Back to Journals » Journal of Blood Medicine » Volume 11
The Need for Comprehensive Care for Persons with Chronic Immune Thrombocytopenic Purpura
Authors Ansteatt KT ![]() , Unzicker CJ, Hurn ML, Olaiya OO, Nugent DJ, Tarantino MD
, Unzicker CJ, Hurn ML, Olaiya OO, Nugent DJ, Tarantino MD
Received 10 November 2020
Accepted for publication 4 December 2020
Published 17 December 2020 Volume 2020:11 Pages 457—463
DOI https://doi.org/10.2147/JBM.S289390
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Kristin T Ansteatt,1 Chanel J Unzicker,1 Marsha L Hurn,1 Oluwaseun O Olaiya,2 Diane J Nugent,3 Michael D Tarantino1
1The Bleeding and Clotting Disorders Institute, Peoria, IL, USA; 2Hematology Oncology, Children’s Mercy Hospital, Kansas City, MO, USA; 3The Center for Inherited Blood Disorders, Santa Ana, CA, USA
Correspondence: Michael D Tarantino
The Bleeding and Clotting Disorders Institute, 9128 North Lindbergh Drive, Peoria, IL 61615, USA
Tel +1 309 692-5337
Fax +1 309 693-3913
Email [email protected]
Abstract: Chronic platelet disorders (CPD), including chronic immune thrombocytopenic purpura (cITP), thrombotic thrombocytopenic purpura (TTP) and platelet function disorders are among the most common bleeding disorders and are associated with morbidity and mortality. The clinical phenotype and complexity of cITP is much like that of hemophilia. In cITP and hemophilia, bleeding is problematic for many, complicating employability, insurability and overall quality-of-life (QoL). While myriad drug therapies are available for cITP and hemophilia, each are variable in their effectiveness, very few (except for clotting factor concentrates for hemophilia) alter the natural history of the disorder and sometimes contribute to specific morbidities and mortality. Like in hemophilia, the management of cITP is not solely based on access to effective treatment but also includes accurate diagnosis and comprehensive care by a multidisciplinary team of specialists trained in the management of bleeding disorders. The model of comprehensive care in Hemophilia Treatment Centers (HTCs) has been recognized as highly effective, improving life expectancy for persons with hemophilia. cITP, and other CPDs, are complex disorders requiring specialized care. However, an integrated care model with a systematic and reliable population-based surveillance program does not exist. Extending the Comprehensive Care model with all its related benefits to the community of persons with cITP is sorely needed. This review will focus on cITP as a prototype chronic platelet disorder that could benefit greatly from the Comprehensive Care model.
Keywords: hemophilia, hemorrhage, idiopathic thrombocytopenic purpura, platelet, primary immune thrombocytopenia, thrombocytopenia
Historical Perspectives of Hemophilia and ITP
The analogous history of cITP and hemophilia cannot be overlooked; with similar advancements in recognition, disease characterization and therapy occurring along comparable timelines. In the nineteenth century, both hemophilia and ITP were recognized; ITP was described as morbus maculosus hemorrhagicus and hemophilia was noted to be a bleeding disorder in males of certain families.1,2 Further advancement was made with the use of plasma therapy for hemophilia and splenectomy for treatment of cITP in the first part of the twentieth century.3,4 In the 1950s–1960s, the distinction between Hemophilia A and B was made and cITP was determined to be an autoimmune disorder.5,6 This led to further progress in therapies with cryoprecipitate used for hemophilia A and corticosteroids as treatment for cITP.7,8 In the 1970s-1980s, the discovery of concentrated plasma derived clotting factor allowed for home treatment of hemophilia.9 However, this advancement was dampened by the transmission of human immunodeficiency virus (HIV), hepatitis C virus (HCV) and hepatitis B virus (HBV) to persons with hemophilia.10 This led to widespread fatalities and subsequent improvement in screening methods for blood donors and pathogen attenuation steps in the manufacturing process. During the same era, intravenous immunoglobulin (IVIG) was shown to be an effective treatment for persons with ITP in the 1980s.11 As IVIG is derived from pooled donor plasma, reports of pathogenic virus transmission (HCV, HBV) emerged between the mid-1980s and the mid-1990s.11 This risk was mitigated with the introduction of pathogen attenuation steps during the production of IVIG.11
Advances in the treatment of hemophilia from the last decade of the twentieth century through the first decade of the twenty-first century largely refined the quality of clotting factor concentrates. Recombinant clotting factors emerged in the mid-1990s and are still considered by many to be the standard of care for persons with hemophilia.12 Most recently, non-factor therapies, that either mimic the effect of factor VIII or “rebalance” the hemostatic defect, have benefited patients, especially those with neutralizing antibodies (inhibitors) to clotting factor.13–15 Gene therapy to correct the inherited defect is the next anticipated treatment advancement.16,17
For cITP, treatment options during the later 1990s, until the late 2000s remained largely unchanged, except for the discovery that a hybridized anti-CD20 monoclonal antibody raised the platelet count in around half of the recipients.18 The 2000s saw affirmation to the longstanding theory that many patients with cITP not only have accelerated platelet destruction but also impaired platelet production.19 The thrombopoietin receptor agonists (TPO-RAs) have subsequently become a vital second-line therapy for adults and children with cITP.20 Today, targeted immunotherapy is a major focus of drug development for persons with cITP21 (see Figure 1).
