")
Back to Journals » Drug Design, Development and Therapy » Volume 12
The mechanism of botulinum A on Raynaud syndrome
Authors Zhou Y, Liu Y, Hao Y, Feng Y, Pan L , Liu W, Li B, Xiao L, Jin L, Nie Z
Received 29 December 2017
Accepted for publication 10 April 2018
Published 26 June 2018 Volume 2018:12 Pages 1905—1915
DOI https://doi.org/10.2147/DDDT.S161113
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Yanwen Zhou,* Ying Liu,* Yunhua Hao, Ya Feng, Lizhen Pan, Wuchao Liu, Bing Li, Libin Xiao, Lingjing Jin, Zhiyu Nie
Department of Neurology, Shanghai Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China
*These authors contributed equally to this work
Background: Botulinum neurotoxin type A (BoNT/A) is emerging as a treatment modality for Raynaud’s phenomenon (RP). However, the mechanism of the role of BoNT/A in antagonizing the constriction of arteriola in RP remains unclear.
Materials and methods: We tested the constriction of arteriole diameter and the distribution of adrenergic receptors on the rat cremaster modle. Moreover, we measured the secretion of norepinephrine (NE), protein level changes and related receptors on cultured rat superior cervical ganglia neurons(SCGNs), a model of sympathetic neuron.
Results: Based on our results, the inhibition of arteriole vasoconstriction was increased with increasing doses of BoNT/A. BoNT/Α, prazosin, and BQ123 treatment can result in significant inhibition of arteriole vasoconstriction with the same electrical stimulation. The inhibition effect of prazosin was equivalent to BoNT/A, while BQ123 has a synergistic effect with BoNT/A. After treating SCGNs using BoNT/A for 30 min, the decrease in fluorescence intensity of FM1-43 slowed down which was correlated with the doses of BoNT/A. Furthermore, release of NE in the supernatant was significantly decreased as measured by enzyme-linked immunosorbent assay, 24 h after a high dose of BoNT/A (25 µ/mL). Cleaved-SNAP-25 was detected by western blotting 24 h following BoNT/A (50 µ/mL) treatment. Moreover, receptor SV2C, GM1, and FGFR3 were detected on sympathetic neurons, similarly to cholinergic neurons.
Conclusion: Our study showed that BoNT/A could significantly inhibit electrical stimulation-induced arteriole vasoconstriction through the sympathetic pathway. The mechanism was similar to the cholinergic one, in which the vesicle release of sympathetic neurons could be inhibited by cleavage of SNAP-25. The end result was blocked vesicle fusion with the presynaptic membrane after BoNT/A treatment, inhibiting the release of the NE.
Keywords: botulinum neurotoxin type A, Raynaud’s phenomenon, α-adrenoceptor, arteriole diameter constrict rate, SNAP-25, SV2C, GM1, FGFR3, sympathetic neuron, vesicle cycle
Introduction
Raynaud’s phenomenon (RP) is a disease in which the digits turn white, then blue accompanied with pain or paresthesia under cold or abnormal emotional condition. These conditions exaggerate a vasospastic response which may participate in the development of the disease.1 The morbidity of RP is about 3%–5% and is closely related to weather and geographical environment.2 It has been defined as a clinical disease for about 150 years, however its etiology and pathogenesis remain unknown.3 Currently, accumulating evidence suggested that upregulated vascular smooth muscle α2C adrenergic receptor may play an important role in RP through inducing hyperfunction of the sympathetic nervous system and causing vasoconstriction.4 Up to now, traditional therapy for RP includes pharmacologic drugs and surgery. The curative effect of drugs is unsatisfactory, and sympathetic trunk transection can cause complications, including compensatory body hydrosis and Horner syndrome. Therefore, a novel replacement therapy is urgently needed.5–8
Botulinum neurotoxin type A (BoNT/A) was first applied in treatment of RP in 2004. Blood flow in the injection site was improved notably by this approach.9 Subsequently, several other clinical trials have also proved that BoNT/A can relieve digital arterial spasm, increase blood flow, promote ulcer healing, and alleviate pain without complications.10–14 Our colleagues reported that the blood flow of a patient’s midfinger increased to 40 cm/s, which could not be detected before BoNT/A injection.15 Furthermore, this kind of therapy has less adverse reactions. As a proposed new therapy, BoNT/A injection has become the hope for refractory RP.
Up to now, the role of BoNT/A in blocking the release of acetylcholine has been reported.23 However, the mechanism of the role of BoNT/A in antagonizing the constriction of arteriolar RP is still under study. To clarify the hypothesis that the vasodilatation caused by BoNT/A is due to the blockage of a norepineprine-mediated sympathetic pathway, we established an electrical stimulation-induced rat cremaster arteriole contraction model. This model allows comparison of the vasodilatation effect between prazosin, Yohimbine, and BQ-123 with BoNT/A. Furthermore, it allows us to investigate the mechanism of BoNT/A on arteriole vasodilation. To define the mechanism, we also clarified the type and distribution of norepinephrine receptors on the cremaster arteriole. Moreover, we cultivated primary sympathetic neurons from superior cervical ganglion neurons (SCGNs) to study the effect of BoNT/A on the sympathetic nerve system and tried to explore its mechanism through the protein level. Then, we also examined whether GM1 (a subtype of ganglioside), SV2C, and FGFR3 existed in cholinergic neurons also presented in the sympathetic nervous system.
Materials and methods
This study was approved by the Institutional Animal Care and Use Committee of Tongji Hospital. All portions of the protocol were performed in accordance with the institutional guidelines. The animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institution of Health and the Policy of Animal Care and Use Committee of Tongji University.
Cremaster arteriole vasoconstriction model establishment
A validated rat cremaster model was used to study the inhibition of BoNT/A on arteriole vasoconstriction.16,17 72 hours before anesthesia, male Sprague Dawley rats (100±10 g; n=96) were injected with BoNT/A or isopycnic 0.9% saline in the proximal and distal rat cremaster muscle. All animals were next anesthetized with 3% pentobarbital (intraperitoneal injection, 30 mg/kg body weight) (Sigma-Aldrich Co., St Louis, MO, USA). They were placed supine on a stage equipped with a reservoir to perfuse the cremaster muscle. After removal of the testicle, the cremaster muscle was exposed, and then stretched over an adjustable, raised pedestal surrounded by a 2-piece reservoir. Regional perfusion was kept with perfusate by the thermostatic circulator at 37°C. Next, we delivered a constant voltage of 30 V with varying frequency (5, 10, 16, 20, and 25 Hz) or a constant frequency of 16 Hz with varying voltage (10, 20, 30, and 40 V). Each electric-impulse stimulation was delivered for 5 s every 5 min to find the ideal vasoconstriction model.
