Back to Journals » The Application of Clinical Genetics » Volume 16
The Many Faces of Arrhythmogenic Cardiomyopathy: An Overview
Authors Tadros HJ, Miyake CY, Kearney DL, Kim JJ, Denfield SW
Received 29 June 2023
Accepted for publication 10 October 2023
Published 1 November 2023 Volume 2023:16 Pages 181—203
DOI https://doi.org/10.2147/TACG.S383446
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Hanna J Tadros,1 Christina Y Miyake,1,2 Debra L Kearney,3 Jeffrey J Kim,1 Susan W Denfield4
1Department of Pediatrics, Section of Pediatric Cardiology, Texas Children’s Hospital, Baylor College of Medicine, Houston, TX, USA; 2Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX, USA; 3Department of Pathology, Texas Children’s Hospital, Baylor College of Medicine, Houston, TX, USA; 4Department of Pediatrics, Division of Pediatric Cardiology, Baylor College of Medicine, Houston, TX, USA
Correspondence: Susan W Denfield, Baylor College of Medicine, One Baylor Plaza, Houston, TX, USA, Email [email protected]
Abstract: Arrhythmogenic cardiomyopathy (AC) is a disease that involves electromechanical uncoupling of cardiomyocytes. This leads to characteristic histologic changes that ultimately lead to the arrhythmogenic clinical features of the disease. Initially thought to affect the right ventricle predominantly, more recent data show that it can affect both the ventricles or the left ventricle alone. Throughout the recent era, diagnostic modalities and criteria for AC have continued to evolve and our understanding of its clinical features in different age groups as well as the genotype to the phenotype correlations have improved. In this review, we set out to detail the epidemiology, etiologies, presentations, evaluation, and management of AC across the age continuum.
Keywords: arrhythmias, arrhythmogenic cardiomyopathy, cardiomyopathy, genotype-to-phenotype correlation, pediatrics
Introduction
Arrhythmogenic cardiomyopathy (AC) is an umbrella term that is a relatively new construct recognizing that electrical function of the heart is as intrinsic to maintaining normal cardiac function as are cardiomyocytes and the scaffolding holding them together. Pathogenic variants leading to disease result in electromechanical uncoupling of cardiomyocytes, triggering histologic changes that create the substate for arrhythmias and dysfunction.1 Life threatening cardiac arrhythmias may occur in the absence of overt cardiac dysfunction while, conversely, cardiac dysfunction can persist and progress even when arrhythmias are controlled. Traditionally, AC was thought to be a primary disorder of the right ventricle (RV) associated with ventricular arrhythmias and fibrofatty infiltration/replacement of the RV cardiomyocytes and was termed arrhythmogenic right ventricular cardiomyopathy (ARVC; OMIM: 107970; for the purposes of this article termed right ventricular dominant AC [RVD-AC]). However, it has become clear that this entity may affect both ventricles (ie termed biventricular arrhythmogenic cardiomyopathy [BiV AC]) or the left ventricle in isolation (for the purposes of this article termed left ventricular dominant AC [LVD-AC]).
These diseases can manifest in different ways across the age spectrum with variable and age dependent penetrance affecting either or both ventricles at variable timepoints throughout the course of the disease, which can make diagnosis challenging. Therefore, it is important for pediatric as well as adult cardiologists, geneticists, and genetic counsellors to recognize age and disease related variability in clinical presentations when assessing and counselling patients regarding implications for them and their relatives.
This article will review what is known about the epidemiology, etiologies (including rare genetic variants and syndromic presentations), clinical presentation, diagnostic evaluation, and special considerations in AC across the age continuum, with a limited discussion on management.
Epidemiology
RVD-AC has a prevalence of 1 in 1000 to 5000 and has been uncommonly reported before 12 years of age or after 60 years of age.2,3 It most often presents in the 2nd–5th decades of life with a median survival of around 60 years of age.4 Initially thought to be almost three fold more prevalent in males, a more recent study suggests the prevalence amongst sexes is similar.1,5 Geographically, Mediterranean regions, particularly Italy, are known to have a higher incidence of RVD-AC. One study found that RVD-AC accounted for 11% of sudden cardiac death (SCD) cases, but it has been reported to be as high as 22.4% in the Veneto Region.6 As the AC literature has grown, differences in geographic distribution of genetic variants have been mapped, showing a high prevalence of PKP2 pathogenic variants in the USA and the Netherlands, while in Italy, Denmark and the UK, variants in other desmosomal protein encoding genes are associated more frequently with the AC phenotype, with DSP variants slightly more common in the UK.7 Pathogenic variants in the desmosomal gene JUP are associated with AC relatively rarely, with the exception of Denmark and Greece/Cyprus.7
Genetic Etiology
The most common mode of inheritance of AC is autosomal dominant. Although the majority are linked to desmosome protein-encoding genes, non-desmosome genes have been implicated. A recent reappraisal of evidence on RVD-AC causing genes by the Clinical Genome Resource Gene Curation Expert Panel found the strongest evidence for disease causation to be due to variants in desmosome genes PKP2, DSP, DSG2, DSC2, and JUP and non-desmosome TMEM43, with moderate evidence for non-desmosome PLN and DES.8 However, the minimum gene set when assessing more broadly for AC recommended by the 2019 HRS expert consensus statement on evaluation, risk stratification and management of arrhythmogenic cardiomyopathy include BAG3, DES, DSC2, DSG2, DSP, FLNC, JUP, LDB3, LMNA, NKX2-5, PKP2, PLN, RBM20, SCN5A, and TMEM43.9 This recommendation will likely include additional genes over time, such as ILK and LEMD2.10,11
Although initially thought to be a result of desmosomal variants, recent evidence suggests that genes outside of the desmosome can cause AC via a “final common pathway” that eventually leads to desmosome dysfunction; virtually all components of the intercalated disc can be involved.9 Intercalated discs are located in the ends of the cardiac myocyte where cell to cell contact and interaction occurs.12 Intercalated discs are composed of three different connections including adherens junctions, desmosomes, and gap junctions. The adherens junctions and desmosomes provide mechanical coupling between cells and help reinforce myocyte structure while the gap junctions are required for the rapid electrical conduction between cells. If these pathways are disrupted, then cardiac dysfunction and arrhythmias may occur.
Since the primary etiology for AC is genetic and the yield of genetic testing is relatively high (ranging around 50–60% overall, but as high as 80% in the pediatric population) genetic testing is warranted when AC is suspected.13–15 In the clinical setting, these tests often utilize next generation sequencing technologies like commercial cardiomyopathy gene panels and exome or genome sequencing. When a pathologic/likely pathologic variant is identified, it should trigger a recommendation for cascade screening to identify genotype-positive members of the family. It is also important to recognize that some pathogenic gene variants may differ between the differing AC phenotypes, with some more likely to present as LVD-AC or progress to BiV AC (Figure 1).
 |
Figure 1 Diagram of non-desmosomal and desmosomal genes associated with arrhythmogenic cardiomyopathy and genotype-phenotype correlations for arrhythmogenic right ventricular cardiomyopathy (ARVC), arrhythmogenic left ventricular cardiomyopathy (ALVC), and biventricular arrhythmogenic cardiomyopathy (BiV AC). |
Desmosome Protein-Encoding Genes
The majority of pathogenic gene variants with definitive associations with AC are found in the desmosome including PKP2, DSP, DSG2, DSC2, and JUP.8 The cardiac desmosome is made up of a set of proteins important for cardiomyocyte adhesion, and include proteins within the cadherin, plakin, and armadillo family of proteins.16 Also, variants within other domains involved in protein interactions critical for desmosome function are implicated and variant type, zygosity, and inheritance all play a role in phenotype and penetrance.
The most common overall gene implicated in RVD-AC is PKP2. The PKP2 protein (plakophilin 2) is part of the desmosome.8 Truncating and missense variants have been reported. Although causative variants may lead to a RVD-AC or BiV AC, a study by Dries et al reported that truncating variants in PKP2 were more likely to present with the RVD-AC phenotype.17 Truncating variants in PKP2 are more enriched overall in RVD-AC patients, but location in the PKP2 gene did not correlate with disease association. By comparison to when missense variants are implicated, sites of enrichment for missense variants are found more localized in regions with known functional domains and the C-terminus.17 Specifically, enrichment was seen in most armadillo repeats and the HR2 domain, which are important functional domains involved in protein interactions.17,18 This is further substantiated by Headrick et al showing pathologic hot-spots in the armadillo repeat domains, ARM7 and ARM8.19 In addition to involving those domains, markers of pathogenicity include variants involving highly conserved residues and radical variants defined as frame-shift insertion and deletions and splice junction variants, as per Kapplinger et al.14 They also found a relatively similar frequency of missense variants in ostensibly unaffected controls and caution that race/ethnicity, mutation location, and sequence conservation should be taken into account when interpreting rare missense variants.14 There is also growing evidence that mode of inheritance and variant burden may play a role in phenotype, with patients with biallelic variants (recessive or compound heterozygous) or those with digenic heterozygosity presenting with earlier onset AC, LV dysfunction, and increased clinical severity compared to monoallelic heterozygous carriers.18,20
DSC2-encoding desmocollin 2 and DSG2-encoding desmoglein 2 are also important causes of AC. Both encode for desmosomal cadherins that link cardiomyocytes. The N-terminal cadherin repeat domains are common sites of pathologic hotspots.14,19 In addition, variants found within the signal peptide and prodomain of DSC2 have also been implicated and may affect intracellular transport of cadherin to the plasma membrane as exhibited in in-vitro studies utilizing cell transfection experiments and confocal microscopy.21 In the study by Syrris et al, the patient cohort was selected due to their clinical criteria based diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Four of the 77 patients were found to have 2 heterozygous variants in DSC2, one being an insertion and the other a deletion. Five of seven individuals in the group had significant left sided involvement with 2 of the 7 having more obvious left sided disease.22 Missense variants in DSC2 also accounted for a RVD-AC phenotype in some patients in the Beffagna et al study.23 A study analyzing a Dutch population that included DSC2 and DSG2 variants suggested variant burden is a phenotype modifier, not just the variant itself, as a higher proportion of patients with biallelic or digenic variants were found in those with definitive RVD-AC compared to a smaller proportion of patients with heterozygous variants who had probable RVD-AC.24 Recessive inheritance was suggested in two index cases, one of whom had a homozygous DSG2 variant and another a hemizygote DSG2 variant with an additional large deletion in DSG2, in the absence of other affected family members.25 Importantly, zygosity has been shown to play a role in both penetrance and phenotype in DSC2 variants with two studies identifying a homozygous missense (D179G) and truncating variant (Q554X) in probands with BiV AC, with little to no phenotype in patients carrying the same variants in a heterozygous fashion.26,27 AC has also been reported to be inherited via uniparental segmental isodisomy in an affected patient who had a homozygous 4-bp deletion in DSC2.28 Homozygous DSC2 variants can lead to cardiocutaneous phenotypes, although some are cardiac restricted.26,27 While some patients with DSG2 variants meet criteria for arrhythmogenic right ventricular cardiomyopathy, the phenotype frequently involves the LV and may occur in isolation as LVD-AC or as BiV AC.29,30 In a study by Hermida et al, a little over 50% of patients harboring a putative DSG2 variant presented with LV dysfunction and were more likely to undergo heart transplant or die from heart failure compared to those with PKP2 variants.31
DSP-encoding desmoplakin encodes the protein that links the intermediate filaments to the desmosome and is found in up to 16% of AC patients.7 DSP harboring patients are also more likely to present with LV dysfunction, both in isolation and as BiV AC.32 Three of the patients in this study with DSP variants were initially diagnosed as dilated cardiomyopathy. Left ventricular noncompaction was also noted in a subset of these patients with DSP variants. Some studies have shown 40% or more having LV changes and compared to other genotypes, increased LV dimensions and reduced LV function.33,34 Similar to previous genes, truncating variants may also increase the clinical severity of the phenotype in regards to both symptoms and LV dimensions and function.34–36 In regards to variant location, the N-terminal region of DSP is a prominent hot-spot and is a domain important in binding to plakophilin and plakoglobin.14 In addition, variants in regions which house the N-terminal region and the central rod domain, essential for interactions with other desmosomal proteins and intermediate filaments, may be associated with earlier ages of presentation and higher clinical severity. LVNC was seen in some of these patients as well as a cardiocutaneous phenotype in some family members.37
The JUP gene encoding for the intercalated disc protein plakoglobin is amongst the rarest of the desmosomal genes implicated in AC and is found predominantly in the Greek/Cyprus regions and Denmark.7 It has been characterized by a homozygous two base deletion leading to a cutaneous phenotype and RVD-AC known as Naxos disease (OMIM: 601214) and the mode of inheritance is autosomal recessive and is discussed in a subsequent subsection in this review.38
Non-Desmosome Protein-Encoding Genes
More recently, non-desmosomal protein encoding genes have been implicated in AC and include genes encoding nuclear envelopes, intermediate filaments, calcium handling, and sarcomeric proteins. Of those identified, only TMEM43 was found to have definitive evidence for the RVD-AC phenotype, while there was moderate evidence for PLN and DES for the RVD-AC phenotype, with most other genes having limited evidence for the “classic phenotype”, per a study by James et al.8 However, these genes may also present with less classic AC phenotypes and are important in identifying genetic etiologies in the “gene elusive” AC patient.
