs from PD patients are significantly higher than in healthy controls.10 Molecular levels of NLRP3 pathway molecules derived from PBMCs positively correlated with plasma IL-1β and α-synuclein concentrations, and showed significant associations with Hoehn-Yahr staging and UPDRS III scores.10 It may serve as a non-invasive biomarker reflecting the progression of motor symptoms in Parkinson’s disease. Additionally, PD patients exhibit significantly reduced peripheral blood lymphocyte counts and elevated neutrophil-to-lymphocyte ratios (NLR), with variations observed across different subtypes (eg, sporadic and GBA-related forms).43 These indicators have been widely adopted in clinical testing and possess a solid foundation for broader.
Combined Detection Strategy: The Integrative Value of NLRP3 and Exosomal α-Synuclein
In recent years, peripheral blood exosome detection has opened new avenues for PD biomarker research. Exosomes are nanoscale vesicles secreted by cells that can carry proteins and nucleic acids from their source cells. Detection of α-synuclein in exosomes derived from the central nervous system indirectly reflects pathological changes within the brain. Its levels are significantly elevated in PD patients and correlate with disease severity. This method combines non-invasiveness with central nervous system specificity, offering new possibilities for early PD diagnosis.
It is noteworthy that the pathological essence of PD lies in a systemic inflammatory network involving “central-peripheral” interactions, and a single biomarker often fails to comprehensively reflect its complex processes. Therefore, a combined detection strategy—integrating NLRP3-associated molecules reflecting peripheral immune activation (such as NLRP3 expression in PBMCs and serum IL-1β levels) with CNS-derived exosomal α-synuclein reflecting central pathological burden—holds significant clinical value. The rationale for this approach lies in the bidirectional relationship between peripheral and central pathways: peripheral inflammatory activation may accelerate central α-synuclein aggregation and dissemination, while central pathological burden can drive peripheral immune dysregulation. Combined detection provides a comprehensive assessment of disease status from both “peripheral immunity” and “central pathology” dimensions, potentially enhancing early PD identification and aiding differentiation from other neurodegenerative diseases. Preliminary studies suggest superior sensitivity and specificity compared to any single biomarker, though large-scale clinical validation remains pending.
Challenges in Clinical Translation
Despite progress, the clinical translation of NLRP3-related biomarkers faces multiple challenges. Insufficient biomarker specificity remains the primary issue. While serum NfL reflects neural damage, it is elevated across various neurological disorders.44 Elevated inflammatory markers such as IL-1β and TNF-α are also observed in other inflammatory diseases, making them inadequate for PD differential diagnosis when used alone. Lack of standardized testing constrains clinical adoption: inconsistencies in assay methods, antibody selection, and cutoff values across studies hinder cross-study comparisons. Disease heterogeneity significantly impacts interpretation: peripheral inflammatory patterns vary across PD subtypes,43 suggesting a single biomarker may not cover all patient populations. Furthermore, most studies employ cross-sectional designs, necessitating large-scale prospective cohort validation to establish longitudinal associations between biomarkers and disease progression.
Overall, detecting NLRP3 pathway molecules in PBMCs, measuring serum inflammatory factor panels, and analyzing peripheral blood exosome α-synuclein represent the most clinically promising approaches at present. These methods feature readily obtainable samples and allow for repeated monitoring, with some indicators already possessing a foundation for clinical testing. Future research should focus on standardizing detection protocols and validating clinical utility through large-scale longitudinal studies. This will advance NLRP3-related biomarkers from research to clinical practice, providing practical tools for early PD identification and disease monitoring.
Therapeutic Strategies Targeting NLRP3 and Their Clinical Translation
Given the central role of NLRP3 inflammasome in neuroinflammation during Parkinson’s disease (as shown in Table 1), targeting this pathway has emerged as a highly promising therapeutic strategy. In recent years, preliminary progress has been made in translating basic research into clinical applications. However, significant variations exist in the translational potential, feasibility, and limitations of different intervention approaches, necessitating rational evaluation. The landscape of therapeutic strategies aiming to modulate the NLRP3 inflammasome pathway in PD is diverse, encompassing direct inhibitors, natural compounds, and combination approaches. For clarity, the key strategies, their mechanisms, and developmental status are summarized in Table 2.