Drug treatment in the vasoconstriction model
A recirculating tissue bath, mimicking physiologic conditions, provided a method of local delivery of drugs at reasonable concentrations. The bath and pump reservoir were filled with buffered modified Krebs solution at room temperature (24°C), recirculating with a peristaltic pump and a pressure servo control (Living Systems Instrumentation, Burlington, VT, USA). All drug solutions were added directly to the tissue bath.
Lyophilized BoNT/A (Botox, 100 U; Allergan, Inc., Irvine, CA, USA) was freshly dissolved with 1 mL of 0.9% saline. Five different doses of BoNT/A were chosen to study: 0.5 U/kg (group 9), 1.5 U/kg (group 10), 5 U/kg (groups 5–8), 15 U/kg (group 11), 50 U/kg (group 12), and 0.9% saline (groups 1–4). The 15 U/kg dose was chosen as the optimum concentration. Vessel diameters were measured 72 hours after toxin addition, and every 5 min thereafter, for a total of 20 min. The concentrations of prazosin, Yohimbine, and BQ-123 were chosen based on clinical doses and reviewed from previously published reports.25,44,45 Krebs solution, prazosin (0.1 μM, selective postsynaptic alpha 1 receptor blocker), Yohimbine (1 μM, selective blocker of presynaptic alpha 2 receptor, promotes the release of norepinephrine), and BQ-123 (1 μM, endothelin A receptor antagonist) were freshly dissolved with 1.0 mL of 0.9% saline, and then added directly to the tissue bath.
Arteriole diameter measurement
Arteriole diameter measurement was obtained with live video microscopy. The vessel’s internal diameters were measured from wall to wall by a video dimension micrometer. The video micrometer was calibrated on-screen with a stage micrometer scale before the start of each experiment. To eliminate the confounding effect of vascular response to the altered metabolic rate by toxin-mediated muscle paralysis, gallamine triethiodide (gallamine, 0.17 nM; Sigma-Aldrich Co.), an acetylcholine receptor antagonist, was added into the perfusate for 10 min. Although gallamine did not cause significant changes in vessel diameter, baseline measurement was done after the addition of gallamine. The electrical stimulation-induced cremaster arteriole vasoconstriction model (30 V, 16 Hz, 5 s) was used to mimic the arteriole vasoconstriction with intervals of 5 min. The diameters of cremaster arteriole were recorded and the arteriole diameter constriction rate was established by the following equation: arteriole diameter shrinkage rate = [diameter of arteriole before electrical stimulation–diameter of arteriole after electrical stimulation]/diameter of arteriole before electrical stimulation ×100%.
Tissue preparation and immunofluorescence
Cremaster arteriole was prepared from Sprague Dawley rats, ages 2–4 months. Tissue sections were incubated with permeabilizing/blocking solution (0.5% Triton X-100 and 10% goat serum in PBS) for 24–48 h at 4°C. The sections were then incubated with α1-AR (1:1,000; Thermo Fisher Scientific, Waltham, MA, USA), α2A-AR (1:1,000; Santa Cruz Biotechnology Inc., Dallas, TX, USA), α2B-AR (1:1,000; Santa Cruz Biotechnology Inc.), and α2c-AR (1:1,000; Santa Cruz Biotechnology Inc.) antibodies, which were dissolved with PBS. Control experiments were done using PBS without antibodies. Samples were next washed with PBS 3 times, and incubated with donkey anti-rabbit biotin (1:1,000; Abcam, Cambridge, UK) for 2 h. Next, they were washed, and incubated with Neutravidin fluorescein isothiocyanate (FITC) (1:1,000; Abcam). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Tissues were imaged on a BX-62 Olympus upright fluorescent microscope using imaging software. Background immunofluorescence was assessed by stains of only secondary antibodies.
Neural cultures and BoNT/A treatment
Sympathetic neurons were collected from the superior cervical ganglia (SCGs) of newborn rats (1–3 days). The procedure for SCGN culturing has been previously described.18 The neurons were grown on glass coverslips or in plastic/glass-bottomed dishes coated with dried rat-tail collagen. To maintain neural growth we used a medium containing neural basal medium A supplemented with B27, nerve growth factor, L-glutamine, penicillin–streptomycin solution, and 5-fluorouracil and uridine. The medium was replaced once every 2 days as needed. After establishing that the cells (cultured for 3 days) were SCGNs through an immunofluorescence assay, mature cells (cultured for 7 days) were exposed to solutions with different concentrations of BoNT/A. BoNT/A was dissolved in stimulation buffer containing 56 mM KCl, 82 mM NaCl, 2 mM CaCl2, and 1 mM MgCl2 to different concentration (5 U/mL, 10 U/mL, 25 U/mL and 50 U/mL).
Immunofluorescence labeling of sympathetic neurons
SCGNs which were grown for 3 days on glass dishes were rinsed three times with PBS and then fixed with intracellular fixation buffer for 20 min, followed by permeabilization for 10 min. After blocking with 1% fetal bovine serum in PBS, the cells were then incubated with a monoclonal anti-mouse antibody against neurofilament-200 (N52) and a polyclonal anti-rabbit antibody against tyrosine hydroxylase (Abcam) diluted to 200× in blocking solution at 4°C overnight.
In experiment 4, the cells were incubated with polyclonal anti-rabbit antibody against tyrosine hydroxylase (Abcam) and monoclonal anti-mouse antibody against SNAP-25 (SMI-81; Biolegend, San Diego, CA, USA) diluted to 200× in blocking solution at 4°C overnight. In experiment 5, the cells were incubated with polyclonal anti-rabbit antibody against SV2C (Abcam), polyclonal anti-rabbit antibody against ganglioside GM1 (Abcam), and monoclonal anti-rabbit antibody against FGFR3 (Abcam). After the cells were washed with PBS 3 times, they were exposed to anti-mouse IgG H&L (tetramethylrhodamine isothiocyanate [TRITC]) and anti-rabbit Alexa Fluor488 (Abcam) for 40 min. Images were captured on a Fluorescence Inverse Microscope Nikon ECLIPSE80i (Nikon corporation, Tokyo, USA).
