")
Back to Journals » Journal of Inflammation Research » Volume 16
The Mechanisms of Resistin-Like Molecule-β-Mediated Airway Inflammation in Chronic Obstructive Pulmonary Disease via Autophagy
Authors Che L , Xie Z, Chen G, Zhang W, Xia T, Lin J, Luo W, Chen L, Yin W, Cai X , Liu S
Received 1 February 2023
Accepted for publication 1 August 2023
Published 31 August 2023 Volume 2023:16 Pages 3853—3870
DOI https://doi.org/10.2147/JIR.S403517
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Li Che,1,2 Zhefan Xie,2,3 Guangshu Chen,4 Wei Zhang,2 Tingting Xia,2,3 Jiaxin Lin,2 Wenzhi Luo,2 Li Chen,2 Wenguang Yin,1 Xingdong Cai,2 Shengming Liu2
1State Key Laboratory of Respiratory Disease, National Clinical Research Center for Respiratory Disease, Guangzhou Institute of Respiratory Health, the First Affiliated Hospital of Guangzhou Medical University, Guangzhou, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China; 3Affiliated Dongguan People’s Hospital, Southern Medical University, Donguan, People’s Republic of China; 4Department of Endocrinology, Guangzhou Red Cross Hospital, The Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China
Correspondence: Shengming Liu; Xingdong Cai, The First Affiliated Hospital of Jinan University, Guangdong, People’s Republic of China, Email [email protected]; [email protected]
Background: The role of irreversible airway inflammatory damage in chronic obstructive pulmonary disease (COPD) progression is evident. Autophagy is an essential process in the cellular material metabolic cycle, and a family of resistant vegetative molecules may be involved in the COPD autophagic process. In this study, we investigated the mechanism of resistin-like molecule β (RELMβ) in COPD smoking-induced autophagy.
Methods: Firstly, the expression differences of RELMβ and autophagy markers between COPD and control groups were analyzed in the Gene Expression Omnibus (GEO) datasets and clinical specimens. Secondly, in vitro and in vivo experiments were conducted using immunoblotting, immunofluorescence, immunohistochemistry, and other methods to investigate the mechanism by which RELMβ promotes airway inflammation through autophagy in a cigarette smoke extract-induced 16HBE cell inflammation model and a cigarette smoke-induced COPD-like mouse model. In addition, immunoprecipitation was used to analyze the binding of RELMβ to the membrane protein TLR4.
Results: The expression of RELMβ and autophagy genes p62 and LC3B in lung tissue of COPD patients was significantly increased. RELMβ can mediate the activation of autophagy in 16HBE cells, and through autophagy, it increases the expression of inflammatory cytokines in a cigarette smoke extract-induced 16HBE cell inflammation model. RELMβ promotes cigarette smoke-induced COPD-like mouse airway inflammation through autophagy, and RELMβ can mediate signal transduction through the cell membrane receptor TLR4.
Conclusion: The RELMβ binds to TLR4 to encourage signal transduction and that RELMβ can promote inflammation in smoky COPD lungs through autophagy.
Keywords: autophagy, COPD, airway inflammation, cigarette smoke extract
Introduction
The high incidence of chronic obstructive pulmonary disease (COPD) and the low control rate in China suggest that COPD will remain an insurmountable problem in China for some time to come1 because the pathogenesis of COPD is still unclear. COPD is a group of respiratory diseases with persistent airflow limitation, and the mechanism of COPD is still inflammatory damage.2,3 Smoking and damage to the airways by harmful particulate matter leads to chronic inflammatory infiltration of the central airways, the proliferation of cupped cells leading to mucus hypersecretion, airway obstruction, and the formation of emphysema.4 There are more studies on the mechanism of COPD. However, none of them are clear, so further in-depth studies on the pathogenesis of COPD, especially the study of chronic airway inflammation, will be essential for diagnosing and treating slow obstructive pulmonary disease.
Autophagy is a normal metabolic process of cells that contributes to the cellular cycle of material metabolism.5 Autophagy engulfs metabolic substances, peptides, and residual organelles within the autophagosomal double membrane layer, which are then delivered to lysosomes for digestion, during which the enclosed substances are broken down.6,7 These enclosed metabolic substances and organelles are also a source of inflammatory signals.8 There is a complex regulatory relationship between inflammation and autophagy in lung diseases. Autophagy is involved in a variety of lung diseases. Autophagic vesicles are significantly more accumulated in patients with COPD.9 Cigarette smoke extract (CSE) in vitro has been shown to stimulate airway epithelial cells to regulate airway inflammation via autophagy and activate high mobility group box 1 (HMGB1)/Rage on the cell membrane, thus promoting signal transduction.10 CSE-mediated autophagy dysfunction was found to lead to intracellular ceramide accumulation and induce emphysema and that antioxidant-induced autophagy attenuates the accumulation of ceramide in autophagic vesicles caused by tobacco smoke extract.11
Resistin-like molecule β (RELM-β) is a newly identified member of the small molecule secretory protein family12 and is a homolog of hypoxia-induced mitogenic factors.13 In hypoxic environments, RELMβ expression is elevated in alveolar epithelial cells and lung fibroblasts, and significant cell proliferation effects can be observed in RELM-β-overexpressing cells.14 The pathological changes of thickened small pulmonary arteries increased the right ventricular hypertrophy index, and increased proper ventricular mean arterial pressure due to hypoxia can be reversed in RELMβ gene-deficient mice.15 In terms of airway inflammation, inflammation is a significant factor in the elevation of RELMβ.16 CSE stimulation mediates elevated airway inflammation RELMβ. Our study found high RELMβ expression in mouse-induced airway inflammation.17 RELMβ is significantly elevated in CSE-stimulated airway epithelial cells, along with the inflammatory factors IL-1β and IL-8, and further studies have shown that recombinant RELMβ protein (rhRELMβ) can regulate inflammatory effects via the p38 mitogen-activated protein kinase (MAPK) signaling pathway.18 However, how RELMβ handles inflammatory effects and whether autophagy is involved in RELMβ-mediated inflammatory regulation have not been reported. Our study first analyzed the expression of RELMβ and autophagy genes in COPD patients from the GEO dataset, verified the mechanism of RELMβ-regulated inflammation via autophagy at the cellular level, and further demonstrated in vivo in animals that RELMβ regulates tobacco smoke-stimulated airway inflammation via autophagy.
Materials and Methods
GEO Database Analysis
We searched for transcriptomic gene expression in COPD patients in the online database Gene Expression Omnibus (GEO) (https://www.aclbi.com/static/index.html/), downloaded the data, and used the R language. The normalize Quantiles function in the preprocessor package was used to normalize the data and analyze the gene expression in COPD patients and control patients.
Human Study Subjects
We collected surgical specimens from the Department of Thoracic Surgery of the First Affiliated Hospital of Jinan University due to surgical resection of lung cancer. COPD diagnosis was made according to the GOLD (Global Initiative for Chronic Obstructive Lung Disease) guidelines. All experiments were performed in accordance with the ethical standards of the Helsinki Declaration.
Cell and Transfection of Small Interfering RNA
We obtained a 16HBE cell line from Sun Yat-sen University. The cell STR analysis using multiplex amplification system (CELL STR IDTM) by Beijing Huake genetechnology co., LTD. The 16HBE-RELMβ-Flag cell line was preserved by our group. The 16HBE cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high sugar medium containing 10% fetal bovine serum (FBS). The cells were passaged when they reached approximately 80–90% confluence. Cells were spread evenly in 6-well plates and transfected overnight when cell confluence reached 50%-70%. The transfection reagent LipoFectMax™ (ABP© Bioscience, Wuhan, China) was mixed with diluted small interfering RNA (siRNA) (GenePharma, Shanghai, China) and left to stand for 20 min. The transfection efficiency was measured after 24 hours of transfection, and the protein expression level was measured after 48 hours.
Preparation of CSE
The cigarette without a filter was passed through 10mL of Dulbecco’s modified Eagle’s medium, filtered through a 0.22-μm filter, and the optical density was measured at 405 nm after filtration. The pH was adjusted to 7.4, and an optical density of 0.2 was considered to represent 100% CSE. These extracts were used for cell preparation within 30 minutes after preparation.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9
Three guide RNAs of the RELMβ gene were designed using CRISPR–Cas9 guide RNA online design software (http://www.e-crisp.org/E-CRISP), and the plasmid was transfected into 293T cells. The viral fluid was collected and transfected into 16HBE cells, and monoclonal cell proliferation was assessed by the limited dilution method after drug screening. The gene was sequenced and validated at the polymerase chain reaction (PCR) and protein levels.
mRFP-GFP-LC3B Transfection of 16HBE Cells with Adenoviral Fluid
The 16HBE cells were spread evenly in a 24-well plate lined with cell slides, changed overnight, and transfected with medium containing 2% FBS. The concentration of the viral solution was diluted at 1:1000 (MDI of 15) and switched to complete fresh medium after the eighth. Fluorescence efficiency was observed after 24 h. Cells were collected after rhRELMβ and chloroquine (CQ inhibited the binding of autophagosomes and lysosomes) stimulation. The cells were fixed with 4% paraformaldehyde, permeabilized with tris-buffered saline with Tween 20 (TBST) (Triton X-100) for 20 min, and then sealed with a blocker containing diamidino-2-phenylindole (DAPI).
