")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
The Mechanism of Sodium-Glucose Cotransporter-2 Inhibitors in Reducing Uric Acid in Type 2 Diabetes Mellitus
Authors Dong M , Chen H , Wen S , Yuan Y, Yang L, Xu D, Zhou L
Received 27 November 2022
Accepted for publication 8 February 2023
Published 14 February 2023 Volume 2023:16 Pages 437—445
DOI https://doi.org/10.2147/DMSO.S399343
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Meiyuan Dong,1,2 Huiling Chen,2 Song Wen,2 Yue Yuan,2 Liling Yang,2 Dongxiang Xu,2 Ligang Zhou1– 3
1Graduate School of Hebei Medical University, Shijiazhuang, People’s Republic of China; 2Department of Endocrinology, Shanghai Pudong Hospital, Fudan University, Shanghai, People’s Republic of China; 3Shanghai Key Laboratory of Vascular Lesions Regulation and Remodeling, Shanghai Pudong Hospital, Fudan University, Shanghai, People’s Republic of China
Correspondence: Ligang Zhou, Department of Endocrinology, Shanghai Pudong Hospital, Fudan University, Shanghai, 201399, People’s Republic of China, Tel +8613611927616, Email [email protected]
Abstract: Hyperuricemia is a common comorbidity in patients with type 2 diabetes mellitus (T2DM), as insulin resistance (IR) or hyperinsulinemia is associated with higher serum uric acid (SUA) levels due to decreased uric acid (UA) secretion, and SUA vice versa is an important risk factor that promotes the occurrence and progression of T2DM and its complications. Growing evidence suggests that sodium-glucose cotransporter 2 inhibitors (SGLT-2i), a novel anti-diabetic drug initially developed to treat T2DM, may exert favorable effects in reducing SUA. Currently, one of the possible mechanisms is that SGLT2i increases urinary glucose excretion, probably inhibiting glucose transport 9 (GLUT9)-mediated uric acid reabsorption in the collecting duct, resulting in increased uric acid excretion in exchange for glucose reabsorption. Regardless of this possible mechanism, the underlying comprehensive mechanisms remain poorly elucidated. Therefore, in the present review, a variety of other potential mechanisms will be covered to identify the therapeutic role of SGLT-2i in hyperuricemia.
Keywords: hyperuricemia, uric acid, type 2 diabetes mellitus, sodium-glucose cotransporter 2 inhibitors
Introduction
Type 2 diabetes mellitus (T2DM) is a metabolic disorder characterized by chronic hyperglycemia and is considered one of the main global causes of morbidity and mortality.1 Serum uric acid (SUA) is the final product of purine metabolism by xanthine oxidoreductase (XO), and hyperuricemia is caused by abnormal purine metabolism. Patients with an increase in SUA concentration are at risk for diabetes, metabolic syndrome (MetS), and other related symptoms. In an observational study, the researchers eventually enrolled 46,561 eligible participants in Taiwan’s Health Examination Registration System from 2013 to 2015, and found that SUA is an important predictor of the risk of MetS, DM and hypertension in adults, especially in men.2 In recent decades, a great deal of studies has demonstrated that hyperuricemia is an independent risk factor for CVD, as well high UA promotes insulin resistance; it is also associated with atrial fibrillation (Malhotra, #26), heart failure (HF), major adverse cardiovascular events (MACE) such as death, infarction, stroke, glomerular damage, tubulointerstitial fibrosis and chronic kidney disease (CKD).3–7 In a meta-analysis based on observational cohort studies, it is demonstrated that SUA is an independent risk factor for CKD, even without diabetes.8 Given the various detrimental complications that could be associated with hyperuricemia, reducing SUA may be beneficial for T2DM patients as they may be at a higher risk of microvascular and cardiovascular diseases. Therefore, reducing SUA levels is necessary.
Sodium-glucose cotransporter 2 (SGLT-2) is predominantly expressed in kidney segments 1 and 2 of the proximal convoluted tubules’ apical membrane.9 Sodium-glucose cotransporter-2 inhibitors (SGLT-2is) are a novel family of medications used to treat T2DM by inhibiting SGLT-2 in the renal proximal tubules, which is responsible for 90% of renal glucose reabsorption.10 This mechanism is glucose-dependent and has nothing to do with the efficacy or availability of insulin. SGLT2I leads to an increase in urine glucose that is accompanied by osmotic diuresis and a decrease in blood pressure.11 In addition to the anti-diabetic effects, weight loss has also been observed in patients receiving SGLT-2i therapy. In the cardiovascular outcome trial (CVOTS), SGLT2i has been shown to reduce major adverse cardiovascular events (MACE) and hospitalization for heart failure, and is associated with slow progression of kidney disease and lower incidence of renal end points, and these effects appear to be independent of hypoglycemia effects.12,13 In addition, the latest clinical trials have shown that SUA levels could be reduced by SGLT2i.10,14 However, the exact mechanism underpin is to be determined. Therefore, in this review, we will talk about the possible mechanisms of SGLT2i in lowering the SUA level, which remains a topic with massive complexity and is still a matter of debate.