Loading and unloading of FM1-43
SCGNs grown for 7 days in plastic-bottomed dishes and optimized for confocal laser scanning microscopy were exposed to fluorescent styryl dye FM1-43 (Molecular Probes, Eugene, OR, USA). The experiment was conducted under static conditions. The dye was dissolved in ice-cold Hank’s balanced salt solution to produce a working staining solution. The staining solution was added into the plastic-bottomed dish with SCGNs grown on it for 5 min. This ensures quick plasma membrane staining. After taking photos, the staining solution was removed, and the stimulation staining solutions with different concentrations of BoNT/A were added to the cultured cells. Photos were captured continuously on a confocal microscopy (model TCS SP5; Leica Microsystems, Wetzlar, Germany) for 30 min. We also interfered with the cells with stimulation staining solutions containing different concentrations of BoNT/A for 1 h. After taking photos, the solutions were removed, and cells were washed with Hank’s balanced salt solution for 3 times. The cells were then stimulated again to capture the changes of vesicle circulation after interfering.
Norepinephrine quantitative analyses
NE was collected from the cell supernatant and stored at −80°C. Enzyme-linked immunosorbent assay (ELISA) kits (Xinqidi Biological Technology Ltd, Wuhan, China) were used to determine the concentrations of NE under various conditions. The sensitivity limits of NE were 5 pg/mL.
Western blot analyses
Cultured SCGNs were washed with PBS. Cells were lysed by radioimmunoprecipitation assay for 10 s on ice, and whole cell lysates were collected and centrifuged (12,000×g, 10 min at 4°C). Samples were incubated for 5 min at 95°C with 5× loading buffer. Next, the proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis on 10% Bis–Tris gels and then transferred to polyvinylidene fluoride membranes. Membranes were incubated in blocking buffer (Tris-buffered saline with Tween 20 [TBST] and 5% skimmed milk powder) for 1 h, followed by incubation with the primary antibodies. The following primary antibodies were used: β-actin (D6A8) rabbit mAb (Cell Signaling Technology, Danvers, MA, USA), 1:1,000, overnight; and purified anti-SNAP-25 antibody (SMI-81; Biolegend, San Diego, CA, USA), 1:1,000, overnight at 4°C. The appropriate secondary antibodies, coupled to horseradish peroxidase, were added at a dilution of 1:1,000 for 1 h at room temperature. Proteins were detected using the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, Billerica, MA, USA) on an AlphaImager HP (Protein Simple, San Jose, CA, USA). Quantification of Western blots was carried out using the gel analysis tool of Alpha View SA (Protein Simple).
Statistics
All data in this research were normally distributed and analyzed by GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA) and SPSS19.0 software (IBM Corporation, Armonk, NY, USA). The data were expressed as mean±standard error of the mean (SEM) using one-way analysis of variance, Fisher’s least significant difference, and Student’s t-test. A two-tailed p<0.05 was considered to be statistically significant.
Results
The effect of different drugs on arteriole constriction
A vasoconstriction model was established and accepted as ideal with the following parameters: electric impulse stimulation for 5 s, voltage of 30 V, and frequency of 16 Hz. To establish the adrenergic and endothelin component of the observed response to BoNT/A, prazosin (alpha 1 receptor blocker), Yohimbine (alpha 2 receptor blocker), and BQ-123 (endothelin A receptor antagonist) were selected. Similar-sized vessels were prepared to study the constriction effect of arterioles under different drugs. Application of Kreb’s solution and gallamine resulted in no change of cremaster arteriole diameter and arteriole diameter constrict rate (p>0.05), and the average arteriole diameter was 59.71±2 μm.
In the absence of pretreatment with BoNT/A, the effects of prazosin (0.1 μM), Yohimbine (1 μM), BQ-123 (1 μM), and BoNT/A (5 U/kg) on the arteriole diameter constriction rate are shown in Figure 1. When compared to Krebs treatment (15.43±2.79), prazosin (6.71±1.04), BQ-123 (7.48±1.06), and BoNT/A (6.51±1.02) can effectively decrease the arteriole diameter constriction rate. The effects of prazosin and BoNT/A treatments are equivalent, while Yohimbine (15.31±0.91) showed no significant effect on arteriole vasoconstriction.
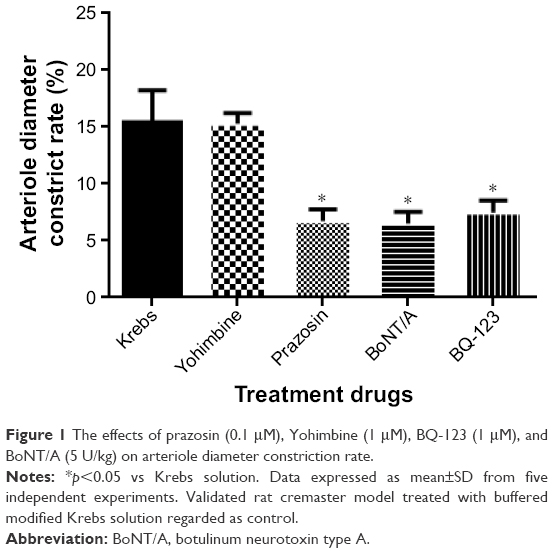 | Figure 1 The effects of prazosin (0.1 μM), Yohimbine (1 μM), BQ-123 (1 μM), and BoNT/A (5 U/kg) on arteriole diameter constriction rate. |
The effect of BoNT/A on arteriole vasodilation is dose dependent
Application of BoNT/A resulted in significant arteriolar vasodilation. After BoNT/A treatment, the arteriole diameters were significantly changed when compared to each control treatment (denatured BoNT/A and saline groups, p<0.01) (Figure 2). Moreover, there was a dose-dependent effect of BoNT/A on arteriole vasodilation. When compared with the saline group, the doses of 0.5 U/kg, 1.5 U/kg, and 5 U/kg had similar roles on arteriole vasodilation, and produced a significantly dilatory effect arteriole. There was no significant difference between these groups. When the concentration increased gradually, the 15 U/kg and 50 U/kg groups had greater effect on the inhibition of arteriole contraction. However, there was no difference between the 15 U/kg group and the 50 U/kg group.