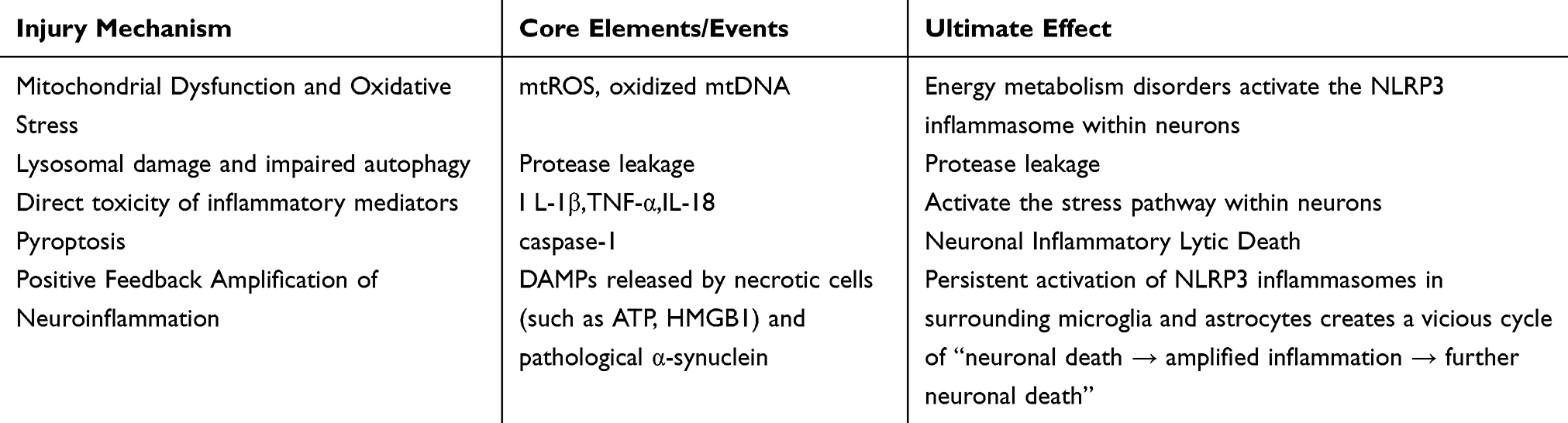 |
Table 1 Neuronal Damage Mechanisms Mediated by NLRP3 Inflammasome Activation |
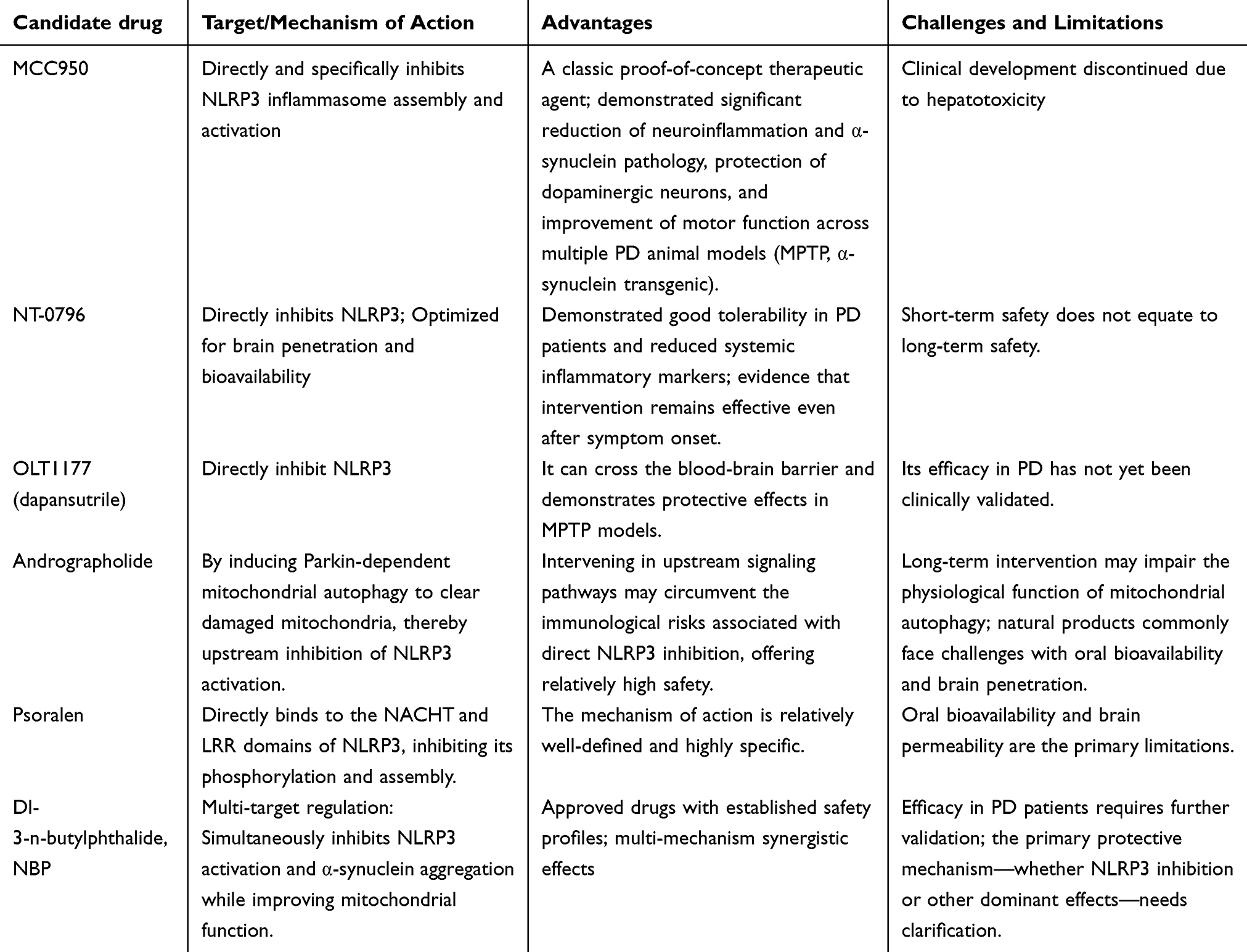 |
Table 2 Key NLRP3-Targeting Approaches for Parkinson’s Disease |
Clinical Research Progress of Small-Molecule Inhibitors
Small-molecule NLRP3 inhibitors represent the fastest-progressing direction in current clinical translation. As a classic inhibitor, MCC950 has been demonstrated to significantly reduce microglial activation, α-synuclein aggregation, and dopaminergic neuron loss in MPTP-induced mouse models and α-synuclein transgenic models, while also improving motor dysfunction.7,30 However, MCC950 did not advance to clinical trials due to liver toxicity. This incident underscores the necessity of long-term safety assessments for NLRP3 inhibitors. NT-0796, developed based on this foundation, has entered early-stage clinical trials. This drug optimizes brain penetration and bioavailability. A 28-day trial in PD patients demonstrated good tolerability and reduced systemic inflammatory markers.8 Notably, this study confirms that NLRP3 inhibitors remain effective when administered after symptom onset. This suggests NLRP3 inhibitors may possess disease-modifying potential beyond early intervention. However, short-term tolerability does not equate to long-term safety. Given NLRP3’s physiological role in antimicrobial immunity, chronic suppression over years or even decades may increase infection risk—a concern that requires resolution through large-scale long-term follow-up studies. OLT1177 has been demonstrated to cross the blood-brain barrier and protect dopaminergic neurons in the MPTP model.45 Having already shown safety in osteoarthritis clinical trials, confirmation of its efficacy in PD could significantly accelerate the clinical translation process. This suggests that the “old drug, new use” strategy holds unique advantages in NLRP3-targeted therapy.