Hematoxylin-Eosin Staining (HE) and Immunohistochemistry (IHC)
The obtained lung tissue specimens were fixed in paraffin, the nucleus was stained with hematoxylin, and then the cytoplasm was eosin-stained. Resin-sealed slices were microscopically observed and counted. Paraffin-fixed lung tissue was sectioned, dewaxed, and hydrated. After antigen repair, the primary antibody working solution was closed overnight, the rabbit secondary antibody was incubated for 2 hours, and the slices were sealed and photographed under microscope observation.
Immunocytochemistry
After treatment, the cells were fixed with 4% PFS, permeabilized with TBST (Triton X-100) for 20 min, closed for 1 h, and incubated with the primary antibody working solution overnight at 4°C. The fluorescent secondary antibody identification was incubated for 1 h at room temperature and protected from light, and blocked with DAPI in a light-protected environment. Microscopic photographs were taken for statistical analysis.
Western Blot (WB) and ELISA
Cell supernatants were used for enzyme-linked immunoassay (ELISA). The supernatants were harvested, and 100µL of antibody dilutions (RELMβ, Abcam, Cambridge, UK; IL-1β, Abcam, Cambridge, UK; IL-8, Abcam, Cambridge, UK) were added to each well and incubated for 1 h. The secondary antibodies were added, and the reaction was terminated with the termination solution. A wavelength of 450 nm was used to measure the absorbance.
Co-Immunoprecipitation (Co-IP) Assay
The supernatant was collected from the lysed cells, and the supernatant was incubated with protein A/G agarose beads (Thermo Fisher Scientific, USA) for 1 hr at 4°C and then incubated with TLR4 antibody (Santa Cruz Biotechnology, USA) overnight at 4°C. The protein A/G agarose beads were eluted with PBS and the precipitated proteins were detected by Western blot analysis.
Reverse Transcription-PCR (RT-PCR) and Real-Time Quantitative PCR (Q-PCR)
After the cells and tissues were collected, the total RNA of the cells and tissues were extracted by adding the appropriate Trizon. Reverse transcription and PCR amplification were performed. The amplification conditions were as follows: preheating at 98°C for 3 min, followed by annealing at 60°C for 10–15 s for 30–35 cycles. The amplification products were detected by agarose gel electrophoresis. GAPDH was used as the loading control. cDNAs were amplified by quantitative PCR using a C.F.X. instrument (BioRad, Hercules, CA, USA).
Animals
Six- to eight-week-old male C57BL/6 mice were purchased from The Southern Medical University Laboratory Animal Center. Retnlb−/− mice were purchased from Nanjing Model Organism (Nanjing, China). Thirty male mice were housed in SPF class mouse holding rooms. The Ethical Committee approved all experimental protocols for Animal Studies at Jinan University.
In vivo CS Exposures and Treatments
Mice were given smoke fumes in stainless steel chambers, 10 cigarettes per session, and each time the smoke was diffused in the room for 30 min, 50 cigarettes per day, 5 days per week, and 2 weeks of smoke exposure. The total particulate matter concentrations in the exposure chamber were between 150–180 mg/m3. The mice were treated with the autophagy inhibitor 3-MA by intra-nasal 2 hours before smoke exposure, placed in air for another 2 weeks, and then started to receive elastase by airway drip after Day 29 for 3 days, 24 hours after the last drip. Mouse elastin peptide was suspended in sterile saline. Each airway drip was 40 µL, for a total of 100 µg per drip. 3-MA was dissolved in saline at 5 mg/mL, 40 µL per drip. 3-MA was administered 2 hours before each smoke stimulation.
Statistical Analysis
Differences between two groups were analyzed using Student’s t-test. Three or more groups were analyzed using one-way analysis of variance (ANOVA) for differences between groups. All data are expressed as the mean ± standard error of the mean (sem). The analyses and graphs were generated using GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA.). A value of P less than 0.05 was considered statistically significant.
Results
RELMβ and Autophagy Markers are Highly Expressed in COPD Patients
We analyzed the correlation between the RELMβ gene and autophagy-related genes in the GSE5058 dataset,19,20 which is a genomic expression analysis of airway and small airway epithelium from 12 patients with normal control, 11 patients with simple smoking, and 16 patients with COPD, by analyzing data from the GEO database COPD dataset. Gene expression was analyzed by downloading the raw data from the GEO database, normalizing the raw data using the R language, and converting the probe ID into a gene symbol. The results showed that RELMβ mRNA expression was higher in COPD patients than in normal controls and the smoking-only group (Figure 1A). The mRNA expression of the autophagy markers LC3B, p62, and BECN1 (Figure 1A) was significantly higher in the COPD group than in the control group and the smoking-only group. We further collected and analyzed the tissues of lung cancer patients with COPD combined with lung cancer removed due to surgery in the thoracic surgery department of our hospital. Preoperative pulmonary function suggests a diagnosis of COPD based on diagnostic criteria.21 All organizations are derived from cancerous tissues greater than 2cm in size in lung cancer tissue. The pathological tissues of lung cancer without COPD were used as the control group. PCR, IHC and WB were used to detect the expression of RELMβ in the tissues. The 20 patients paraffin tissue sections were used for immunohistochemistry staining, and patient information is shown in Table 1. The 8 fresh patient tissues were used for total RNA and total protein extraction, and patient information is shown in Table 2. The results showed that the expression of RELMβ was significantly increased both at the mRNA level (Figure 1B) and protein level (Figure 1D) as well as at the tissue level (Figure 1D). We further analyzed the expression of the autophagy markers LC3BI/II and p62, and the results showed that the accumulation of p62 was significantly increased in COPD patients at the mRNA level (Figure 1B) and protein level (Figure 1D). The mRNA level showed a significant increase in LC3B expression (Figure 1B), and the tissue level showed a significant increase in COPD patients (Figure 1C).