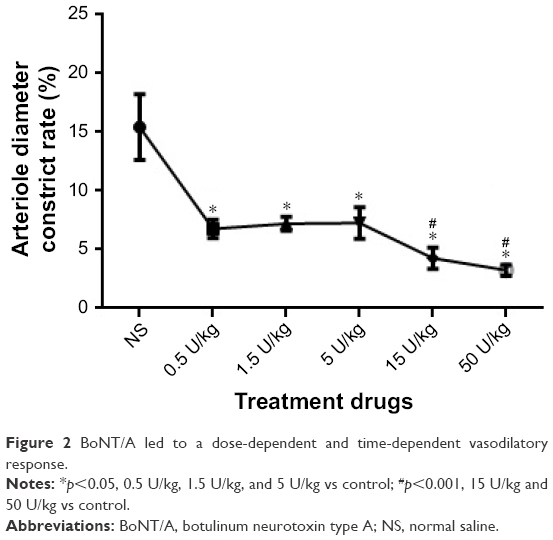 | Figure 2 BoNT/A led to a dose-dependent and time-dependent vasodilatory response. |
The interaction effect between BoNT/A and other drugs
Pretreatment experiments with BoNT/A (5 U/kg) and the functional consequences of prazosin (0.1 μM), Yohimbine (1 μM), and BQ-123 (1 μM) treatments on arteriole diameter constriction rate are shown in Figure 3. When compared to Krebs treatment alone (6.51±1.02), BQ-123 (4.11±0.63, p<0.01) treatment has a synergistic effect on the arteriole diameter constrict rate with BoNT/A (5 U/kg) at pretreatment administration. However, prazosin (6.54±1.11, p>0.05) and Yohimbine (6.67±1.12, p>0.05) treatments did not have an interaction effect with BoNT/A.
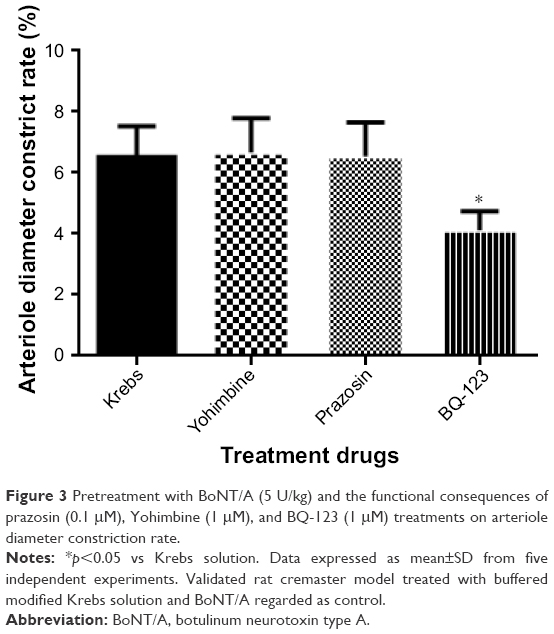 | Figure 3 Pretreatment with BoNT/A (5 U/kg) and the functional consequences of prazosin (0.1 μM), Yohimbine (1 μM), and BQ-123 (1 μM) treatments on arteriole diameter constriction rate. |
Distribution of α1 and α2 receptors on the rat cremaster arteriole
The immunofluorescence microscope was used to analyze the distribution of adrenergic receptors on the rat cremaster arteriole. In our study, we found that α1 and α2A receptors were coexpressed on rat cremaster arteriole. However, α2B and α2C receptors were absent (Figure 4).
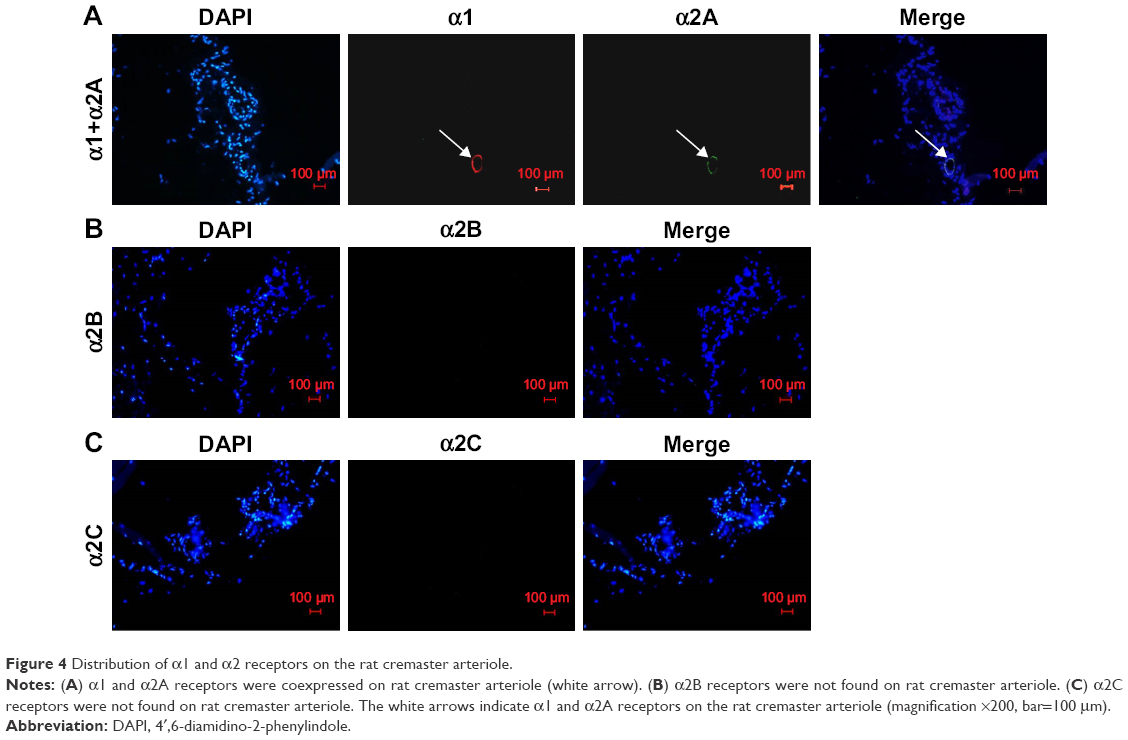 | Figure 4 Distribution of α1 and α2 receptors on the rat cremaster arteriole. |
Identification of primary sympathetic neurons
To confirm the purity of cells, SCGNs cultured for 3 days were immunostained with NF-200 antibody and TH antibody.19 As shown in Figure 5, immunofluorescence analyses showed that the cells cultured in our study were positive for both NF-200 and TH antibodies. This finding proves that the cells assessed were sympathetic neurons with catecholamine secreting function. Moreover, the primary sympathetic neurons had high purity.