Regulatory Effects of Natural Products and Traditional Chinese Medicines
Natural products offer a diverse source with relatively high safety profiles, providing a rich library of candidate molecules for NLRP3-targeted therapies. However, research in this field often prioritizes phenomena over mechanisms, with most studies confined to cellular and animal models, resulting in limited clinical evidence. Based on their mechanisms of action, these compounds can be categorized as follows: 1. Mitochondrial autophagy-promoting agents: Andrographolide clears damaged mitochondria by inducing Parkin-dependent mitochondrial autophagy, thereby inhibiting NLRP3 activation.46 The advantage of this strategy lies in its ability to intervene in NLRP3 upstream signaling, theoretically avoiding the immune risks associated with direct NLRP3 inhibition. However, given the extensive physiological functions of mitochondrial autophagy, long-term intervention may carry unknown consequences. 2. Direct NLRP3 Assembly Inhibitors: Psoralen binds to NLRP3’s NACHT and LRR domains, inhibiting its phosphorylation.47 These drugs exhibit high specificity, but oral bioavailability and brain penetration remain common limitations of natural products. 3. Multi-target Regulation Category: Dl-3-n-butylphthalide (NBP) is a domestically developed Class I novel drug in China, currently used clinically for ischemic stroke treatment. It has been demonstrated in MPTP and 6-OHDA models to simultaneously inhibit NLRP3 activation and α-synuclein aggregation while improving mitochondrial function.48 NBP’s clinical availability positions it as a prime candidate for extension into PD therapy, though its efficacy in PD patients requires clarification—whether it stems from NLRP3 inhibition or other dominant mechanisms.
Overall, despite their diverse structures, most natural products exert their effects by modulating NLRP3 upstream signaling, demonstrating a “multi-target, multi-pathway” characteristic. However, natural product research still faces multiple hurdles—including dose conversion, bioavailability, and metabolic stability—to bridge the gap from “in vitro efficacy” to “in vivo efficacy” and ultimately to “clinical efficacy”.
Rational Design of Combination Therapy Strategies
Combination therapy aims to enhance efficacy through multi-target synergistic intervention, and its design should be based on clear mechanistic complementarity rather than blindly stacking agents. 1. The mechanism underlying combination with levodopa is that levodopa not only replenishes dopamine, but dopamine itself can inhibit NLRP3 activation in microglia via D1/D2 receptors.9 Therefore, combining levodopa with NLRP3 inhibitors may achieve dual objectives of “symptom control” and “inflammation suppression”. Observations of comparable efficacy in MPTP models25 suggest potential overlapping effects, necessitating further research to optimize combination regimens. 2. Strategies combining autophagy inducers are based on a dual intervention logic of “inhibiting activation + promoting clearance”. Autophagy inducers such as andrographolide promote NLRP3 degradation,46 and their combination with NLRP3 inhibitors synergistically suppresses inflammation at multiple levels. However, autophagy is a double-edged sword; excessive activation may induce autophagic cell death, necessitating precise definition of the safety window. 3. The concept of combining NLRP3 inhibitors with neurotrophic support remains theoretical. While NLRP3 inhibitors can mitigate inflammatory damage, they struggle to directly promote neuronal repair. Theoretically, pairing them with neurotrophic factors or stem cell therapies could address these limitations. However, the complexity of such combined regimens would significantly increase the difficulty of clinical validation.
It is worth noting that while combination therapy holds theoretical advantages, it also presents practical challenges such as drug interactions, cumulative side effects, and complex clinical validation. Therefore, combination strategies should be based on addressing inherent limitations rather than simply piling on more drugs under the assumption that more is better.
The Blood-Brain Barrier and Practical Considerations for Long-Term Safety
Blood-brain barrier penetration remains a core challenge that cannot be avoided in NLRP3-targeted therapies. Although compounds such as NT-0796 and OLT1177 have demonstrated BBB penetration capabilities,8,45 most candidate molecules still rely on delivery systems. The rabies virus glycopeptide (RVG)-modified siRNA delivery system (CMV-RVG-9dR-siRNLRP3) enables brain targeting,27 but its complexity and cost limit clinical adoption. Non-invasive routes like intranasal administration can bypass the BBB, yet precise control over dosage and intracerebral distribution remains challenging.
A more fundamental challenge lies in defining the therapeutic window for NLRP3 inhibitors. On one hand, NLRP3 participates in anti-pathogen immunity, and long-term suppression may increase infection risk—a concern particularly noteworthy in elderly PD patients. On the other hand, NLRP3 performs physiological functions in both central and peripheral systems, and complete inhibition could lead to unknown consequences. Existing short-term clinical trials are insufficient to address these questions; future long-term follow-up and real-world data accumulation are needed to establish safety evidence. Another question worthy of deep consideration is: Who is suitable for NLRP3-targeted therapy? Preclinical studies indicate that NLRP3 activation mechanisms differ across PD models (eg, LRRK2 mutant, sporadic),43 suggesting not all PD patients may benefit equally from NLRP3 inhibition. Future efforts should integrate biomarkers (eg, NLRP3 expression levels in PBMCs, serum IL-1β) to identify “inflammatory” PD subgroups, enabling precision medicine rather than a one-size-fits-all approach.
In summary, therapeutic strategies targeting NLRP3 are at a critical juncture in their transition from basic research to clinical application. The most promising avenues for near-term translation include: Clinical validation of brain-penetrant small-molecule inhibitors (eg, NT-0796). Exploring repurposing of approved drugs (eg, dibenzoylmethane) for new indications; and biomarker research to identify patient populations benefiting from NLRP3 inhibition based on inflammatory markers like NLR and IL-1β. These directions collectively point to a core challenge: how to select the “right treatment” for the “right patients” at the “right time” within the highly heterogeneous disease of PD. Addressing this question may hold greater clinical significance than developing yet another novel drug.
Mechanistic Controversies and Practical Considerations for Clinical Translation
Although the role of NLRP3 inflammasome in Parkinson’s disease neuroinflammation has been extensively demonstrated, several unresolved controversies and practical challenges remain in the transition from basic research to clinical application. Rationally examining these issues helps avoid unrealistic expectations for targeted therapies and guides future research directions.