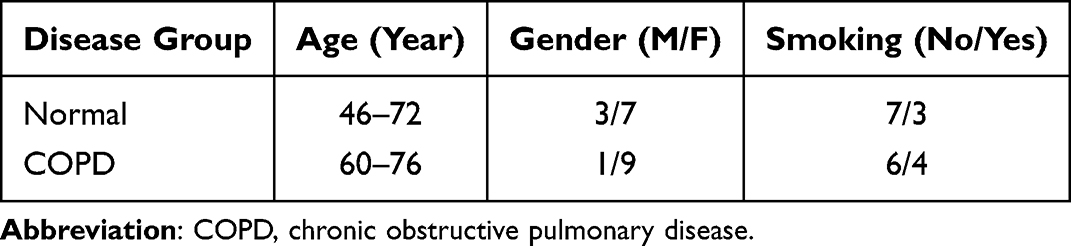 |
Table 1 Demorgraphics on the Patients |
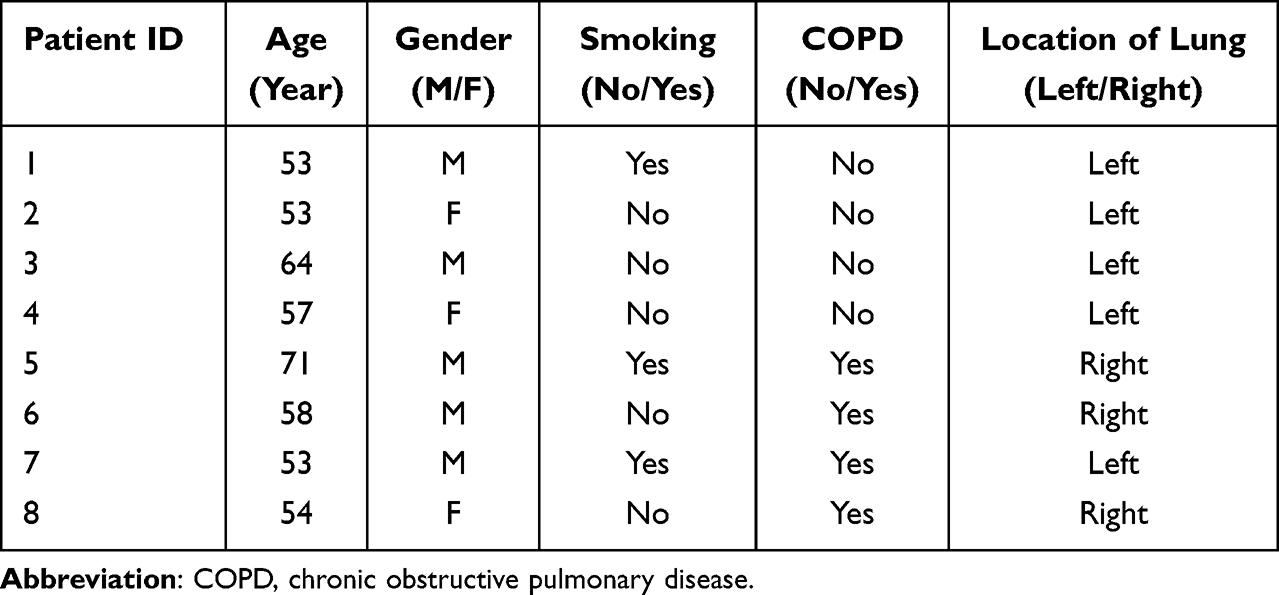 |
Table 2 The Information of Patients |
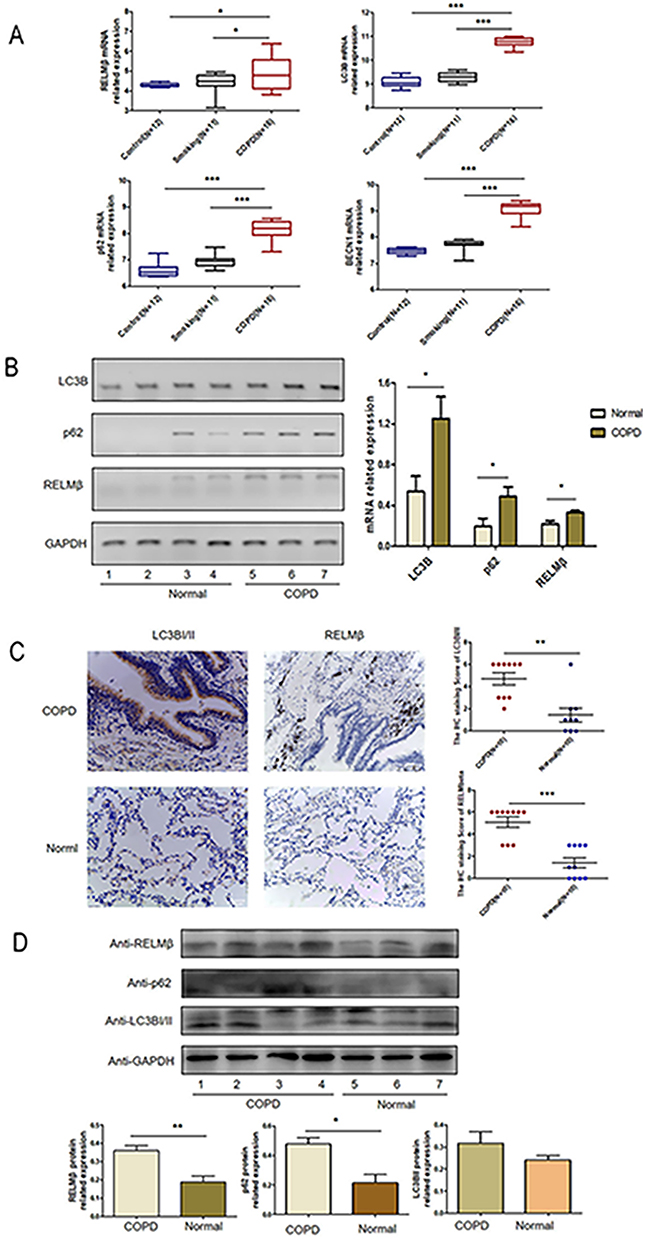 |
Figure 1 The expression of RELMβ and autophagy genes is elevated in COPD patients. (A), Analysis of RELMβ and autophagy gene expression in the GEO database; (B), RT-PCR detection of RELMβ and autophagy gene mRNA levels in the COPD and control groups; (C), Immunohistochemical detection of RELMβ and autophagy genes in the COPD and control groups. The expression at the tissue level in the control group; (D), Western blot detection of the expression of RELMβ and autophagy genes at the protein level in the COPD and control groups. (*p < 0.05, **p < 0.01, ***p < 0.001.). |
RELMβ Promotes the Expression of Autophagy Markers in vitro
To verify that RELMβ affects autophagy, autophagic markers were detected by culturing 16HBE cells in vitro at different concentrations and times. 16HBE cells were verified by STR and no cross-contamination of other human cell lines was found. The results showed that at a concentration of 100 ng/mL rhRELMβ, with the extension of stimulation time, the expression of the autophagic marker LC3BI/II was evident at the 24th hour of rhRELMβ (Figure 2A). The accumulation of p62 started to appear at the 3rd hour and was evident at the 24th hour (Figure 2A). The BECN protein showed increased expression at the 6th hour (Figure 2A), and thereafter, there was always a significant increase in expression. We further analyzed the expression of autophagy markers stimulated at different concentrations in the presence of rhRELMβ for 24 h. The results showed that the expression of LC3BI/II started to appear significantly increased at rhRELMβ 50 ng/mL (Figure 2B), the accumulation of p62 was significant at rhRELMβ 100 ng/mL (Figure 2B), and BECN protein showed increased expression at rhRELMβ 100 ng/mL (Figure 2B). From this, we know that rhRELMβ promotes the expression of autophagy in 16HBE cells, and the most pronounced autophagy was observed at an action concentration of 100 ng/mL and 24 h stimulation time. In addition, we also analyzed the intracellular localization of the autophagy marker LC3BI/II using the immunofluorescence technique. The analysis showed that LC3BI/II was expressed outside the nucleus, and no apparent intranuclear localization was observed (Figure 2C). Mrfp-GFP-LC3B adenovirus solution transfected with 16HBE cells showed that rhRELMβ alone promoted autophagic vesicles in 16HBE cells, and the accumulation of autophagic vesicles was more pronounced when the autophagy-lysosome inhibitor CQ was present (Figure 2D). Our results suggest that RELMβ promotes early autophagy in 16HBE cells and autophagosome accumulation in 16HBE cells.
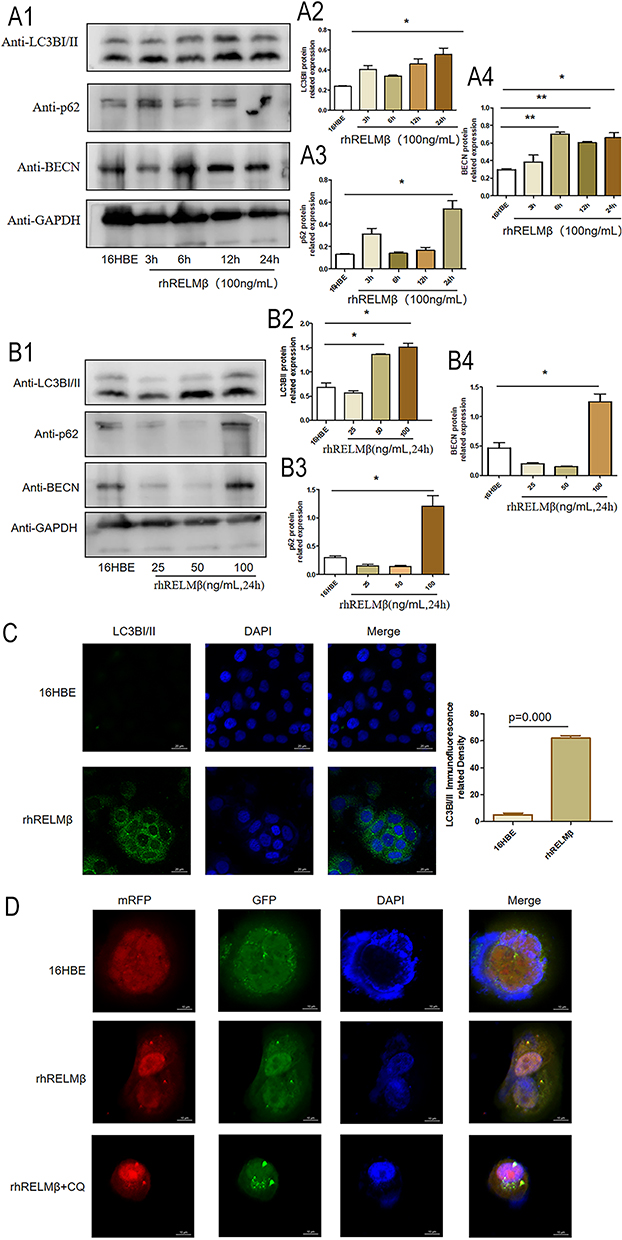 |
Figure 2 RELMβ promotes autophagic effects. (A), rhRELMβ promotes autophagy protein expression in 16HBE cells in a time-dependent manner (A1), Western blot analysis of the expression of autophagy proteins p62, LCB3I/II, and BECN in 16HBE cells; (A2–4), Densitometric analysis of the corresponding protein grayscale values). (B), rhRELMβ promoted the expression of autophagy proteins in 16HBE cells in a concentration-dependent manner (B1), Western blot analysis of the expression of autophagy proteins p62, LCB3I/II, and BECN in 16HBE cells; (B2–4), Densitometric analysis of the corresponding protein grayscale values). (C), Immunofluorescence detection of autophagy protein accumulation around the nucleus of 16HBE cells. (D), mRFP-GFP-LC3B double-fluorescence infection with 16HBE to detect autophagosome aggregation. (*p < 0.05, **p < 0.01). |
RELMβ-Mediated Accumulation of Autophagy Promotes the Effects of Inflammation
We further investigated the rhRELMβ-mediated autophagic effect of 16HBE to further investigate the autophagic effect of rhRELMβ. We further analyzed the time-dependent autophagic vesicle accumulation of rhRELMβ protein over time, and the results showed that the accumulation of LC3BI/II started at 6 h and was significant at 24 h (Figure 3A); the collection of p62 began at 12 h and was effective at 24 h (Figure 3A). When cells were pretreated with chloroquine (CQ) for 2 hours, there was a significant increase in autophagic vesicle accumulation, indicating that rhRELMβ could activate early autophagy. We also further analyzed the effect of autophagy inhibitors on inflammatory development. The results showed that the expression of the inflammatory factors IL-1β and IL-8 was increased in the presence of increased autophagosome accumulation, and IL-1β (Figure 3B) and IL-8 (Figure 3B) at the mRNA and protein levels (Figure 3B) were significantly increased. In the presence of the autophagy initiation inhibitor 3-MA, IL-8 expression was significantly decreased both at the mRNA level (Figure 3B) and at the protein level (Figure 3C). The expression of IL-1β was reduced at both the mRNA and protein levels, indicating that the autophagic process influences rhRELMβ-mediated inflammatory effects in bronchial epithelial cells and suggesting that the regulation of autophagy exerts an important influence on inflammation.