TMEM43-encoding transmembrane protein 43, a nuclear envelope protein which interacts with the lamina proteins emerin and lamin A/C, has been implicated in AC.39 The founder variant, S358KL, was identified in a population from Newfoundland with a likely European origin, but since has been identified in individuals outside of the founder population.39–41 The variant is highly penetrant and although primarily presenting as RVD-AC, almost half of the patients also have LV involvement.41 Further, it is also associated with a high rate of SCD, approaching 40%, and SCD was more common in males compared to females.41 Using dermal fibroblasts and force indentation experiments, it is theorized that introduction of this missense variant leads to increased stiffness and elasticity in the cell nucleus, which may lead to stochastic death of mechanically active cells, like cardiomyocytes.39
Animal models have been developed to try to understand the molecular mechanism(s) of TMEM43 variant associated AC, including transgenic mouse models and cell lines from zebra fish, with some variability in phenotypes seen in the mouse models.42–44 In a TMEM43 knock-in mouse model carrying the p.S358L variant, fibrofatty infiltration was seen, but cardiac function was preserved.42 They also found that the TMEM43-S358L variant enhanced the NF-kappaB-TGFbeta signal cascade contributing to the fibrotic changes seen.42 In another transgenic mouse model, cardiac-specific overexpression of the human TMEM43-p.S358L variant produced a more severe cardiac phenotype closely resembling the human form of AC with changes seen on ECG and echocardiography, early death, and fibrofatty replacement of cardiac myocytes.43 Additional mechanistic studies were performed showing that TMEM43 localized at the nuclear membrane, interacting with emerin and β-actin.43 TMEM43-S358L partially delocalized to the cytoplasm, reducing interaction with emerin and β-actin, and activating glycogen synthase kinase-3β (GSK3β).43 The GSK3β signaling pathway previously has been shown to play a role in AC pathogenesis. The Padron-Barthe et al study also reported that treatment of TMEM43 mutant mice with a GSK3β inhibitor improved cardiac function.43 Alterations in cardiac structure and function have also been shown in a transgenic zebrafish model when TMEM43 variants (c.1073C>T, p.S358L; c.332C>T, p.P111L) were expressed.44 Both variants displayed cardiac morphological defects at juvenile stages and ultrastructural changes within the myocardium, accompanied by dysregulated gene expression profiles in adulthood.44 These studies emphasize the importance of bedside to bench for further mechanistic elucidation and hopefully translation back to the bedside to improve patient outcomes.
PLN-encoding phospholamban, is a protein important in maintaining calcium homeostasis.45 A study by Van der Zwaag et al reported on a group of patients in the Netherlands with a phospholamban R14del mutation who were diagnosed with DCM or RVD-AC.45 The variant was found in 12% of the RVD-AC patients and 15% of the DCM patients. The DCM patients with the deletion had a higher frequency of appropriate implantable cardiac defibrillator (ICD) discharges, family history of SCD at a young age and cardiac transplantation compared to the other DCM patients but similar to their RVD-AC patient cohort consistent with an arrhythmogenic cardiomyopathy.45 A majority of those with the predominantly RVD-AC phenotype and the R14del, were found to have a reduced plakoglobin signal at the intercalated disks by immunohistochemistry, suggesting that impairment of non-desmosomal proteins like phospholamban, may affect a final common pathway leading to desmosomal instability in the RVD-AC phenotype.45 However this was an uncommon finding in the DCM phenotype patients with the same variant, suggesting this finding is associated with the RVD-AC phenotype and not the genotype. Other reported differences in those with predominantly LVD-AC with the PLN variant include differing ECG patterns with more commonly found low voltage ECGs, T wave inversion in the lateral chest leads, more LV structural and functional changes and differing patterns of fibrosis on histologic specimens from cardiac explants or autopsies.46,47
Key in cardiomyocyte structure and cytoskeleton organization is the cardiac intermediate filament, of which DES-encoded desmin is a key subunit. Desmin is made up of a central rod domain flanked by a terminal head and tail domain.48 One particular genetic hotspot is within the 1A subdomain in the central rod domain, some variants functionally disrupt filament assembly.48,49 Pathogenic variants affecting the desmin protein cause “desminopathies” leading to a variety of clinical phenotypes which may include skeletal and/or cardiomyopathies. One of the first descriptions of a DES variant related to RVD-AC was in a patient with no clinical skeletal myopathy.50 Other variants have since been identified in patients with LVD-AC and BiV AC in a highly penetrant and arrhythmogenic phenotype without an apparent skeletal myopathy.50–52 However, patients with desminopathy related cardiomyopathy may have onset of skeletal muscle disease after the onset of the cardiomyopathy; therefore continued monitoring for the development of skeletal muscle disease is important. Also of note, other forms of cardiomyopathy are more frequent than AC in desminopathies including DCM and less frequently restrictive cardiomyopathy (RCM) and hypertrophic cardiomyopathy (HCM).53 Left ventricular noncompaction has also been rarely reported.53
FLNC-encoding filamin C and its link to AC has been more recently identified. Filamin C is important for mechanotransduction in cardiomyocytes and localizes to the intercalated disk.54 This again supports the theory of a final common pathway that can disrupt cell to cell adhesion and communication from multiple integrated sites. Immunohistochemistry staining has demonstrated reduced desmoplakin signals in ventricular samples in some patients with an FLNC-variant AC. The phenotype often overlaps with DCM with a high arrhythmia burden and sudden death risk.55–58 Most variants described are truncating and the penetrance is quite variable, reported from 9% in one study to 97% in individuals >40 years of age in another study.56,57 This wide range is likely due to case ascertainment differences with the Carruth et al study identifying patients through whole exome sequencing of a general clinical population (reported 9%), while the Ortiz-Genga et al study reported the 97% in a population identified using next-generation sequencing with 28 unrelated probands identified with truncating variants in FLNC. Their relatives were then cascade tested which likely contributed to the high penetrance reported over time. As with desminopathies, FLNC variants have also been associated with HCM and RCM, typically with high arrhythmia burdens.54,59,60
Other variants in non-desmosomal genes that have been implicated in AC include ILK-encoding integrin linked kinase which has been implicated in the clinical setting as well as in in-vitro and in-vivo models and LEMD2-encoding LEM domain containing protein that has been associated with LVD-AC.10,11,61 Additional implicated AC genes include SCN5A, CTNNA3, SCN5A, BAG3, RBM20, CDH2, TGFB3, TTN, and LMNA, amongst others. However, data regarding some of these genes and their link to AC is more limited and warrants further investigation. These findings highlight the potential clinical/phenotypic overlap of many gene variants as is evident in the sarcomeric protein variants that can cause differing phenotypes even within first degree relatives. Therefore, if a targeted gene panel is used to try to identify the common variants for a cardiomyopathy in a proband, it is important to expand the search for genes that are less commonly associated with the particular phenotype. One of the major “tip offs” to look for AC genes is a heavy arrhythmia burden in those with an “atypical” phenotype such as DCM.
Clinical Features
The classic clinical features of AC have been primarily related to ventricular arrhythmias and sudden cardiac death. The natural history of RVD-AC was classically defined in four stages: 1) the concealed phase where the patient is or appears to be free of significant symptoms or arrhythmias but structural changes are starting to occur and may be seen on echocardiography if performed. The presentation in this phase still can be SCD or aborted SCD, 2) the overt phase where the patient develops symptoms, typically arrhythmias, syncope, palpitations, and more overt structural changes, 3) end-stage disease leading to right ventricular dilation and dysfunction, and 4) biventricular failure mimicking dilated cardiomyopathy (Figure 2).62 However, more recent studies have shown that patients may initially present at any stage and may have primary LV involvement or biventricular disease at presentation, which may manifest as heart failure with a worse prognosis.