Causality Debate in NLRP3 Activation: Driving Factor or Concomitant Phenomenon?
Whether LRP3 inflammasome activation in PD serves as the primary driver of neurodegeneration or merely a secondary concomitant phenomenon following neuronal injury remains a central controversy. This distinction directly informs therapeutic strategy positioning: if it is the driver, early intervention holds significant promise; if it is merely a concomitant phenomenon, targeted therapies may yield limited efficacy, potentially addressing symptoms rather than the root cause.
Key evidence supporting its role as a driving factor comes from animal models: In MPTP-induced mouse models, NLRP3 gene knockout significantly mitigates dopaminergic neuron loss and motor deficits, whereas microglia-specific expression of an active NLRP3 mutant exacerbates the pathological phenotype.37 Clinical correlation studies further demonstrate elevated expression of NLRP3, ASC, and caspase-1 in peripheral blood mononuclear cells, along with increased plasma IL-1β levels in PD patients, positively correlated with disease severity,10 providing indirect support for the driver hypothesis.
However, Mendelian randomization studies found no significant association between genetic expression alterations of NLRP3, IL-1β, or IL-18 and PD risk or progression.49 This finding poses a substantial challenge to the “driver” hypothesis—if the NLRP3 pathway were an upstream driver, its genetic variants should influence disease risk, yet this is not the case. This suggests NLRP3 is more likely an “amplifier” of disease progression rather than an “initiator”.
A more plausible integrated explanation is that in the early stages of the disease, initial pathological events such as peripheral inflammation and α-synuclein aggregation activate NLRP3. This complex then amplifies the inflammatory response through mechanisms like IL-1β release and pyroptosis induction, accelerating neuronal damage. Subsequently, the damaged neurons release more DAMPs, further activating NLRP3 and creating a vicious cycle. In this model, NLRP3 serves both as a “responder” to initial injury and a “driver” of chronic progression. Clinically, this insight suggests that NLRP3-targeted therapies may be more effective for patients already in the inflammatory amplification phase, rather than being universally applicable to all PD patients. It also implies that simply inhibiting NLRP3 is unlikely to halt disease onset but holds promise for slowing disease progression.
Cell-Specific Effects and the Complexity of Regulatory Networks
The activation of NLRP3 inflammasomes is not uniformly distributed across all cell types, and this cell specificity adds further complexity to their role in PD. Studies indicate that NLRP3 activation primarily occurs in microglia,30,50 but NLRP3 inflammasomes expressed by dopaminergic neurons themselves may also directly contribute to neurodegeneration.51 The relative contributions of these two sources of NLRP3 activation—the “parainflammation” from immune cells and the “autoinflammation” from neurons—to disease progression remain unclear and require analysis using cell-specific intervention models (eg, microglia-specific Nlrp3 knockout mice). If neuronal NLRP3 activation constitutes the primary pathway for neuronal death, therapeutic strategies targeting microglia may yield limited efficacy. Conversely, if microglial NLRP3 activation drives inflammatory amplification, targeting microglia becomes a more rational therapeutic approach.
The regulatory network governing NLRP3 activity exhibits multi-layered complexity with numerous nodes. Upstream regulatory mechanisms include: ubiquitin-mediated degradation (Parkin-mediated degradation defects exacerbate inflammation32), autophagy/ mitophagy (mitophagy defects promote NLRP3 activation,46 while quercetin inhibits NLRP3 by promoting autophagy52), transcription factors (eg, C/EBPβ participates in NLRP3 transcriptional regulation53), microRNAs (eg, miR-7,54 miR-188-3p55 regulate NLRP3 expression at the post-transcriptional level), ion channels (eg, Kv1.3 participates in NLRP3 activation in microglia6), and neurotransmitter regulation (dopamine inhibits NLRP3 via D1/D2 receptors, and this negative feedback fails with neuronal loss6,9). These regulatory nodes intertwine to form a complex signaling network, where abnormalities in any node may alter NLRP3 activation thresholds and intensity.
The clinical implication of this complexity is that simply inhibiting NLRP3 itself may struggle to block the continuous influx of multiple upstream activation signals. Conversely, intervening in upstream common pathways—such as enhancing autophagy or protecting mitochondria—might offer more comprehensive suppression of NLRP3 activation. However, such interventions could also trigger broader biological effects, making the safety window harder to define.
Mechanistic Heterogeneity Among Different PD Models: The “Reproducibility Crisis” in Preclinical Research
The activation mechanisms and functions of NLRP3 exhibit significant heterogeneity across different PD models. This phenomenon warrants careful consideration and partially explains why certain interventions that demonstrate remarkable efficacy in animal models may fail in human subjects.
Regarding the activation mechanism of α-synuclein, human microglia exhibit weaker dependence on caspase-1 enzyme activity compared to mouse microglia,5 suggesting that findings based on mouse-derived cells may overestimate the efficacy of certain interventions in humans. More notably, β-hydroxybutyrate inhibits ATP-induced NLRP3 activation but fails to block α-synuclein fibril activation.29 This implies that inhibitors effective in PD-specific pathological contexts may be missed by conventional ATP-based screening systems. It also suggests that NLRP3 inhibitor strategies from other disease areas cannot be readily applied to PD.
Model type also influences outcomes: In Parkin mutation models, USP9X inhibition improves NLRP3 activation.56 Conversely, NLRP3 deficiency confers protective effects in MPTP models.37 In the LRRK2-G2019S mutation model, NLRP3 exhibits spontaneous activation even without stimulation.57 This heterogeneity strongly suggests that PD patients with different etiological subtypes may respond differently to NLRP3-targeted therapies. This implies that distinct intervention strategies may be required for genetic versus sporadic forms, as well as for early-onset versus late-onset PD.