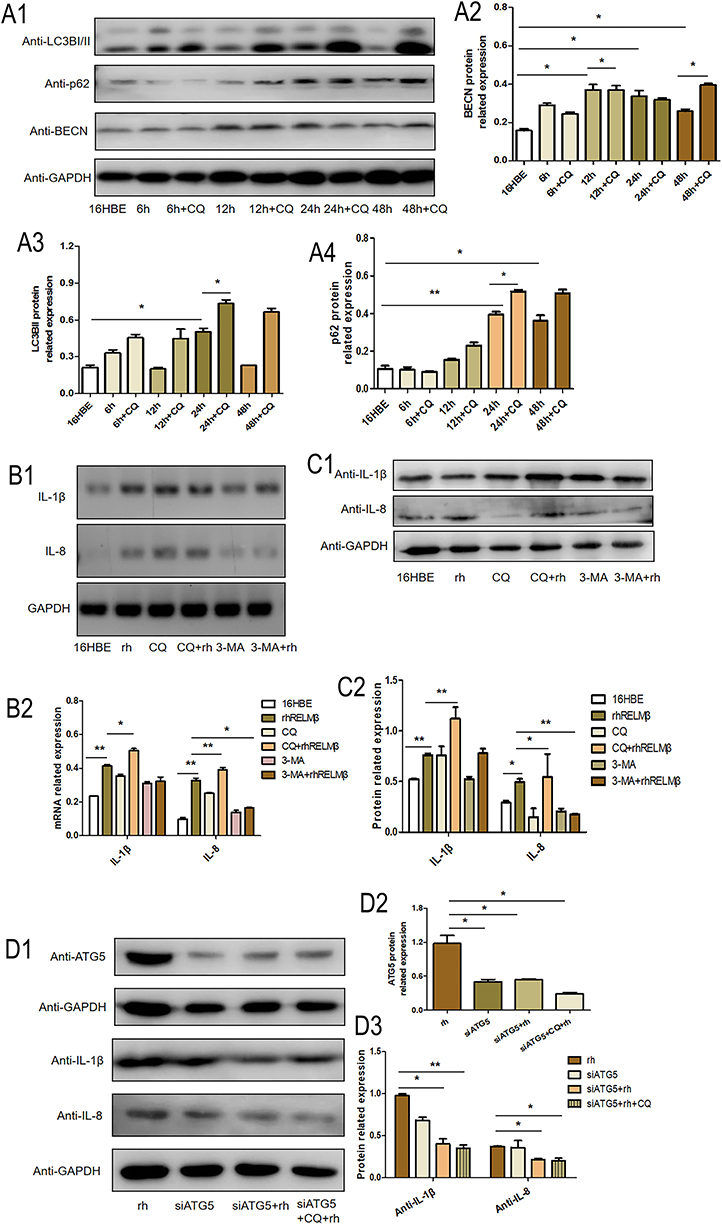 |
Figure 3 RELMβ promotes 16HBE inflammatory effects via autophagy. (A), The effect of autophagy became more pronounced in a time-dependent manner (A1), Western blot analysis of autophagy proteins p62, LCB3I/II, and BECN expression in 16HBE cells; (A2–4), Densitometric analysis of the corresponding protein grayscale values). (B and C), RELMβ promotes the expression of IL-1β and IL-8 in 16HBE cells via autophagy (B1), PCR analysis of autophagy proteins p62, LCB3I/II, and BECN expression in 16HBE cells; (B2), PCR densitometric analysis; (C1), Western blot analysis of autophagy proteins IL-1β and IL-8 expression in 16HBE cells; (C2), Densitometric analysis of the corresponding protein grayscale values). (D), Replenishment experiments confirmed that RELMβ promotes the expression of IL-1β and IL-8 in 16HBE cells via autophagy (D1), Western blot analysis of autophagy proteins IL-1β and IL-8 expression in 16HBE cells; (D2 and 3), Densitometric analysis of the corresponding protein grayscale values). (*p < 0.05, **p < 0.01). |
We performed backfill experiments to further investigate the regulation of inflammation involved in autophagy. Using small interfering RNA, an essential gene in the autophagic process, ATG5 interfered with the expression of ATG5. The results of the analysis of inflammatory factors showed that the expression of IL-1β (Figure 3D) and IL-8 (Figure 3D) showed a significant decrease compared to rhRELMβ alone, even with the intervention of CQ IL-1β (Figure 3D) and IL-8 (Figure 3D) expression remained significantly reduced, suggesting that rhRELMβ mediates inflammation, both in the early and late stages of autophagy and that the effect of autophagy on inflammation is present. Our analysis indicates that autophagic mechanisms regulate RELMβ-mediated inflammation in bronchial epithelial cells.
Tobacco Smoke Promotes COPD Airway Inflammatory Expression via RELMβ-Mediated Autophagy
To further analyze the effect of the inflammatory autophagy-regulated impact of RELMβ, we edited the expression of the RELMβ gene using the CRISPR Cas9 guide-RNA technique. We observed the inflammatory effects in the 16HBE cell model with CSE intervention in the presence of a small interfering RNA technique to interfere with the ATG5 gene. The results showed that CSE alone promoted RELMβ gene expression. In contrast, when the RELMβ gene was edited using CRISPR Cas9 guide-RNA gene-editing technology, RELMβ was expressed at the mRNA (Figure 4A), and the protein levels (Figure 4A) were almost undetectable We also interfered with the expression of the ATG5 gene using the small interfering RNA technique. The results showed that only CSE alone stimulated significant expression of the 16HBE inflammatory factors IL-1β (Figure 4B) and IL-8 (Figure 4B), while both the RELMβ gene and the ATG5 gene expression were edited. IL-1β (Figure 4B) and IL-8 (Figure 4B) were significantly decreased. We found that when the RELMβ gene was edited with or without ATG5 gene intervention, the expression of IL-8 (Figure 4B) was further decreased. IL-1β showed a decreasing trend, but the difference was not significant. Our experiments further confirmed that autophagic mechanisms are involved in RELMβ-mediated airway inflammation in tobacco smoke sensitization.
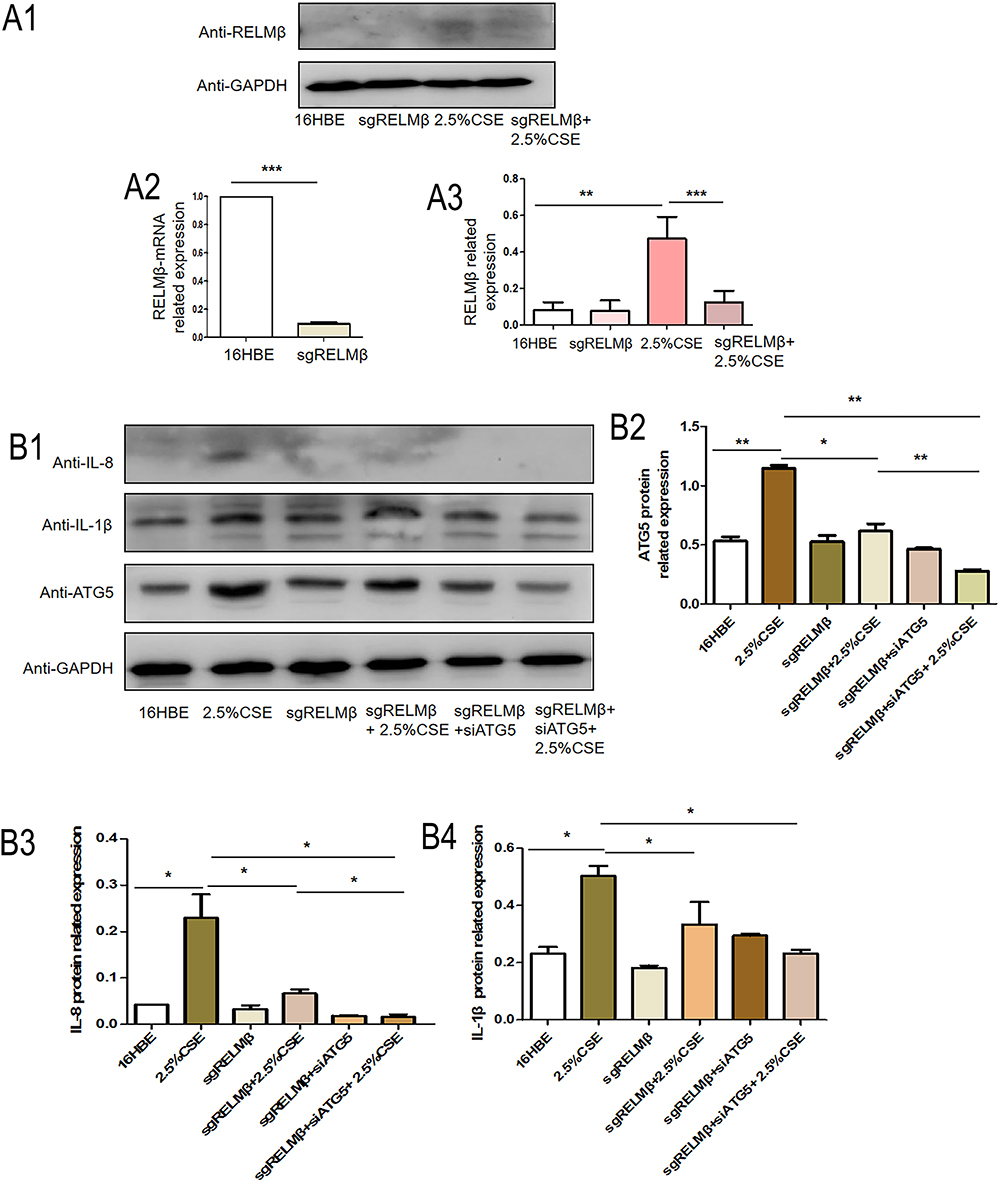 |