 |
Figure 1 Historical comparison of hemophilia and cITP. Abbreviations: cITP, chronic immune thrombocytopenic purpura; IVIG, intravenous immunoglobulin; HIV, human immunodeficiency virus; HBV, hepatitis B virus; HCV, hepatitis C virus; TPO, thrombopoietin; Syk, spleen tyrosine kinase; mAbs, monoclonal antibodies. |
Epidemiology
Hemophilia is almost always caused by hereditary factor VIII or factor IX deficiency that creates a lifelong bleeding disorder. Hemophilia affects approximately 400,000 persons worldwide with more recent estimates suggesting the prevalence is over 1 million.22,23 In the United States, the prevalence of hemophilia is estimated to be approximately 20,000, based on expected births and deaths since 1994.24 Hemophilia A occurs in 1 in 5000 live male births and is four times as common as hemophilia B.23,24 The overall life expectancy of persons with hemophilia is now comparable to that of the general population.25
cITP is an acquired autoimmune disorder that causes isolated thrombocytopenia and related bleeding. The prevalence of cITP in the United States is approximately 60,000 persons; nearly three times more prevalent than hemophilia.26 The diagnosis of cITP is based primarily on the exclusion of other causes of isolated thrombocytopenia and there are no clinical or laboratory parameters to establish its diagnosis.27 The incidence of cITP is difficult to establish due to limited studies and variable symptomology, the mildest of which falls short of the threshold for platelet count testing.28 However, the strongest estimate of the incidence of acute ITP in children is between 1.9 and 6.4 per 100,000 children/year and in adults, is between 1.6 and 3.9 per 100,000 adults/year.29
Shared Clinical Challenges in Hemophilia and cITP
Hemophilia and cITP are bleeding disorders with a spectrum of phenotypes and much clinical overlap. The clinical phenotype of hemophilia is directly related to the severity of the factor deficiency, but with large variability in the bleeding phenotype among patients with any severity of hemophilia.30 The clinical manifestations of hemophilia include spontaneous bleeding as well as delayed bleeding after trauma. Beginning in infancy, patients may present with a cephalohematoma, subgaleal or intracerebral hemorrhage.31 As they grow into toddlers and then children, they often present with hemarthrosis, intramuscular hemorrhage, soft tissue hematomas, hematuria, and, less commonly, hematemesis, hematochezia and intracranial hemorrhage.31 Intracranial hemorrhage (ICH) remains the main cause of fatal bleeding in persons with hemophilia.