This phenomenon offers insights for clinical translation: no single drug can be suitable for all PD patients. Future clinical trials should incorporate inflammation-related biomarkers into their inclusion criteria, prioritizing the selection of the “NLRP3-activated” patient subgroup rather than targeting the entire PD population indiscriminately. This also implies that the beneficiary population for NLRP3-targeted therapies may be narrower than anticipated, necessitating a rational assessment of their commercial development value.
Safety Concerns Regarding Long-Term Suppression of NLRP3
Preclinical studies indicate that short-term use of NLRP3 inhibitors (such as MCC950 and OLT1177) can reduce neuroinflammation and improve motor function.7,8,45 The oral inhibitor NT-0796 demonstrated good tolerability in a 28-day trial involving PD patients.26 However, short-term safety does not equate to long-term safety. As a chronic progressive disease, PD requires sustained intervention over years or even decades, necessitating vigilance regarding the following risks. Immune defense impairment is the primary concern. NLRP3 participates in anti-pathogen immunity, and its prolonged suppression may increase infection risk.15,28
1. Most PD patients are elderly with inherently compromised immune function. Infectious complications such as community-acquired pneumonia are already common causes of death in advanced patients; the risk may multiply when compounded by drug-induced immunosuppression.
2. Compensatory activation of other inflammatory pathways may diminish therapeutic efficacy. Following NLRP3 inhibition, alternative inflammatory pathways (such as NLRC4 and AIM2) may undergo compensatory activation,58 leading to reduced therapeutic response. This phenomenon has been suggested in animal studies, though its long-term implications remain unclear. Should compensatory activation indeed occur, it implies that NLRP3 inhibition alone may prove insufficient for sustained inflammation control, necessitating combined intervention targeting additional pathways.
3. The potential for NLRP3 inhibitors to interfere with cellular clearance mechanisms cannot be overlooked. NLRP3 degradation relies on autophagy-lysosomal pathways (such as Parkin-mediated ubiquitin-dependent degradation).32 Prolonged NLRP3 inhibition may disrupt cellular clearance mechanisms, thereby exacerbating the accumulation of pathological proteins like α-synuclein. This presents a potential paradox: suppressing inflammation may simultaneously weaken the cell’s capacity to clear pathological proteins.
4. Additionally, dopamine itself can inhibit NLRP3 activation in microglia through D1/D2 receptors,9 forming an endogenous regulatory network. Long-term exogenous intervention may disrupt this balance, leading to unknown consequences. For example, following exogenous NLRP3 inhibition, would dopamine’s endogenous inhibitory effect be downregulated compensatorily? This question remains unanswered.
Moreover, most current animal studies span only weeks to months,59 lacking systematic evaluation of long-term side effects.25 Given the absence of long-term safety data, NLRP3 inhibitors should be introduced into clinical practice with caution. Crucially, clinical trial designs must incorporate infection events and tumor incidence as secondary endpoints for long-term follow-up.
From Controversy to Clinical Practice: Several Issues Worth Considering
Timing Considerations: What is the optimal intervention window for NLRP3-targeted therapy? Animal models suggest that early suppression of NLRP3 yields superior outcomes.7 While administration after symptom onset may still improve neurological function,8 its efficacy may fall short of early intervention. For patients in the middle-to-late stages with significant neuronal loss, whether inflammation suppression alone can deliver clinical benefits remains to be validated. Theoretically, once neuronal loss exceeds a threshold, functional deficits may be irreversible even with complete inflammation blockade. This suggests NLRP3 inhibitors may be better suited for early-stage patients, yet this conflicts with the reality of challenging early PD diagnosis.
Population Issues: Who is most likely to benefit from NLRP3-targeted therapy? Screening “inflammatory” PD patients based on peripheral blood NLRP3 expression levels or serum IL-1β concentrations may serve as a reasonable starting point. However, it should be noted that peripheral inflammation does not fully parallel central inflammation, and whether peripheral markers accurately reflect central NLRP3 activation status remains to be validated. A more fundamental question is: even if NLRP3 activation is present, its differential distribution across cell types (glia vs. neurons) will influence therapeutic strategy selection.
Advantages and Disadvantages: What advantages do NLRP3 inhibitors offer compared to existing anti-inflammatory drugs (such as NSAIDs)? Clinical trial results for NSAIDs in PD have been inconsistent, with most showing negative outcomes, suggesting that non-specific anti-inflammatory approaches may not be effective strategies. NLRP3 inhibitors exhibit higher specificity, but this also implies a narrower scope of application, necessitating precise selection of beneficiary populations. The question is: Does the benefit gained from this “precision” sufficiently justify the clinical advantage and commercial value of NLRP3 inhibitors relative to existing therapies?
Endpoint Issues: How Should Clinical Trials Evaluate Efficacy? NLRP3 inhibitors are classified as “disease-modifying therapies”, and their efficacy assessment cannot rely solely on motor symptom improvement, as such improvements may stem from the interference of symptomatic treatment with dopaminergic medications. Ideally, a comprehensive assessment should integrate imaging markers (eg, DAT-PET), biomarkers (eg, plasma NfL), and long-term disease progression (eg, time to Hoehn-Yahr staging progression). This undoubtedly increases the complexity and cost of clinical trials while imposing higher demands on trial design.
In summary, the NLRP3 inflammasome serves as a central coordinator of PD neuroinflammation, driving disease progression through a vicious cycle with α-synuclein pathology. Targeting this pathway holds significant disease-modifying therapeutic potential, but current translational bottlenecks must be overcome by: deciphering cell-specific mechanisms; understanding the complexity of regulatory networks; defining precise intervention timings; developing brain-targeted delivery systems; integrating multi-omics-guided personalized strategies; and conducting rigorous clinical trials. Therefore, before advancing new drugs into clinical translation, it is imperative to first clarify the target patient population, optimal intervention timing, and therapeutic strategies. Only then can NLRP3 inhibitors truly transition from the laboratory to the clinic, delivering tangible benefits to suitable PD patients rather than becoming yet another case of “effective in animal models, disappointing in humans”.