Figure 4 CSE promotes 16HBE inflammatory effects via RELMβ-mediated autophagy. (A), CRISPR-Cas9 technology edits the RELMβ gene, and Western blot detects the expression of RELMβ in 16HBE cells stimulated by tobacco smoke extract (A1), Western blot analysis of RELMβ protein expression in 16HBE cells; (A2), QPCR analysis of RELMβ mRNA expression level; (A3), Densitometric analysis of corresponding protein grayscale values). (B), When the RELMβ gene is edited, combined with small interfering RNA technology to detect the expression of the inflammatory factors IL-8 and IL-1β (B1), Western blot analysis of protein expression of ATG5, IL-1β, and IL-8 in 16HBE cells; (B2–4), Densitometric analysis of corresponding protein grayscale values). (*p < 0.05, **p < 0.01, ***p < 0.001). |
RELMβ Promotes Airway Inflammatory Effects via Autophagy in Smoked Mice
To further clarify the real-world effects of the RELMβ gene on autophagy, we verified the altered airway inflammation in mice by tobacco smoke combined with elastase in a C57BL/6J mouse slow-onset lung-like model with or without intervention with the autophagy inhibitor 3-MA using CRISPR Cas9 technology to edit RELMβ. The WB (Figure 5A) and IHC (Figure 5B) detection showed almost no expression of RELMβ in gene knockout mice. HE staining showed that tobacco smoke fumigation combined with elastase resulted in a significant increase in the airway and alveolar inflammatory infiltrates in mice, with a slight rise in alveolar septa compared to wild mice (Figure 5C). The airway inflammation was significantly better in mice when airway drops of the autophagy inhibitor 3-MA were administered. There was a trend of reduced inflammation in smoked C57BL/6JRELMβ-/- mice compared with C57BL/6JWT mice. In the case of C57BL/6JRELMβ-/- mice that smoked with airway drops of the autophagy inhibitor 3-MA, we found a significant reduction in airway inflammation. Our study showed an important regulatory role of RELMβ in tobacco smoke-sensitized subpulmonary inflammation and autophagy in RELMβ-mediated tobacco smoke-sensitized airway inflammation.
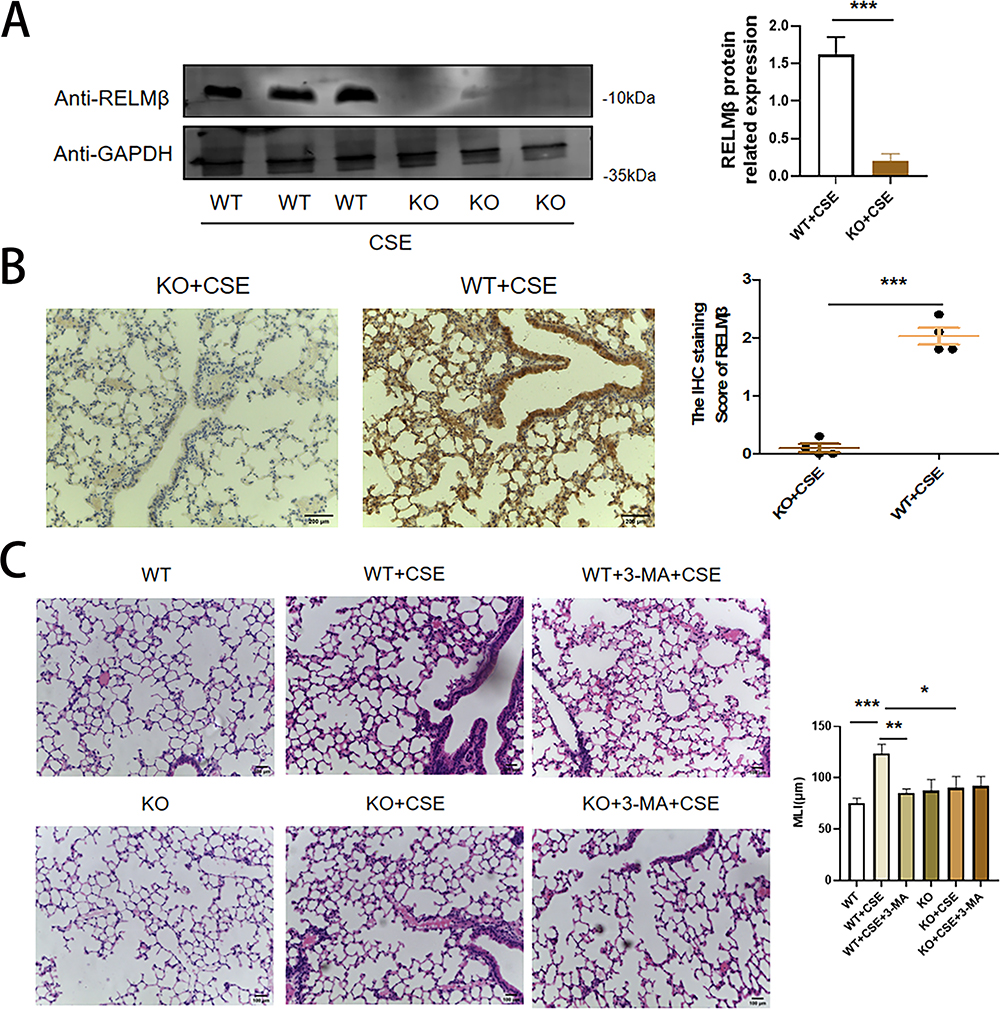 |
Figure 5 Validation of the C57BL/6JRELMβ-/- knockout COPD mouse model. (A), Western blot verification of RELMβ knockout in C57BL/6J mice. (B), IHC staining verification of RELMβ knockout in smoked C57BL/6 J RELMβ-/- mice and smoked C57BL/6J mice. (C), HE staining verified that the COPD model was successfully established in C57BL/6J mice. (*p < 0.05, **p < 0.01, ***p < 0.001). |
We further analyzed RELMβ promotes COPD airway inflammation in C57BL/6J mice via autophagy. BALF total cell counts and neutrophil percentages (Figure 6B1) were lower in the smoked C57BL/6JWT group pretreated with 3-MA than C57BL/6JWT smoked mice. Moreover, the results showed that the expression of IL-8 and IL-1β (Figure 6B2) in mice BALF was significantly inhibited in smoked C57BL/6JWT mice pretreated with 3-MA. Meanwhile, the expression of the mice lung inflammatory factors IL-1β (Figure 6B3) and IL-8 (Figure 6B3) was decreased in smoked C57BL/6JWT mice pretreated with 3-MA. The expression of the autophagy markers LC3BI/II (Figure 6B3) and p62 (Figure 6B3) was also decreased, indicating that autophagy can promote inflammatory effects in mice. The IHC stain of p62 was also showed 3-MA inhibited airway inflammation induced by cigarette smoking in C57BL/6JWT mice (Figure 6A).