The Physiology and Pathophysiology of Uric Acid Metabolism
Metabolism of UA
UA (2,6,8-trihydroxypurine, C5H4N4O3) is the ultimate product of exogenous and endogenous purine. The production and metabolism of UA is a complex process involving various factors that regulate the production of the liver and the excretion of the compound in the kidneys and intestines.15 The excretion of UR is controlled by postglomerular reabsorption and secretion. UA is reabsorbed and secreted in the proximal tubule, with around 90% of it being reabsorbed into the blood mostly by the transport of proteins on the renal tubules.16 In human body, purine nucleotides convert to inosine monophosphate (IMP) by deaminase, following IMP be phosphorylated into adenosine monophosphate (AMP) or guanosine monophosphate (GMP), which can be respectively transformed into xanthine. At length, xanthine is metabolized to the terminal product UA by xanthine oxidase enzymatic reaction15,17 (Figure 1). XO is abundant in liver and small intestine, but low in endothelial cells, brain, kidney and other cell types, and it is key enzyme in urate metabolism. The functional pKa of SUA is 5.75, while at physiological pH it exists mainly in the form of urate anion.18
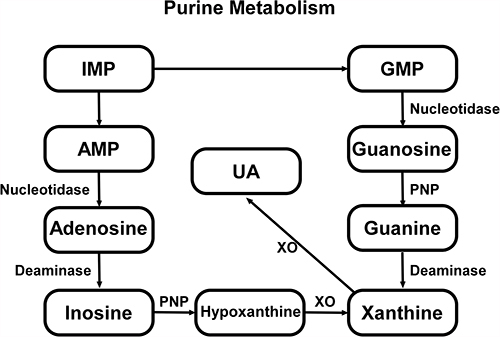 |
Figure 1 Exogenous and endogenous purine is deaminated to form inosine monophosphate (IMP), which can convert to adenosine monophosphate (AMP) or guanosine monophosphate (GMP) and terminally turn into xanthine through two metabolic pathways. Part of AMP is dephosphorylated to adenosine by nucleotidase, and subsequently adenosine is deaminated to inosine. Similarly, GMP is converted to guanosine by nucleotidase. Inosine and guanosine are further converted to hypoxanthine and guanine by purine nucleoside phosphorylase (PNP) respectively, followed by hypoxanthine being oxidized while guanine deaminated, both to form xanthine. Eventually, uric acid is produced through xanthine oxidized reaction by xanthine oxidase (XO). |
Urate Excretion and Absorption
About 70% UA is excreted accompanied with urine, whereas the rest UA is excreted through the intestinal tract.19 In human kidneys, urate excrement involves urate glomerular filtration, followed by a series of complex reabsorption and secretion mechanisms in the proximal tubules. There are two main categories of UA transporters: one is urate reabsorption transporter, and another is UA excretory transporter. The urate reabsorption pathway involves UA reabsorption transporter 1 (URAT1), organic anion transporter 4(OAT4), and glucose transporter 9 (GLUT9) (long isoform GLUT9-L located in the basal membrane of the proximal tubule; short isoform GLUT9-S located in the apical membrane of the collecting duct); the excretion pathway includes OAT1, OAT3, multi-drug resistance protein 4 (MRP4/ABCC4), ATP binding cassette transporter G-2 (ABCG-2) and sodium/phosphate cotransporters (NPT1/NPT4).20,21 (Figure 2) All these transporters or proteins significantly affect UA levels in the body, which may cause metabolic disorders such as hyperuricemia when they are dysfunctional or damaged.