Bleeding associated with hemophilia and cITP are very similar, with the exception that persons with cITP rarely have bleeding in their joints (Table 1). Mucocutaneous, gastrointestinal and genitourinary bleeding are common, fortunately CNS hemorrhage is less common, especially in the young.32,33 The severity of thrombocytopenia correlates directly with the bleeding risk but not entirely, many patients with very low platelet counts have only mild bleeding such as easy bruising.28,34 Comparatively, persons with severe bleeding most often have platelet counts less than 10,000–20,000/µL.35
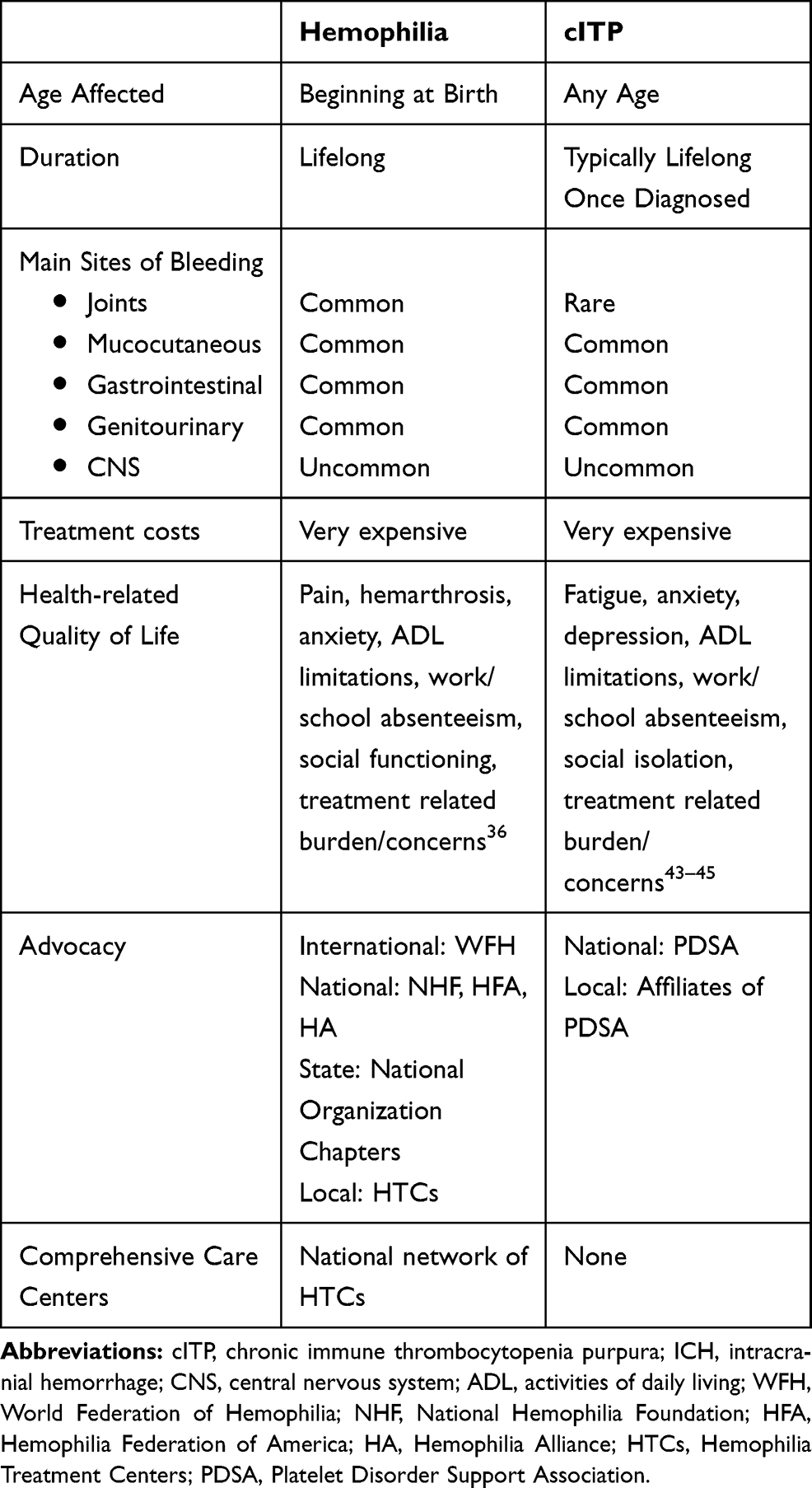 |
Table 1 Comparison of Clinical and Non-Clinical Aspects of Hemophilia and cITP |
Like hemophilia, the main cause of fatal bleeding in patients with ITP is intracranial hemorrhage (ICH), the incidence of which is 0.1–0.5% in children, 1% in adults with acute ITP, and 5% in adults with cITP.32 Severe and life-threatening bleeding affects a subset of persons with hemophilia and cITP which may alter the lifespan of persons with either disorder.33 When thrombocytopenia related to cITP is left untreated, a 70-year-old woman loses 5 quality-adjusted life years (QALY), whereas a 25-year-old woman will lose approximately 15 QALY of potential life expectancy.33
Improving Quality of Life for Persons with cITP
At an early age, persons with hemophilia must learn to adapt to the essential therapies and the overall complexity of the disorder. As treatments and therapies continue to advance, there has been an emphasis on health-related quality of life (HRQoL) as an outcome measure.36 The long-term complications of hemophilia have had effects on HRQoL, revealing that persons with hemophilia experience depression and anxiety more often than the general population.36 In addition, the presence of one or more target joints has a major impact on HRQoL for persons with severe hemophilia.36
Like in hemophilia, it is important to understand how cITP can impact HRQoL (Table 1). The effects of cITP on a person’s HRQoL should not be overlooked; including emotional and functional health, work life, social and leisure activities, and reproductive health.37 Patients characterize the emotional toll by depression, anxiety and report it affecting their overall mental health. In addition, fatigue, fear of bleeding, and the need to limit daily activities are pervasive sequalae of cITP.38 cITP during childhood may negatively impact the HRQoL of the child with cITP and their parents.38 However, when children receive platelet-enhancing therapy to maintain a platelet count in the hemostatic range, HRQoL in children and parental burden improve.39 Although studies of HRQoL have helped in our understanding of the clinical and sociological burden for persons with cITP, more short- and long-term studies are needed to fully appreciate the struggle experienced by many patients.