 |
Figure 2 Four phases classically described in arrhythmogenic right ventricular cardiomyopathy and the associated findings in the 1) concealed phase where no structural changes are seen on imaging, 2) overt phase where arrhythmias may manifest and right ventricular (RV) changes may be seen on echocardiogram (ECHO) or cardiac magnetic resonance imaging (CMR), 3) end-stage disease phase where RV dilation and dysfunction is seen, and 4) biventricular disease where the left ventricle (LV) becomes involved and a dilated cardiomyopathy phenotype may manifest. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). |
Arrhythmias
Arrhythmias have been the hallmark of AC and although primarily manifested as ventricular tachycardia, arrhythmias of atrial origin may also occur. Ventricular arrhythmias have led to the diagnosis of AC in a large proportion of patients.63 Ventricular arrhythmias can range from frequent premature ventricular complexes to ventricular electrical storms, defined as ≥3 separate episodes of sustained ventricular tachycardia within a 24-hour period. Ventricular electrical storms were seen in approximately 22% of patients at a median follow up of 4 years after ICD placement.64 Additionally, ventricular arrhythmia morphology may correlate with underlying genetic findings. PKP2 variants were more commonly associated with left bundle branch block morphology ventricular tachycardia (VT) or both left and right bundle brunch block morphology VT. The right bundle branch pattern when present should raise suspicion for significant left sided involvement as well. Right bundle branch block morphology VT has also been associated with DSP variants again raising suspicion for significant left sided disease.65 Atrial fibrillation has been reported in up to 14% of patients, typically presenting in adults and was associated with more RV structural abnormalities and greater LA volumes, suggesting this is a finding in more advanced disease.66
Sudden Cardiac Death
Sudden cardiac death is the most feared outcome in AC, some of which is not preventable as SCD may be the first presentation of the disease, highlighting the importance of screening asymptomatic first degree relatives. AC is a cause of SCD in approximately 11−13% of cases in Europe and the UK, compared to only 2.7% of cases in North America, following the known epidemiology of AC.67,68 SCD may occur during exercise or at rest.67,68 The disease does appear to progress more rapidly in those who exercise heavily and increases the risk of sudden death during athletics.9,67 Although early studies suggested isolated RVD-AC in SCD cases, recent studies have shown that LV involvement in SCD cases is common, and that most cases had biventricular involvement.68–70 Genetic testing of available specimens is extremely important in the SCD patient, particularly without an obvious post mortem cause or in which the pathology does not fit classic cardiomyopathy criteria.71 In SCD cases, clinically actionable variants are most often seen in cardiomyopathy-associated genes and are overrepresented in cases with subdiagnostic structural changes.71 Of these, AC-related genes are commonly found and may lead to an earlier diagnosis in a large proportion of previously unrecognized genotype-positive relatives.71
Syndromic and Extra-Cardiac Manifestations
Syndromic and extra-cardiac manifestations may be seen in AC patients with desmosomal variants.71,72 Dermatological findings may be an indicator of underlying cardiac disease. Both Naxos disease, and a variant of this disease called Carvajal syndrome, can have cardiac manifestations of AC. The cutaneous phenotype usually involves woolly hair and palmoplantar keratosis, which is an epidermal thickening of the palms and soles of the feet.72 The skin and hair findings are typically present from infancy, but the presence and timing of the development of AC varies. Naxos disease was first described in a Greek family and involves a recessive, truncating deletion in JUP, which leads to a frameshift and premature termination at the C-terminal domain.72 These patients typically present as the RVD-AC phenotype, although LV involvement was also noted.72 Carvajal syndrome was found in an Ecuadorian family with a recessive, truncating deletion in DSP, resulting in a premature termination, leading to absence of the C-terminus.72 The prominent cardiac manifestation, although arrhythmogenic, overlaps with a dilated cardiomyopathy phenotype.72 In Carvajal syndrome and the dilated phenotype, cardiac morbidity tends to occur earlier, in patients as young as 7 to 8 years old, and includes ventricular arrhythmias, heart failure, and cardiac death.
Since these descriptions, knowledge regarding the cardio-cutaneous phenotype has evolved. A systematic review reported that three combinations of dermatological and cardiac phenotypes are seen in order of prevalence: 1) palmoplantar keratoderma, hair shaft anomalies, and no skin fragility which was always associated with cardiac disease and associated with recessive variants in JUP, DSP, and DSC2, 2) palmoplantar keratoderma, hair shaft anomalies, and skin fragility, which was not associated with cardiac disease and commonly associated with recessive PKP1, DSP, and JUP variants, and 3) palmoplantar keratoderma, hair shaft anomalies, and skin fragility, which was associated with cardiac disease and associated with homozygous or compound heterozygous DSP variants.73 This suggests that palmoplantar keratoderma and hair shaft anomalies in combination should warrant cardiac investigations. Common “hot-spots” for cutaneous phenotypes were identified in exon 14 (c.2157del2) in JUP, known to cause Naxos disease, and exons 23 and 24 in DSP.73 Although traditionally associated with recessive DSP variants, heterozygous, loss of function DSP variants can also lead to a highly penetrant cutaneous phenotype and the presence of cutaneous findings may lead to the earlier diagnosis of underlying cardiac disease in DSP+ carriers if appropriately screened.74,75 Desmoplakin variants have also been associated with autosomal dominant palmoplantar keratoderma, recessive skin fragility/woolly hair syndrome, and autosomal dominant RVD-AC without a cutaneous phenotype.72
Diagnosis
The diagnosis of AC is challenging and can require multiple modalities. A detailed family history of at least 3 generations is paramount, the diagnosis is usually made with an additional combination of findings from an electrocardiogram (ECG), echocardiogram or cardiac MRI (CMR), genetic testing, and histological findings on tissue specimens in some cases, either ante or postmortem. There is also more recent research on positron emission tomography (PET) imaging and speckle echocardiography, although these have not been added to consensus diagnostic criteria.76,77
Consensus Criteria
Consensus statements have been described in the literature with the goal of providing sensitive and specific AC diagnostic criteria. The first Task Force Criteria (TFC) were described in 1994, and were made up of a set of major and minor criteria involving global and/or regional RV structural and functional abnormalities, histologic findings in the RV, ECG abnormalities including repolarization and depolarization/conduction abnormalities, arrhythmias, and family history.78 The consensus was that either 2 major, 1 major and 2 minor, or 4 minor criteria met the diagnosis of RVD-AC.78 Furthermore, the criteria stipulated the absence of LV disease.78 Recognizing a vast increase in AC knowledge, lack of sensitivity of the original criteria, and the existence of LV or BiV AC, the TFC were revised in 2010. The criteria introduced new echocardiographic, cardiac magnetic resonance imaging (CMR), and angiography findings vital in identifying RV disease, stipulated specific histological findings in tissue samples, built on more recent ECG findings important in AC including terminal activation durations in V1-V3 and filtered QRS durations, reduced the number of ventricular extrasystoles from 1000 to 500 per 24 hours, and introduced the genetic finding of a pathogenic variant as a major criterion, amongst other smaller changes (Table 1).79 Also importantly, they removed the stipulation of the absence of LV disease.79 Even with these changes, the 2010 TFC may not be suitable for all AC patients, especially those with BiV or predominantly LV disease, whose heart failure symptoms may be more prominent or at least as prominent as arrhythmias, and the pediatric population.80,81 To address this, the Padua criteria were proposed in 2020 to describe diagnostic criteria specifically for the LVD-AC phenotype, including echocardiography, CMR, ECG, and arrhythmia-related findings indicative of LV disease.82 For the Padua criteria those with BiV disease must fulfill the criteria for RVD-AC and have evidence of left sided involvement. A major criterion for left sided involvement is contrast enhanced CMR (CE-CMR) based, demonstrating a scar by late gadolinium enhancement (stria pattern) of ≥1 “Bull’s Eye” segment(s) (in 2 orthogonal views) of the free wall (subepicardial or midmyocardial), septum, or both (excluding septal junctional LGE). Actual LV dysfunction, regional or global, is not required as fibrosis can be present while function is preserved until the disease becomes more extensive. However, to diagnose LVD-AC with no apparent RV involvement both the CE-CMR major criterion and a pathogenic gene mutation in an AC gene is required because the CE-CMR appearance is not pathognomonic for LVD-AC. Other causes of late gadolinium enhancement, including other genetic causes can result in a similar appearance on CMR. The proposed Padua criteria may improve diagnostic accuracy in non-classic RVD-AC presentations, such as those with isolated left sided disease and in the pediatric population.83 The proposed Padua criteria also recommend some modification to the criteria for RVD-AC to include tissue characterization by contrast enhanced CMR as a major criterion to further demonstrate RV disease and to eliminate late potentials on signal averaged ECGs as a criteria since they are nonspecific. Epsilon waves and terminal delay of the S-wave upstroke of ≥55 ms in the right precordial leads (particularly if followed by negative T waves) on ECG would be considered minor criteria (Figure 3).82
 |
Figure 3 Electrocardiogram from an RVD-AC patient found to have an Epsilon wave in lead V1 and denoted by the black arrow. |
 |
Table 1 Modified Task Force Criteria from 2010 |
Electrocardiogram Findings
ECG findings may aid in AC diagnosis and broadly include repolarization, depolarization, and conduction abnormalities. The 2010 TFC ECG findings that are considered major criteria for RVD-AC include T wave inversion in V1-V3 in those older than 14 years of age in the absence of complete right bundle branch block or Epsilon waves.79 The prevalence of these changes vary. Epsilon waves are infrequently seen in the pediatric age range and T wave inversion in leads V1-V3 is normal in the prepubertal child making these criteria less useful in that age range.
T-wave inversions are a hallmark repolarization abnormality in the right chest leads in RVD-AC when seen in those >14 years of age and were associated with increased underlying RV volumes and decreased RV function in a study by DeLazzari et al.84 That study also found that more lateral inversion was associated with more severe RV dysfunction while low voltages in the limb leads were a hallmark of more left sided disease based on LV late gadolinium enhancement (LGE), a method of assessing myocardial fibrosis, seen on contrast enhanced-CMR (CE-CMR).84 However, the Casella study found more left sided disease in those with T wave inversion in the inferolateral leads and left precordial leads.30 Regardless, this finding appears to suggest a more extensive disease.
Epsilon waves are useful when present, but their presence is highly variable ranging from 0.9–25% depending on the study.85 The Epsilon wave is a positive deflection between the QRS and T waves in the right precordial leads, which is a marker of delayed depolarization. The presence of this ECG finding was associated with RVOT motion abnormalities and its duration and extension into V3 correlated with increased RVOT diameter.86 It was also associated with sustained VT episodes.86 Epsilon waves are usually a marker of more advanced disease, when other signs of disease are already present and the disease is diagnosable without the finding of an Epsilon wave, which has called in to question its usefulness as a diagnostic criterion.82,85
The signal averaged ECG (SAECG) is done over multiple recorded cardiac cycles in an attempt to maximize the ECG signal to noise ratio and identify ECG features undetectable by standard techniques, including late potentials.87 The 2010 TFC includes SAECG as a minor criterion if at least 1 of 3 parameters are met including: filtered QRS duration ≥114 ms, duration of terminal QRS <40 µV ≥38 ms, and a root-mean-square voltage of terminal 40 ms of ≤20 µV.79 An abnormal SAECG is more often seen in severe disease and is associated with underlying structural abnormalities.87 In this study by Pearman et al, genotype positive and minimal phenotype patients had SAECG findings similar to healthy subjects, indicating that abnormalities are more likely seen when structural abnormalities are detectable and disease has progressed.87 Similar to Epsilon waves, other criteria to diagnose AC are usually present by the time the SAECG findings become abnormal.
Although not included in the 2010 TFC, low voltage ECGs can be seen in subsets of patients, including the pediatric population and those with PLN or FLNC variant AC; however low voltages are not seen in all cases and other genetic variants may also have low voltage ECGs.88–90 In the study by de Brouwer et al, of PLN-variant AC, males with low voltage ECGs had the lowest ventricular arrhythmia (VA)-free survival.88 Low QRS voltages in the limb leads has also correlated with presence and degree of LV LGE.84
Imaging
Imaging modalities are vital to the diagnosis of AC. Conventional 2-D echocardiography and CMR are first-line imaging modalities used in AC. The 2010 TFC describe a list of major and minor criteria including RV wall motion abnormalities or aneurysms, RV outflow tract (RVOT) abnormalities, and/or RV dilation or dysfunction on 2D echocardiography or CMR, which are the imaging bases of AC diagnosis.
Echocardiogram
In addition to the standard echocardiographic parameters for assessing ventricular abnormalities, newer echocardiographic modalities, including speckle-tracking echocardiography, aid in identifying more subtle regional wall deformation or contractility abnormalities before the eye detects regional wall motion abnormalities or more gross functional parameter changes, such as a decline in the ejection fraction. There is increasing evidence that regional or global LV longitudinal strain by echocardiography is not only abnormal in AC, but may aid in early detection of AC in otherwise non-diagnostic stages and that epicardial longitudinal strain may be abnormal prior to the endocardial strain.91 In another study utilizing the speckle tracking technique that assessed RV deformation pattern and RV dispersion (a measure of heterogenous contraction of the ventricle), Kirkels et al found that deformation abnormalities and a greater degree of RV dispersion were associated with ventricular arrhythmias and may carry prognostic significance.76 RV global longitudinal strain and average segmental peak end-systolic strain utilizing intracardiac echocardiography may also help in detecting underlying functional and substrate changes, potentially prior to some CMR findings.92 The authors reported that using this method they were able to distinguish patients with AC compared to those with non-AC PVCs or atrial fibrillation. The patients were already undergoing cardiac catheterization for electrophysiology study with catheter ablation for arrhythmia management. At present there are no consensus recommendations for assessment by intracardiac echocardiography nor recommendations for inclusion of transthoracic speckle tracking in the assessment for AC.