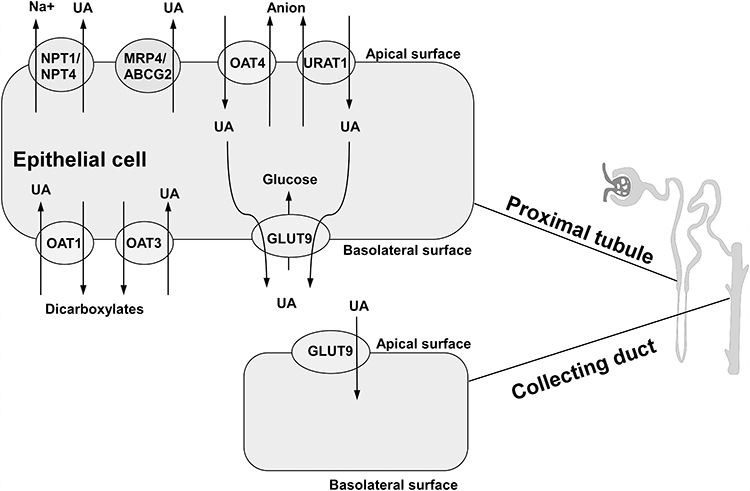 |
Figure 2 Transporters and proteins regulating uric acid transport in renal proximal tubule and collecting duct. OAT1 and OAT3, located on basolateral surface, are referred to as major multi-specific influx transporters that transfer uric acid from the blood to proximal tubule cells. Urate reabsorption pathway involves OAT4 and URAT1. Intracellular UA, moved by OAT4 and URAT1 from lumen, is transported through GLUT9 on basolateral membrane. ABCG-2, MRP4 and NPT1/4 on apical membrane mediate the excretion of uric acid. |
Influence of Pathologically Increased UA: Promoting Diabetic Development
Causing Inflammation
Evidence shows that UA may be involved in the development of atherosclerosis and other vascular disease through pro-inflammatory pathway. Soluble UA could activate NLRP3 inflammasome. In human studies, the increase of UA level in blood promotes the expression of many inflammatory factors such as interleukin-1β(IL-1 β),22 tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6).23 UA up-regulates the expression of C-reactive protein (CRP) in cultured human vascular smooth muscle cells and endothelial cells.24 In addition, Toll-like receptor 4 (TLR4) is expressed in renal tubular epithelial cells and participates in the process of promoting production of proinflammatory cytokines, inducing insulin resistance and cardiovascular damage in patients with obesity and T2DM.25 TLR activation increases the expression of several inflammatory cytokines and chemokines, such as TNF-α, that are associated with the pathogenesis of diabetic nephropathy.26
Causing Endothelial Dysfunction
Due to the effects of hyperglycemia and neurohormone activation, UA levels could be independently related to endothelial dysfunction in animals and humans, thus could promote hypertension and other cardiovascular disease.7 Decreased nitric oxide (NO) level of endothelial cells leads to endothelial dysfunction. UA is the most abundant antioxidant in plasma, which increases and reacts irreversibly with NO, resulting in the production of 6-aminouracil, and NO depletion.27 Research suggests that high UA inhibits the production of NO in aortic endothelial cells stimulated by vascular endothelial growth factors.28 In addition, UA can down-regulate the expression of nitric oxide synthase 1 (NOS1) to inhibit the production of NO through activating renin-angiotensin system (RAAS).29 Interestingly, UA-induced endothelial dysfunction is associated with decreased mitochondrial mass and ATP generation. In rats with hyperuricemia, it was observed that the increase in mitochondrial DNA damage was related to the increase in uric acid and oxidative stress in the kidneys.30
Causing Oxidative Stress
Excessive UA can lead to increased production of reactive oxygen clusters (ROS), which can cause vascular inflammation and dysfunction.31 XO is regarded as a crucial enzyme in uric acid metabolism and a major generator of reactive oxygen species, particularly superoxide anions, which rapidly interact with endothelial-derived nitric oxide to produce peroxynitrite and initiate a cascade of damaging oxygen-free radical effects.32 Besides, oxidative stress is an important pathogenic factor of UA-mediated renal tubular cells, vascular endothelial cells and smooth muscle cells (SMC), hepatocytes and adipocytes.33 Studies have shown that uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the RAAS, whereas angiotensin II induces an increase in the activities of NOX and XO, ultimately resulting in oxidative damage.34 Oxidative stress is associated with T2DM, as it can result in decreased insulin secretion and insulin resistance by interfering with the genetic expression of insulin.35
The Mechanisms of SGLT2i in Lowering SUA Levels
SGLT2i effectively induces urinary glucose excretion by inhibiting the activity of SGLT2 in renal proximal tubule S1 segment and reduces the reabsorption of glucose by tubular epithelial cells, exerting a hypoglycemic effect.36 In addition to its glucose-lowering effect, large randomized clinical trials show that SUA levels can be continuously lowered by SGLT2i.37–40 In the study of uric acid metabolism analysis of T2DM patients taking SGLT2i orally, it is confirmed that the increase of UA excretion rate leads to the decrease of SUA, which is related to the increase of urine glucose excretion rate. The treatment with SGLT2i leads to the increase of glucose excretion in urine, which eventually leads to the increase of UA exchange in the apical membrane of tubular cells and the increase of UA release from blood to urine, hence decreasing the SUA level.41 Besides, a substantial body of evidence shows that SGLT2i can also reduce the incidence rate of gout.42,43 A meta-analysis of randomized controlled trial about the effect of SGLT2i on SUA level showed that SUA was significantly decreased by all SGLT2is (dapagliflozin, canagliflozin, tofogliflozin, empagliflozin, luseogliflozin or ipragliflozin), especially empagliflozin exerted a superior SUA-lowering effect, and there was a dose-dependent manner (from 5 to 50 mg) in treat with dapagliflozin.44