The Evolution of Comprehensive Care for Hemophilia
Comprehensive Care for persons with hemophilia is defined as the continuous supervision of all medical and psychological aspects affecting the patient and their family.40 Early detection, diagnosis, prevention, treatment, along with psychosocial and educational support are all factors needed to provide optimal management for patients with bleeding disorders.41 Recognized since the 1960s, comprehensive clinical management of hemophilia and related inherited bleeding disorders require a multidisciplinary integrated approach at specialized establishments called Hemophilia Treatment Centers (HTCs).42 Over the past 60 years, this model has been expanded to include patients with other rare inherited or acquired bleeding disorders such as von Willebrand disease, Hereditary Hemorrhagic Telangiectasia, and acquired hemophilias, to name a few.
The approach implemented by Comprehensive Care clinics includes focused education incorporating the basics of hemophilia and other bleeding disorders. These include training about bleeding prevention, managing complications, QoL improvement and self-advocacy.41 In addition, advocacy organizations exist on the local, state, national, and international levels; providing education, empowering individuals, and aiding in financial burdens by actively pursuing long-term relationships with individuals and organizations that align with the tenets of Comprehensive Care.43,44 For patients with hemophilia, HTCs provide multidisciplinary expertise in bleeding disorders, individualized management plans, readily available access to multiple medical disciplines, with a focus on preventative medicine.45 Collectively, this design optimizes effectiveness and efficiency of health care management.
The model of care in comprehensive HTCs has become the standard for persons with hemophilia worldwide. It has improved access to care for many thousands of patients while providing the opportunity for family involvement which improves therapy adherence.40 The HTCs have advocated progress through basic, translational and clinical research. In addition, care at HTCs reduces health care costs, by decreasing emergency room visits, decreasing absenteeism from work or school and preventing life-altering sequalae of hemophilia.45 In another CDC-sponsored study, HTCs were shown to reduce mortality among persons with hemophilia by 40% as well as decrease healthcare resource utilization.45,46 Comprehensive care is one of the essential principles in the model for the development of a national healthcare program for patients with hemophilia.31 HTCs have been established to guarantee patients and families access to all services essential for the management of their bleeding disorder.31 Within these comprehensive HTCs, the core team members typically include hematologists trained in the management of bleeding disorders, nurses, clinical specialists, nurse practitioners, laboratory staff, physical therapists, medical social workers, and may include dentists, genetic counselors, dieticians, clinical research coordinators, psychologists as well as other specialists that aid in the coordination of care.