It is also important to note that several studies have reported that echocardiograms of some AC gene positive patients demonstrate left ventricular noncompaction and some patients have also had congenital heart disease.90,93 These are atypical findings in AC, but it suggests that AC genes should be part of additional genetic screening in patients with an atypical phenotype with a high arrhythmia burden and cardiac dysfunction in the absence of other positive genes.
Cardiac Magnetic Resonance Imaging
Generally, CMR abnormalities in AC include structural and functional anomalies. These criteria were initially determined by RVD-AC phenotypes. Functional anomalies include RV dysfunction and regional wall abnormalities. In RVD-AC, these abnormalities are classically seen in an area bordered by the inflow, apex, and outflow tract of the RV, termed the “triangle of dysplasia”.5 A subsequent study reported that the RV apex is primarily involved in more severe phenotypes and suggested a biventricular triangle for AC involving the RV basal anterior wall, basal inferior wall, and LV posterolateral wall based on CMR and electrophysiologic mapping.94 Structural abnormalities include RV dilation, aneurysms and high T1 signals within the RV myocardium indicative of fat. Higher RV end-diastolic dimensions and lower RV ejection fractions were seen in RVD-AC in patients meeting TFC criteria when compared to controls.95 Tandri et al also found that compared to other structural abnormalities like wall thinning and RV aneurysms, RV trabecular disarray was more common. Trabecular disarray was defined as densely trabeculated RVs with thick Y-shaped trabecula.95 The 2010 TFC used the CMR data available at the time to define CMR criteria supporting the diagnosis of RVD-AC.79
At the time of the writing of the 2010 TFC, CMR structural and functional criteria were defined, but CE-CMR criteria were not included. CE-CMR utilizes gadolinium enhancement, and LGE has a high correlation with histological evidence of fatty infiltration and myocardial fibrosis.96 At the time of the last TFC, CE-CMR was not as reliable as it is now for detection of LGE/fibrosis/fat in the RV free wall, so LGE criteria were not included, but it has become an important adjunct for diagnosis.82 CE-CMR may also have the potential for identifying disease earlier in its natural history and in potentially suggesting certain underlying genotypes, where older TFC criteria may lack accuracy.82,97 Patients carrying desmosomal variants more often had RV LGE, although this did not quite reach statistical significance in the study by Segura-Rodriguez et al.98 That study also showed that the p.Glu401Asp variant in DES (desmin) typically had a subepicardial circumferential pattern of LGE distribution, that was less commonly seen with other variants. A study by Augusto et al, of patients with a DCM phenotype who had a variety of known gene variants including AC genes and more typical DCM genes, found that those with DSP or FLNC variants were more likely to have subepicardial LV LGE in a ring-like pattern affecting at least three contiguous segments in a short axis compared to the non AC gene mutation carriers with DCM.99
CMR is superior to traditional echocardiography in identifying AC mimics like myocarditis or sarcoidosis and may lead to a change in diagnosis in up to 20% of adult age patients referred for concern for AC.100 However, it must be kept in mind that AC can also mimic myocarditis with signs of inflammation on CMR and myocardial enzyme elevations in the blood indicative of myocardial injury, but neither of these findings are specific for the type of injury or inflammation and must be considered in the clinical context. CMR detected LV LGE and higher native T1 values (correlating with fibrosis), T2 values (correlating with edema and inflammation), and extracellular volumes are associated with a poorer prognosis with heart failure related events in the Chun et al study; an increased risk of the combined endpoint of sudden cardiac death, appropriate implantable cardioverter-defibrillator intervention, and aborted cardiac arrest was also noted in the Aquaro et al study.101,102
Positron Emission Tomography
Positron emission tomography (PET) is another newer modality, being used to evaluate AC patients, that assesses for inflammation in the heart.77 Protonotarios et al found that some patients with confirmed AC had evidence of myocardial inflammation by PET scan. A “Hot Phase” of AC has been well described and mimics myocarditis in a subset of patients, which will be discussed in another section.103 Again, the importance of “putting the whole clinical picture” together cannot be over emphasized as differing etiologies of cardiac dysfunction may mimic each other in presentation and imaging. Thus, an emphasis on family history and genetic testing is paramount while recognizing that some genes remain elusive at this time.
Histological Diagnosis
The histologic diagnosis of AC can be made from endomyocardial biopsy during cardiac catheterization, at the time of ventricular assist device placement in those with severe heart failure, or cardiac explant at the time of cardiac transplant or autopsy. While endomyocardial biopsy can be done safely at very experienced centers, there needs to be surgical back up, as the thin wall of the diseased right ventricle is easier to perforate than other forms of cardiomyopathy, particularly in smaller, younger children. Typically, the ventricular septum is the preferred area to biopsy as it tends to be thicker and a perforation is usually intracardiac rather than through to the pericardial space, but the septum is less likely to demonstrate disease than other areas of the right ventricle and is not a preferred site in AC. The left ventricular side of the septum may be involved but LV biopsies are done less frequently.104,105 Also, because the disease can be patchy, a negative biopsy does not rule out AC. Therefore, all efforts should be made to confirm the diagnosis by other criteria when possible.
The findings seen in AC include fibrofatty replacement of native myocardium and ventricular atrophy (Figure 4). However, fibrofatty replacement may be found to some degree in normal adults and in patients with Duchenne muscular dystrophy, although clinically this would not result in confusion with AC.106 Because fibrofatty replacement can be seen in other contexts, to be counted as a major criterion, the 2010 TFC stipulate that <60% of residual cardiomyocytes be identified in specimens by morphometric analysis, secondary to fibrous replacement of the RV free wall, with or without fatty replacement (or less than 50% if estimated).79 Minor histologic criterion include residual cardiomyocytes 60% to 75% by morphometric analysis (or 50% to 65% if estimated). Fibrofatty replacement usually starts at the epicardium and progresses to the endocardium, leading to a transmural appearance in last stage disease.5,69 Original studies reported that these changes are often seen in the RV free wall, the inflow, the outflow, and apex of the RV, with aneurysms seen in the inflow and outflow.5,69 These are findings that can also be found in the LV, in LVD-AC and BiV AC phenotypes. Another important histological finding is mononuclear inflammatory infiltrates seen in the ventricular walls.104,105 This can be seen in a large subset of patients and is a sign of ongoing myocardial damage.104 Additional non-classic features to be aware of are the histologic appearance of left ventricular noncompaction in association with AC (Figures 5 and 6) and congenital heart disease (Figure 6).
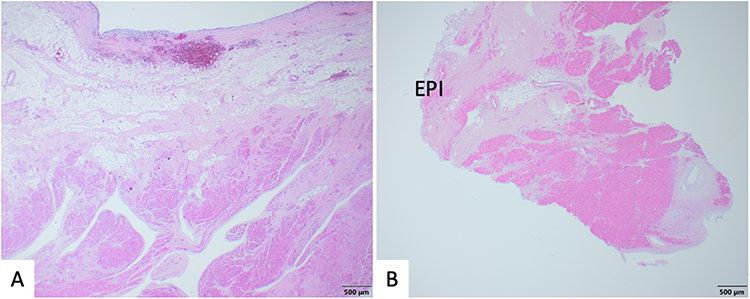 |
Figure 4 Histological images from a subject with biventricular arrhythmogenic cardiomyopathy. (A) Right ventricular free wall with fibrofatty myocardial replacement and relative preservation of myocardium within trabeculations. (B) LV apical core from assist device placement with accentuated fibrofatty myocardial replacement towards the epicardium (EPI). |
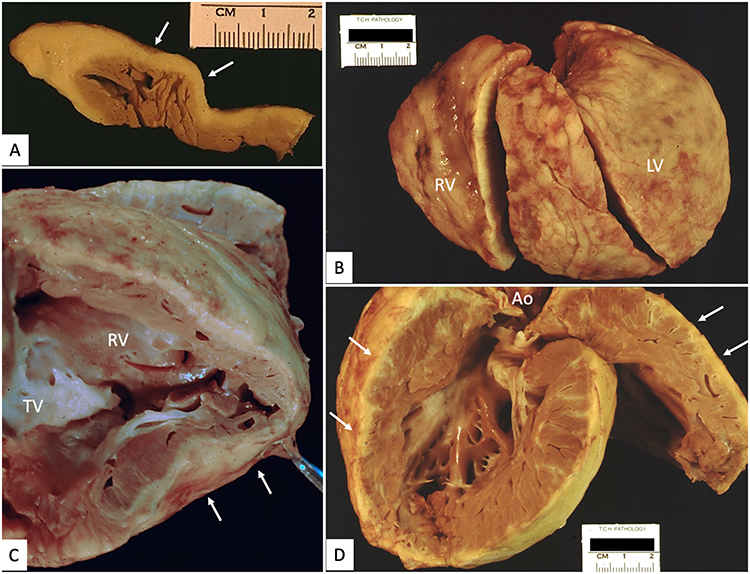 |
Figure 5 Cardiac explant with arrhythmogenic cardiomyopathy and left ventricular noncompaction. (A) Cross-section of right ventricular free wall with pale adipose tissue (white arrows) extending from epicardium, focally replacing all but subendocardial trabeculations. (B) External view of right (RV) and left (LV) ventricles with abundant epicardial adipose tissue. (C) Septal surface of right ventricle (RV) with extensive fibrofatty replacement (white arrows) of large portions of the free wall. (D) Left ventricle with extensive, deeply trabeculated myocardium and segmental areas of subepicardial fatty infiltration (white arrows). |
 |
Figure 6 Cardiac explant with tricuspid valve atresia and left ventricular non-compaction. (A) View of right (RA) and left (LA) atria from above with imperforate, atretic TV (*). (B) Hypertrophied and dilated left ventricle with marked hypertrabeculation of free wall and apical half of ventricular septum (between the white arrows). (C and D) Opened hypoplastic right ventricle (RV) with extensive fatty replacement of the myocardium, blending imperceptibly with epicardial fat (white arrows). Different images for this patient were used in reference.90 |
Special Considerations in AC
The Left Ventricular and Biventricular Presentation
The LVD or BiV presentations are significant challenges in AC diagnosis due to their less characteristic features with significant left sided disease early in the course. This includes T wave inversion more commonly seen in the inferolateral and left chest leads, rather than the right chest leads as is typically seen with RVD-AC. In addition the ventricular tachycardia typically has a right bundle branch morphology, rather than the left bundle branch morphology typically seen in RVD-AC.30,107 Practically, determining bundle branch morphology for VT is difficult to do in the clinical setting since this would require an ECG at the time of VT. Since the 2010 TFC is geared for RVD-AC diagnosis and not LV disease, less than half of patients with LVD-AC meet definite or borderline diagnostic criteria.30 Commonly those with LVD-AC or BiV are misdiagnosed as DCM, viral myocarditis, or idiopathic VT, and may not be diagnosed until after transplant or autopsy.90,107,108 In addition to LGE in the LV on MRI these patients frequently have LV dysfunction by imaging or may have heart failure symptoms at presentation. Often, what sets these patients apart from the common misclassification of cardiomyopathy type is the high ventricular arrhythmia burden.30,90,108 Sudden cardiac death remains a common presentation in this subset of patients as well, based on a large autopsy study of patients with sudden death in the UK.70