Despite these challenges, targeting the NLRP3 inflammasome pathway remains a highly promising direction for disease-modifying therapies in Parkinson’s disease. Future research should focus on: (1) precisely identifying “inflammatory-type” beneficiaries using peripheral and central biomarkers; (2) optimizing intervention timing within the disease progression; (3) Developing safe, efficient, and specific brain-targeted delivery strategies; (4) Exploring mechanism-complementary combination therapies to maximize efficacy. Through this multidimensional, precision-driven R&D pathway, the theoretical potential of the NLRP3 pathway may be translated into clinical reality that tangibly improves Parkinson’s disease progression.
Data Sharing Statement
Any additional data from this study are available from the corresponding author upon reasonable request.
Acknowledgments
We wish to acknowledge Chengdu Medical College-Chengdu No. 7 People’s Hospital Joint Scientific Research Fund Project (23LHQYZ-01) AND 2025 Sichuan Provincial Health Commission Medical Science and Technology Project (No. 25LCYJ45) for their contribution to this study.
Author Contributions
Keting Liu: Conceptualization, Methodology, Investigation, Writing - Original Draft, Visualization. Li‑hao Zhang and Ze‑min He: Conceptualization, Methodology, Investigation, Visualization. Yong‑ming Wu: Conceptualization, Supervision, Writing - Review & Editing. Li Xiao: Conceptualization, Writing - Review & Editing, Funding Acquisition, Supervision. All authors have read and approved the final version of the paper submitted for publication. All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Chengdu Medical College-Chengdu No. 7 People’s Hospital Joint Scientific Research Fund Project (23LHQYZ-01) AND 2025 Sichuan Provincial Health Commission Medical Science and Technology Project (No. 25LCYJ45).
Disclosure
The authors declare no conflicts of interest.
References
1. Tansey MG, Wallings RL, Houser MC, et al. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol. 2022;22(11):657–17. doi:10.1038/s41577-022-00684-6
2. Xu W, Huang Y, Zhou R. NLRP3 inflammasome in neuroinflammation and central nervous system diseases. Cell Mol Immunol. 2025;22(4):341–355. doi:10.1038/s41423-025-01275-w
3. Paik S, Kim JK, Shin HJ, et al. Updated insights into the molecular networks for NLRP3 inflammasome activation. Cell Mol Immunol. 2025;22(6):563–596. doi:10.1038/s41423-025-01284-9
4. Li Y, Xia Y, Yin S, et al. Targeting Microglial α-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front Immunol. 2021;12:719807. doi:10.3389/fimmu.2021.719807
5. Pike AF, Varanita T, Herrebout M, et al. α-Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia. 2021;69(6):1413–1428. doi:10.1002/glia.23970
6. Pike AF, Szabò I, Veerhuis R, et al. The potential convergence of NLRP3 inflammasome, potassium, and dopamine mechanisms in Parkinson’s disease. NPJ Parkinsons Dis. 2022;8(1):32. doi:10.1038/s41531-022-00293-z
7. Grotemeyer A, Fischer JF, Koprich JB, et al. Inflammasome inhibition protects dopaminergic neurons from α-synuclein pathology in a model of progressive Parkinson’s disease. J Neuroinflammation. 2023;20(1):79. doi:10.1186/s12974-023-02759-0
8. Albornoz EA, Mardon K, Bhalla R, et al. PET-MRI biomarkers reveal efficacy of a novel NLRP3 inhibitor in Parkinson’s disease models. Brain. 2025. doi:10.1093/brain/awaf372
9. Pike AF, Longhena F, Faustini G, et al. Dopamine signaling modulates microglial NLRP3 inflammasome activation: implications for Parkinson’s disease. J Neuroinflammation. 2022;19(1):50. doi:10.1186/s12974-022-02410-4
10. Fan Z, Pan YT, Zhang ZY, et al. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation. 2020;17(1):11. doi:10.1186/s12974-019-1670-6
11. Harms AS, Ferreira SA, Romero-Ramos M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021;141(4):527–545. doi:10.1007/s00401-021-02268-5
12. Wang L, Hauenstein AV. The NLRP3 inflammasome: mechanism of action, role in disease and therapies. Mol Aspects Med. 2020;76:100889. doi:10.1016/j.mam.2020.100889
13. Paik S, Kim JK, Silwal P, et al. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18(5):1141–1160. doi:10.1038/s41423-021-00670-3
14. McKee CM, Coll RC. NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J Leukoc Biol. 2020;108(3):937–952. doi:10.1002/JLB.3MR0720-513R
15. Feng YS, Tan ZX, Wu LY, et al. The involvement of NLRP3 inflammasome in the treatment of neurodegenerative diseases. Biomed Pharmacother. 2021;138:111428. doi:10.1016/j.biopha.2021.111428
16. Hurtado-Navarro L, Angosto-Bazarra D, Pelegrín P, et al. NLRP3 inflammasome and pyroptosis in liver pathophysiology: the emerging relevance of Nrf2 inducers. Antioxidants. 2022;11(5):870. doi:10.3390/antiox11050870
17. Yao J, Wang Z, Song W, et al. Targeting NLRP3 inflammasome for neurodegenerative disorders. Mol Psychiatry. 2023;28(11):4512–4527. doi:10.1038/s41380-023-02239-0
18. Li J, Xu P, Hong Y, et al. Lipocalin-2-mediated astrocyte pyroptosis promotes neuroinflammatory injury via NLRP3 inflammasome activation in cerebral ischemia/reperfusion injury. J Neuroinflammation. 2023;20(1):148. doi:10.1186/s12974-023-02819-5