SGLT-2i Blocks SGLT-2 Affecting GLUT9 and URAT-1
The transporter URAT1, encoded by SLC22A12 gene, is located on the basolateral surface of the proximal tubule, and is responsible for urate reabsorption after glomerular filtration. The accumulation of organic anions in cells is conducive to the reabsorption of urate in exchange for these anions transferred to the lumen, providing the basis for hyperuricemia associated with elevated levels of these anions.45,46 Glucose transporter 9 (GLUT9) appears to be involved in the efflux of UA.47 GLUT9, encoded by the SLC2A9 gene, is a fructose transporter that mediates UA reabsorption from renal tubular cells into circulation.48 The current hypothesis about the effect of SGLT2i on reducing UA is that glucose promotes UA excretion in the urine, involving transporters URAT1 and GLUT9.49 Studies have shown that luseogliflozin inducing excretion of glucose may increase urinary excretion rate of UA, as glycosuria influences activity of urate transport rather than activity of any other transporters.50 The glucose level in the beginning proximal tubule may be higher due to SGLT-2i blocking the SGLT-2 located on S1 segment. SGLT-2i increases intraluminal glucose concentration, stimulate UA excretion in proximal tubules mediated by GLUT9 or any other transporter.50 There is research demonstrating that SGLT-2i suppresses UA reabsorption in collecting ducts mediated by GLUT9.51 Another mechanism may be that SGLT-2i reduces the reabsorption of UA via urate reabsorption protein 1 (URAT1) thereby reducing the concentration of serum insulin. Figure 3 shows the concrete mechanism.
 |
Figure 3 SGLT-2i blocks SGLT-2 on apical surface resulting in increase of urinary glucose, and glycosuria subsequently stimulates excretion of uric acid transported by apical GLUT9, NPT1/NPT4 and MRP4/ABCG2. In contrast, elevated urine glucose suppresses UA reabsorption by URAT1 on apical membrane at proximal tubule and apical GLUT9 at collecting duct. In addition, SGLT-2i inhibit uric acid reabsorption by URAT1 through reducing serum insulin concentration. |
SGLT-2i Enhances Sirtuin-1
Sirtuin (SIRT) is a member of the silencing information regulator 2 family. SIRT1 plays a cytoprotective role through a variety of mechanisms. It has the effects of anti-apoptosis, anti-oxidation and anti-inflammation. Hyperglycemia disrupts activation/dynamic interaction between SIRT1 and AMPK, decreasing the expression of SIRT1 and AMPK.52 Basolateral high glucose stimulates SGLT-2 expression. Correlation between SIRT1 and SGLT-2 expression under diabetic conditions is negative, the over-expression of SGLT-2 leads to the increase of glucose from lumen into proximal tubule cells, reducing the expression of SIRT1.53 Xanthine oxidase levels in plasma and tissue increase in metabolic disorder-related inflammatory states (obesity, metabolic syndrome and type 2 diabetes)54,55 and chronic heart failure.56 Activation of SIRT1 downregulates the activity of xanthine oxidase.57 Research shows that SGLT-2i, blocking reabsorption of glucose by the SGLT-2 on proximal tubule apical to reduce serum glucose, stimulate activation of sirtuin-1, which can inhibit xanthine oxidase to reduce UA production.58 It can be expected that SIRT1 reduces the activation of xanthine oxidase, thus reducing the formation of UA. In addition, the activation of SIRT1 may inhibit xanthine oxidase through direct action. Another possibility is that SIRT1 promotes intestinal UA excretion by activating ABCG2 through the peroxisome proliferator-activated receptor γ /co-activator α (PGC-1α/PPARγ) pathway59 (Figure 4)
 |
Figure 4 Pathways by which SGLT-2i enhance SIRT1 to reduce SUA. Different arrows represent three dependent pathways. |
SGLT2i Attenuates Activation of NLRP3
Urate crystallization and soluble UA are proved to activate pyrin domain-containing 3 (NLRP3) inflammasome and induce the production of IL-1β.60 NLRP3 inflammasome is a significant factor to induce acute gout.61 ABCG2 is a high-volume urate exporter that promotes the secretion of UA in the intestinal tract, decreased ABCG2 function leading to decreased UA excretion, could cause hyperuricemia.62 However, several research demonstrated that soluble UA upregulates the expression of ABCG2 to promote the excretion of intestinal cells by activating TLR4-NLRP3 inflammasome and PI3K/Akt signal pathway, which may be a feedback regulation.63 SGLT-2i can attenuate the activation of NLRP3 inflammasome in T2DM patients with high-risk of hyperuricemia by increasing circulating β-hydroxybutyrate (BHB) levels. Besides, SGLT-2i inhibits the SGLT-2 resulting in reduced serum glucose and insulin levels to attenuate the activation of NLRP3 inflammasome, which decrease the risk of acute gout.64