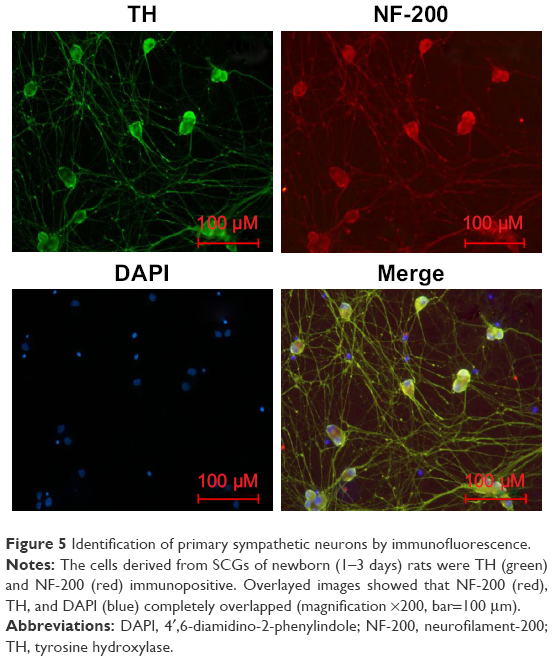 | Figure 5 Identification of primary sympathetic neurons by immunofluorescence. |
The inhibition of vesicle cycle by BoNT/A
To investigate the vesicle cycle in sympathetic neurons and the effect that BoNT/A on it, FM1-43, a membrane dye, was used in this study.20 Exposure of sympathetic neurons to FM1-43 for 5 min in the absence of depolarization stimuli resulted in small fluorescent spots in various sizes on the cell bodies (Figure 6A). Direct bath application of 56 mM K+ increased the intensity of FM1-43-labeled puncta on the cell instantly when compared to the control. The intensity of FM dye-labeled puncta gradually decreased during the 30-min observation time (Figure 6A, control). This finding suggests an increase in endocytosis and exocytosis in the presence of an external stimuli. Subsequent washout of the dye reduced the intensity of puncta to a lower level. The changes of laboratory dye intensity were examined in the presence of different doses of BoNT/A (5 U/mL, 10 U/mL, and 25 U/mL) for 30 min. The intensity of FM dye-labeled puncta decreased gradually with increasing reaction time. BoNT/A infusion to the cells delayed the intensity of puncta size in the cell stomata. Furthermore, as the dose of BoNT/A increased, the rate of dye fading also decreased. There was no significant difference in the change of the intensity of FM dye-labeled puncta per minute between the 10 U/mL BoNT/A group treatment and the 25 U/mL BoNT/A group treatment (p>0.05).
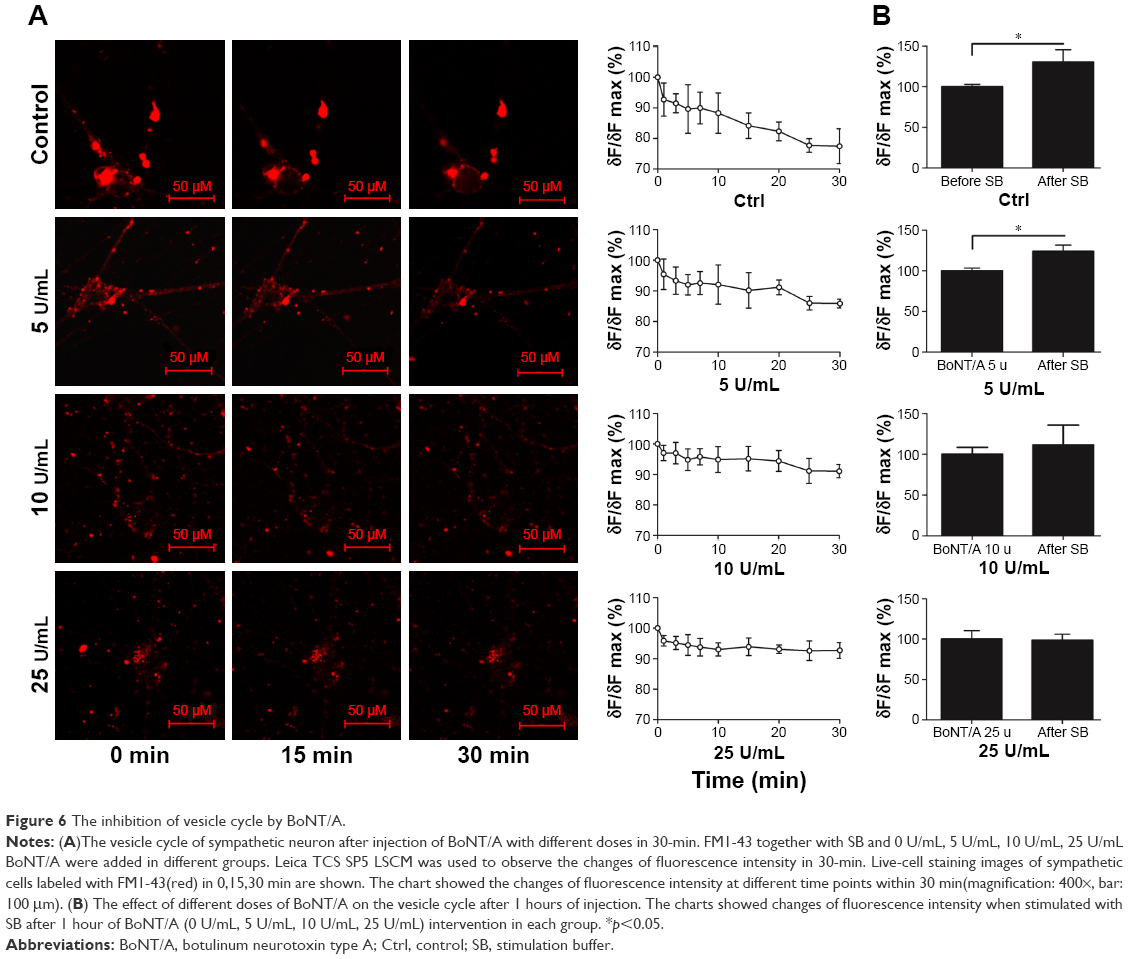 | Figure 6 The inhibition of vesicle cycle by BoNT/A. |
The vesicle cycle of cultured sympathetic neurons after BoNT/A interfering
To further test the effect of BoNT/A on sympathetic neurons, we treated the targeted cells with different doses of BoNT/A (0 U/mL, 5 U/mL, 10 U/mL, and 25 U/mL) for 1 h. The cells were then stained with FM1-43 in bath application containing 56 mM K+. The high K+ induced high fluorescence intensity in different doses of BoNT/A-treated samples. Furthermore, the fluorescence intensity was enhanced rapidly and remarkably (p<0.05) after the infusion of 56 mM K+ in the control (Figure 6B) and the 5 U/mL treated group (Figure 6B, 5 U/mL). A dose increment of BoNT/A decreased the fluorescence intensity after high K+ infusion. However, the enhancement of fluorescence intensity was not significant (p>0.05) in the 10 U/mL and 25 U/mL BoNT/A-treated groups (Figure 6B, 10 U/mL and 25 U/mL).