19. Drinkall S, Lawrence CB, Ossola B, et al. The two pore potassium channel THIK-1 regulates NLRP3 inflammasome activation. Glia. 2022;70(7):1301–1316. doi:10.1002/glia.24174
20. Wang Y, Cao C, Zhu Y, et al. TREM2/β-catenin attenuates NLRP3 inflammasome-mediated macrophage pyroptosis to promote bacterial clearance of pyogenic bacteria. Cell Death Dis. 2022;13(9):771. doi:10.1038/s41419-022-05193-x
21. Wang D, Wu Y, Sun S, et al. NLRP3 inflammasome-mediated pyroptosis involvement in cadmium exposure-induced cognitive deficits via the Sirt3-mtROS axis. Sci Total Environ. 2023;903:166478. doi:10.1016/j.scitotenv.2023.166478
22. Liu H, Zhan X, Xu G, et al. Cryptotanshinone specifically suppresses NLRP3 inflammasome activation and protects against inflammasome-mediated diseases. Pharmacol Res. 2021;164:105384. doi:10.1016/j.phrs.2020.105384
23. Anderson FL, Biggs KE, Rankin BE, et al. NLRP3 inflammasome in neurodegenerative disease. Transl Res. 2023;252:21–33. doi:10.1016/j.trsl.2022.08.006
24. Moonen S, Koper MJ, Van Schoor E, et al. Pyroptosis in Alzheimer’s disease: cell type-specific activation in microglia, astrocytes and neurons. Acta Neuropathol. 2023;145(2):175–195. doi:10.1007/s00401-022-02528-y
25. Huang P, Zhu Z, Li W, et al. rTMS im demonstrates dysphagia by inhibiting NLRP3 inflammasome activation and caspase-1 dependent pyroptosis in PD mice. NPJ Parkinsons Dis. 2024;10(1):156. doi:10.1038/s41531-024-00775-2
26. Clarke N, Thornton P, Reader V, et al. Anti-neuroinflammatory and anti-inflammatory effects of the NLRP3 inhibitor NT-0796 in subjects with Parkinson’s disease. Mov Disord. 2025;40(10):2199–2208. doi:10.1002/mds.30307
27. Lin L, Li Z, Su F, et al. An autonomous siRNA delivery system targeting NLRP3: implications in Parkinson’s disease. BMC Med. 2026;24(1). doi:10.1186/s12916-026-04688-0
28. Harrison D, Billinton A, Bock MG, et al. Discovery of clinical candidate NT-0796, a brain-penetrant and highly potent NLRP3 inflammasome inhibitor for neuroinflammatory disorders. J Med Chem. 2023;66(21):14897–14911. doi:10.1021/acs.jmedchem.3c01398
29. Deora V, Albornoz EA, Zhu K, et al. The ketone body β-hydroxybutyrate does not inhibit synuclein mediated inflammasome activation in microglia. J Neuroimmune Pharmacol. 2017;12(4):568–574. doi:10.1007/s11481-017-9754-5
30. Gordon R, Albornoz EA, Christie DC, et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. 2018;10(465). doi:10.1126/scitranslmed.aah4066
31. Panicker N, Kam TI, Wang H, et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron. 2022;110(15):2422–2437.e9. doi:10.1016/j.neuron.2022.05.009
32. Yan YQ, Zheng R, Liu Y, et al. Parkin regulates microglial NLRP3 and represses neurodegeneration in Parkinson’s disease. Aging Cell. 2023;22(6):e13834. doi:10.1111/acel.13834
33. Zhao N, Chen QG, Chen X, et al. Intestinal dysbiosis mediates cognitive impairment via the intestine and brain NLRP3 inflammasome activation in chronic sleep deprivation. Brain Behav Immun. 2023;108:98–117. doi:10.1016/j.bbi.2022.11.013
34. Pellegrini C, Antonioli L, Calderone V, et al. Microbiota-gut-brain axis in health and disease: is NLRP3 inflammasome at the crossroads of microbiota-gut-brain communications? Prog Neurobiol. 2020;191:101806. doi:10.1016/j.pneurobio.2020.101806
35. Wen C, Wei S, Zong X, et al. Microbiota-gut-brain axis and nutritional strategy under heat stress. Anim Nutr. 2021;7(4):1329–1336. doi:10.1016/j.aninu.2021.09.008
36. Wang H, Chen H, Jin J, et al. Inhibition of the NLRP3 inflammasome reduces brain edema and regulates the distribution of aquaporin-4 after cerebral ischaemia-reperfusion. Life Sci. 2020;251:117638. doi:10.1016/j.lfs.2020.117638
37. Lee E, Hwang I, Park S, et al. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019;26(2):213–228. doi:10.1038/s41418-018-0124-5
38. Qiao C, Zhang Q, Jiang Q, et al. Inhibition of the hepatic Nlrp3 protects dopaminergic neurons via attenuating systemic inflammation in a MPTP/p mouse model of Parkinson’s disease. J Neuroinflammation. 2018;15(1):193. doi:10.1186/s12974-018-1236-z
39. Anderson FL, von Herrmann KM, Andrew AS, et al. Plasma-borne indicators of inflammasome activity in Parkinson’s disease patients. NPJ Parkinsons Dis. 2021;7(1):2. doi:10.1038/s41531-020-00147-6
40. Marinescu AM, Machado V, Pagano G, et al. Cerebrospinal fluid biomarkers of NLRP3 pathway, immune dysregulation, and neurodegeneration in parkinson’s disease: a meta-analysis. Mov Disord. 2025;41:466–477. doi:10.1002/mds.70103
41. Eidson LN, Kannarkat GT, Barnum CJ, et al. Candidate inflammatory biomarkers display unique relationships with alpha-synuclein and correlate with measures of disease severity in subjects with Parkinson’s disease. J Neuroinflammation. 2017;14(1):164. doi:10.1186/s12974-017-0935-1
42. Nissen SK, Ferreira SA, Nielsen MC, et al. Soluble CD163 Changes Indicate Monocyte Association With Cognitive Deficits in Parkinson’s Disease. Mov Disord. 2021;36(4):963–976. doi:10.1002/mds.28424