SGLT2i Influences RAAS and Sympathetic Nervous System
Recent studies have demonstrated that blocking XO for four weeks in rats with cerebral infarction depresses sympathetic innervation. This suggests that sympathetic nerve activity may contribute to the production of uric acid and that UA may inhibit sympathetic nervous system (SNS) activity via a superoxide-dependent mechanism.65 Accumulating evidence suggests that SGLT2i could down-regulate sympathetic activity, as demonstrated by decreases in SNS indicators such as norepinephrine (NE), which may be a potential mechanism for lowering SUA.66 In addition, activation of RAAS interacting with UA metabolism induces a series of inflammatory damage.29,34 Recently, emerging evidence suggests that SGLT2i could improve redox state and oxidative stress, thereby reducing oxidative damages to ameliorate IR, by regulating the activity of the renin-angiotensin system (RAAS), down-regulating the pro-oxidant enzymes and enhancing mitochondrial function.67,68
Conclusion
Patients with T2DM can benefit from the lower incidence of gout caused by SGLT-2 inhibitors. Numerous clinical trials and fundamental research have established a relationship between T2DM and hyperuricemia. Considering the systemic diseases associated with hyperuricemia, decreasing serum uric acid in T2DM patients is undoubtedly beneficial. Due to its effectiveness in reducing serum uric acid, the SGLT2 inhibitor will be recommended over the other oral anti-diabetic medications. In this review, we examined the potential processes by which SGLT2 inhibitors reduce uric acid: 1. SGLT-2i may improve urinary glucose excretion by blocking renal tubular uric acid transporters, such as URAT1, which increase UA secretion in response to glucose reabsorption, resulting in higher UA excretion and decreased SUA. In addition, SGLT-2i inhibit UA reabsorption by URAT1 by decreasing serum insulin levels. 2. SGLT-2i inhibits the entry of intracellular glucose into proximal renal tubular cells, restores the production of sirtuin-1, and sirtuin-1 inhibits the main enzyme xanthine oxidase (OX) in uric acid metabolism, resulting in a decreased UA level. SIRT1 may also stimulate the intestinal excretion of UA. 3. SGLT-2i inhibits greatly the activation of inflammatory bodies, the production of IL-1 and NLRP3, and the activation of NLRP3, hence alleviating gout. Through RAAS and SNS, SGLT2i is able to downregulate the activity of XO and suppress the inflammatory response to decrease SUA formation. In addition to the aforementioned probable mechanisms, there are a significant number of other potential mechanisms that will require extensive experimental and clinical research in the future.
Funding
This work was supported by the Project of Key Medical Discipline of Pudong Hospital of Fudan University (Zdxk2020-11), Project of Key Medical Specialty and Treatment Center of Pudong Hospital of Fudan University (Zdzk2020-24), Integrative Medicine special fund of Shanghai Municipal Health Planning Committee (ZHYY- ZXYJHZX-2-201712), Special Department Fund of the Pudong New Area Health Planning Commission (PWZzk2017-03), Outstanding Leaders Training Program of Pudong Health Bureau of Shanghai (PWR12014-06), Pudong New Area Clinical Plateau Discipline Project (PWYgy-2021-03), the Natural Science Foundation of China (21675034), National Natural Science Foundation of China (81370932), Shanghai Natural Science Foundation (19ZR1447500), Fudan Zhangjiang Clinical Medicine Innovation Fund Project (KP0202118), and Education Funding in Wenzhou Medical University (JG2021197).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14:88–98. doi:10.1038/nrendo.2017.151
2. Chen YY, Kao TW, Yang HF, et al. The association of uric acid with the risk of metabolic syndrome, arterial hypertension or diabetes in young subjects- An observational study. Clin Chim Acta. 2018;478:68–73. doi:10.1016/j.cca.2017.12.038
3. Kodama S, Saito K, Yachi Y, et al. Association between serum uric acid and development of type 2 diabetes. Diabetes Care. 2009;32:1737–1742. doi:10.2337/dc09-0288
4. Chang -C-C, Wu C-H, Liu L-K, et al. Association between serum uric acid and cardiovascular risk in nonhypertensive and nondiabetic individuals: the Taiwan I-Lan Longitudinal Aging Study. Sci Rep. 2018;8:5234.