The “Hot Phase” Presentation
An inflammatory component of AC has been well described and inflammatory infiltrates are noted often on AC histological specimens.90,109 A “hot-phase” presentation in AC has been reported in 4–5% of subjects and represents a myocarditis-like episode with signs and symptoms typical of myocarditis (ie chest pain, diaphoresis, palpitations) and increases in troponin in blood samples and injury-related ECG changes.103 This presentation tends to occur more commonly in the younger population, with a large component in the pediatric population in children as young as 2 years of age.110 Underlying genetic variants were most often found in DSP, PKP2 and DSG2.110,111 The majority of these patients had LV LGE on CMR and endomyocardial biopsies with virus negative myocarditis.103,112 A family history of cardiomyopathy or SCD was common in these cohorts, and carried a high positive predictive value for an underlying genetic variant, again highlighting the importance of a comprehensive family history of at least 3 generations.112 Similar to other atypical presentations, AC misclassification was common with only around half of patients receiving an AC diagnosis at the time of the “hot phase” presentation, with other common diagnoses being viral myocarditis and DCM.110
Exercise
It has been demonstrated that competitive or regular strenuous endurance exercise is likely to contribute to the progression of RVD-AC.9 These data are based predominantly on carriers of PKP2 variants, with data on exercise and disease progression less clear for other desmosomal and nondesmosomal AC genetic variants. Strenuous endurance exercise in RVD-AC is associated with a younger age of presentation, higher likelihood of meeting TFC criteria for RVD-AC, and a lower lifetime survival free of VA and heart failure.113,114 Survival following a first time episode of VT or ventricular fibrillation was also lowest amongst those who exercised most.114 There is also a dose dependent effect of exercise, with those with the highest intensity endurance exercise having earlier first major arrhythmic events and occurrence of severe RV dysfunction, compared to those with moderate or low intensity exercise.113,115 An increased risk of VA or death was not seen in RVD-AC patients who participated in recreational sports when compared to inactive RVD-AC patients, suggesting lower levels of exercise may be tolerated.116 As such, in a recent consensus statement, it was suggested that participation in up to 150 minutes of low-intensity exercise per week was recommended for all patients and in low-to moderate-intensity recreational exercise in patients with no history of unexplained syncope, cardiac arrest or ventricular arrhythmias, and minimal structural cardiac abnormalities.117
Imaging findings also correlate with exercise, with athletes and their gene positive athletic family members having diminished RV and LV function and strain in the form of RV fractional area change, global longitudinal strain, and ejection fraction.118 These studies demonstrate increased gene penetrance in athletes and their athletic family members and have important implications for exercise recommendations. It is recommended that phenotype-genotype positive individuals with RVD-AC should not participate in competitive or frequent high intensity endurance exercise.9 Their phenotype negative-genotype positive relatives should be counseled that participating in competitive or frequent high intensity endurance exercise increases their risk of developing overt RVD-AC and ventricular arrhythmias.9 This should be done under the umbrella of a shared decision making model to ensure that families are aware of the evidence behind the recommendations, but that their personal interests and desires are also taken into account. This is in contradistinction to family members of HCM patients who are genotype positive, but phenotype negative in whom it is deemed reasonable to allow them to play, as exercise may have different effects in regards to disease development in different cardiomyopathies.119 Exercise may play a significant role in the pathogenesis of RVD-AC in some patients with no known mutation and no family history of RVD-AC.120 Sawant et al reported that “gene-elusive” RVD-AC patients, compared to patients with desmosomal pathogenic variants, were more likely to have exercised at a higher intensity before presentation and were more likely to have a negative family history than the desmosomal positive RVD-AC patients. The gene elusive patients were also younger at presentation, further supporting the fact that intense exercise plays a role in expressing the RVD-AC phenotype.120
Pediatric AC
The pediatric presentation in AC is understudied and often presents atypically, particularly in pre- and early teenage years. This likely results in the current diagnostic criteria being less sensitive, such that Deshpande et al recommended modified pediatric criteria.80 Deshpande et al emphasized that the typical ECG findings in young and older adults are not typically found or are normal in the younger children, virtually eliminating some major and minor criteria for this age group. Cicenia et al reported that the Padua criteria improved diagnostic accuracy in children compared to TFC guidelines.83 Pediatric studies are summarized in Table 2. The LVD and BiV AC phenotypes can be as prevalent as 22% and 31%, respectively, and are more commonly encountered in pre-pubertal patients compared to post-pubertal patients where RVD-AC was the more common phenotype.80,93,121 In the DeWitt et al study, 50% of their cohort had either LVD-AC or BiV-AC, 19% had evidence of myocardial inflammation, and 31% underwent cardiac transplantation.93 Deshpande et al reported on 16 children, 6 (38%) of whom required cardiac transplantation,80 while Surget et al reported that 37% of their prepubertal cohort presented in heart failure.121 In a study analyzing pediatric transplant patients, all patients at time of presentation had BiV AC and 80% were misclassified as other types of cardiomyopathies prior to transplant.90 In addition, congenital heart disease was seen in cases that progressed to transplant in two separate studies, suggesting that AC should not be overlooked as plausible causes of heart failure in younger children even in the congenital heart disease patient (Figure 6).90,93 The LVNC phenotype has also been reported occasionally in pediatric as well as adult studies of patients with AC; therefore genetic testing for AC genes should be considered in these patients as well if other genetic studies do not reveal a cause (Figures 5 and 6).90,93,107 Although genetic findings in pediatric studies varied, the yield of genetic studies was relatively high (Table 2).
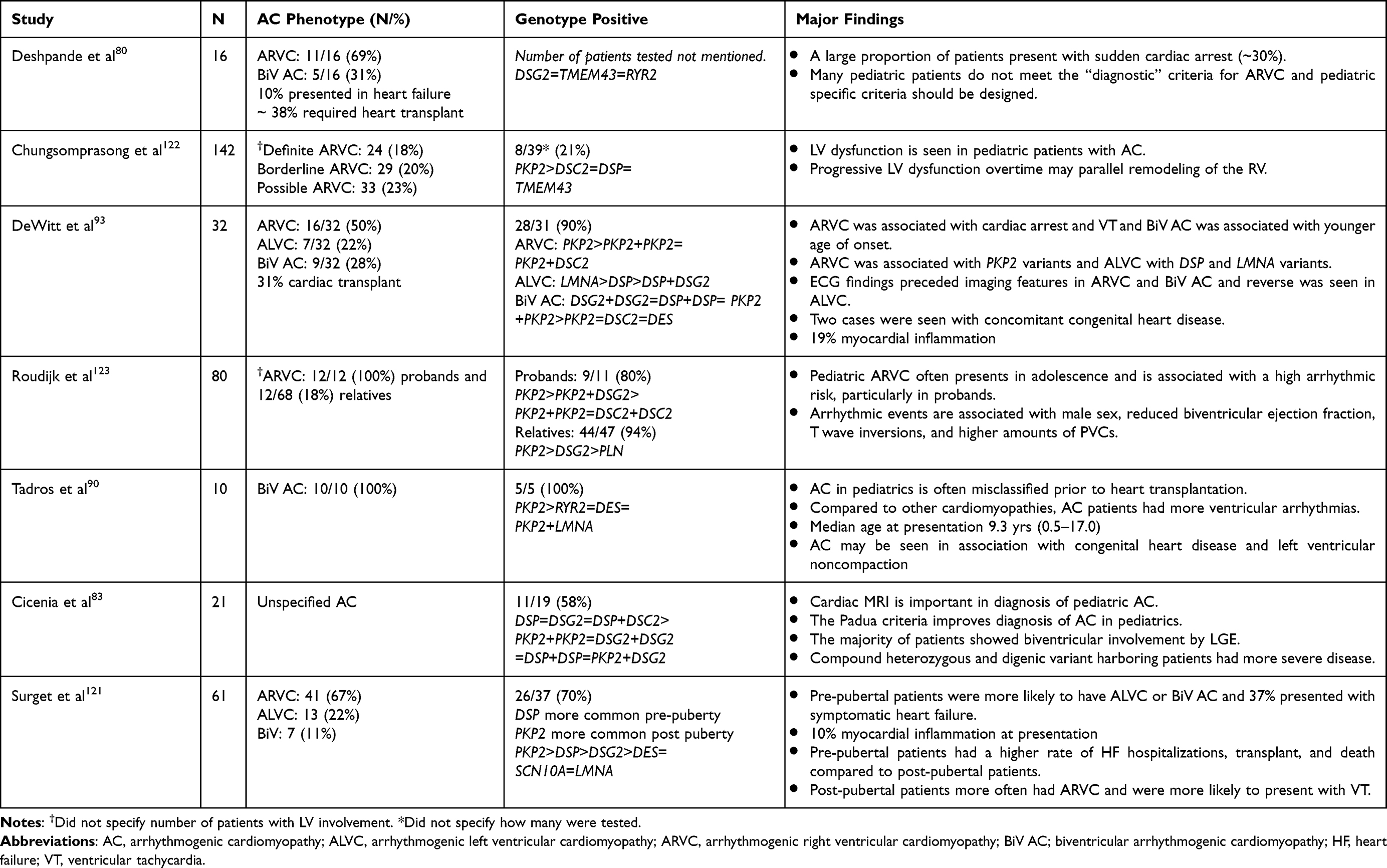 |
Table 2 Pediatric Studies on Arrhythmogenic Cardiomyopathy |
Genetic testing in children raises special consideration as the legal age of consent for testing in the US is 18 years.13 Young children or those with significant intellectual impairment cannot give assent (agree to) or consent (legally agree to) whereas older children may at least be able to give assent when the pros and cons of testing are discussed. The issue of genetic testing is less controversial when the child being tested is the proband, showing signs of clinical disease as their management/treatment will be directly impacted; cascade testing is more controversial particularly for diseases with variable penetrance and those that typically do not manifest until late teenage years or adulthood.13 Cascade testing should be offered to first degree family members of gene positive probands as this may impact their management even if currently phenotype negative, particularly in regards to exercise/competitive sports recommendations. This should be done in the setting of a multidisciplinary team with comprehensive pretest counseling, discussing the genetic testing process and the implications of the results for patients and their family members including, social, lifestyle, insurance and psychological impacts, again emphasizing the importance of shared decision-making in this process. The 2019 consensus statement recommends offering cascade testing when the gene found is pathogenic or likely pathogenic, but not for variants of unknown significance (VUS) and those that are likely benign or benign. Finding VUS’s in potentially disease causing genes can be a vexing issue. In our experience, many parents still wish to pursue VUS testing in their other seemingly unaffected children. Pursuing gene testing in first degree relatives of probands with a VUS may be considered on a case by case basis with the pros and cons clearly discussed with the family. Certainly, in the proband for whom there is concern for an evolving cardiomyopathy or overt cardiomyopathy ongoing follow up is required, as would be done even if the patient were gene negative because there is a clinical indication. In families with a phenotype positive child the recommended practice is to screen all first degree relatives at diagnosis of the proband with an echocardiogram and ECG. If or when genetic testing results are available, first degree relatives can then be screened for the pathologic or likely pathologic gene and if negative, do not require ongoing follow up as long as the variant fits the phenotype of the proband. If the test result is a VUS or no variant is found, then follow ups approximately every 2–3 years with echocardiogram and ECG are recommended for the first degree family members due to variable timing of onset of cardiomyopathy, even within a nuclear family, or sooner if a family member develops symptoms. More frequent echocardiogram and ECG screening may be indicated during puberty or if the sibling is a competitive athlete.