NE release decreased after BoNT/A treatment
Cell supernatants were collected on day 3, 5, and 7 before BoNT/A treatment and on day 8 after 24 h BoNT/A treatment. As shown in Figure 7, the concentration of NE decreased remarkably in the BoNT/A-treated group. Moreover, the concentration of NE declined from 148.965±5.192 (mean±SEM) to 78.708±0.805 pg/mL in the 25 U/mL treated group and from 131.441±3.720 to 68.460±3.130 pg/mL in the 50 U/mL treated group (p<0.01 for both). These in vitro results indicate that the ability of sympathetic neurons to release NE transmitter was suppressed by BoNT/A.
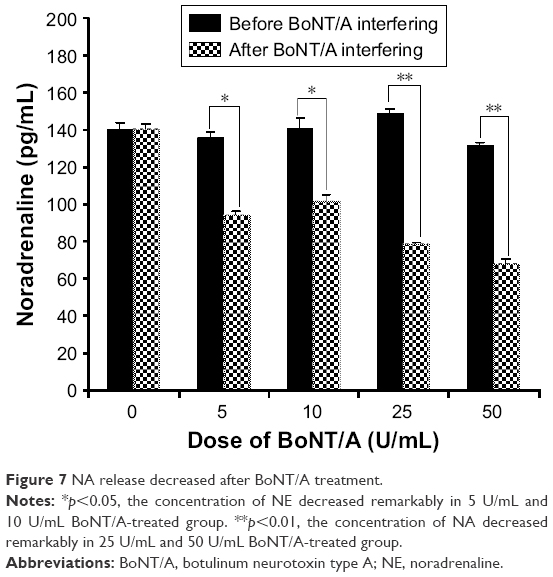 | Figure 7 NA release decreased after BoNT/A treatment. |
The target protein of BoNT/A in sympathetic neurons is SNAP-25
To confirm that the release of NA in sympathetic neurons was also related to SNAP-25, we studied the distribution of SNAP-25 in sympathetic neurons by immunofluorescence. As shown in Figure 8A, SNAP-25 was detected in SCGN.
 | Figure 8 The target protein of BoNT/A in sympathetic neurons is SNAP-25. |
After confirming the presence of SNAP-25 in sympathetic neurons, we examined protein levels by Western blot. After a high-dose treatment of BoNT/A (50 U/mL) for 24 h, cleaved SNAP-25 protein was detected. This result indicates that the functional role of BoNT/A was similar to cholinergic neurons. In contrast, cleaved SNAP-25 was not detected in the control group, and the 5 U/mL and 10 U/mL BoNT/A-treated groups (Figure 8B).
GM1, SV2C, and FGFR3 are located on sympathetic neurons
It has been previously shown that receptors like GM1, SV2C, and FGFR3 are the binding sites utilized by BoNT/A to enter the cholinergic neurons.24 In our study, we could see from Figure 9 that the neurons were immunopositive for GM1, SV2C, and FGFR3. The relationships between BoNT/A and these receptors need further study.
 | Figure 9 GM1, SV2C, and FGFR3 are located on sympathetic neurons. |
Discussion
In this study, we demonstrated the effect and possible mechanisms of BoNT/A on RP. Through the electrical stimulation-induced cremaster arteriole vasoconstriction model, we found that BoNT/A dose-dependently restrained the vasoconstriction of arteriole. The effect was similarly observed with prazosin, an α1-andrenoceptor antagonist. We also studied the impact of BoNT/A on sympathetic neurons, including the release of NE and the vesicle cycle in primary cultured superior cervical sympathetic neurons. The BoNT/A inhibition effect may be mechanistically related to the cleavage of SNAP-25 which is also observed on cholinergic nerves. Moreover, the receptors involved in BoNT/A transportation were also detected in our cell model.
In the cremaster arteriole vasoconstriction model, we investigated and compared the role of BoNT/A in combination with endothelin receptor antagonist BQ-123, α1 adrenoceptor antagonist prazosin, and α2 adrenoceptor antagonist Yohimbine. We found that BoNT/A, prazosin, and BQ-123 have an effect on decreasing the arteriole diameter constrict rate. However, no effect was seen with Yohimbine treatment, which was similar to Stone et al’s report,25 indicating that BoNT/A application, in a dose-dependent manner, resulted in significant vasodilation of observed cremaster arterioles. In our study, we also found that the effects of prazosin and BoNT/A were also equivalent. Pretreatment with BoNT/A followed by BQ-123 treatment had a synergistic effect on arteriole vasoconstriction, while no beneficial result was observed from prazosin and Yohimbine, suggesting that BoNT/A can effectively abolish arteriole vasoconstriction by the “prazosin way”, and the endothelin-A receptor antagonist can enhance the role of BoNT/A, indicating BoNT/A might abolish arteriole vasoconstriction through the α1 adrenoceptor way.
Our results showed that the sympathetic adrenoceptors α1 and α2A were coexpressed on rat cremaster arteriole. However, α2B and α2C adrenoceptors were not detected by immunofluorescence. As reported in previous studies, α-adrenoceptors were subclassified into α1 and α2 subcategories. α1 adrenoceptors were predominately distributed on the postsynaptic membrane, while α2 receptors can be divided into Α, B, and C subtypes distributed on both presynaptic and postsynaptic membranes.26–28 The lower expression of α2 adrenoceptors can perhaps explain the absence of Yohimbine effect.