5. Bombelli M, Quarti-Trevano F, Tadic M, et al. Uric acid and risk of new-onset metabolic syndrome, impaired fasting glucose and diabetes mellitus in a general Italian population: data from the Pressioni Arteriose Monitorate E Loro Associazioni study. J Hypertens. 2018;36:1492–1498. doi:10.1097/HJH.0000000000001721
6. Cicero AFG, Fogacci F, Giovannini M, et al. Serum uric acid predicts incident metabolic syndrome in the elderly in an analysis of the Brisighella Heart Study. Sci Rep. 2018;8. doi:10.1038/s41598-018-29955-w
7. Ali N, Mahmood S, Islam F, et al. Relationship between serum uric acid and hypertension: a cross-sectional study in Bangladeshi adults. Sci Rep. 2019;9:9061. doi:10.1038/s41598-019-45680-4
8. Li L, Yang C, Zhao Y, et al. Is hyperuricemia an independent risk factor for new-onset chronic kidney disease? A systematic review and meta-analysis based on observational cohort studies. BMC Nephrol. 2014;15:1–12. doi:10.1186/1471-2369-15-122
9. Chen J, Williams S, Ho S, et al. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010;1:57–92. doi:10.1007/s13300-010-0006-4
10. Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: basic physiology and consequences. Diab Vasc Dis Res. 2015;12:78–89. doi:10.1177/1479164114561992
11. Ferrannini E, Solini A. SGLT2 inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol. 2012;8:495–502. doi:10.1038/nrendo.2011.243
12. Khat DZ, Husain M. Molecular mechanisms underlying the cardiovascular benefits of SGLT2i and GLP-1RA. Curr Diab Rep. 2018;18:45. doi:10.1007/s11892-018-1011-7
13. Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393:31–39. doi:10.1016/S0140-6736(18)32590-X
14. Zelniker TA, Braunwald E. Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J Am Coll Cardiol. 2020;75:422–434. doi:10.1016/j.jacc.2019.11.031
15. Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol. 2016;213:8–14. doi:10.1016/j.ijcard.2015.08.109
16. Bobulescu IA, Moe OW. Renal transport of uric acid: evolving concepts and uncertainties. Adv Chronic Kidney Dis. 2012;19:358–371. doi:10.1053/j.ackd.2012.07.009
17. Xiong Q, Liu J, Xu Y. Effects of uric acid on diabetes mellitus and its chronic complications. Int J Endocrinol. 2019;2019:9691345. doi:10.1155/2019/9691345
18. Murea M, Tucker BM. The physiology of uric acid and the impact of end-stage kidney disease and dialysis. Semin Dial. 2019;32:47–57. doi:10.1111/sdi.12735
19. Lipkowitz MS. Regulation of uric acid excretion by the kidney. Curr Rheumatol Rep. 2012;14:179–188. doi:10.1007/s11926-012-0240-z
20. Nigam SK, Bhatnagar V. The systems biology of uric acid transporters. Curr Opin Nephrol Hypertens. 2018;27:305–313. doi:10.1097/MNH.0000000000000427
21. Xu L, Shi Y, Zhuang S, Liu N. Recent advances on uric acid transporters. Oncotarget. 2017;8:100852–100862. doi:10.18632/oncotarget.20135
22. Mulla MJ, Myrtolli K, Potter J, et al. Uric acid induces trophoblast IL-1beta production via the inflammasome: implications for the pathogenesis of preeclampsia. Am J Reprod Immunol. 2011;65:542–548. doi:10.1111/j.1600-0897.2010.00960.x
23. Ruggiero C, Cherubini A, Ble A, et al. Uric acid and inflammatory markers. Eur Heart J. 2006;27:1174–1181. doi:10.1093/eurheartj/ehi879
24. Kanellis J, Kang DH. Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin Nephrol. 2005;25:39–42. doi:10.1016/j.semnephrol.2004.09.007
25. Garibotto G, Carta A, Picciotto D, Viazzi F, Verzola D. Toll-like receptor-4 signaling mediates inflammation and tissue injury in diabetic nephropathy. J Nephrol. 2017;30:719–727. doi:10.1007/s40620-017-0432-8
26. Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol. 2016;12:13–26. doi:10.1038/nrneph.2015.175
27. Gersch C, Palii SP, Kim KM, et al. Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucleic Acids. 2008;27(8):967–978. doi:10.1080/15257770802257952
28. Khosla UM, Zharikov S, Finch JL, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005;67:1739–1742. doi:10.1111/j.1523-1755.2005.00273.x