Current standard of care for patients with hemophilia now provides safe and effective treatment, allowing growth into the adult life with minimal disability, a normal lifestyle and employment opportunities.21,45 In the 1990s, approximately 67% of persons with hemophilia received care at HTCs at least once a year.45 More recently, in one US HTC, approximately 81.7% of persons with hemophilia receive care at least once a year.46 Those that benefit from care at HTCs have a reduced rate of emergency department (ED) utilization and an increased rate of prophylaxis and self-infusion compared to those receiving care outside of the HTC network.46
Potential Value of Comprehensive Care for cITP
Despite advances in medical therapy, the long-term morbidity and mortality of cITP remains. In the United States, a limited number of hematologists have focused expertise in cITP. To improve health outcomes and HRQoL, a network of medical centers that focuses on the diagnosis, management and prevention of complications of cITP is needed. The contributing centers would adopt or extend the Comprehensive Care model within an existing Hemophilia Treatment Center. This would include disease-specific education, outreach and advocacy programs, and conduct longitudinal surveillance on the complications of treatment. The Comprehensive Care model utilized in persons with hemophilia is well-suited for patients with platelet disorders. This model already incorporates integrated care for patients with inherited platelet disorders, including disorders of platelet number and/or function. This includes Bernard Soulier syndrome, Glanzmann Thrombasthenia, MYH9 disorders and Platelet Storage Pool Defect, to name a few.
The core members of the Comprehensive Care cITP team should be the same as those in the Comprehensive Care model for hemophilia: hematologists, nurse practitioners, nurses, dentists, laboratory staff, physical therapists, medical social workers, and clinical psychologists, but may include immunologists, dieticians, and clinical research coordinators. Patients with cITP may be broadly affected by the disease, specifically the clinical consequences of bleeding episodes, fear of bleeding, fatigue, decreased emotional health, social isolation, and reproductive health issues (see Table 1).37,47
The model of Comprehensive Care in HTCs has been recognized to improve many aspects of health, including life expectancy. An expert team would be equally as valuable to persons with cITP. However, an integrated care model with a reliable population-based surveillance program does not exist for cITP. Although advocacy groups, like Platelet Disorder Support Association (PDSA), have provided an invaluable resource for persons with cITP for the past 20 years, integration with the site of clinical care is usually lacking.48 The PDSA would be a logical liaison to the ITP treatment centers for patient education and advocacy, similar to the relationship of the National Hemophilia Foundation (NHF) to HTCs. Given the similarities between cITP and hemophilia and the success of comprehensive care centers in caring for persons with hemophilia, the Comprehensive Care model for hemophilia should be extended to persons with cITP.
In late 2017, the Bleeding and Clotting Disorders Institute (BCDI) in Peoria, Illinois launched the first ever Comprehensive Care Clinic in the US for persons with cITP. Beyond the Comprehensive Clinic team (hematologist, nurse practitioner, nurse coordinator, physical therapist, pharmacist, dentist, and medical social worker), each cITP patient is also seen by an immunologist and dietician. The addition of the immunologist has proven to be particularly beneficial and has led to the diagnosis of additional immunological disorders in some patients, such as common variable immune deficiency (CVID) and various autoimmune disorders.
The ITP program at BCDI provides care for approximately 200 patients from 16 US states and Mexico. Of these, more than fifty have been served on at least one occasion in the Comprehensive Care Clinic. The implementation of comprehensive care transcends the formal clinic encounter and, in some cases results in more than 200 interactions per year between the patient and various members of the care team. The response to the Comprehensive Care Clinic for cITP at BCDI has been tremendously positive and patients have reported an overall improvement in ease of their activities of daily living. Formal studies of clinical, psychosocial and economic outcomes are needed and in process at BCDI.
Conclusions
The complex phenotype of cITP is much like that of hemophilia, necessitating specialized care. A better understanding of the natural history of cITP, including its impact on HRQoL and drug safety in the treatment of cITP is urgently needed. To accomplish this, the Comprehensive Care model used for persons with hemophilia should be extended to include adults and children with cITP by adapting the infrastructure used for HTCs in the US to serve this patient population.
Disclosure
Michael Tarantino reports The Bleeding and Clotting Disorders Institute - salaried position, CEO, CMO, personal fees from Amgen, Principia, Genentech, Grifols, HemaBiologics, Novo Nordisk, Octapharma, Takeda, and Spark Therapeutics, outside the submitted work. The authors report no other potential conflicts of interest for this work.
References
1. Tarantino MD, Goldsmith G. Treatment of acute immune thrombocytopenic purpura. Sem Hem. 1998;35(1):28–35.
2. Jones HW, Tocantins LM. The history of purpura hemorrhagica. Ann Med Hist. 1933;5:349–359.
3. Frommeyer WB, Epstein RD, Taylor FHL. Refractoriness in hemophilia to coagulation-promoting agents: whole blood and plasma derivatives. Blood. 1950;5(5):401–420. doi:10.1182/blood.V5.5.401.401
4. Kasnelson P. Verschwinden der hamorrhagischen diathese bei einem falle von “essentieller thrombopenia” nach Milzexstirpation: splenogene thrombolytische purpura. Wien Klin Wochenschr. 1916;29:1451–1454.