Management Strategies
Once diagnosed the treatment of AC is typically multipronged and aimed at preventing and treating potentially life-threatening ventricular arrhythmias, heart failure and other sequelae of structural heart disease. This may include antithrombotic therapy in the setting of ventricular aneurysms or depressed ventricular function or anticoagulation in the setting of atrial fibrillation or known thrombosis or thromboembolism. It is important to monitor for atrial arrhythmias and conduction disturbances. The 2019 Heart Rhythm Society Expert Consensus Statement on AC has made extensive recommendations for AC management.9 In most cases arrhythmia related symptoms bring the patient to medical attention and therapies are directed towards preventing and managing arrhythmias and their complications. In those presenting with aborted sudden death or hemodynamically significant ventricular tachycardia or otherwise stratified as high risk then implantable cardiac defibrillators (ICDs) are recommended as they are the only therapy that clearly reduces the risk of mortality.9 Additionally, antiarrhythmic medications are usually recommended to reduce arrhythmia related symptoms and appropriate and inappropriate ICD shocks. Antiarrhythmic therapies may include beta blockers, sotalol (particularly in younger patients where the risks of amiodarone toxicity long term are greater), amiodarone, and potentially flecainide. At times, combinations of these drugs are used. In those who continue to have frequent ICD shocks endo and epicardial catheter ablation of the arrhythmia focus may be considered although the recurrence risk of arrhythmias is relatively high given the ongoing myocardial pathology.124 As discussed previously, for those who exercise strenuously or competitively, cessation or modification of strenuous exercise is recommended to minimize the risk of sudden death with exercise and to slow the structural changes that can lead to heart failure. As ventricular function declines or if patients present with heart failure symptoms, goal directed heart failure therapies are recommended as they are in other forms of cardiomyopathy and include the use of beta blockers, angiotensin converting enzyme inhibitors, angiotensin receptor blockers, aldosterone receptor antagonists and newer recommended agents such as an angiotensin/neprilysin inhibitor and ivabradine for heart rate control.125 Device therapies may include cardiac resynchronization therapy to try to decrease dyssynchronous contraction as dysynchrony can contribute to cardiac dysfunction. If the heart failure is severe enough and has not responded to the first line therapies, a ventricular assist device can be implanted to serve as a “mechanical pump.” However, patients with severe right ventricular dysfunction may be harder to support with a ventricular assist device than those with primarily left ventricular dysfunction. Cardiac transplant is required in a subset of AC patients due to end stage heart failure or occasionally due to treatment refractory arrhythmias.
The 2019 Heart Rhythm Society Expert Consensus Statement on AC also suggests additional treatment considerations for patients with nondesmosomal variants.9 Leveraging studies that considered risk factors for SCD and VA, the statement suggests gene-directed Class IIa indications that ICD implantation is reasonable in: 1) patients with PLN AC with LVEF <45% or nonsustained VT (NSVT), 2) patients with LMNA AC with at least 2 risk factors including male sex, NSVT, and/or LVEF <45%, and 3) patients with FLNC AC with an LVEF <45%.9
Recent advances may provide novel avenues for AC management in the future. For instance, in AC mice and patient samples, glycogen synthase kinase 3 beta (GSK3beta) is re-distributed to the intercalated disks compared to cytoplasmic localization in non-AC tissue, suggesting aberrant GSK3beta signaling may play a role in pathogenesis.126 Chelko et al found that inhibiting this molecule leads to improved survival and LV function in AC mice and leads to normalization of distribution of GSK3beta, suggesting this may be a molecule to target in AC patients.126 Additionally, with improving gene therapy techniques like CRISPR/Cas9 gene editing, the ability to inactivate dysfunctional proteins or restore protein function is becoming a reality. In a recent study on humanized mice with the PLN R14del variant, disruption of the hPLN-R14del allele by AAV9-CRISPR/Cas9 reduced susceptibility to VT and improved cardiac function, suggesting that this may be a plausible avenue to explore in AC patients.127 Also, as diagnosis and awareness of AC continues to improve, trials focusing on AC are gaining traction, as recently seen with the Flecainide trial.128
Conclusion
Knowledge of arrhythmogenic cardiomyopathy is growing rapidly. It has progressed from the initial consensus that it is a RV dominant disease to more recent data showing that it can affect both ventricles or the LV in isolation. It can also occasionally be complicated with association with left ventricular noncompaction and/or congenital heart disease. This has important implications for genetic testing which may need to be broadened when typical candidate genes are not found, as some of the currently less common genes are more likely to cause an LVD-AC or BiV-AC, or if a patient with DCM has a high arrhythmia burden but none of the usual genes are found. Genetic testing should also be considered in those with myocarditis like episodes with no associated virus found and/or a protracted arrhythmia burden or slow or no recovery of function, particularly if there is a family history of sudden death or nonischemic cardiomyopathies. Young children may also have atypical presentations with heart failure being a relatively common presentation. Finally, with expanding next generation technologies, the likelihood of VUS identification is rising. It is important to continue to closely monitor the proband with the VUS who has a clinical disease and family members in order to be able to reliably re-evaluate VUSs regarding their pathogenicity as more evidence accumulates.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Choudhary N, Tompkins C, Polonsky B, et al. Clinical presentation and outcomes by sex in arrhythmogenic right ventricular cardiomyopathy: findings from the North American ARVC Registry. J Cardiovasc Electrophysiol. 2016;27(5):555. doi:10.1111/JCE.12947
2. Calkins H, Corrado D, Marcus F. Risk Stratification in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2017;136(21):2068–2082. doi:10.1161/CIRCULATIONAHA.117.030792
3. Peters S, Trümmel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2004;97(3):499–501. doi:10.1016/j.ijcard.2003.10.037
4. Dalal D, Nasir K, Bomma C, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112(25):357–359. doi:10.1161/CIRCULATIONAHA.105.542266
5. Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384–398. doi:10.1161/01.CIR.65.2.384
6. Thiene G, Basso C. Arrhythmogenic right ventricular cardiomyopathy: an update. Cardiovasc Pathol. 2001;10(3):109–117. doi:10.1016/S1054-8807(01)00067-9
7. Jacob KA, Noorman M, Mgpj C, Groeneweg JA, Hauer RNW, van der Heyden MAG. Geographical distribution of plakophilin-2 mutation prevalence in patients with arrhythmogenic cardiomyopathy. Netherlands Heart J. 2012;20(5):234. doi:10.1007/S12471-012-0274-X
8. James CA, Jongbloed JDH, Hershberger RE, et al. International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the clinical genome resource framework. Circ Genomic Precis Med. 2021;14(3):E003273. doi:10.1161/CIRCGEN.120.003273
9. Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16(11):e301–e372. doi:10.1016/J.HRTHM.2019.05.007
10. Brodehl A, Rezazadeh S, Williams T, et al. Mutations in ILK, encoding integrin linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl Res. 2019;208:15. doi:10.1016/J.TRSL.2019.02.004
11. Abdelfatah N, Chen R, Duff HJ, et al. Characterization of a unique form of arrhythmic cardiomyopathy caused by recessive mutation in LEMD2. JACC Basic to Transl Sci. 2019;4(2):204. doi:10.1016/J.JACBTS.2018.12.001
12. Goodwin RL, Koestler JL, Trask AJ, Lucchesi PA. Development of myocardial structure and function. In: Shaddy RE, Penny DJ, Feltes TF, editors. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents.
13. Landstrom AP, Kim JJ, Gelb BD, et al. Genetic testing for heritable cardiovascular diseases in pediatric patients: a scientific statement from the American Heart Association. Circ Genomic Precis Med. 2021;14(5):e000086. doi:10.1161/HCG.0000000000000086
14. Kapplinger JD, Landstrom AP, Salisbury BA, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57(23):2317–2327. doi:10.1016/J.JACC.2010.12.036
15. Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5(5):258. doi:10.1038/NCPCARDIO1182
16. Kowalczyk AP, Green KJ. Structure, function and regulation of Desmosomes. Prog Mol Biol Transl Sci. 2013;116:95. doi:10.1016/B978-0-12-394311-8.00005-4
17. Dries AM, Kirillova A, Reuter CM, et al. The genetic architecture of Plakophilin 2 cardiomyopathy. Genet Med. 2021;23(10):1961–1968. doi:10.1038/S41436-021-01233-7
18. Chen X, Bonné S, Hatzfeld M, van Roy F, Green KJ. Binding and Functional Characterization of Plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem. 2002;277(12):10512–10522. doi:10.1074/jbc.m108765200
19. Headrick AT, Rosenfeld JA, Yang Y, et al. Incidentally identified genetic variants in arrhythmogenic right ventricular cardiomyopathy‐associated genes among children undergoing exome sequencing reflect healthy population variation. Mol Genet Genomic Med. 2019;7(6):593. doi:10.1002/MGG3.593
20. Xu T, Yang Z, Vatta M, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55(6):587. doi:10.1016/J.JACC.2009.11.020
21. Pohl GM, Göz M, Gaertner A, et al. Cardiomyopathy related desmocollin-2 prodomain variants affect the intracellular cadherin transport and processing. Front Cardiovasc Med. 2023;10:1127261. doi:10.3389/fcvm.2023.1127261
22. Syrris P, Ward D, Evans A, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79(5):978–984. doi:10.1086/509122
23. Beffagna G, De Bortoli M, Nava A, et al. Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med Genet. 2007;8. doi:10.1186/1471-2350-8-65
24. Bhuiyan ZA, Jongbloed JDH, Van Der Smagt J, et al. Desmoglein-2 and Desmocollin-2 Mutations in dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients. Circ Cardiovasc Genet. 2009;2(5):418–427. doi:10.1161/CIRCGENETICS.108.839829