Our findings suggested that BoNT/A might inhibit sympathetic nerves to mediate vasodilation. To further elaborate the impact of BoNT/A on sympathetic neurons we measured the vesicle cycle and the release of NE. Shone and Melling30 found that the release of NE decreased by 20% within 4 days when compared to the control group which decreased by 60%–65% by day 8 in PC12 cells. However, the PC12 cell model, which has sympathetic neuronal property, is differentiated from a rat adrenal pheochromocytoma clonal cell line by nerve growth factor (NGF).30 These cells cannot fully reflect the characteristics of sympathetic neurons. Therefore, in this study, primary cultures of neonatal rat superior cervical sympathetic ganglion neurons were established to further verify the reliability of previous results.
The vesicle cycle completion was measured by endocytosis and exocytosis, detected by FM1-43, which has been widely used in vesicle research.20–22 In our experiments, we found that the decrease of FM1-43 intensity was delayed in the presence of BoNT/A, indicating that the vesicle cycle of sympathetic neurons could be inhibited by BoNT/A. However, this finding could not address whether the release of NE was affected. In an earlier study, Habermann et al29 found that BoNT/A could inhibit the release of NE in fetal rat brain stem neurons. Our results showed a significant decrease of the NE concentration after BoNT/A intervention, especially in high concentrations of BoNT/A (50 U/mL), suggesting that the release of NE is dose-dependently blocked by BoNT/A, through inhibition of the vesicle cycle.
Studies on motor neurons have demonstrated that BoNT/A could inhibit formation of the SNARE complex and thereby block the quantization release of Ach,23 causing chemical denervation of the motor end plates. In this study, we found that the inhibitory effect of BoNT/A on NE release in sympathetic neurons might be related to SNAP-25 as well. However, cleaved SNAP-25 was only detected under 50 U/mL BoNT/A treatment after 24 h. In a previous study, our team had found that BoNT/As produced by different countries retain distinct potencies and various durations of effect after intramuscular administration.43 In other studies, natural BoNT/A which contained neither chelating protein nor stabilizer was used to explore the mechanism of BoNT/A in influencing motor neurons. The commercial one we used might influence the efficiency of toxin when compared to the purified one used in other studies.36 What is more, a different new target protein might exist. Besides, BoNT/A might also function through other SNARE complex-dependent pathways in neurons and nonneuronal cells.31–35
Until recently, some studies indicated that BoNT/A could combine with specific receptors located on the presynaptic membrane of motor neurons in a double-receptor model.37,38 The binding of BoNT/A with ganglioside launches the initial link step.39 It then combines with vesical transmembrane protein SV2 which has three isoforms, SV2A, SV2B, and SV2C. SV2C is believed to be the main receptor involved in BoNT/A binding in vitro.40–42 In addition, receptor FGFR3 is also involved. In our study, we were able to visualize the existence of ganglioside GM1, SV2C, and FGFR3 on the sympathetic neurons. However, the specific connection mode between BoNT/A and these three receptors is still under investigation.
Conclusion
Our study showed that BoNT/A can inhibit arteriole vasoconstriction in a dose-dependent manner by cleaving SNAP-25 in sympathetic neurons. This action inhibited vesicle fusion with the presynaptic membrane and blocked the release of NE. Through the study of the mechanism of BoNT/A affecting the sympathetic pathway in the treatment of Raynaud’s syndrome, this research provides a theoretical basis for the treatment of small vessel diseases caused by sympathetic dysfunction.
Acknowledgments
The work was supported by the National Natural Science Foundation of China (81500970 and 81470582), Shanghai Education Development Foundation and Shanghai Municipal Education Commission (14SG21), Priority of Shanghai Key Discipline of Medicine (2017ZZ02020), Shanghai Municipal Medical and Health Excellent Academic Leaders Training Program (2017BR029), and Shanghai Putuo District High Level Talents Scientific Research Innovation Project (2014-A-21).
Disclosure
The authors report no conflicts of interest in this work.
References
Herrick AL. The pathogenesis, diagnosis and treatment of Raynaud phenomenon. Nat Rev Rheumatol. 2014;8:469–479. | ||
Pope JE. Raynaud’s phenomenon (primary). BMJ Clin Evid. 2013;10:1119. | ||
Pope JE. The diagnosis and treatment of Raynaud’s phenomenon: a practical approach. Drugs. 2007;67(4);517–525. | ||
Herrick AL. Pathogenesis of Raynaud’s phenomenon. Rheumatology. 2005;44:587–596. | ||
Levien TL. Advances in the treatment of Raynaud’s phenomenon. Vasc Health Risk Manag. 2010;6:167–177. | ||
Herrick AL. Contemporary management of Raynaud’s phenomenon and digital ischaemic complications. Curr Opin Rheumatol. 2011;23(6):555–561. | ||
Herrick AL. Treatment of Raynaud’s phenomenon: new insights and developments. Curr Rheumatol Rep. 2003;5(2):168–174. | ||
Mannava S, Plate JF, Stone AV, Smith TL, Smith BP, Koman LA, Tuohy CJ. Recent advances for the management of Raynaud phenomenon using botulinum neurotoxin A. J Hand Surg Am. 2011;36(10):1708–1710. | ||
Sycha T, Graninger M, Auff E, Schnider P. Botulinum toxin in the treatment of Raynaud’s phenomenon: a pilot study. Eur J Clin Invest. 2004;34:312–313. | ||
Van Beek AL, Lim PK, Gear AJ, Pritzker MR. Management of vasospastic disorders with botulinumtoxin A. Plast Reconstr Surg. 2007;119(1):217–226. | ||
Fregene A, Ditmars D, Siddiqui A. Botulinum toxin type A: a treatment option for digital ischemia in patients with Raynaud’s phenomenon. J Hand Surg Am. 2009;34(3):446–452. | ||