5. Aggeler PM, White SG, Glendening MB, et al. Plasma thromboplastin component (PTC) deficiency: a new disease resembling hemophilia. Proc Soc Exp Biol Med. 1952;79:692–694. doi:10.3181/00379727-79-19488
6. Shulman NR, Marder VJ, Weinrach RS. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura. Physiologic, serologic and isotopic studies. Ann N Y Acad Sci. 1965;124:499–542. doi:10.1111/j.1749-6632.1965.tb18984.x
7. Pool JG. Cryoprecipitate in the treatment of hemophilia. Calif Med. 1970;113(2):66–67.
8. Dameshek W, Rubio F, Mahoney JP, et al. Treatment of idiopathic thrombocytopenic purpura (ITP) with prednisone. JAMA. 1958;166:1805–1815. doi:10.1001/jama.1958.02990150001001
9. Kasper CK. Judith Graham Pool and the discovery of cryoprecipitate. Haemophilia. 2012;18:833–835. doi:10.1111/hae.12042
10. Kernoff PB, Lee CA, Karayiannis P, et al. High risk of non-A non-B hepatitis after a first exposure to volunteer or commercial clotting factor concentrates: effects of prophylactic immune serum globulin. Br J Haematol. 1985;60(3):469–479. doi:10.1111/j.1365-2141.1985.tb07444.x
11. Imbach P. Treatment of immune thrombocytopenia with intravenous immunoglobulin and insights for other disease. Swiss Med Wkly. 2012;142(w13593):1–10.
12. Aledort L, Mannucci MP, Schramm W, Tarantino MD. Factor VIII replacement is still the standard of care in haemophilia A. Blood Transfusion. 2019;17:479–486.
13. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377:809–818. doi:10.1056/NEJMoa1703068
14. Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab prophylaxis in patients with hemophilia A without inhibitors. N Engl J Med. 2018;379(9):811–822. doi:10.1056/NEJMoa1803550
15. Pasi JK, Rangarajan S, Georgiev P, et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377:819–828. doi:10.1056/NEJMoa1616569
16. Spencer HT, Riley BE, Doering CB. State of the art: gene therapy of haemophilia. Haemophilia. 2016;22(Suppl. 5):66–71. doi:10.1111/hae.13011
17. Perrin GQ, Herzog RW, Markusic DM. Update on clinical gene therapy for hemophilia. Blood. 2019;133(5):407–414. doi:10.1182/blood-2018-07-820720
18. Stasi R, Pagano A, Stipa E, et al. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood. 2001;98(4):952–957. doi:10.1182/blood.V98.4.952
19. Nugent D, McMillan R, Nichol JL, Slichter SJ. Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol. 2009;146:585–596. doi:10.1111/j.1365-2141.2009.07717.x
20. Tarantino MD, Chalmers S. Therapeutic thrombopoietin mimetics. In: Gresele P, Kleiman NS, Lopez JA, Page CP, editors. Platelets in Thrombotic and Non-Thrombotic Disorders: Pathophysiology, Pharmacology and Therapeutics.
21. Bussel J, Arnold DM, Grossbard E, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: results of two Phase 3, randomized, placebo-controlled trials. Am J Hematol. 2018;93:921–930. doi:10.1002/ajh.25125
22. Stonebraker JS, Bolton-Maggs PHB, Soucie M, et al. A study of variations in the reported haemophilia A prevalence around the world. Haemophilia. 2009;16(1):20–32. doi:10.1111/j.1365-2516.2009.02127.x
23. Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Int Med. 2019;171(8):1–9. doi:10.7326/M19-1208
24. Centers for Disease Control and Prevention. Data & statistics on hemophilia; 2019. Para 1–2. Available from: https://www.cdc.gov/ncbddd/hemophilia/data.html.
25. Biere-Rafi S, Baarslag MA, Peters M, et al. Cardiovascular risk assessment in haemophilia patients. Thromb Haemost. 2011;105:274–278. doi:10.1160/TH10-07-0460
26. Segal JB, Powe NR. Prevalence of immune thrombocytopenia: analyses of administrative data. J Throm Haemost. 2006;4(11):2377–2383. doi:10.1111/j.1538-7836.2006.02147.x
27. Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;2(22):3780–3817.