25. Brodehl A, Meshkov A, Myasnikov R, et al. Hemi‐ and homozygous loss‐of‐function mutations in dsg2 (Desmoglein‐2) cause recessive arrhythmogenic cardiomyopathy with an early onset. Int J Mol Sci. 2021;22(7):3786. doi:10.3390/IJMS22073786
26. Lorenzon A, Pilichou K, Rigato I, et al. Homozygous Desmocollin-2 mutations and arrhythmogenic cardiomyopathy. Am J Cardiol. 2015;116(8):1245–1251. doi:10.1016/J.AMJCARD.2015.07.037
27. Gerull B, Kirchner F, Chong JX, et al. Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ Cardiovasc Genet. 2013;6(4):327–336. doi:10.1161/CIRCGENETICS.113.000097
28. Brodehl A, Weiss J, Debus JD, et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J Mol Cell Cardiol. 2020;141:17–29. doi:10.1016/J.YJMCC.2020.03.006
29. Pilichou K, Nava A, Basso C, et al. Mutations in Desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113(9):1171–1179. doi:10.1161/CIRCULATIONAHA.105.583674
30. Casella M, Gasperetti A, Sicuso R, et al. Characteristics of patients with arrhythmogenic left ventricular cardiomyopathy: combining genetic and histopathologic findings. Circ Arrhythmia Electrophysiol. 2020;13(12):E009005. doi:10.1161/CIRCEP.120.009005
31. Hermida A, Fressart V, Hidden-Lucet F, et al. High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Heart Fail. 2019;21(6):792–800. doi:10.1002/EJHF.1423
32. López-Ayala JM, Gómez-Milanés I, Muñoz JJ, et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: characterizing a phenotype. EP Eur. 2014;16(12):1838–1846. doi:10.1093/EUROPACE/EUU128
33. Reza N, de Feria A, Chowns JL, et al. Cardiovascular characteristics of patients with genetic variation in Desmoplakin (DSP). Cardiogenetics. 2022;12(1):24–36. doi:10.3390/CARDIOGENETICS12010003
34. Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115(13):1710–1720. doi:10.1161/CIRCULATIONAHA.106.660241
35. Castelletti S, Vischer AS, Syrris P, et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: genotype-phenotype correlation. Int J Cardiol. 2017;249:268–273. doi:10.1016/J.IJCARD.2017.05.018
36. Singh SM, Casey SA, Berg AA, et al. Autosomal-dominant biventricular arrhythmogenic cardiomyopathy in a large family with a novel in-frame DSP nonsense mutation. Am J Med Genet A. 2018;176(7):1622–1626. doi:10.1002/AJMG.A.38719
37. Hoorntje ET, Burns C, Marsili L, et al. Variant location is a novel risk factor for individuals with arrhythmogenic cardiomyopathy due to a Desmoplakin (DSP) truncating variant. Circ Genomic Precis Med. 2022;16:e003672. doi:10.1161/CIRCGEN.121.003672
38. McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet (London, England). 2000;355(9221):2119–2124. doi:10.1016/S0140-6736(00)02379-5
39. Milting H, Klauke B, Christensen AH, et al. The TMEM43 Newfoundland mutation p.S358L causing ARVC-5 was imported from Europe and increases the stiffness of the cell nucleus. Eur Heart J. 2015;36(14):872–881. doi:10.1093/EURHEARTJ/EHU077
40. Merner ND, Hodgkinson KA, Haywood AFM, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82(4):809–821. doi:10.1016/J.AJHG.2008.01.010
41. Dominguez F, Zorio E, Jimenez-Jaimez J, et al. Clinical characteristics and determinants of the phenotype in TMEM43 arrhythmogenic right ventricular cardiomyopathy type 5. Heart Rhythm. 2020;17(6):945–954. doi:10.1016/J.HRTHM.2020.01.035
42. Zheng G, Jiang C, Li Y, et al. TMEM43-S358L mutation enhances NF-κB-TGFβ signal cascade in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Protein Cell. 2019;10(2):104. doi:10.1007/S13238-018-0563-2
43. Padrón-Barthe L, Villalba-Orero M, Gómez-Salinero JM, et al. Severe cardiac dysfunction and death caused by arrhythmogenic right ventricular cardiomyopathy type 5 are improved by inhibition of glycogen synthase kinase-3β. Circulation. 2019;140(14):1188–1204. doi:10.1161/CIRCULATIONAHA.119.040366
44. Zink M, Seewald A, Rohrbach M, et al. Altered expression of TMEM43 causes abnormal cardiac structure and function in zebrafish. Int J Mol Sci. 2022;23(17):9530. doi:10.3390/ijms23179530
45. Van Der Zwaag PA, Van Rijsingen IAW, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14(11):1199–1207. doi:10.1093/EURJHF/HFS119
46. Sepehrkhouy S, Gho JMIH, van Es R, et al. Distinct fibrosis pattern in desmosomal and phospholamban mutation carriers in hereditary cardiomyopathies. Heart Rhythm. 2017;14(7):1024–1032. doi:10.1016/J.HRTHM.2017.03.034
47. Groeneweg JA, Van Der Zwaag PA, Olde Nordkamp LRA, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am J Cardiol. 2013;112(8):1197–1206. doi:10.1016/J.AMJCARD.2013.06.017
48. Brodehl A, Hedde PN, Dieding M, et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated Desmin mutants. J Biol Chem. 2012;287(19):16047. doi:10.1074/JBC.M111.313841
49. Brodehl A, Holler S, Gummert J, Milting H. The N-Terminal Part of the 1A domain of desmin is a hot spot region for putative pathogenic DES mutations affecting filament assembly. Cells. 2022;11(23):3906. doi:10.3390/CELLS11233906
50. Klauke B, Kossmann S, Gaertner A, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010;19(23):4595–4607. doi:10.1093/HMG/DDQ387
51. Protonotarios A, Brodehl A, Asimaki A, et al. The Novel Desmin Variant p.Leu115Ile is associated with a unique form of biventricular arrhythmogenic cardiomyopathy. Can J Cardiol. 2021;37(6):857–866. doi:10.1016/J.CJCA.2020.11.017
52. Bermúdez-Jiménez FJ, Carriel V, Brodehl A, et al. Novel Desmin Mutation p.Glu401Asp impairs filament formation, disrupts cell membrane integrity, and causes severe arrhythmogenic left ventricular cardiomyopathy/dysplasia. Circulation. 2018;137(15):1595–1610. doi:10.1161/CIRCULATIONAHA.117.028719
53. Maggi L, Mavroidis M, Psarras S, Capetanaki Y, Lattanzi G. Skeletal and cardiac muscle disorders caused by mutations in genes encoding intermediate filament proteins. Int J Mol Sci. 2021;22(8):4256. doi:10.3390/IJMS22084256
54. Brun F, Gigli M, Graw SL, et al. FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2020;57(4):254. doi:10.1136/JMEDGENET-2019-106394
55. Gigli M, Stolfo D, Graw SL, et al. Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by Filamin C truncating variants. Circulation. 2021;144(20):1600–1611. doi:10.1161/CIRCULATIONAHA.121.053521
56. Ortiz-Genga MF, Cuenca S, Dal Ferro M, et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68(22):2440–2451. doi:10.1016/J.JACC.2016.09.927
57. Carruth ED, Qureshi M, Alsaid A, et al. Loss-of-function FLNC variants are associated with arrhythmogenic cardiomyopathy phenotypes when identified through exome sequencing of a general clinical population. Circ Genomic Precis Med. 2022;15(4):278–286. doi:10.1161/CIRCGEN.121.003645
58. Hall CL, Akhtar MM, Sabater-Molina M, et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int J Cardiol. 2020;307:101–108. doi:10.1016/J.IJCARD.2019.09.048
59. Tucker NR, McLellan MA, Hu D, et al. Novel mutation in FLNC (Filamin C) causes familial restrictive cardiomyopathy. Circ Cardiovasc Genet. 2017;10(6). doi:10.1161/CIRCGENETICS.117.001780
60. Brodehl A, Ferrier RA, Hamilton SJ, et al. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum Mutat. 2016;37(3):269–279. doi:10.1002/HUMU.22942
61. Le QK, Maguy A, Qi XY, et al. Loss of cardiomyocyte integrin-linked kinase produces an Arrhythmogenic cardiomyopathy in mice. Circ Arrhythmia Electrophysiol. 2015;8(4):921–932. doi:10.1161/CIRCEP.115.001668
62. Thiene G, Nava A, Angelini A, Daliento L, Scognamiglio R, Corrado D. Anatomoclinical aspects of arrhythmogenic right ventricular cardiomyopathy. Adv Cardiomyopathies. 1990;397–408. doi:10.1007/978-3-642-83760-9_41
63. Rootwelt-Norberg C, Lie ØH, Dejgaard LA, et al. Life-threatening arrhythmic presentation in patients with arrhythmogenic cardiomyopathy before and after entering the genomic era; a two-decade experience from a large volume center. Int J Cardiol. 2019;279:79–83. doi:10.1016/J.IJCARD.2018.12.066
64. Zhai L, Hu Y, Li X, et al. Incidence, predictors and clinical impact of ventricular electrical storm in Arrhythmogenic Cardiomyopathy patients with an implantable cardioverter-defibrillator: a single-center report with medium-term follow-up. Int J Gen Med. 2021;14:10055–10063. doi:10.2147/IJGM.S345872
65. Belhassen B, Laredo M, Roudijk RW, et al. The prevalence of left and right bundle branch block morphology ventricular tachycardia amongst patients with arrhythmogenic cardiomyopathy and sustained ventricular tachycardia: insights from the European Survey on Arrhythmogenic Cardiomyopathy. Europace. 2022;24(2):285–295. doi:10.1093/EUROPACE/EUAB190
66. Baturova MA, Haugaa KH, Jensen HK, et al. Atrial fibrillation as a clinical characteristic of arrhythmogenic right ventricular cardiomyopathy: experience from the Nordic ARVC Registry. Int J Cardiol. 2020;298:39–43. doi:10.1016/J.IJCARD.2019.07.086
67. D’Ascenzi F, Valentini F, Pistoresi S, et al. Causes of sudden cardiac death in young athletes and non-athletes: systematic review and meta-analysis: sudden cardiac death in the young. Trends Cardiovasc Med. 2022;32(5):299–308. doi:10.1016/J.TCM.2021.06.001
68. Finocchiaro G, Papadakis M, Robertus JL, et al. Etiology of sudden death in sports: insights from a United Kingdom Regional Registry. J Am Coll Cardiol. 2016;67(18):2108–2115. doi:10.1016/J.JACC.2016.02.062
69. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318(3):129–133. doi:10.1056/NEJM198801213180301
70. Miles C, Finocchiaro G, Papadakis M, et al. Sudden death and left ventricular involvement in Arrhythmogenic Cardiomyopathy. Circulation. 2019;139(15):1786–1797. doi:10.1161/CIRCULATIONAHA.118.037230
71. Isbister JC, Nowak N, Yeates L, et al. Concealed cardiomyopathy in autopsy-inconclusive cases of sudden cardiac death and implications for families. J Am Coll Cardiol. 2022;80(22):2057–2068. doi:10.1016/J.JACC.2022.09.029
72. Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2004;13(4):185–194. doi:10.1016/J.CARPATH.2004.03.609
73. Polivka L, Bodemer C, Hadj-Rabia S. Combination of palmoplantar keratoderma and hair shaft anomalies, the warning signal of severe arrhythmogenic cardiomyopathy: a systematic review on genetic desmosomal diseases. J Med Genet. 2016;53(5):289–295. doi:10.1136/JMEDGENET-2015-103403
74. Hylind R, Beauséjour-Ladouceur V, Plovanich ME, et al. Cardiocutaneous features of autosomal dominant Desmoplakin-associated arrhythmogenic cardiomyopathy. Circ Genomic Precis Med. 2020;13(6):E003081. doi:10.1161/CIRCGEN.120.003081
75. Maruthappu T, Posafalvi A, Castelletti S, et al. Loss of function desmoplakin I and II mutations underlie dominant arrhythmogenic cardiomyopathy with a hair and skin phenotype. Br J Dermatol. 2019;180(5):1114. doi:10.1111/BJD.17388