43. Muñoz-Delgado L, Macías-García D, Periñán MT, et al. Peripheral inflammatory immune response differs among sporadic and familial Parkinson’s disease. NPJ Parkinsons Dis. 2023;9(1):12. doi:10.1038/s41531-023-00457-5
44. Mollenhauer B, Dakna M, Kruse N, et al. Validation of serum neurofilament light chain as a biomarker of parkinson’s disease progression. Mov Disord. 2020;35(11):1999–2008. doi:10.1002/mds.28206
45. Amo-Aparicio J, Daly J, Højen JF, et al. Pharmacologic inhibition of NLRP3 reduces the levels of α-synuclein and protects dopaminergic neurons in a model of Parkinson’s disease. J Neuroinflammation. 2023;20(1):147. doi:10.1186/s12974-023-02830-w
46. Ahmed S, Kwatra M, Ranjan Panda S, et al. Andrographolide suppresses NLRP3 inflammasome activation in microglia through induction of parkin-mediated mitophagy in in-vitro and in-vivo models of Parkinson disease. Brain Behav Immun. 2021;91:142–158. doi:10.1016/j.bbi.2020.09.017
47. Zhu RX, Han RX, Chen YH, et al. Inactivation of NLRP3 inflammasome by dephosphorylation at Serine 658 alleviates glial inflammation in the mouse model of Parkinson’s disease. Mol Neurodegener. 2025;20(1):27. doi:10.1186/s13024-025-00818-z
48. Que R, Zheng J, Chang Z, et al. Dl-3-n-butylphthalide rescues dopaminergic neurons in parkinson’s disease models by inhibiting the NLRP3 inflammasome and ameliorating mitochondrial impairment. Front Immunol. 2021;12:794770. doi:10.3389/fimmu.2021.794770
49. Senkevich K, Liu L, Alvarado CX, et al. Lack of genetic evidence for NLRP3 inflammasome involvement in Parkinson’s disease pathogenesis. NPJ Parkinsons Dis. 2024;10(1):145. doi:10.1038/s41531-024-00744-9
50. Cyr B, Vontell RT, Hadad R, et al. IC100 blocks inflammasome activation induced by α-synuclein aggregates and ASC specks. NPJ Parkinsons Dis. 2025;11(1):92. doi:10.1038/s41531-025-00963-8
51. von Herrmann KM, Anderson FL, Martinez EM, et al. Slc6a3-dependent expression of a CAPS-associated Nlrp3 allele results in progressive behavioral abnormalities and neuroinflammation in aging mice. J Neuroinflammation. 2020;17(1):213. doi:10.1186/s12974-020-01866-6
52. Han X, Sun S, Sun Y, et al. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: implications for Parkinson disease. Autophagy. 2019;15(11):1860–1881. doi:10.1080/15548627.2019.1596481
53. Wang J, Li Y, Xia Y. C/EBPβ as a master regulator of inflammasome signaling in neurodegenerative diseases: mechanisms and therapeutic implications. Front Immunol. 2025;16:1656165. doi:10.3389/fimmu.2025.1656165
54. Zhou Y, Lu M, Du RH, et al. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener. 2016;11:28. doi:10.1186/s13024-016-0094-3
55. Li Q, Wang Z, Xing H, et al. Exosomes derived from miR-188-3p-modified adipose-derived mesenchymal stem cells protect Parkinson’s disease. Mol Ther Nucleic Acids. 2021;23:1334–1344. doi:10.1016/j.omtn.2021.01.022
56. Guo Y, Qi X, Wang A, et al. USP9X suppression attenuates NLRP3 inflammasome activation and ameliorates neuroinflammatory phenotypes with motor function recovery in murine models. Brain Behav Immun. 2026;132:106215. doi:10.1016/j.bbi.2025.106215
57. Ballotto L, Baratta T, Winterberg H, et al. Nuclear ASC speck formation in microglia is associated with inflammasome priming and is exacerbated in LRRK2-G2019S Parkinson’s disease. Neurobiol Dis. 2026;218:107237. doi:10.1016/j.nbd.2025.107237
58. Heinisch O, Zeyen T, Goldmann T, et al. Erythropoietin abrogates post-ischemic activation of the NLRP3, NLRC4, and AIM2 inflammasomes in microglia/macrophages in a TAK1-dependent manner. Transl Stroke Res. 2022;13(3):462–482. doi:10.1007/s12975-021-00948-8
59. Sadier NS, Hazimeh IA, Khazaal W, et al. Exploring the therapeutic potential of NLRP3 inhibitors in Parkinson’s Disease: a systematic review of in-vivo studies. Inflammopharmacology. 2025;33(5):2657–2677. doi:10.1007/s10787-025-01733-x
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Pyroptosis: A Novel Intervention Target in the Progression of Osteoarthritis
Chang X, Kang Y, Yang Y, Chen Y, Shen Y, Jiang C, Shen Y
Journal of Inflammation Research 2022, 15:3859-3871
Published Date: 11 July 2022
Characterization of Cathepsin B in Mediating Silica Nanoparticle-Induced Macrophage Pyroptosis via an NLRP3-Dependent Manner
Ma L, Han Z, Yin H, Tian J, Zhang J, Li N, Ding C, Zhang L
Journal of Inflammation Research 2022, 15:4537-4545
Published Date: 8 August 2022
Inhibition of Dectin-1 Alleviates Neuroinflammatory Injury by Attenuating NLRP3 Inflammasome-Mediated Pyroptosis After Intracerebral Hemorrhage in Mice: Preliminary Study Results
Ding Z, Zhong Z, Wang J, Zhang R, Shao J, Li Y, Wu G, Tu H, Yuan W, Sun H, Wang Q
Journal of Inflammation Research 2022, 15:5917-5933
Published Date: 19 October 2022
Lactobacillus Plantarum Promotes Wound Healing by Inhibiting the NLRP3 Inflammasome and Pyroptosis Activation in Diabetic Foot Wounds
Wang X, Li X, Liu J, Tao Y, Wang T, Li L
Journal of Inflammation Research 2024, 17:1707-1720
Published Date: 16 March 2024
The Impact of NLRP3 Inflammasome on Osteoblasts and Osteogenic Differentiation: A Literature Review
Yang Z, Xu J, Kang T, Chen X, Zhou C
Journal of Inflammation Research 2024, 17:2639-2653
Published Date: 29 April 2024