28. Boral LI, Monohan GP, Moirangthem V. Overview of adult immune thrombocytopenia. Pathol Lab Med. 2016;1(1):21–31.
29. Terrell DR, Beebe LA, Vesely SK. The incidence of immune thrombocytopenic purpura in children and adults: a critical review of published reports. Am J Hematol. 2010;85(3):174–180.
30. Van Den Berg HM, De Groot PHG, Fischer K. Phenotypic heterogeneity in severe hemophilia. J Thromb Haemost. 2007;5(S1):151–156. doi:10.1111/j.1538-7836.2007.02503.x
31. World Federation of Hemophilia. Guidelines for the Management of Hemophilia.
32. Cortelazzo S, Finazzi G, Buelli M, et al. High risk of severe bleeding in aged patients with chronic idiopathic thrombocytopenia purpura. Blood. 1991;77(1):31–33. doi:10.1182/blood.V77.1.31.31
33. Cohen YC, Djulbegovic B, Shamai-Lubovitz O, Mozes B. The bleeding risk and natural history of idiopathic thrombocytopenic purpura in patients with persistent low platelet counts. Arch Intern Med. 2000;160:1630–1638. doi:10.1001/archinte.160.11.1630
34. Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost. 2010;8:1372–1382. doi:10.1111/j.1538-7836.2010.03830.x
35. Psaila B, Petrovic A, Page LK, Menell J, Schonholz M, Bussel JB. Intracranial hemorrhage (ICH) in children with immune thrombocytopenic purpura (ITP): study of 40 cases. Blood. 2009;114:4777–4783. doi:10.1182/blood-2009-04-215525
36. O’Hara J, Walsh S, Camp C, et al. The impact of severe haemophilia and the presence of target joints on health-related quality-of-life. Health Qual Life Outcomes. 2018;16(1):1–8. doi:10.1186/s12955-018-0908-9
37. Mathias SD, Gau SK, Miller KL. Impact of immune thrombocytopenic purpura (ITP) on health-related quality of life: a conceptual model starting with the patient perspective. Health Qual Life Outcomes. 2008;6(1):1–14. doi:10.1186/1477-7525-6-13
38. Efficace F, Mandeilli F, Fazi P. Health-related quality of life and burden of fatigue in patients with primary immune thrombocytopenia by phase of disease. Am J Hem. 2016;91(10):995–1001. doi:10.1002/ajh.24463
39. Mathias SD, Li X, Eisen M, Carpenter N, Crosby RD, Blanchette VS. A phase 3, randomized, double-blind, placebo-controlled study to determine the effect of romiplostim on health-related quality of life in children with primary immune thrombocytopenia and associated burden in their parents. Pediatr Blood Cancer. 2016;63:1232–1237. doi:10.1002/pbc.25984
40. Evatt BL, Black C, Batorova A, Street A, Srivastava A. Comprehensive care for haemophilia around the world. Haemophilia. 2004;10(Suppl s4):9–13. doi:10.1111/j.1365-2516.2004.01010.x
41. Ruiz-Sa´ez A. Comprehensive care in haemophilia. Hematology. 2012;17(sup1):S141–S143. doi:10.1179/102453312X13336169156492
42. Bolton-Maggs P. Optimal haemophilia care versus the reality. Br J Haematol. 2005;132(6):671–682. doi:10.1111/j.1365-2141.2005.05952.x
43. National Hemophilia Foundation; n.d. Available from: https://www.hemophilia.org/.
44. World Federation of Hemophilia. Vision and mission; n.d. Available from: https://www.wfh.org/en/about/vision-and-mission.
45. Soucie M, Nuss R, Evatt B, et al. Mortality among males with hemophilia: relations with source of medical care. Blood. 2000;96(2):437–442.
46. Okolo AI, Soucie JM, Grosse SD, et al. Population-based surveillance of haemophilia and patient outcomes in Indiana using multiple data sources. Haemophilia. 2019;25(3):456–463. doi:10.1111/hae.13734
47. Tarantino MD, Mathias SD, Snyder CF, et al. Impact of ITP on physician visits and workplace productivity. Cur Med Res Opin. 2010;26(2):319–328. doi:10.1185/03007990903451298
48. Platelet Disorder Support Association; n.d. Available from: https://www.pdsa.org/.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.