29. Wang XD, Liu J, Zhang YC, et al. Correlation between the elevated uric acid levels and circulating renin-angiotensin-aldosterone system activation in patients with atrial fibrillation. Cardiovasc Diagn Ther. 2021;11:50–55. doi:10.21037/cdt-20-830
30. Sanchez-Lozada LG, Lanaspa MA, Cristobal-Garcia M, et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol. 2012;121:e71–e78. doi:10.1159/000345509
31. Ko J, Kang HJ, Kim DA, et al. Uric acid induced the phenotype transition of vascular endothelial cells via induction of oxidative stress and glycocalyx shedding. FASEB J. 2019;33:13334–13345. doi:10.1096/fj.201901148R
32. Doehner W, Landmesser U. Xanthine oxidase and uric acid in cardiovascular disease: clinical impact and therapeutic options. Semin Nephrol. 2011;31:433–440. doi:10.1016/j.semnephrol.2011.08.007
33. Lanaspa MA, Sanchez-Lozada LG, Choi YJ, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem. 2012;287:40732–40744. doi:10.1074/jbc.M112.399899
34. Landmesser U, Spiekermann S, Preuss C, et al. Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler Thromb Vasc Biol. 2007;27:943–948. doi:10.1161/01.ATV.0000258415.32883.bf
35. Hurrle S, Hsu WH. The etiology of oxidative stress in insulin resistance. Biomed J. 2017;40:257–262. doi:10.1016/j.bj.2017.06.007
36. Ni L, Yuan C, Chen G, Zhang C, Wu X. SGLT2i: beyond the glucose-lowering effect. Cardiovasc Diabetol. 2020;19:98. doi:10.1186/s12933-020-01071-y
37. McDowell K, Welsh P, Docherty KF, et al. Dapagliflozin reduces uric acid concentration, an independent predictor of adverse outcomes in DAPA-HF. Eur J Heart Fail. 2022;24(6):1066–1076. doi:10.1002/ejhf.2433
38. Hao Z, Huang X, Shao H, Tian F. Effects of dapagliflozin on serum uric acid levels in hospitalized type 2 diabetic patients with inadequate glycemic control: a randomized controlled trial. Ther Clin Risk Manag. 2018;14:2407–2413. doi:10.2147/TCRM.S186347
39. Ferreira JP, Inzucchi SE, Mattheus M, et al. Empagliflozin and uric acid metabolism in diabetes: a post hoc analysis of the EMPA-REG OUTCOME trial. Diabetes Obes Metab. 2022;24:135–141. doi:10.1111/dom.14559
40. Davies MJ, Trujillo A, Vijapurkar U, Damaraju CV, Meininger G. Effect of canagliflozin on serum uric acid in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2015;17:426–429. doi:10.1111/dom.12439
41. Suijk DL, van Baar MJ, van Bommel EJ, et al. SGLT2 inhibition and uric acid excretion in patients with type 2 diabetes and normal kidney function. Clin J Am Soc Nephrol. 2022;17:663–671. doi:10.2215/CJN.11480821
42. Lund LC, Hojlund M, Henriksen DP, Hallas J, Kristensen KB. Sodium-glucose cotransporter-2 inhibitors and the risk of gout: a Danish population based cohort study and symmetry analysis. Pharmacoepidemiol Drug Saf. 2021;30:1391–1395. doi:10.1002/pds.5252
43. Fralick M, Chen SK, Patorno E, Kim SC. Assessing the risk for gout with sodium-glucose cotransporter-2 inhibitors in patients with type 2 diabetes: a population-based cohort study. Ann Intern Med. 2020;172:186–194. doi:10.7326/M19-2610
44. Zhao Y, Xu L, Tian D, et al. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: a meta-analysis of randomized controlled trials. Diabetes Obes Metab. 2018;20:458–462. doi:10.1111/dom.13101
45. Mancikova A, Krylov V, Hurba O, et al. Functional analysis of novel allelic variants in URAT1 and GLUT9 causing renal hypouricemia type 1 and 2. Clin Exp Nephrol. 2016;20:578–584. doi:10.1007/s10157-015-1186-z
46. Toyoda Y, Kawamura Y, Nakayama A, et al. Substantial anti-gout effect conferred by common and rare dysfunctional variants of URAT1/SLC22A12. Rheumatology. 2021;60:5224–5232. doi:10.1093/rheumatology/keab327
47. Caulfield MJ, Munroe PB, O’Neill D, et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008;5:e197. doi:10.1371/journal.pmed.0050197