76. Kirkels FP, Lie ØH, Cramer MJ, et al. Right ventricular functional abnormalities in arrhythmogenic cardiomyopathy: association with life-threatening ventricular arrhythmias. JACC Cardiovasc Imaging. 2021;14(5):900–910. doi:10.1016/J.JCMG.2020.12.028
77. Protonotarios A, Wicks E, Ashworth M, et al. Prevalence of 18F-fluorodeoxyglucose positron emission tomography abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2019;284:99–104. doi:10.1016/J.IJCARD.2018.10.083
78. McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71(3):215–218. doi:10.1136/HRT.71.3.215
79. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Eur Heart J. 2010;31(7):806. doi:10.1093/EURHEARTJ/EHQ025
80. Deshpande SR, Herman HK, Quigley PC, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D): review of 16 pediatric cases and a proposal of modified pediatric criteria. Pediatr Cardiol. 2016;37(4):646–655. doi:10.1007/S00246-015-1327-X
81. Gilotra NA, Bhonsale A, James CA, et al. Heart failure is common and under-recognized in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Heart Fail. 2017;10(9). doi:10.1161/CIRCHEARTFAILURE.116.003819
82. Corrado D, Perazzolo Marra M, Zorzi A, et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int J Cardiol. 2020;319:106–114. doi:10.1016/j.ijcard.2020.06.005
83. Cicenia M, Cantarutti N, Adorisio R, et al. Arrhythmogenic cardiomyopathy in children according to “Padua criteria”: single pediatric center experience. Int J Cardiol. 2022;350:83–89. doi:10.1016/J.IJCARD.2022.01.008
84. De Lazzari M, Zorzi A, Cipriani A, et al. Relationship between electrocardiographic findings and cardiac magnetic resonance phenotypes in arrhythmogenic cardiomyopathy. J Am Heart Assoc. 2018;7(22). doi:10.1161/JAHA.118.009855
85. Platonov PG, Svensson A. Epsilon waves as an extreme form of depolarization delay: focusing on the arrhythmogenic right ventricular cardiomyopathy/dysplasia. Curr Cardiol Rev. 2021;17(1):17–23. doi:10.2174/1573403X16666200810105029
86. Protonotarios A, Anastasakis A, Tsatsopoulou A, et al. Clinical significance of epsilon waves in arrhythmogenic cardiomyopathy. J Cardiovasc Electrophysiol. 2015;26(11):1204–1210. doi:10.1111/JCE.12755
87. Pearman CM, Lee D, Davies B, et al. Incremental value of the signal-averaged ECG for diagnosing arrhythmogenic cardiomyopathy. Heart Rhythm. 2023;20(2):224–230. doi:10.1016/J.HRTHM.2022.10.005
88. de Brouwer R, Meems LMG, Verstraelen TE, et al. Sex-specific aspects of phospholamban cardiomyopathy: the importance and prognostic value of low-voltage electrocardiograms. Heart Rhythm. 2022;19(3):427–434. doi:10.1016/J.HRTHM.2021.11.009
89. Celeghin R, Cipriani A, Bariani R, et al. Filamin-C variant-associated cardiomyopathy: a pooled analysis of individual patient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2022;19(2):235–243. doi:10.1016/J.HRTHM.2021.09.029
90. Tadros HJ, Choudhry S, Kearney DL, et al. Arrhythmogenic cardiomyopathy is under-recognized in end-stage pediatric heart failure: a 36-year single-center experience. Pediatr Transplant. 2022. doi:10.1111/PETR.14442
91. Réant P, Hauer AD, Castelletti S, et al. Epicardial myocardial strain abnormalities may identify the earliest stages of arrhythmogenic cardiomyopathy. Int J Cardiovasc Imaging. 2016;32(4):593–601. doi:10.1007/S10554-015-0813-9
92. Chen S, Ren JF, Jiang C, Marchlinski FE. Intracardiac echocardiography-derived right ventricular myocardial strain deformation patterns with segmental dyssynchrony in arrhythmogenic cardiomyopathy. J Am Soc Echocardiogr. 2021;34(1):100–102. doi:10.1016/J.ECHO.2020.10.001
93. DeWitt ES, Chandler SF, Hylind RJ, et al. Phenotypic manifestations of arrhythmogenic cardiomyopathy in children and adolescents. J Am Coll Cardiol. 2019;74(3):346–358. doi:10.1016/J.JACC.2019.05.022
94. Riele ASJM T, James CA, Philips B, et al. Mutation positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol. 2013;24(12):1311. doi:10.1111/JCE.12222
95. Tandri H, Calkins H, Nasir K, et al. Magnetic resonance imaging findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2003;14(5):476–482. doi:10.1046/J.1540-8167.2003.02560.X
96. Perazzolo Marra M, Cipriani A, Rizzo S, et al. Myocardial tissue characterization in arrhythmogenic cardiomyopathy: comparison between endomyocardial biopsy and cardiac magnetic resonance. JACC Cardiovasc Imaging. 2021;14(8):1675–1678. doi:10.1016/J.JCMG.2021.02.015
97. Sen-Chowdhry S, Prasad SK, Syrris P, et al. Cardiovascular magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited: comparison with task force criteria and genotype. J Am Coll Cardiol. 2006;48(10):2132–2140. doi:10.1016/J.JACC.2006.07.045
98. Segura-Rodríguez D, Bermúdez-Jiménez FJ, Carriel V, et al. Myocardial fibrosis in arrhythmogenic cardiomyopathy: a genotype-phenotype correlation study. Eur Heart J Cardiovasc Imaging. 2020;21(4):378–386. doi:10.1093/EHJCI/JEZ277
99. Augusto JB, Eiros R, Nakou E, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Heart J Cardiovasc Imaging. 2020;21(3):326–336. doi:10.1093/EHJCI/JEZ188
100. Amadu AM, Baritussio A, Dastidar AG, et al. Arrhythmogenic right ventricular cardiomyopathy (ARVC) mimics: the knot unravelled by cardiovascular MRI. Clin Radiol. 2019;74(3):228–234. doi:10.1016/J.CRAD.2018.12.002
101. Chun KH, Oh J, Hong YJ, et al. Prognostic cardiac magnetic resonance markers of left ventricular involvement in arrhythmogenic cardiomyopathy for predicting heart failure outcomes. J Am Heart Assoc. 2022;11(6):23167. doi:10.1161/JAHA.121.023167
102. Aquaro GD, De Luca A, Cappelletto C, et al. Prognostic value of magnetic resonance phenotype in patients with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2020;75(22):2753–2765. doi:10.1016/J.JACC.2020.04.023
103. Bariani R, Cipriani A, Rizzo S, et al. ‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy. EP Eur. 2021;23(6):907–917. doi:10.1093/EUROPACE/EUAA343
104. Corrado D, Basso C, Thiene G, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30(6):1512–1520. doi:10.1016/S0735-1097(97)00332-X
105. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94(5):983–991. doi:10.1161/01.CIR.94.5.983
106. Magrath P, Maforo N, Renella P, Nelson SF, Halnon N, Ennis DB. Cardiac MRI biomarkers for Duchenne muscular dystrophy. Biomark Med. 2018;12(11):1271–1289. doi:10.2217/BMM-2018-0125
107. Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52(25):2175–2187. doi:10.1016/J.JACC.2008.09.019
108. Rimac G, Poulakos N, Beaulieu-Shearer A, et al. Clinical and echocardiographic evolution of patients with arrhythmogenic cardiomyopathy before heart transplantation. Clin Transplant. 2023;37(2). doi:10.1111/CTR.14869
109. Tavora F, Zhang M, Franco M, et al. Distribution of biventricular disease in arrhythmogenic cardiomyopathy: an autopsy study. Hum Pathol. 2012;43(4):592–596. doi:10.1016/J.HUMPATH.2011.06.014
110. Bariani R, Rigato I, Cipriani A, et al. Myocarditis-like episodes in patients with arrhythmogenic cardiomyopathy: a systematic review on the so-called hot-phase of the disease. Biomolecules. 2022;12(9):1324. doi:10.3390/BIOM12091324
111. Ollitrault P, Al Khoury M, Troadec Y, et al. Recurrent acute myocarditis: an under-recognized clinical entity associated with the later diagnosis of a genetic arrhythmogenic cardiomyopathy. Front Cardiovasc Med. 2022;9. doi:10.3389/FCVM.2022.998883
112. Piriou N, Marteau L, Kyndt F, et al. Familial screening in case of acute myocarditis reveals inherited arrhythmogenic left ventricular cardiomyopathies. ESC Heart Fail. 2020;7(4):1520–1533. doi:10.1002/EHF2.12686
113. Ruiz Salas A, Barrera Cordero A, Navarro-Arce I, et al. Impact of dynamic physical exercise on high-risk definite arrhythmogenic right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2018;29(11):1523–1529. doi:10.1111/JCE.13704
114. James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62(14):1290–1297. doi:10.1016/J.JACC.2013.06.033
115. Lie ØH, Dejgaard LA, Saberniak J, et al. Harmful Effects of Exercise Intensity and Exercise Duration in Patients With Arrhythmogenic Cardiomyopathy. JACC Clin Electrophysiol. 2018;4(6):744–753. doi:10.1016/J.JACEP.2018.01.010
116. Ruwald AC, Marcus F, Estes NAM, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36(27):1735–1743. doi:10.1093/EURHEARTJ/EHV110
117. Pelliccia A, Sharma S, Gati S, et al. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur Heart J. 2021;42(1):17–96. doi:10.1093/EURHEARTJ/EHAA605
118. Saberniak J, Hasselberg NE, Borgquist R, et al. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur J Heart Fail. 2014;16(12):1337–1344. doi:10.1002/EJHF.181
119. Members WC, Ommen SR, Mital S, et al. 2020 AHA/ACC Guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy. Circulation. 2020;105(2):207–209. doi:10.1161/CIR.0000000000000937
120. Sawant AC, Bhonsale A, te Riele ASJM, et al. Exercise has a disproportionate role in the pathogenesis of arrhythmogenic right ventricular dysplasia/cardiomyopathy in patients without desmosomal mutations. J Am Heart Assoc. 2014;3(6). doi:10.1161/JAHA.114.001471
121. Surget E, Maltret A, Raimondi F, et al. Clinical presentation and heart failure in children with arrhythmogenic cardiomyopathy. Circ Arrhythm Electrophysiol. 2022;15(2):e010346. doi:10.1161/CIRCEP.121.010346
122. Chungsomprasong P, Hamilton R, Luining W, Fatah M, Yoo SJ, Grosse-Wortmann L. Left ventricular function in children and adolescents with arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2017;119(5):778–784. doi:10.1016/J.AMJCARD.2016.11.020
123. Roudijk RW, Verheul L, Bosman LP, et al. Clinical characteristics and follow-up of pediatric-onset arrhythmogenic right ventricular cardiomyopathy. JACC Clin Electrophysiol. 2022;8(3):306–318. doi:10.1016/J.JACEP.2021.09.001
124. Ermakov S, Scheinman M. Arrhythmogenic right ventricular cardiomyopathy – antiarrhythmic therapy. Arrhythmia Electrophysiol Rev. 2015;4(2):86. doi:10.15420/AER.2015.04.02.86
125. van der Meer P, Gaggin HK, Dec GW. ACC/AHA versus ESC Guidelines on Heart Failure: JACC Guideline Comparison. J Am Coll Cardiol. 2019;73(21):2756–2768. doi:10.1016/J.JACC.2019.03.478
126. Chelko SP, Asimaki A, Andersen P, et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 2016;1(5):85923. doi:10.1172/JCI.INSIGHT.85923
127. Dave J, Raad N, Mittal N, et al. Gene editing reverses arrhythmia susceptibility in humanized PLN-R14del mice: modelling a European cardiomyopathy with global impact. Cardiovasc Res. 2022;118(15):3140–3150. doi:10.1093/CVR/CVAC021
128. NCT03685149. Pilot Randomized Trial With Flecainide in ARVC Patients; 2018. Available from: https://clinicaltrials.gov/show/NCT03685149.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.