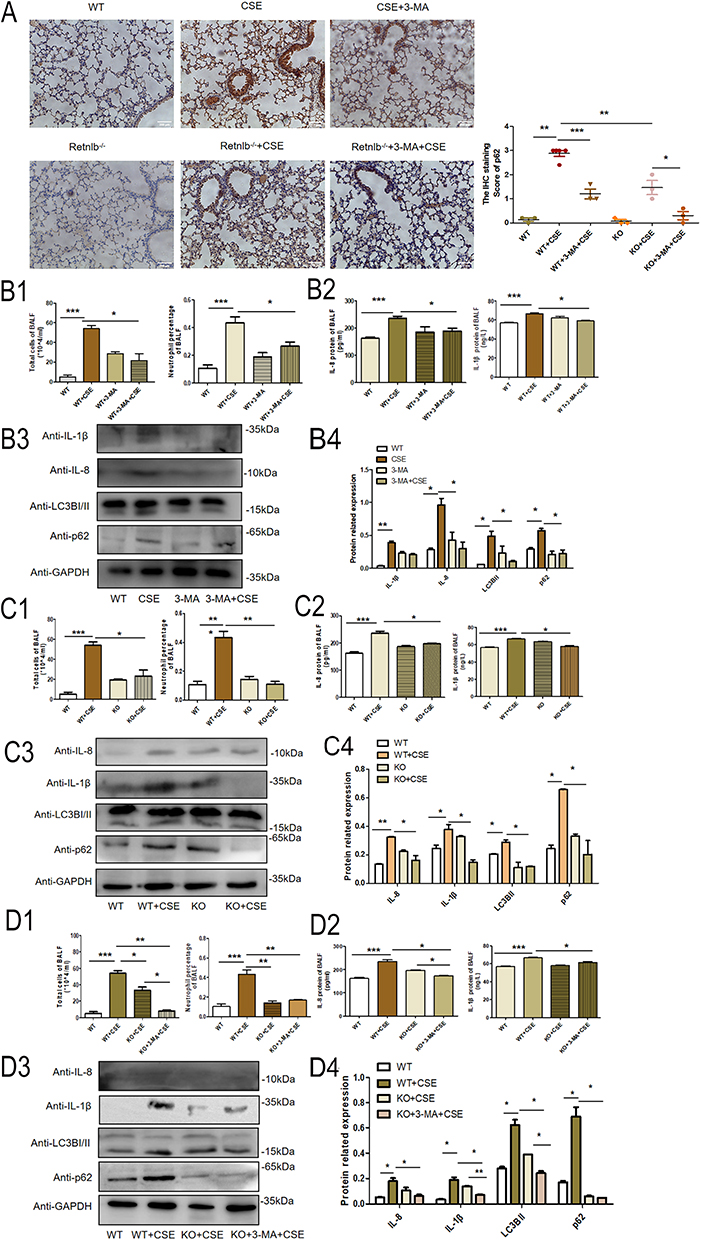 |
Figure 6 RELMβ promotes COPD airway inflammation in C57BL/6J mice via autophagy. (A), IHC verification of p62 expression in C57BL/6J mice. (B1), Mouse BALF total cell count and percentage of neutrophils in 3-MA pretreated C57BL/6J mice; (B2) Mouse BALF IL- 8 and IL-1βsecretion by ELISA in 3-MA pretreated C57BL/6J mice; (B3) The expression of IL-8, IL-1β and autophagy protein in mouse lung tissue; (B4) Densitometric analysis of the protein grayscale values. (C1), Mouse BALF total cell count and percentage of neutrophils in C57BL/6JRELMβ-/- mice; (C2) mouse BALF IL- 8 and IL-1βsecretion by ELISA in C57BL/6JRELMβ-/- mice; (C3) The expression of IL-8 and IL-1β and autophagy protein in C57BL/6JRELMβ-/- mice lung tissue; (C4) Densitometric analysis of the protein grayscale values. (D1), Mouse BALF total cell count and percentage of neutrophils in 3-MA pretreated C57BL/6J RELMβ-/- mice; (D2) Mouse BALF IL- 8 and IL-1βsecretion by ELISA in 3-MA pretreated C57BL/6J RELMβ-/- mice; (D3) The expression of IL-8 and IL-1β and autophagy protein in 3-MA pretreated C57BL/6J RELMβ-/- mice; (D4) Densitometric analysis of the protein grayscale values. (*p < 0.05, **p < 0.01, ***p < 0.001). |
We also analyzed the expression of inflammatory factors in C57BL/6J RELMβ-/- mice. Our analysis showed that total cell counts (Figure 6C1) and neutrophil percentages (Figure 6C1) were lower in the BALF of the smoked C57BL/6JRELMβ-/- group than in the BALF of the C57BL/6JWT smoked mice. Meanwhile, in smoked C57BL/6JRELMβ-/- mice, the expression of IL-8 (Figure 6C2) and IL-1β (Figure 6C2) was significantly decreased compared with that in smoked C57BL/6JWT mice BALF. We also examined the changes inflammatory effects in lung tissues by WB, the expression of the inflammatory factors IL-1β (Figure 6C3) and IL-8 (Figure 6C3) was significantly downregulated in smoked C57BL/6J RELMβ-/- mice compared with smoked C57BL/6JWT mice. The expression of the autophagy markers LC3BI/II (Figure 6C3) and p62 (Figure 6C3) were significantly lower in smoked C57BL/6J RELMβ-/- mice pretreated with 3-MA for 2 h than in smoked C57BL/6JRELMβ-/- and C57BL/6JWT mice alone. The IHC stain of p62 was significantly decreased in C57BL/6JWT cigarette smoking mice (Figure 6A). The results showed that RELMβ gene loss has a protective effect on airway inflammation in mice.
While, after concomitant airway dripping of 3-MA in C57BL/6JRELMβ-/- mice, total cell counts (Figure 6D1) in BALF were significantly reduced in C57BL/6JRELMβ-/- smoked mice with 3-MA pretreated than C57BL/6JRELMβ-/- smoked mice. In contrast, in C57BL/6J RELMβ-/- mice, there was a more pronounced expression of the inflammatory factor IL-8 (Figure 6D2) and a decrease in IL-1β expression in C57BL/6J RELMβ-/- smoked mice when airway drops of 3-MA were administered 2 hours prior to tobacco smoke fumigation. Nevertheless, there was no significant difference (Figure 6D2). The expression and inflammatory effect of IL-8 (Figure 6D3) were significantly decreased in mice lung, and IL-1β was slightly decreased, but the differences were not statistically significant (Figure 6D3) in smoked C57BL/6 J RELMβ-/- mice pretreated with 3-MA. The IHC stain of p62 was significantly decreased in smoked C57BL/6 J RELMβ-/- mice pretreated with 3-MA (Figure 6A). Our results further confirm that autophagy intervention can alleviate airway inflammation in C57BL/6JRELMβ-/- mice.
RELMβ Binds to the Cell Membrane Receptor TLR4 to Promote Signal Transduction
RELMβ is a cytoplasmic secretory protein, and its expression was significantly elevated in extracellular plasma and serum in inflammatory states. To further analyze whether RELMβ mediates signaling into the cell and thus promotes signaling, we verified the binding of RELMβ to TLR4 by cellular assays. The results showed that rhRELMβ stimulated increased TLR4 gene expression at the mRNA (Figure 7A) and protein levels (Figure 7B). In comparison, TLR4 expression was increased at the mRNA (Figure 7A) and protein levels (Figure 7B) in the presence of increased accumulation of autophagic vesicles under the action of the autophagy inhibitor CQ. To clarify whether RELMβ binds to the TLR4 cell membrane receptor, we analyzed TLR4 expression with RELMβ by immunocytochemistry, and the immunofluorescence results showed that TLR4 expression was also significantly increased in the RELMβ-overexpressing cell line (Figure 7C). Additionally, immunoprecipitation results showed that the expression of 16HBE cell lines with Flag tags overexpressing the RELMβ gene was also increased using antigen-antibody complexes after incubation with TLR4 antibody (Figure 7D). Our results suggest that RELMβ activates the increased expression of TLR4 receptors on the membranes of 16HBE cells while binding to TLR4 and thus mediating signaling.
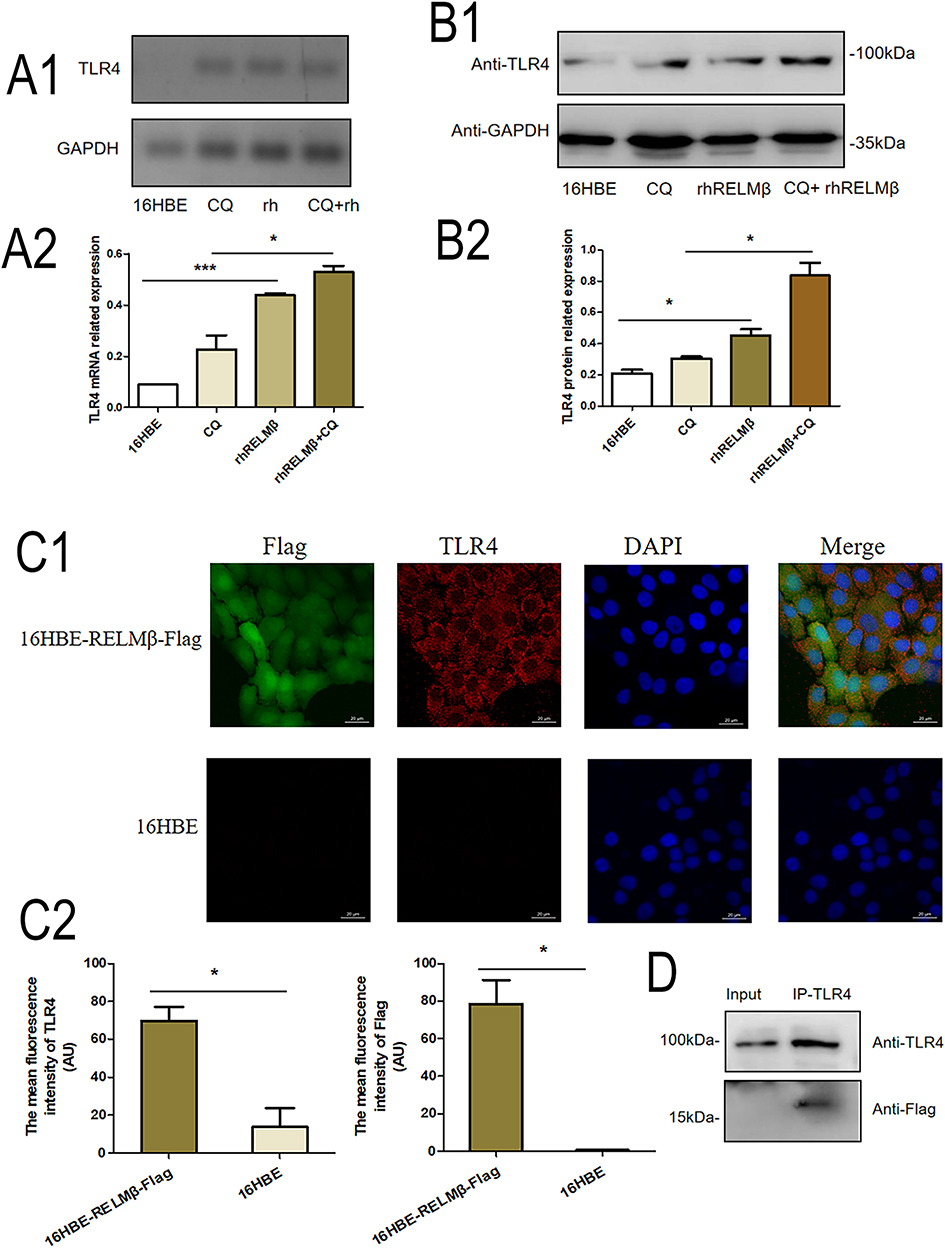 |
Figure 7 RELMβ and cell membrane TLR4 receptors promote signal transduction. (A1), RT-PCR to verify rhRLMEβ activation of TLR4 in 16HBE cells; (A2) Densitometric analysis of (A1). (B1), Western blot to verify rhRLMEβ activation of TLR4 levels in 16HBE cells; (B2) Densitometric analysis of the protein grayscale values. (C1), Immunofluorescence to detect TLR4 binding to Flag of 16HBE-RELMβ; (C2) Mean fluorescence density analysis. (D), Immunoprecipitation analysis of TLR4 binding to Flag of 16HBE-RELMβ. (*p < 0.05, ***p < 0.001). |
Discussion
We analyzed the results by searching the GEO database and showed that in COPD patients with increased RELMβ, the expression of autophagy markers was also increased, suggesting a role for autophagy in COPD patients. Although RELMβ expression is elevated in tumors,22–24 we analyzed RELMβ expression in cancerous tissue located over 2cm away from the tumor, and the results showed increased expression of autophagy markers in pathological tissues of COPD patients at the mRNA and protein levels as well as at the tissue level. The study of autophagy in COPD is a current research hotspot, and immunohistochemical analysis of lung tissues from patients with COPD showed a significant increase in the accumulation of p62 protein in patients with severe COPD compared to regular nonsmoking patients.25,26 Increased p62 protein accumulation leads to increased secretion of inflammatory factors, such as increased expression of γ-IFN, which is involved in the inflammatory response,27 suggesting that RELMβ is associated with autophagy regulation. Studies on the RELM family in autophagy have focused on RELMα, a cytokine highly expressed under hypoxia, and researchers have found significant accumulation in alveolar macrophages in a model of pulmonary hypertension. These macrophages also overexpress high mobility protein 1 and late glycosylation end-product receptors,28 blocked by related antibodies. The expression of LC3B-II and Beclin1 was decreased in cells when relevant antibodies blocked these proteins. These results suggest that RELMα may mediate the regulation of LC3B-II and Beclin1 expression in hypoxic or inflammatory states. A previous researcher indicated29 that the cumulative expression of the autophagy marker p62 increased in lung mouse smooth muscle cells in a hypoxic environment. Nevertheless, in mice with the deletion of the RELMα gene,24 the accumulation of the autophagy marker p62 was reduced, suggesting the existence of hypoxia-induced RELMα-mediated autophagy. In contrast, researchers observed that treating human pulmonary vascular smooth muscle using human resistin epithelial cells affected the expression of the autophagy markers LC3BI/II and p62. The above studies suggest a role for the RELM family in airway inflammatory diseases. Our results confirm that RELMβ also has a facilitative role in COPD autophagic mechanisms. However, there are some drawbacks to our data. The expression of the autophagy marker LC3BI/II was not statistically significant at the protein level, considering the inconsistent autophagy levels in different patients with chronic obstructive pulmonary disease. Lung specimens from paracancerous tissue, where the level of inflammation affects the carcinogenesis of the tissue, have been shown to further affect the expression of autophagy levels.30