Neumeister MW, Chambers CB, Herron MS, et al. Botox therapy for ischemic digits. Plast Reconstr Surg. 2009;124(1):191–201. | ||
Neumeister MW. Botulinum toxin type A in the treatment of Raynaud’s phenomenon. J Hand Surg Am. 2010;35:2085–2092. | ||
Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012;41(4):599–603. | ||
Zhang XL, Hu Y, Nie Z, Song Y, Pan Y, Liu Y, Jin L. Treatment of Raynaud’s phenomenon with botulinum toxin type A. Neurol Sci. 2015;36(7):1225–1231. | ||
Baez S. An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res. 1973;5(3):384–394. | ||
Grant RT. The effects of denervation on skeletal muscle blood vessels (rat cremaster). J Anat. 1966;100(Pt2):305–316. | ||
Mains RE, Patterson PH. Primary cultures of dissociated sympathetic neurons. I. Establishment of long-term growth in culture and studies of differentiated properties. J Cell Biol. 1973;59(2 Pt 1):329–345. | ||
Takahashi H, Ishikawa H, Tanaka A. Regenerative medicine for Parkinson’s disease using differentiated nerve cells derived from human buccal fat pad stem cells. Hum Cell. 2017;30(2):60–71. | ||
Fuenzalida LC, Keen KL, Terasawa E. Colocalization of FM1-43, Bassoon, and GnRH-1: GnRH-1 release from cell bodies and their neuroprocesses. Endocrinology. 2011;152(11):4310–4321. | ||
Henkel AW, Lübke J, Betz WJ. FM1-43 dye ultrastructural localization in and release from frog motor nerve terminals. Proc Natl Acad Sci U S A. 1996;93(5):1918–1923. | ||
Amaral E, Guatimosim S, Guatimosim C. Using the fluorescent styryl dye FM1-43 to visualize synaptic vesicles exocytosis and endocytosis in motor nerve terminals. Methods Mol Biol. 2011;689:137–148. | ||
Blasi J, Chapman ER, Link E, et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 1993;365(6442):160–163. | ||
Jacky BP, Garay PE, Dupuy J, et al. Identification of fibroblast growth factor receptor 3 (FGFR3) as a protein receptor for botulinum neurotoxin serotype A (BoNT/A). PLoS Pathog. 2013;9(5):e1003369. | ||
Stone AV, Koman LA, Callahan MF, Eckman DM, Smith BP, Plate JF, Smith TL. The effect of botulinum neurotoxin-A on blood flow in rats: a potential mechanism for treatment of Raynaud phenomenon. J Hand Surg Am. 2012;37(4):795–802. | ||
Piascik MT, Hrometz SL, Edelmann SE, Guarino RD, Hadley RW, Brown RD. Immunocytochemical localization of the alpha-1B adrenergic receptor and the contribution of this and the other subtypes to vascular smooth muscle contraction: analysis with selective ligands and antisense oligonucleotides. J Pharmacol Exp Ther. 1997;283(2):854–868. | ||
Piascik MT, Smith MS, Soltis EE, Perez DMl. Identification of the mRNA for the novel alpha 1D-adrenoceptor and two other alpha 1-adrenoceptors in vascular smooth muscle. Mol Pharmacol. 1994;46(1):30–40. | ||
Ruffolo RR Jr. Distribution and function of peripheral alpha-adrenoceptors in the cardiovascular system. Pharmacol Biochem Behav. 1985;22(5):827–833. | ||
Habermann E, Miiller H, Hudel M. Tetanus toxin and botulinum A and C neurotoxins inhibit noradrenaline release from cultured mouse brain. J Neurochem. 1988;51(2):522–527. | ||
Shone CC, Melling J. Inhibition of calcium-dependent release of noradrenaline from PC12 cells by botulinum type-A neurotoxin. Long-term effects of the neurotoxin on intact cells. EurJ Biochem. 1992;207(3):1009–1016. | ||
Matak I, Lacković Z. Botulinum toxin A, brain and pain. Prog Neurobiol. 2014;119–120:39–59. | ||
Shimizu T, Shibata M, Toriumi H, et al. Reduction of TRPV1 expression in the trigeminal system by botulinum neurotoxin type-A. Neurobiol Dis. 2012;48(3):367–378. | ||
Ray P, Ishida H, Millard CB, Petrali JP, Ray R. Phospholipaise A2 and arachidonic acid-mediated mechanism of neuroexocytosis: a possible target of botidinum neurotoxin A other then SNAP-25. J Appl Toxicol. 1999;19 Suppl 1:S27–S28. | ||
Ishida H, Zhang X, Erickson K, Ray P. Botulinum toxin type A targets Rho B to inhibit lysophosphatidic acid-stimulated actin reorganization and acetylcholine release in nerve growth factor-treated PC12 cells. J Pharmacol Exp Ther. 2004;310:881–889. | ||
Karsenty G, Rocha J, Chevalier S, et al. Botulinum toxin type A inhibits the growth of LNCaP human prostate cancer cells in vitro and in vivo. Prostate. 2009;69(11):1143–1150. | ||
Lawrence GW, Ovsepian SV, Wang J, Aoki KR, Dolly JO. Therapeutic effectiveness of botulinum neurotoxin A: potent blockade of autonomic transmission by targeted cleavage of only the pertinent SNAP-25. Neuropharmacology. 2013;70:287–295. | ||
Montecucco C. How do tetanus and botulinum toxins bind to neuronal membranes? Trends Biochem Sci. 1986;11(8):314–317. | ||
Montecucco C, Rossetto O, Schiavo G. Presynaptic receptor arrays for clostridial neurotoxins. Trends Microbiol. 2004;12(10):442–446. | ||
Yowler BC, Schengrund CL. Botulinum neurotoxin A changes conformation upon binding to ganglioside GT1b. Biochemistry. 2004;43(30):9725–9731. | ||
Mahrhold S, Rummel A, Bigalke H, Davletov B, Binz T. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006;580(8):2011–2014. | ||
Dong M, Liu H, Tepp WH, Johnson EA, Janz R, Chapman ER. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol Biol Cell. 2008;19(12):5226–5237. | ||
Baldwin MR, Barbieri JT. Association of botulinum neurotoxins with synaptic vesicle protein complexes. Toxicon. 2009;54(5):570–574. | ||
Feng Y, Liu WC, Pan LZ, et al. Comparison of neurotoxic potency between a novel chinbotulinumtoxinA with onabotulinumtoxinA, incobotulinumtoxinA and lanbotulinumtoxinA in rats. Drug Des Devel Ther. 2017;11:1927–1939. | ||
Kaufmann J, Martinka P, Moede O, et al. Noradrenaline enhances angiotensin II responses via p38 MAPK activation after hypoxia/reoxygenation in renal interobar ateries. Acta Physiologica. 2015;213(4):920–932. | ||
Arrick DM, Mayhan WG. Inhibition of endothelin-1 receptors improves impaired nitric oxide synthase-dependent dilation of cerebral arterioles in type 1 diabetic rats. Microcirculation. 2010;17(6):439–446. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.