48. Matsuo H, Chiba T, Nagamori S, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet. 2008;83:744–751. doi:10.1016/j.ajhg.2008.11.001
49. Novikov A, Fu Y, Huang W, et al. SGLT2 inhibition and renal urate excretion: role of luminal glucose, GLUT9, and URAT1. Am J Physiol Renal Physiol. 2019;316:F173–F185. doi:10.1152/ajprenal.00462.2018
50. Chino Y, Samukawa Y, Sakai S, et al. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos. 2014;35:391–404. doi:10.1002/bdd.1909
51. Kimura T, Takahashi M, Yan K, Sakurai H, Kanzaki M. Expression of SLC2A9 isoforms in the kidney and their localization in polarized epithelial cells. PLoS One. 2014;9(1):e84996. doi:10.1371/journal.pone.0084996
52. Clarke NE, Belyaev ND, Lambert DW, Turner AJ. Epigenetic regulation of angiotensin-converting enzyme 2 (ACE2) by SIRT1 under conditions of cell energy stress. Clin Sci. 2014;126:507–516. doi:10.1042/CS20130291
53. Umino H, Hasegawa K, Minakuchi H, et al. High basolateral glucose increases sodium-glucose cotransporter 2 and reduces sirtuin-1 in renal tubules through glucose transporter-2 detection. Sci Rep. 2018;8:6791. doi:10.1038/s41598-018-25054-y
54. Battelli MG, Bortolotti M, Polito L, Bolognesi A. The role of xanthine oxidoreductase and uric acid in metabolic syndrome. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2557–2565. doi:10.1016/j.bbadis.2018.05.003
55. Washio KW, Kusunoki Y, Murase T, et al. Xanthine oxidoreductase activity is correlated with insulin resistance and subclinical inflammation in young humans. Metabolism. 2017;70:51–56. doi:10.1016/j.metabol.2017.01.031
56. Ahmed MI, Gladden JD, Litovsky SH, et al. Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction >60%. J Am Coll Cardiol. 2010;55:671–679. doi:10.1016/j.jacc.2009.08.074
57. Huang XF, Li HQ, Shi L, et al. Synthesis of resveratrol analogues, and evaluation of their cytotoxic and xanthine oxidase inhibitory activities. Chem Biodivers. 2008;5:636–642.
58. Packer M. Uric acid is a biomarker of oxidative stress in the failing heart: lessons learned from trials with allopurinol and SGLT2 inhibitors. J Card Fail. 2020;26:977–984. doi:10.1016/j.cardfail.2020.08.015
59. Wang J, Zhu XX, Liu L, et al. SIRT1 prevents hyperuricemia via the PGC-1alpha/PPARgamma-ABCG2 pathway. Endocrine. 2016;53:443–452. doi:10.1007/s12020-016-0896-7
60. Braga TT, Forni MF, Correa-Costa M, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep. 2017;7:39884. doi:10.1038/srep39884
61. McDermott M, Kingsbury S, Conaghan PG. The role of the NLRP3 inflammasome in gout. J Inflamm Res. 2011;39–49. doi:10.2147/JIR.S11330
62. Matsuo H, Takada T, Ichida K, et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Transl Med. 2009;1:5ra11. doi:10.1126/scitranslmed.3000237
63. Chen M, Lu X, Lu C, et al. Soluble uric acid increases PDZK1 and ABCG2 expression in human intestinal cell lines via the TLR4-NLRP3 inflammasome and PI3K/Akt signaling pathway. Arthritis Res Ther. 2018;20. doi:10.1186/s13075-018-1512-4
64. Kim SR, Lee SG, Kim SH, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. 2020;11:2127. doi:10.1038/s41467-020-15983-6
65. Lee TM, Lin SZ, Chang NC. Effects of urate-lowering agents on arrhythmia vulnerability in post-infarcted rat hearts. J Pharmacol Sci. 2016;131:28–36. doi:10.1016/j.jphs.2016.03.009
66. Herat LY, Matthews J, Azzam O, Schlaich MP, Matthews VB. Targeting features of the metabolic syndrome through sympatholytic effects of SGLT2 inhibition. Curr Hypertens Rep. 2022;24:67–74. doi:10.1007/s11906-022-01170-z
67. Yaribeygi H, Atkin SL, Butler AE, Sahebkar A. Sodium-glucose cotransporter inhibitors and oxidative stress: an update. J Cell Physiol. 2019;234:3231–3237. doi:10.1002/jcp.26760
68. Osorio H, Coronel I, Arellano A, et al. Sodium-glucose cotransporter inhibition prevents oxidative stress in the kidney of diabetic rats. Oxid Med Cell Longev. 2012;2012:542042. doi:10.1155/2012/542042
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.