There is an inherently complex regulatory relationship between autophagy and inflammation. Mice with deletion of the autophagy-associated gene Atg7 lysosome-specific gene mediated the development of lung inflammation, with significantly increased expression of the inflammatory factors TNFα, IL-6, and Cxcl1 and significantly increased neutrophil counts and dendritic cell counts.31,32 Microscopic pathology showed that lung tissue inflammatory cells in mice with deletion of the Atg7 gene or Atg5 gene aggregated in the submucosa increased, along with an increase in collagen content and cuprite chemotaxis.33 The role of autophagy in alveolar macrophage-mediated airway inflammation is crucial for sputum secretion and airway mucus secretion by rhabdocytes,31,32 and airway mucosal rhabdocytes are significantly proliferated in Atg16L1 gene-deficient mice in inflammatory states. Second, it has been shown in the literature34,35 that mice deficient in the autophagy genes Hdac6 and Pink1 inhibit the development of emphysema and mucosal secretion capacity, but the exact mechanism is unclear. We verified the relationship between the regulation of inflammation by RELMβ via autophagy at the cellular and animal levels. The results showed that at the cellular level, rhRELMβ promoted the inflammatory effect of 16HBE cells. This effect could be limited by inhibiting ATG5 gene expression, and the greater the accumulation of autophagic vesicles, the more pronounced the inflammatory effect. In tobacco smoke combined with elastase-induced COPD-like model mice, we further observed that both autophagic and inflammatory effects were differentially inhibited in autophagy inhibitor-pretreated smoked mice. Moreover, in C57BL/6JWT mice, lung inflammation was partially alleviated, and autophagy marker expression was significantly decreased in the group of smoked mice with 3-MA pretreatment. Recent studies have shown that inhibition of autophagy in an acute COPD-like mouse model reduces the counts of neutrophils, macrophages, etc., in BALF,9 which is consistent with our experimental results. The above data suggest that autophagy has a significant role in promoting airway inflammation in COPD. In C57BL/6JRELMβ-/- mice, this airway inflammation relief was even more pronounced. In conclusion, our experiments further demonstrate that RELMβ promotes airway inflammation progression via autophagy in COPD mice.
RELMβ, as a secreted protein, is less studied in signal transduction. TLR4 is a member of the cell membrane receptor molecule family. TLR4 is highly expressed in patients with COPD,36 and cigarette smoke promotes TLR4 expression in peripheral blood CD8+ T cells. The expression of TLR4 correlates with lung function, especially the degree of obstructive ventilatory dysfunction, which is negatively correlated with the presentation of TL4.36 TLR4-mediated autophagy has been studied more frequently. Recently, in a mouse model of LPS-mediated ALI, LPS was shown to activate the expression of the autophagy marker LC3I/II via TLR4. In porcine pancreatic elastin- and LPS-stimulated interferon-regulated protein 3-deficient mice, alveolar septa were reduced, the alveolar lumen was enlarged, and the expression of LC3BI/II was significantly increased in extracted lung neutrophils after chloroquine stimulation.37,38 These studies show that TLR4 and its related signaling pathways are closely associated with the occurrence of autophagic mechanisms in COPD. We stimulated 16HBE cells with rhRELMβ and showed that TLR4 expression was significantly increased when autophagic vesicle accumulation was increased, indicating that rhRELMβ activates the upregulation of TLR4. We further analyzed whether rhRELMβ binds to TLR4 by immunofluorescence and immunoprecipitation. Our analysis showed that RELMβ could attach to the cell membrane TL4R receptor, thereby mediating the signaling pathway transduction activated by RELMβ. Since RELMβ binds to and activates TLR4, it is possible that RELMβ activates cellular or immune regulation and thus is involved in inflammatory effects. In fact, early in the study of intestinal diseases, we found that RELMβ was closely associated with intestinal mucosal immunity.39 In the intestine of RELMβ-deficient mice, the parasite load was higher, and intestinal immune function was significantly reduced.40 Therefore, the role of RELMβ in airway mucosal immunity is unknown and deserves in-depth analysis.
In conclusion, our study shows that RELMβ, as a secretory protein, can bind to TLR4 receptors on the cell membrane to promote signal transduction and that RELMβ promotes inflammation in tobacco smoke-stimulated bronchial epithelial cells that can be regulated by autophagy. Chloroquine, an autophagy inhibitor that we use at the cellular level, is commonly used as an autophagy-based agent in acute and chronic inflammation, especially in novel coronavirus infections that have emerged in recent years. CQ is a substantial basis for a therapeutic agent in this disease to support a significant ameliorative effect on the condition.41–43 In COPD in a three-dimensional airway organoid COPD model, CQ has been shown to inhibit the differentiation of airway basal cells to mucous and ciliated cells and suppress the ciliated function of the airway.44 In contrast, the autophagy-inducing agent fisetin restored phagocytosis in tobacco smoke-stimulated mice with impaired airway autophagy.45 However, whether it can be further applied to clinical patients is still unknown and needs to be further investigated.
Ethics Statement
The ethical approval for the human specimen research involved in this study was granted by the Ethics Committee of the First Affiliated Hospital of Jinan University (KY-2021-051). Written informed consent was obtained from all participants. The 16HBE bronchial epithelial cells was also approved by an institutional review board from the First Affiliated Hospital of Jinan University (Kyk-2021-017). The animal experimental protocol ethics in this study has been reviewed and approved by the Animal Experimentation Ethics Committee of Jinan University (IACUC-20220506-11). All the applied procedures followed the Chinese guidelines for the welfare of the laboratory animals (GB/T 35823-2018).
Funding
Funding by Science and Technology Projects in Guangzhou (202102010064).
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Wang C, Xu J, Yang L, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet. 2018;391(10131):1706–1717.
2. Halpin D, Criner G, Papi A, et al. Global initiative for the diagnosis, management, and prevention of chronic obstructive lung disease. The 2020 GOLD science committee report on COVID-19 and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2021;203(1):24–36. doi:10.1164/rccm.202009-3533SO
3. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
4. Lu Z, Van Eeckhoutte HP, Liu G, et al. Necroptosis signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2021;204(6):667–681. doi:10.1164/rccm.202009-3442OC
5. Morishita H, Mizushima N. Diverse Cellular Roles of Autophagy. Annu Rev Cell Dev Biol. 2019;35:453–475.
6. Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: from membrane growth to closure. J Cell Biol. 2020;219(6). doi:10.1083/jcb.202002085
7. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. 2021;54(3):437–453. doi:10.1016/j.immuni.2021.01.018
8. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14(2):243–251. doi:10.1080/15548627.2017.1402992
9. Huang HQ, Li N, Li DY, et al. Autophagy promotes cigarette smoke-initiated and elastin-driven bronchitis-like airway inflammation in mice. Front Immunol. 2021;12:594330. doi:10.3389/fimmu.2021.594330
10. Mehta P, McAuley DF, Brown M, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. doi:10.1016/S0140-6736(20)30628-0
11. Bodas M, Pehote G, Silverberg D, et al. Autophagy augmentation alleviates cigarette smoke-induced CFTR-dysfunction, ceramide-accumulation and COPD-emphysema pathogenesis. Free Radic Biol Med. 2019;131:81–97. doi:10.1016/j.freeradbiomed.2018.11.023
12. Steppan C, Brown E, Wright C, et al. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci USA. 2001;98(2):502–506. doi:10.1073/pnas.98.2.502
13. Holcomb I, Kabakoff R, Chan B, et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000;19(15):4046–4055. doi:10.1093/emboj/19.15.4046
14. Renigunta A, Hild C, Rose F, et al. Human RELMbeta is a mitogenic factor in lung cells and induced in hypoxia. FEBS Lett. 2006;580(3):900–903. doi:10.1016/j.febslet.2006.01.012
15. Tian H, Liu L, Wu Y, et al. Resistin-like molecule β acts as a mitogenic factor in hypoxic pulmonary hypertension via the Ca(2+)-dependent PI3K/Akt/mTOR and PKC/MAPK signaling pathways. Respir Res. 2021;22(1):8. doi:10.1186/s12931-020-01598-4
16. Yamaji-Kegan K, Su Q, Angelini D, et al. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. J Immunol. 2010;185(9):5539–5548. doi:10.4049/jimmunol.0904021
17. Lin C, Chen L, Huang Z, et al. Effect of cigarette smoke extraction on the expression of found in inflammatory zone 1 in rat lung epithelial L2 cells. Chin Med J. 2014;127(12):2363–2367.
18. Che L, Yu C, Chen G, et al. The inflammatory response induced by RELMβ upregulates IL-8 and IL-1β expression in bronchial epithelial cells in COPD. Int J Chron Obstruct Pulmon Dis. 2021;16:2503–2513. doi:10.2147/COPD.S321877
19. Carolan BJ, Heguy A, Harvey BG, et al. Up-regulation of expression of the ubiquitin carboxyl-terminal hydrolase L1 gene in human airway epithelium of cigarette smokers. Cancer Res. 2006;66(22):10729–10740. doi:10.1158/0008-5472.CAN-06-2224
20. Global initiative for chronic obstructive lung disease global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease; 2021. Aailable from: https://goldcopd.org/.
21. Hogg JCJL. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364(9435):709–721. doi:10.1016/S0140-6736(04)16900-6
22. Zheng LD, Tong QS, Weng MX, et al. Enhanced expression of resistin-like molecule beta in human colon cancer and its clinical significance. Dig Dis Sci. 2009;54(2):274–281. doi:10.1007/s10620-008-0355-2
23. Zheng L, Weng M, He J, et al. Expression of resistin-like molecule beta in gastric cancer: its relationship with clinicopathological parameters and prognosis. Virchows Arch. 2010;456(1):53–63. doi:10.1007/s00428-009-0861-4
24. Zheng L, Tong Q, Weng M, et al. Expression of resistin-like molecule beta in Barrett’s esophagus: a novel biomarker for metaplastic epithelium. Dig Dis Sci. 2010;55(1):32–39. doi:10.1007/s10620-009-0719-2
25. Bodas M, Patel N, Silverberg D, et al. Master autophagy regulator transcription factor EB regulates cigarette smoke-induced autophagy impairment and chronic obstructive pulmonary disease-emphysema pathogenesis. Antioxid Redox Signal. 2017;27(3):150–167. doi:10.1089/ars.2016.6842
26. Kimura T, Isaka Y, Yoshimori T. Autophagy and kidney inflammation. Autophagy. 2017;13(6):997–1003. doi:10.1080/15548627.2017.1309485
27. Prabakaran T, Bodda C, Krapp C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018;37(8). doi:10.15252/embj.201797858
28. Lin Q, Fan C, Skinner JT, et al. RELMα licenses macrophages for damage-associated molecular pattern activation to instigate pulmonary vascular remodeling. J Immunol. 2019;203(11):2862–2871. doi:10.4049/jimmunol.1900535
29. Lin Q, Fan C, Gomez-Arroyo J, et al. HIMF (Hypoxia-Induced Mitogenic Factor) signaling mediates the HMGB1 (High Mobility Group Box 1)-dependent endothelial and smooth muscle cell crosstalk in pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2019;39(12):2505–2519. doi:10.1161/ATVBAHA.119.312907
30. Xu X, Lei Y, Chen L, et al. Phosphorylation of NF-κBp65 drives inflammation-mediated hepatocellular carcinogenesis and is a novel therapeutic target. J Exp Clin Cancer Res. 2021;40(1):253. doi:10.1186/s13046-021-02062-x
31. Kanayama M, He YW, Shinohara ML. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. J Immunol. 2015;194(11):5465–5471. doi:10.4049/jimmunol.1403249
32. Abdel Fattah E, Bhattacharya A, Herron A, et al. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J Immunol. 2015;194(11):5407–5416. doi:10.4049/jimmunol.1402277
33. Dickinson JD, Alevy Y, Malvin NP, et al. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. 2016;12(2):397–409. doi:10.1080/15548627.2015.1056967
34. Mizumura K, Cloonan SM, Nakahira K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD.The. J Clin Invest. 2014;124(9):3987–4003. doi:10.1172/JCI74985
35. Lam HC, Cloonan SM, Bhashyam AR, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2020;130(11):6189. doi:10.1172/JCI143863
36. Di Stefano A, Ricciardolo FLM, Caramori G, et al. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur Respir J. 2017;49(5):1602006. doi:10.1183/13993003.02006-2016
37. Ishii T, Hosoki K, Nikura Y, et al. IFN regulatory factor 3 potentiates emphysematous aggravation by lipopolysaccharide. J Immunol. 2017;198(9):3637–3649. doi:10.4049/jimmunol.1601069
38. Zou Y, Bhat OM, Yuan X, et al. Release and actions of inflammatory exosomes in pulmonary emphysema: potential therapeutic target of acupuncture. J Inflamm Res. 2021;14:3501–3521. doi:10.2147/JIR.S312385
39. Wang ML, Keilbaugh SA, Cash-Mason T, et al. Immune-mediated signaling in intestinal goblet cells via PI3-kinase- and AKT-dependent pathways.Am. J Physiol Gastrointest Liver Physiol. 2008;295(5):G1122–30. doi:10.1152/ajpgi.90430.2008
40. Chen G, Wang S, Jang J, et al. Comparison of RELMα and RELMβ single- and double-gene-deficient mice reveals that RELMα expression dictates inflammation and worm expulsion in hookworm infection. Infect Immun. 2016;84(4):1100–1111. doi:10.1128/IAI.01479-15
41. Axfors C, Schmitt AM, Janiaud P, et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat Commun. 2021;12(1):2349. doi:10.1038/s41467-021-22446-z
42. Vouri SM, Thai TN, Winterstein AG. An evaluation of co-use of chloroquine or hydroxychloroquine plus azithromycin on cardiac outcomes: a pharmacoepidemiological study to inform use during the COVID19 pandemic. Res Social Adm Pharm. 2021;17(1):2012–2017. doi:10.1016/j.sapharm.2020.04.031
43. Sivapalan P, Ulrik CS, Bojesen RD, et al. Proactive prophylaxis with azithromycin and hydroxyChloroquine in hospitalised patients with COVID-19 (ProPAC-COVID): a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21(1):513. doi:10.1186/s13063-020-04409-9
44. Chen SL, Chou HC, Lin KC, et al. Investigation of the role of the autophagic protein LC3B in the regulation of human airway epithelium cell differentiation in COPD using a biomimetic model. Mater Today Bio. 2022;13:100182. doi:10.1016/j.mtbio.2021.100182
45. Pehote G, Bodas M, Brucia K, et al. Cigarette smoke exposure inhibits bacterial killing via TFEB-mediated autophagy impairment and resulting phagocytosis defect. Mediators Inflamm. 2017;2017:3028082. doi:10.1155/2017/3028082
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.